

Abstract
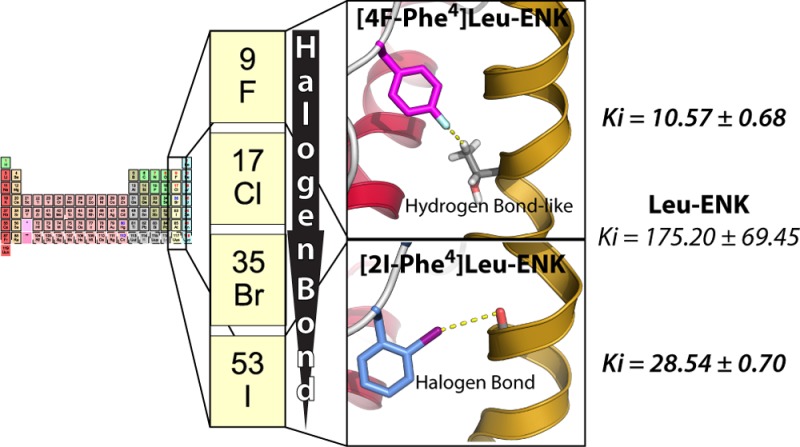
Systematic halogenation of two native opioid peptides has shown that halogen atoms can modulate peptide–receptor interactions in different manners. First, halogens may produce a steric hindrance that reduces the binding of the peptide to the receptor. Second, chlorine, bromine, or iodine may improve peptide binding if their positive σ-hole forms a halogen bond interaction with negatively charged atoms of the protein. Lastly, the negative electrostatic potential of fluorine can interact with positively charged atoms of the protein to improve peptide binding.
Keywords: Halogen bond, drug design, G protein-coupled receptors, opioid receptors, halogenated peptides, endomorphin-1, Leu-enkephalin, neuropeptides
Halogenation has traditionally been used in medicinal chemistry to modulate both potency and ADME properties of drugs.1,2 For many years halogens were only considered for their hydrophobic properties and as Lewis bases due to their electronegativities. However, theoretical studies have shown that, on the van der Waals surface of the halogen atom, fluorine (F) remains entirely electronegative, whereas chlorine (Cl), bromine (Br), and iodine (I) show a small positively charged surface (named σ-hole) on the opposite side of the C—X axis (X is Cl, Br, I).3,4 The size and intensity of the σ-hole increases with the charge of the σ-hole (I > Br > Cl). Thus, fluorines are most likely to act as hydrogen bond acceptors (F···H—D, D is O, N, S, C).5 In contrast, the other three halogens can act as electron acceptors (C—X···B, B is O, N, S), via the σ-hole, forming the so-called halogen bond, a Lewis acid–base interaction in which halogens act as Lewis acids.6 The strength of the halogen bond depends on the halogen atom and the Lewis base involved in the interaction, but it is comparable in magnitude to hydrogen bond interactions.7 A survey of protein crystal structures shows that halogen bonds are the prevalent interactions between halogenated ligands and target proteins.2,6,8−10 These halogen bonds are preferentially formed between the halogen atom of the ligand and the carbonyl oxygen of the protein backbone, which is the most abundant Lewis base in proteins.7 In these cases the halogen atom shares its donor oxygen with the backbone hydrogen of the amide bond of the protein (the backbone C=O···H–N hydrogen bond forms the secondary structure of proteins) in such a manner that the halogen bond tends to be geometrically perpendicular to the shared hydrogen bond.8
The evolution and structural integration of fluorine into drugs introduced over the past 50 years have been recently evaluated.11 We have extended this analysis to the other halogen atoms (Figure 1) by surveying their presence in compounds deposited in DrugBank. Out of the 1584 FDA-approved drugs, 10.4% of them contain fluorine, 14.6% chlorine, 1.5% bromine, and 1.4% iodine. The largest database of commercially available compounds, the ZINC database, shows similar trends. Out of the almost 30 million compounds deposited in ZINC, 16.5% contain fluorine, 14% chlorine, 5.2% bromine, and 0.3% iodine. Similar percentages (F, 12.3%; Cl, 16.5%; Br, 1.7%; I, 2.1%) are found in the analysis of the IUPHAR/BPS database of ligands for the G protein-coupled receptor (GPCR) superfamily. Clearly, halogens have a key role in drug development.
Figure 1.
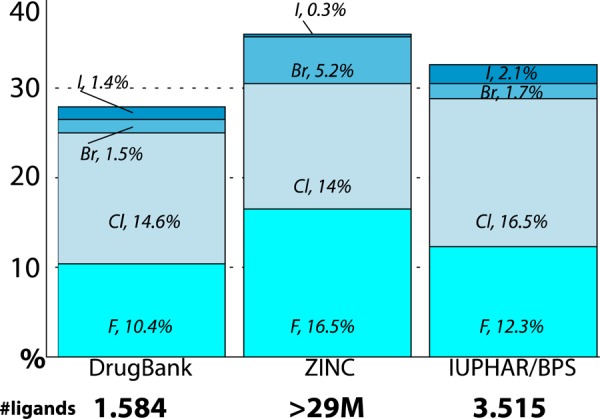
Presence of halogens in ligands deposited in the DrugBank (FDA-approved), ZINC (commercially available), and IUPHAR/BPS (Class A GPCR ligands) databases.
Therefore, in this study we aimed to test the effect of halogen substitution in endogenous peptides of opioid receptors, which belong to the GPCR superfamily.12 Like other GPCRs,13 the structures of the μ-, δ-, and κ-opioid receptors14−16 are formed by seven membrane-spanning α-helical segments (TMs 1–7), connected by extracellular (ECL 1–3) and intracellular loops. It is known that chlorination of aromatic residues (i.e., 4-Cl-Phe4) of certain opioid peptides ([d-Pen2, d-Pen5]enkephalin (DPDPE); biphalin) increases their lipophilicity and hence cell permeability and blood–brain barrier passage.17,18 Other peptides like DPDPE, metkephamid, and dermorphin/deltorphin analogues when halogenated at Phe4 or Phe3 increased their affinity for δ-opioid receptors.19−21 More recently, chlorination or fluorination of Phe4 residues on a series of Dmt-substituted enkephalin-like tetrapeptides have led to the development of nonselective potent opioid agonists for μ- and δ-opioid receptors.22 Due to the absence of systematic halogenation of Phe residues to produce homologous series (F, Cl, Br, I), in the present work we wanted to examine the possibility to induce stabilizing ligand–receptor interactions by the systematic halogenation of two native opioid ligands, endomorphin-1 and Leu-enkephalin.
Three homologous series of halogenated analogues have been prepared. The first series corresponds to ortho-Phe4 halogenated endomorphin-1 ([4-X-Phe4]Endo-1), while the other two are based on ortho-Phe4 ([2-X-Phe4]Leu-ENK) and para-Phe4 ([4-X-Phe4]Leu-ENK) halogenated Leu-ENK. Binding affinities of these three series of halogenated peptides were determined by competition experiments on the μ-opioid receptor (μ-OR) from zebrafish23 and on two duplicate δ-opioid receptors (δ-ORs), dre-δ1a24 and dre-δ1b,25 using [3H]-diprenorphine as radioligand. All tested ligands displaced [3H]-diprenorphine binding, and the experimental data fitted better to one-site displacement model. Nonspecific binding was determined in the presence of 10 μM of the nonspecific antagonist naloxone. Results are shown in Table 1 (see also SI). Halogenation of the selective Endo-1 peptide reduces affinity for μ-OR in a linear manner along the series (Table 1), which suggests an increasing steric hindrance in the ligand–receptor interaction proportional to the bulkiness of the halogen atom. In contrast, this effect along the -Cl, -Br, and -I derivatives is positive for δ-ORs (more significant in δ1a than in δ1b, Table 1). An interesting result of the combination of these opposite effects of enhanced binding for δ-ORs and less binding for μ-OR is a modification of peptide selectivity. The selectivity of Endo-1, between μ-OR and δ1a-OR gradually decreases from 258 to 5 times (Table 1). In the second and third series we constructed ortho-Phe4- and para-Phe4-halogenated Leu-ENK peptides with different effects (Table 1).
Table 1. Summary of the Ki Values (Mean ± SEM) Found for the Halogenated Series of Endo-1 and Leu-ENK when Tested on the μ and δ Opioid Receptorsa.
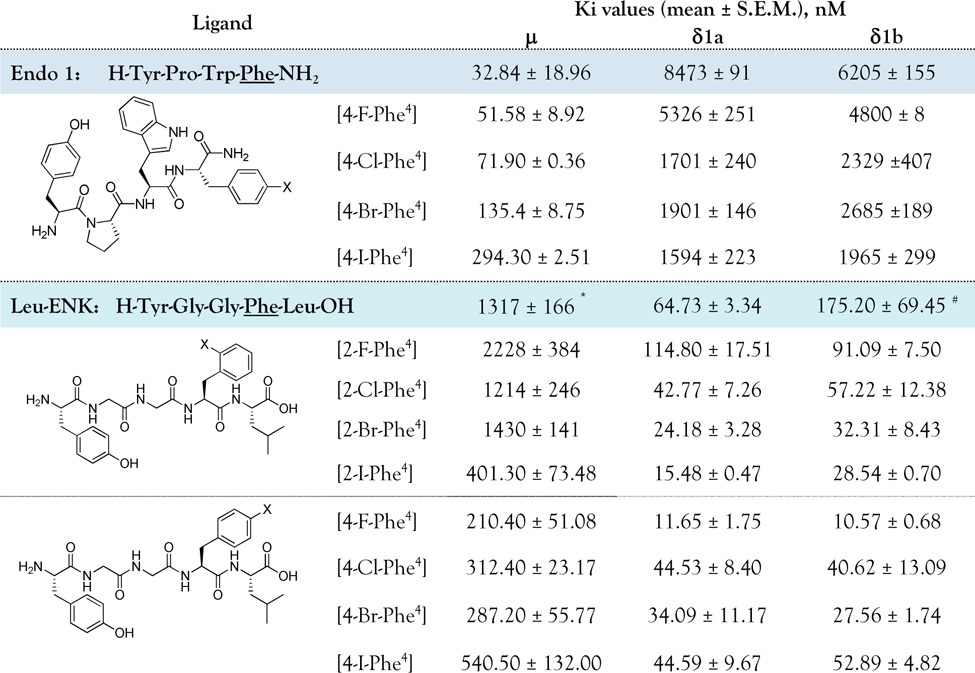
The binding pattern of the [2-X-Phe4]-Leu-ENK series to both types of δ receptors indicates the formation of a halogen bond interaction. There is a linear (R2 of 0.94 and 0.97 for δ1a and δ1b, respectively) decrease of Ki values with an increase of the halogen volume, as measured by the effective van der Waals radii for phenyl substitution (H, 1.20 Å; Cl, 1.77 Å; Br, 1.92 Å; I, 2.06 Å).27 In the [4-X-Phe4]-Leu-ENK series, the -Cl, -Br, and -I substitutions reduce affinity for δ-OR, compared to F-substitution, which markedly improves binding affinity for both δ-ORs, leading to the most potent peptide tested in this work.
In order to understand the nature of the repulsive interaction between the halogen atom of [4-X-Phe4]Endo-1 and the μ-OR (steric hindrance), and the attractive interaction between the -Cl, -Br, and -I atom of [2-X-Phe4]-Leu-ENK and δ-ORs (halogen bond donor) and the -F atom of [4-F-Phe4]-Leu-ENK and δ-ORs (hydrogen bond acceptor), we performed a combined NMR and modeling study.28 Indeed, in their free state in water solution, and independently of the substitution, no medium-range or long-range NOEs were detected. These data indicate that these molecules behave similarly to other endogenous enkephalin peptides and analogues, with a remarkable flexibility and major extended conformations.29−32 The halogen substitution does not modify the natural presentation of these peptides, and therefore, the observed different binding affinities are likely to be due to intermolecular interactions with the key receptors.
Endo-1 and Leu-ENK peptides were docked in extended conformations, as suggested by the NMR experiments, into homology models of μ- and δ1b-opioid receptors, respectively, in such a manner that the observed binding modes in crystal structures of nonpeptidic ligands to these receptors11 are mimicked (see Experimental Procedures). In the modeled complexes, the message part33 of Endo-1 and Leu-ENK (NH3+-Tyr1) interact with μ- and δ-opioid receptors, respectively, in a similar manner. The protonated N-terminus amine of the peptide forms an ionic interaction with Asp3.32, whereas Tyr1 interacts with His6.52 via a conserved water molecule, present in all released crystal structures of opioid receptors (Figure S1). In contrast, the address part (Pro2Trp3Phe4 in Endo-1 and Gly2Gly3Phe4Leu5 in Leu-ENK), responsible for selectivity, forms slightly different interactions. In the complex between Endo-1 and μ-OR, Pro2 induces a tight turn of the backbone to facilitate an aromatic–aromatic interaction between Trp3 and Tyr2.64, and Phe4 is positioned in a hydrophobic pocket formed by Thr2.56, Phe2.59, Val3.38, and a conserved Trp127 aromatic residue (the WxFG motif in ECL 1,13 and the −CONH2 C-terminus of the peptide hydrogen bonds Gln2.60 and Asn2.63) (Figure S1). In the complex between Leu-ENK and δ-ORs, the flexible Gly2Gly3 spacer mimics the tight turn of Pro2 in Endo-1, positioning Phe4 in the same hydrophobic pocket (but with slightly different orientation, see below) shaped by Thr2.56, Phe2.59, Val3.38, and Trp118 in ECL 1, Leu5 forms hydrophobic interactions with Ile202 in ECL 2, and the -CONH2 C-terminus of the peptide hydrogen bonds with the exposed backbone of ECL2. These binding modes explain the observed selectivity of Endo-1 for μ-OR and Leu-ENK for δ-ORs. Asn2.63 in μ-OR (Lys2.63 in δ-ORs) provides selectivity of Endo-1 for μ-OR, whereas Ile 202 in ECL 2 of δ-ORs (Asp 213 in μ-OR) provides selectivity of Leu-ENK for δ-ORs (Figure S2). These are the only nonconserved/nonsimilar residues in the binding pocket of both receptors. This is in agreement with previous work suggesting that the amino acid at position 2.63 is responsible for the selectivity of the enkephaline DAMGO between rat μ- and δ-opioid receptors.34,35
Molecular dynamics (MD) simulations were used to study the observed effects of halogen substitution in the peptide–receptor interaction. Importantly, the presence of both a positively charged σ-hole and an electronegative crown,3 as depicted in electrostatic potential surfaces (Figure S3) calculated at the B3LYP level of theory (see Methods), impedes the proper modeling of halogen atoms by the standard single point charge. Thus, halogen atoms (with the exception of the entirely electronegative F) are modeled using two point charges following the scheme proposed by Ibrahim36 (see Experimental Procedures and SI). Halogenation at the para position of Phe4 in Endo-1 (steric hindrance) and Leu-ENK (fluorine acts as hydrogen bond acceptor) has different effects (see above) despite Phe4 is located at the same hydrophobic pocket. The MD simulations reproduce slightly different orientations of Phe4 in Endo-1 (the preceding amino acid before the C-terminus) and Leu-ENK (two amino acids before the C-terminus) in the cavity (Figures 2 and S1). The formation of the interactions between the amidated C-terminus Endo-1 and Gln2.60 and Asn2.63 (Figures 2A and S1) profoundly restrains Phe4 to adapt inside this small hydrophobic cavity, formed by the β-branched Thr2.56 and Val3.28 residues. Thus, halogenation of Endo-1 ([4-X-Phe4]Endo-1) is repulsive due to a steric hindrance (Figure 2A). In contrast, Phe4 in Leu-ENK is positioned two amino acids before the amidated C-terminus, which confer significant flexibility due to a less restrictive interaction of Leu5, the following amino acid, with Ile202 in ECL 2 (Figure S1). Thus, Leu-ENK benefits from fluorine substitution in para ([4-F-Phe4]-Leu-ENK), yielding a peptide with high affinity for δ-opioid receptors (Table 1). para-Fluorinated phenylalanine is characterized by a slightly negative electrostatic potential that is distributed as a hollow ring around the tip of the opposite side of the C–F axis, whereas this trend is the opposite (positive σ-hole) in the other three halogen atoms (Figure S3). Thus, the facts that fluorine substitution benefits peptide binding whereas chlorine, bromine, and iodine substitution is detrimental for peptide binding (Table 1) suggest that the halogen atom at the para position acts as hydrogen bond acceptor. MD simulations of [4-F-Phe4]-Leu-ENK in complex with the δ-opioid receptor show that the slightly negative electrostatic potential of fluorine interacts with the slightly positive charge of the methyl group of Thr2.56, forming a F···H–C hydrogen bond (Figure 2B). Noteworthy, interactions with hydrophobic residues account for almost 40% of the observed complexes of fluorinated ligands in a recent survey of the PDB database.9
Figure 2.
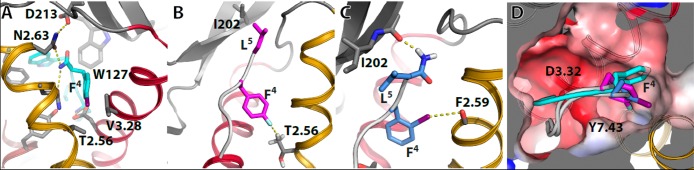
Computational models of the complexes between halogenated Endo-1 and Leu-ENK peptides in complex with μ- and δ1b-opioid receptors. (A) [4-I-Phe4]Endo-1 (in cyan) in complex with μ-OR. Phe4 is placed in a small cavity formed by Thr2.56 and Val3.28, making halogenation (iodine is shown) at the para position repulsive (steric hindrance). (B) [4-F-Phe4]Leu-ENK (in magenta) in complex with δ1b-OR. The negative electrostatic potential of fluorine forms a F···H–C hydrogen bond with the methyl group of Thr2.56. (C) [2-I-Phe4]-Leu-ENK-1 (in light blue) in complex with δ1b-OR. The positive σ-hole of iodine forms a halogen bond with the backbone carbonyl oxygen of Phe2.59. (D) The orientation of Phe4 in Endo-1 (cyan sticks), [4-F-Phe4]Leu-ENK (magenta), and [2-I-Phe4]Leu-ENK (light blue) inside the small cavity located between TMs 2 and 7. The negatively charged area (red) created by Asp3.32 is shown. The color code of the helices is TMs 1 in white, 2 in yellow, 3 in red, 4 in gray, 5 in green, 6 in blue, and 7 in brown.
The orientation of Phe4, in MD simulations of the complex between [2-I-Phe4]-Leu-ENK with the δ-opioid receptor (Figure 2C), shows that halogen substitution at the ortho position forms a halogen bond interaction with the oxygen atom of the backbone carbonyl of F2.59. In accordance, fluorine substitution at this ortho position is repulsive while chlorine, bromine, and iodine substitution increases peptide binding linearly with the charge of the σ-hole (Table 1). In our computer simulations the halogen bond remains stable and almost geometrically perpendicular to the backbone carbonyl group (Figure S4). Notably, the chlorinated ligand bound to OX2 orexin receptor, whose structure has been released recently,37 forms halogen bond with the same carbonyl oxygen as postulated for [2-I-Phe4]-Leu-ENK, in a similar position (Figure S4). Figure 2D shows the different orientation of Phe4 in [Phe4]-Leu-ENK, [2-I-Phe4]-Leu-ENK, and [4-F-Phe4]-Leu-ENK, illustrating the flexibility of Phe4 inside the hydrophobic cavity.
Here, we have shown that halogen atoms can modulate peptide–receptor interactions in different manners. First, substitution of a hydrogen atom by bulkier fluorine, chlorine, bromine, or iodine atoms, in small constraint cavities, might produce steric hindrances that reduce the binding of the peptide to the receptor (proportional to the bulkiness of the halogen atom) as shown in the [4-X-Phe4]Endo-1 series. In contrast, substitution of a hydrogen atom by chlorine, bromine, or iodine might be beneficial (proportional to the charge of the σ-hole) if the positive σ-hole of the halogen forms a halogen bond interaction with negatively charged atoms of the protein (i.e., backbone carbonyls) as shown in the [2-X-Phe4]-Leu-ENK series. We believe this interaction is purely σ-hole halogen bonding and does not involve lipophilicity. Substitution of hydrogen by fluorine stabilizes the ligand–receptor complex if the negative electrostatic potential of fluorine interacts with positively charged atoms of the proteins (i.e., hydrophobic CH groups) as shown in the [2-X-Phe4]-Leu-ENK series. In conclusion, our data show how halogen atoms can be used to fine-tune ligand–protein interactions in different protein environments. Each halogen atom can sensibly affect ligand binding as well as ligand selectivity.
Experimental Procedures
Chemistry
Peptides have been prepared by standard stepwise solid phase peptide synthesis techniques using Nα-Fmoc chemistry. Halogen substitutions have been introduced on the peptides from appropriate commercially available Fmoc-Phe building blocks. Crude peptides were purified by RP-HPLC to afford >98% pure compounds. Characterization of compounds was effected by ESI-UPLC methods for the determination of the peptide exact mass (see SI for full description).
Binding Studies to Opioid Receptors
Stably transfected HEK293 cells expressing the μ, δ1a, or δ1b receptor from zebrafish were grown to 80% confluence, harvested, and collected by centrifugation. Membrane preparation, radioligand binding, and data analysis were performed as previously described26 (SI for details). Naloxone was purchased from Sigma-Aldrich (Madrid, Spain) and [3H]-diprenorphine (50 Ci/mmol) from PerkinElmer (Boston, MA).
Conformation in Solution of Peptides by NMR
Samples were prepared in H2O/D2O (90:10), and spectra were recorded on a Bruker Avance 600 MHz instrument equipped with a triple-channel cryoprobe and at 278 K. NMR assignments were reached using standard 2D TOCSY experiments at 20 and 60 ms mixing times and 2D NOESY experiments at 300 ms. The resonance of 3-(trimethylsilyl) propionic-2,2,3,3-d4 acid (TSP) was used as a chemical shift reference in the 1H NMR experiments [δ(TSP) = 0 ppm] (see SI for spectra).
Molecular Models of Peptide–Receptor Complexes
Modeler was used to model zebrafish μ- and δ1b-opioid receptors using the structure of mouse μ- (PDB 4DKL)14 and δ- (PDB 4N6H)15 opioid receptors, respectively, as templates. Endo-1 and Leu-ENK, in the extended conformation as determined by NMR, were docked into the homology models using the Autodock Vina tool. All docking solutions were visually inspected, and the poses in which the N-terminus amine of the peptide mimicked the mode of interaction observed in crystal structures of opioid receptors in complex with nonpeptidic ligands were selected for energy minimization and MD simulations in a explicit lipid bilayer (see SI for details) with the GROMACS software using the protocol previously described.38 Cl, Br, and I were modeled by two point charges (a negative charge centered on the halogen atom to reproduce the electronegative crown,3 and a positive charge centered on a dummy atom on the opposite side of the C—X axis in order to reproduce the positively charged σ-hole).
Glossary
ABBREVIATIONS
- Endo-1
endomorphin-1
- dre
Danio rerio zebrafish
- Dmt
2,6-dimethyltyrosine
- Leu-ENK
Leu-enkephalin
- GPCR
G-protein coupled receptor
- SEM
standard error of the mean
- OR
opioid receptor
- TM
transmembrane domain
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.5b00126.
Experimental details for the synthesis and characterization of individual halogenated endomorphin-1 and Leu-enkephalin analogues, additional figures, competition binding studies, details for computational studies, and 2D NMR spectra of Leu-enkephalin analogues.
Author Contributions
∇ These authors (M.R. and G.C.) contributed equally to this work.
The work was supported by a grant from the Fundació Marató de TV3 (Pain, 070430-31-32-33), MINECO (SAF2010-18597; SAF2013-48271-C2-2-R) and Junta de Castilla y León (B1039/SA25/10). L.P. participates in the European COST Action CM1207 (GLISTEN). M.R. acknowledges a fellowship of Formación del Profesorado Universitario (AP2009-2534) from MINECO.
The authors declare no competing financial interest.
Supplementary Material
References
- Hernandes M. Z.; Cavalcanti S. M.; Moreira D. R.; de Azevedo W. F. Jr.; Leite A. C. Halogen atoms in the modern medicinal chemistry: hints for the drug design. Curr. Drug Targets 2010, 11, 303–314. 10.2174/138945010790711996. [DOI] [PubMed] [Google Scholar]
- Wilcken R.; Zimmermann M. O.; Lange A.; Zahn S.; Boeckler F. M. Using halogen bonds to address the protein backbone: a systematic evaluation. J. Comput.-Aided Mol. Des. 2012, 26, 935–945. 10.1007/s10822-012-9592-8. [DOI] [PubMed] [Google Scholar]
- Auffinger P.; Hays F. A.; Westhof E.; Ho P. S. Halogen bonds in biological molecules. Proc. Natl. Acad. Sci. U. S. A. 2004, 1, 16789–16794. 10.1073/pnas.0407607101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark T.; Hennemann M.; Murray J. S.; Politzer P. Halogen bonding: the σ-hole. J. Mol. Model. 2007, 13, 291–296. 10.1007/s00894-006-0130-2. [DOI] [PubMed] [Google Scholar]
- Dalvit C.; Invernizzi C.; Vulpetti A. Fluorine as a Hydrogen-Bond Acceptor: Experimental Evidence and Computational Calculations. Chem. - Eur. J. 2014, 20, 11058–11068. 10.1002/chem.201402858. [DOI] [PubMed] [Google Scholar]
- Xu Z.; Yang Z.; Liu Y.; Lu Y.; Chen K.; Zhu W. Halogen bond: its role beyond drug-target binding affinity for drug discovery and development. J. Chem. Inf. Model. 2014, 54, 69–78. 10.1021/ci400539q. [DOI] [PubMed] [Google Scholar]
- Xu Z.; Liu Z.; Chen T.; Chen T.; Wang Z.; Tian G.; Shi J.; Wang X.; Lu Y.; Yan X.; Wang G.; Jiang H.; Chen K.; Wang S.; Xu Y.; Shen J.; Zhu W. Utilization of halogen bond in lead optimization: a case study of rational design of potent phosphodiesterase type 5 (PDE5) inhibitors. J. Med. Chem. 2011, 54, 5607–5611. 10.1021/jm200644r. [DOI] [PubMed] [Google Scholar]
- Voth A. R.; Khuu P.; Oishi K.; Ho P. S. Halogen bonds as orthogonal molecular interactions to hydrogen bonds. Nat. Chem. 2009, 1, 74–79. 10.1038/nchem.112. [DOI] [PubMed] [Google Scholar]
- Parisini E.; Metrangolo P.; Pilati T.; Resnati G.; Terraneo G. Halogen bonding in halocarbon-protein complexes: a structural survey. Chem. Soc. Rev. 2011, 40, 2267–2678. 10.1039/c0cs00177e. [DOI] [PubMed] [Google Scholar]
- Sirimulla S.; Bailey J. B.; Vegesna R.; Narayan M. Halogen interactions in protein-ligand complexes: implications of halogen bonding for rational drug design. J. Chem. Inf. Model. 2013, 53, 2781–2791. 10.1021/ci400257k. [DOI] [PubMed] [Google Scholar]
- Ilardi E. A.; Vitaku E.; Njardarson J. T. Data-mining for sulfur and fluorine: an evaluation of pharmaceuticals to reveal opportunities for drug design and discovery. J. Med. Chem. 2014, 57, 2832–2842. 10.1021/jm401375q. [DOI] [PubMed] [Google Scholar]
- Filizola M.; Devi L. A. Grand opening of structure-guided design for novel opioids. Trends Pharmacol. Sci. 2013, 34, 6–12. 10.1016/j.tips.2012.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez A.; Cordomí A.; Caltabiano G.; Pardo L. Impact of Helix Irregularities on Sequence Alignment and Homology Modeling of G Protein-Coupled Receptors. ChemBioChem 2012, 13, 1393–1399. 10.1002/cbic.201200189. [DOI] [PubMed] [Google Scholar]
- Manglik A.; Kruse A. C.; Kobilka T. S.; Thian F. S.; Mathiesen J. M.; Sunahara R. K.; Pardo L.; Weis W. I.; Kobilka B. K.; Granier S. Crystal structure of the μ-opioid receptor bound to a morphinan antagonist. Nature 2012, 485, 321–326. 10.1038/nature10954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granier S.; Manglik A.; Kruse A. C.; Kobilka T. S.; Thian F. S.; Weis W. I.; Kobilka B. K. Structure of the delta-opioid receptor bound to naltrindole. Nature 2012, 485, 400–404. 10.1038/nature11111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H.; Wacker D.; Mileni M.; Katritch V.; Han G. W.; Vardy E.; Liu W.; Thompson A. A.; Huang X. P.; Carroll F. I.; Mascarella S. W.; Westkaemper R. B.; Mosier P. D.; Roth B. L.; Cherezov V.; Stevens R. C. Structure of the human kappa-opioid receptor in complex with JDTic. Nature 2012, 485, 327–332. 10.1038/nature10939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber S. J.; Greene D. L.; Sharma S. D.; Yamamura H. I.; Kramer T. H.; Burks T. F.; Hruby V. J.; Hersh L. B.; Davis T. P. Distribution and analgesia of [3H][D-Pen2, D-Pen5]enkephalin and two halogenated analogs after intravenous administration. J. Pharmacol. Exp. Ther. 1991, 259, 1109–1117. [PubMed] [Google Scholar]
- Abbruscato T. J.; Williams S. A.; Misicka A.; Lipkowski A. W.; Hruby V. J.; Davis T. P. Blood-to-central nervous system entry and stability of biphalin, a unique double-enkephalin analog, and its halogenated derivatives. J. Pharmacol. Exp. Ther. 1996, 276, 1049–1057. [PubMed] [Google Scholar]
- Vaughn L. K.; Knapp R. J.; Toth G.; Wan Y. P.; Hruby V. J.; Yamamura H. I. A high affinity, highly selective ligand for the delta opioid receptor: [3H]-[D-Pen2, pCl-Phe4, d-Pen5]encephalin. Life Sci. 1989, 45, 1001–1008. 10.1016/0024-3205(89)90154-9. [DOI] [PubMed] [Google Scholar]
- Gesellchen P. D.; Shuman R. T.. Substitution on the Phe3 aromatic ring in cyclic.delta. opioid receptor-selective dermorphin/deltorphin tetrapeptide analogs: electronic and lipophilic requirements for receptor affinity. Pharmacologically active peptides. US Patent US4265808, 1981.
- Heyl D. L.; Mosberg H. I. Substitution on the Phe3 aromatic ring in cyclic.delta. opioid receptor-selective dermorphin/deltorphin tetrapeptide analogs: electronic and lipophilic requirements for receptor affinity. J. Med. Chem. 1992, 35, 1535–1541. 10.1021/jm00087a006. [DOI] [PubMed] [Google Scholar]
- Lee Y. S.; Kulkarani V.; Cowell S. M.; Ma S. W.; Davis P.; Hanlon K. E.; Vanderah T. W.; Lai J.; Porreca F.; Vardanyan R.; Hruby V. J. Development of potent μ and δ opioid agonists with high lipophilicity. J. Med. Chem. 2011, 54, 382–386. 10.1021/jm100982d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrallo A.; Gonzalez-Sarmiento R.; Alvar F.; Rodriguez R. E. ZFOR2, a new opioid receptor-like gene from the teleost zebrafish (Danio rerio). Mol. Brain Res. 2000, 84, 1–6. 10.1016/S0169-328X(00)00152-2. [DOI] [PubMed] [Google Scholar]
- Rodriguez R. E.; Barrallo A.; García-Malvar F.; Mcfadyen I. J.; Gonzalez-Sarmiento R.; Traynor J. R. Characterization of ZFOR1, a putative delta-opioid receptor from the teleost zebrafish (Danio rerio). Neurosci. Lett. 2000, 288, 207–210. 10.1016/S0304-3940(00)01239-8. [DOI] [PubMed] [Google Scholar]
- Pinal-Seoane N.; Martin I. R.; Gonzalez-Nunez V.; de Velasco E. M.; Alvarez F. A.; Sarmiento R. G.; Rodriguez R. E. Characterization of a new duplicate delta-opioid receptor from zebrafish. J. Mol. Endocrinol. 2006, 37, 391–403. 10.1677/jme.1.02136. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Nunez V.; Jiménez-Gonzalez A.; Barreto-Valer K.; Rodriguez R. E. In vivo regulation of the mu opioid receptor: role of the endogenous opioid agents. Mol. Med. 2013, 19, 7–17. 10.2119/molmed.2012.00318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bondi A. van der Waals volumes and radii. J. Phys. Chem. 1964, 68, 441–451. 10.1021/j100785a001. [DOI] [Google Scholar]
- Arsequell G.; Rosa M.; Mayato C.; Dorta R. L.; Gonzalez-Nunez V.; Barreto-Valer K.; Marcelo F.; Calle L. P.; Vázquez J. T.; Rodríguez R. E.; Jiménez-Barbero J.; Valencia G. Synthesis, biological evaluation and structural characterization of novel glycopeptide analogues of nociceptin N/OFQ. Org. Biomol. Chem. 2011, 9, 6133–6142. 10.1039/c1ob05197k. [DOI] [PubMed] [Google Scholar]
- Wieberneit F.; Korste A.; Albada H. B.; Metzler-Nolte N.; Stoll R. Structural and biological implications of the binding of Leu-enkephalin and its metal derivatives to opioid receptors. Dalton Trans. 2013, 42, 9799–9802. 10.1039/c3dt50635e. [DOI] [PubMed] [Google Scholar]
- Naito A.; Nishimura K. Conformational analysis of opioid peptides in the solid states and the membrane environments by NMR spectroscopy. Curr. Top. Med. Chem. 2004, 4, 135–145. 10.2174/1568026043451645. [DOI] [PubMed] [Google Scholar]
- Hu X. G.; Thomas D. S.; Griffith R.; Hunter L. Stereoselective fluorination alters the geometry of a cyclic peptide: exploration of backbone-fluorinated analogues of unguisin A. Angew. Chem., Int. Ed. 2014, 53, 6176–6179. 10.1002/anie.201403071. [DOI] [PubMed] [Google Scholar]
- Wang Y. C.; Wu Y. C.; Yeh C. C.; Hwang C. C. Structure-activity relationships of Leu-Enkephalin analog with (4-Carboxamido)phenylalanine substituted for tyrosine: a molecular dynamics study. Biopolymers 2007, 86, 231–239. 10.1002/bip.20728. [DOI] [PubMed] [Google Scholar]
- Portoghese P. S.; Sultana M.; Takemori A. E. Design of peptidomimetic delta opioid receptor antagonists using the message-address concept. J. Med. Chem. 1990, 33, 1714–1720. 10.1021/jm00168a028. [DOI] [PubMed] [Google Scholar]
- Fukuda K.; Terasako K.; Kato S.; Mori K. Identification of the Amino Acid Residues Involved in Selective Agonist Binding in the First Extracellular Loop of the delta- -and mu-Opioid Receptors. FEBS Lett. 1995, 373, 177–181. 10.1016/0014-5793(95)01034-C. [DOI] [PubMed] [Google Scholar]
- Minami M.; Nakagawa T.; Seki T.; Onogi T.; Aoki Y.; Katao Y.; Katsumata S.; Satoh M. A single residue, Lys108, of the delta-opioid receptor prevents the mu-opioid-selective ligand [D-Ala2,N-MePhe4,Gly-ol5]enkephalin from binding to the delta-opioid receptor. Mol. Pharmacol. 1996, 50, 1413–1422. [PubMed] [Google Scholar]
- Ibrahim M. A. A. Molecular mechanical study of halogen bonding in drug discovery. J. Comput. Chem. 2011, 32, 2564–2574. 10.1002/jcc.21836. [DOI] [PubMed] [Google Scholar]
- Yin J.; Mobarec J. C.; Kolb P.; Rosenbaum D. M. Crystal structure of the human OX2 orexin receptor bound to the insomnia drug suvorexant. Nature 2015, 519, 247–250. 10.1038/nature14035. [DOI] [PubMed] [Google Scholar]
- Cordomí A.; Caltabiano G.; Pardo L. Membrane Protein Simulations Using AMBER Force Field and Berger Lipid Parameters. J. Chem. Theory Comput. 2012, 8, 948–958. 10.1021/ct200491c. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.