

Abstract
The influenza virus RNA-dependent RNA polymerase complex (RdRp), a heterotrimeric protein complex responsible for viral RNA transcription and replication, represents a primary target for antiviral drug development. One particularly attractive approach is interference with the endonucleolytic “cap-snatching” reaction by the RdRp subunit PA, more precisely by inhibiting its metal-dependent catalytic activity which resides in the N-terminal part of PA (PA-Nter). Almost all PA inhibitors (PAIs) thus far discovered bear pharmacophoric fragments with chelating motifs able to bind the bivalent metal ions in the catalytic core of PA-Nter. More recently, the availability of crystallographic structures of PA-Nter has enabled rational design of original PAIs with improved binding properties and antiviral potency. We here present a coupled pharmacophore/docking virtual screening approach that allowed us to identify PAIs with interesting inhibitory activity in a PA-Nter enzymatic assay. Moreover, antiviral activity in the low micromolar range was observed in cell-based influenza virus assays.
Keywords: Influenza virus PA endonuclease, polymerase, metal chelation, pharmacophore−structure virtual screening, PA inhibitors (PAIs), dihydroxy-1H-indole-2-carboxamides
Seasonal influenza A and B virus infections are a worldwide concern, causing each year 3–5 million severe infections and 250000–500000 fatalities.1 The current influenza vaccines are only partially effective in some populations2 and require annual updating. Also, antiviral therapy is not fully satisfactory because only two classes of antiviral drugs are available. Resistance is already widespread for the M2 blockers and increasingly recognized for the neuraminidase inhibitors.3,4 Hence, there is an urgent need for new anti-influenza drugs.
The influenza virus genome consists of eight negative-sense RNA segments which encode at least 17 viral proteins. Transcription and replication of viral RNA (vRNA) is carried out by the viral RNA-dependent RNA polymerase (RdRp).5 The crystal structure of the large (∼250 kDa) RdRp complex was reported very recently.6,7 It is composed of three subunits, PB1, PB2, and PA, which are highly conserved among influenza A and B viruses. During vRNA transcription, the RdRp cleaves host pre-mRNAs at a distance of 10–15 nucleotides from their 5′-capped terminus.8 While cap binding is performed by PB2, the endonuclease activity resides in the N-terminal domain of PA (PA-Nter; containing residues 1 to ∼195).9,10 After endonuclease cleavage, the short 5′-capped RNA serves as primer for viral mRNA synthesis by the PB1 unit and, subsequently, the viral mRNAs are translated by the host cell machinery.
Inhibition of the PA endonuclease appears a powerful strategy to suppress influenza virus replication.9−12 In the last two decades, several small molecule PA inhibitors (PAIs) have been discovered.13−21,24−26 Structurally diverse classes of potential PAIs have been identified (Figure 1) such as flutimide and derivatives,15,17N-hydroxamic acids and N-hydroxyimides,16 and epigallocatechin gallate (EGCG).21 Neither of these have comparable antiviral potency as L-742,00114 and closely related DKAs. More recently, a series of hydroxypyridazinones and hydroxypyri(mi)dinones18,20 were identified with particularly strong activity toward the PA-Nter enzyme.
Figure 1.

Representative influenza virus endonuclease inhibitors. The putative metal-chelating chemotype is marked in bold.
The catalytic core of PA-Nter contains a (P)DXN(D/E)XK motif formed by D108, E119, a proline (influenza A) or alanine (influenza B) at position 107, and K134 or K137.9,10 It comprises a histidine (H41) and a cluster of three acidic residues (E80, D108, E119), conserved in all influenza viruses, which coordinate (together with I120) one,10 two,9 or three18 divalent metal ions (Mg2+ or Mn2+, with Mg2+ being the probable cofactor in vivo22). To date, 35 crystal structures related to the influenza virus PA endonuclease have been deposited in the RCSB Protein Data Bank,23 and more than 20 are in complex with an inhibitor. In combination with biochemical studies, these structural studies support the assumption that all PAIs thus far identified inhibit the PA enzyme through chelation of its metal cofactor(s) within the catalytic core. The availability of these complementary PA-Nter crystal structures has created the opportunity to rationally design PAIs with novel chelating structures and enhanced enzyme binding properties to improve antiviral activity in cell culture.18,20,24−26
Indeed, together with other traditional strategies, virtual screening (VS) is recognized as a powerful tool in drug discovery,27,28 as previously explored by us to identify some novel and potent metalloenzyme inhibitors.29 To be effective, the VS method should have a proper balance between predictability and time consumption. With regard to the PA enzyme, only a few examples of computer-aided inhibitor design have thus far been reported, in which molecular diversity was explored to recognize unique pharmacophores different from the DKA scaffold.30,31 Herein, we present a coupled pharmacophore/docking virtual screening approach that allowed us to identify novel PAIs with interesting inhibitory activity in a PA-Nter enzymatic assay, as well as antiviral activity in cell-based influenza virus yield and vRNP reconstitution assays.
The outline of the experimental plan was as follows. First, a hybrid library of roughly 5 million compounds was built by merging the Clean Lead Database retrieved from ZINC and an in-house database of compounds bearing metal chelating functionalities, hence having the potential to inhibit PA-Nter on the basis of their previously evaluated activities against other metalloenzymes such as HIV-1 integrase and carbonic anhydrases.
Second, a suitable pharmacophore model was obtained. Generation of consistent models depends on the quality of both training and testing sets mainly in terms of structural diversity. As already shown by Parkes32 and Kim,33 the minimal pharmacophore motif is composed of two or three donor (i.e., oxygen or nitrogen) atoms capable of chelating the two metal ions. Besides, the spatial disposition of these metal ligator (ML) moieties is critical to achieve effective inhibition of the influenza virus PA endonuclease. In particular, the oxygens should be displaced at the vertices of a triangle with dimensions of 2.60–2.80, 2.60–2.80, and 4.50–5.50 Å for the model proposed by Parkes,32 and of 2.56–2.87, 2.22–2.62, and 3.49–4.51 Å for that put forward by Kim.33 Our best three pharmacophore models shown in Figure 2 (PH4-2, PH4-3, and PH4-9) were in nice agreement with these requirements (i.e., ML1-ML2-ML3 interfeature distances are 2.75, 2.96, and 4.46 Å). Moreover, together with these three coordinating functionalities, our pharmacophore models combine one or two aromatic or hydrophobic regions that allow for additional stabilizing interactions of PAIs within the catalytic site (see Figure 2). Among these three models, model PH4-3 was found to properly distinguish between inactive and active compounds when applied to a test set of 50 structures (of which 10 were known to be active PAIs).
Figure 2.
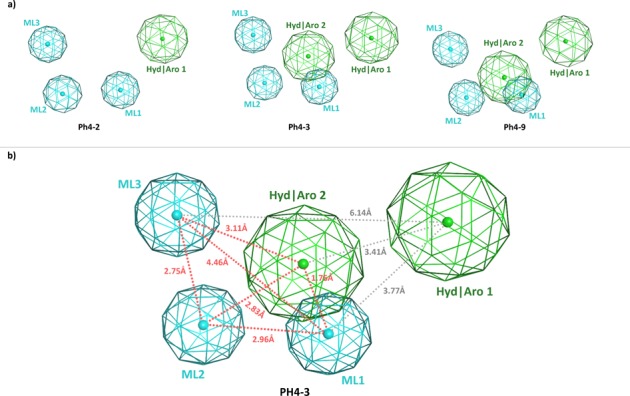
Influenza virus PA endonuclease pharmacophore models generated by MOE: (a) three-dimensional arrangement of the best three models; (b) representation of pharmacophore model PH4-3 that was selected for further study. Distances between centroids of the pharmacophore features are indicated as red or gray dashed lines.
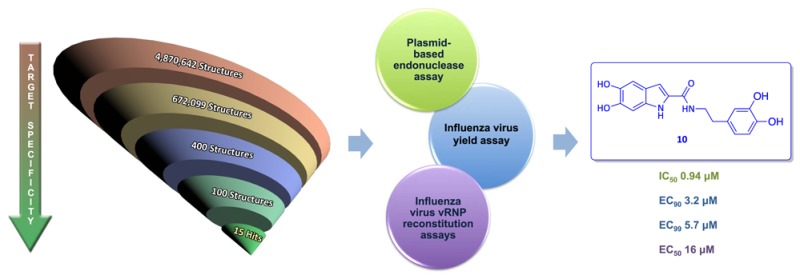
In the third step, a VS procedure on the above-mentioned database by means of combined pharmacophore-filtration and structure-based docking procedures was carried out. To speed up the process, the database was partitioned into 36 sublibraries that were processed in a parallel way using the software platform MOE.34 Each library was first filtered using the Pharmacophore Search implemented in MOE. The derived five-points pharmacophore model PH4-3 was chosen as query, and all structures that matched at least four pharmacophore features where stored, thus realizing a ∼7-fold reduction in the number of structures (see Figure 3). In the last steps of our in silico studies, the MOE Docking protocol was applied to the resulting libraries. After running the docking process, the best 400 hits from all libraries were collected, and the top-ranked energy hits (about 100 molecules) with immediate availability were selected for the AutoDock refinement.
Figure 3.
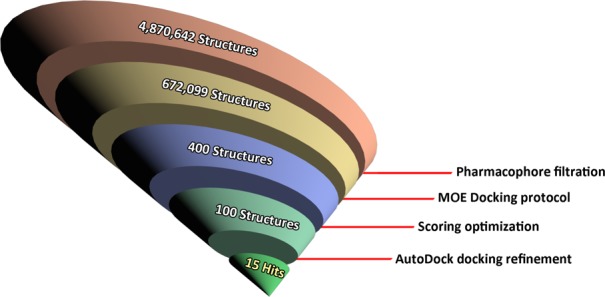
Scheme of the virtual screening approach.
Once these molecules were scored, clusterization by scaffold similarity was done, and compounds 1–15 (Chart 1) were finally selected for subsequent biological evaluation by three complementary methods, i.e., the enzymatic plasmid-based endonuclease assay with influenza virus PA-Nter and cell-based influenza vRNP reconstitution and virus yield assays.
Chart 1. Chemical Structures of the Hit Compounds Identified by the Virtual Screening Procedure.

To enable biological testing, compounds 1–8 were purchased while compounds 9–15 were retrieved from our collection.35−37 Compounds 10, 11, and 15 have been resynthesized in our laboratory using a previously described (and slightly modified) procedure,35 which is depicted in Scheme 1. 10, 11, and 15 were obtained in moderate yields (58%, 46%, and 65%, for 10, 11, and 15, respectively) by deprotection of the catechol moiety of the respective intermediates 21–23, with boron tribromide in dichloromethane at low temperature (Scheme 1). Amides 21–23 were prepared by conversion of 2-carboxylic indole 16 to the acyl chloride 17, and next coupling with the appropriate amines. The key synthone 16 was easily obtained using a previously validated three-steps synthetic route.35
Scheme 1. Synthesis of Compounds 10, 11, and 15.
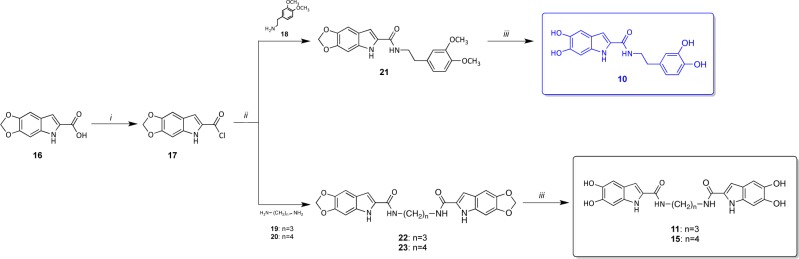
Reagents and conditions: (i) PCl5, diethyl ether, rt, 2 h; (ii) diethyl ether, rt, 2 h; (iii) 1 M BBr3 solution in CH2Cl2, −70 °C to −40 °C (for 10), or −70 °C to −0 °C (for 11 and 15), 4 h.
Among the 15 test compounds evaluated in the PA-Nter enzymatic assay (see Table 1), only compounds 10, 11, and 15 demonstrated inhibitory activity, with IC50 values of 0.94, 65, and 7.0 μM for 10, 11, and 15, respectively. Interestingly, 10 was only 2-fold less active than the prototype PAI L-742,001 (IC50: 0.48 μM), which is one of the more active PAIs reported thus far. Compounds 10, 11, and 15 possess a similar dihydroxyindole scaffold structure which thus appears to be an important structural determinant for PA-Nter inhibitory activity. However, (pseudo) dimerization of this scaffold to obtain 11 and 15 leads to a significant reduction in activity. The dihydroxyindole scaffold of all three active compounds fits well within the pharmacophore model PH4-3 (see Supporting Information, Figure 1).
Table 1. Activity of the 15 Test Compounds in the Plasmid-Based Enzymatic Assay with PA-Nter.
| compd | IC50 (μM)a | compd | IC50 (μM)a |
|---|---|---|---|
| 1 | >500 | 9 | >500 |
| 2 | >500 | 10 | 0.94 |
| 3 | >500 | 11 | 65 |
| 4 | >500 | 12 | >500 |
| 5 | >500 | 13 | >500 |
| 6 | >500 | 14 | >500 |
| 7 | >500 | 15 | 7.0 |
| 8 | >500 | L-742,001 | 0.48 |
IC50: 50% inhibitory concentration, calculated using nonlinear regression analysis. Values are the result of at least three independent experiments.
As far as antiviral activity in cell culture is concerned, all three compounds 10, 11, and 15 inhibited virus replication in a virus yield assay in influenza virus-infected MDCK cells, with EC90 and EC99 values of 3.2 and 5.7 μM for 10, 32 and 73 μM for 11, and 6.3 and 12 μM for 15 (see Table 2). It is remarkable that derivatives 10 and 15 had a potency comparable to that of the reference compound L-742,001 (which had EC90 and EC99 values of 5.4 and 8.4 μM, respectively). Moreover, 10 was 2-fold more active than ribavirin, a broad antiviral molecule that was included as a reference molecule. In the vRNP reconstitution assay, compounds 10, 11, and 15 reached EC50 values of 16, 64, and 24 μM, respectively, while the reference compounds L-742,001 and ribavirin had EC50 values of 3.4 and 8.4 μM, respectively. Hence, compounds 10 and 15 are relevant candidates for further lead optimization and antiviral/mechanistic studies.
Table 2. Anti-Influenza Virus Activity of Selected Compounds 10, 11, and 15 in Cell-Based Influenza Virus Assays.
| virus
yield assay in MDCK cellsa |
vRNP
reconstitution assay in HEK293T cellsa |
||||
|---|---|---|---|---|---|
| compd | EC90 (μM)b | EC99 (μM)b | CC50 (μM)c | EC50 (μM)d | CC50 (μM)c |
| L-742,001 | 5.4 ± 0.3 | 8.4 ± 0.3 | 181 | 3.4 | >100 |
| 10 | 3.2 ± 0.9 | 5.7 ± 1.6 | ≥50 | 16 | 110 |
| 11 | 32 ± 7 | 73 ± 6 | >200 | 64 | >200 |
| 15 | 6.3 ± 1.5 | 12 ± 3 | >200 | 24 | >200 |
| ribavirin | 6.8 ± 0.5 | 11 ± 1 | >200 | 8.4 | >200 |
MDCK, Madin–Darby canine kidney cells; HEK293T cells, human embryonic kidney 293T cells.
Compound concentration (μM) causing 1-log10 (EC90) or 2-log10 (EC99) reduction in virus yield at 24 h pi, as determined by real time RT-PCR. Values shown are the mean ± SEM of at least four experiments.
CC50, 50% cytotoxic concentration determined by MTS cell viability assay at 24 h.
EC50: 50% effective concentration, i.e. compound concentration producing 50% reduction in vRNP-driven firefly reporter signal, estimated at 24 h after transfection, and calculated by nonlinear regression analysis from data of 3 independent experiments.
In the last stage, we performed docking using AutoDock 4.2 to predict the PA-Nter binding mode of the three active molecules, i.e., 10, 11, and 15 (Figure 4a). Our results indicate that their common dihydroxyindole moiety is directed toward the two catalytic metal ions. The orientation of the metal-chelating hydroxyl groups appears more favorable for 10 and 11 compared to 15. For 10 and 11, both hydroxyl groups chelate metal ion B (MB2+) and only one of the hydroxyls interacts with metal ion A (MA2+). The opposite is seen with 15 because both its hydroxyl groups are predicted to chelate MA2+, while only one hydroxyl can interact with MB2+. Because MB2+ is generally considered to be bound with higher affinity compared to MA2+ (at least when no substrate is present in PA-Nter),10,22 this slight difference in orientation may be the basis for the 7-fold higher potency of 10 compared to 15. A striking discrepancy between 11 on the one hand and 10 and 15 on the other hand involves the compounds’ disposition in the cavities surrounding the active site. 10 and 15 engage opposite pockets compared to 11. The catechol functionality of 10 and the second dihydroxyindole ring of 15 orientate toward the pocket lined by Val122, Arg124, and Tyr 130 (in blue, Figure 4a). In contrast, 11 binds via its second dihydroxyindole functionality in the pocket surrounded by Ala20, Tyr24, and Gly81 (Figure 4a, in green and red). The relevance of the pocket delimited by Val122, Arg124, and Tyr130 was previously proposed in our mutational analysis of the binding pockets of L-742,001 (see Figure 4b).19 Likewise, this pocket also proved to be of critical importance for the binding of three recently identified PAIs with strong inhibitory activity, as demonstrated in PA-Nter cocrystallization experiments.18,20 Taken together, our docking results suggest that the superior PA-Nter inhibitory activity of 10 (IC50 = 0.94 μM) is related to its optimal orientation for metal chelation, combined with its engagement into the Val122–Arg124–Tyr130 cavity. Compound 11 (IC50: 65 μM) has a similar metal-chelating binding mode yet does not occupy the Val122–Arg124–Tyr130 pocket. The compound with intermediate activity, i.e.,15 (IC50: 7.0 μM), is able to occupy the Val122–Arg124–Tyr130 pocket but, compared to 10, has a less favorable orientation of the metal-chelating functionality. The nice correlation between the results from the enzymatic assay and cell-based (i.e., virus yield and vRNP reconstitution) methods supports our hypothesis that the antiviral activity in cell culture is related to inhibition of PA-Nter. Mechanistic studies are underway to verify this assumption.
Figure 4.
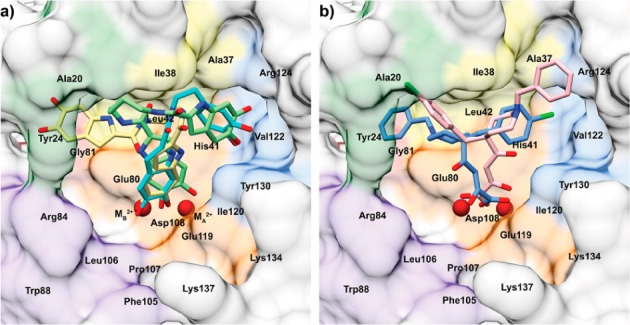
Comparison between the predicted poses of 10, 11, 15 (a) and L-742,001 (b) obtained by docking into the published9 structure of inhibitor-free PA-Nter (PDB entry 2W69). The protein structures are shown as surfaces and in the same orientation after structural alignment using the DALI server. The active site metal ions are colored dark red. (a) Superimposition of the best pharmacophore-fitting docking poses obtained for compounds 10 (cyan), 11 (yellow), and 15 (green). (b) Disposition of L-742,001 in PA-Nter as predicted by docking: the conformer representing the most favorable binding energies (in blue) and that representing the most diffuse population of conformers (in pink).19
To summarize, a large database of roughly 5 million structures was screened to identify novel influenza virus endonuclease inhibitors by applying pharmacophore and structure-based docking procedures. Fifteen hits were then evaluated in a PA-Nter enzymatic assay, and three compounds bearing an original bis-dihydroxy-1H-indole-2-carboxamide scaffold demonstrated interesting inhibitory activity, with compounds 10 and 15 having IC50 values in the low micromolar range. Both prototypes also showed antiviral activity in cell-based assays and had comparable potency compared to the reference PAI L-742,001 and the nucleoside analogue inhibitor ribavirin. Follow-up studies are warranted to further assess the full potential of the bis-dihydroxy-1H-indole-2-carboxamide scaffold to develop new influenza PAIs with preclinical relevance.
Acknowledgments
The authors thank Dr. Andrea Brancale for the use of MOE program. A.S. and L.N. acknowledge Wim van Dam and Ria Van Berwaer for fine technical assistance, and Meehyein Kim (Korea Research Institute of Chemical Technology, Daejeon, South Korea) for generous donation of the vRNP reconstitution plasmids.
Glossary
Abbreviations
- RdRp
RNA-dependent RNA polymerase complex
- PA-Nter
N-terminal part of PA
- PAI
PA inhibitor
- vRNA
viral RNA
- DKA
β-diketoacid
- VS
virtual screening
Supporting Information Available
Synthetic and computational procedures. Influenza plasmid-based endonuclease, virus yield, and vRNP reconstitution assays. The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.5b00109.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript. N.P. and A.S. contributed equally.
We thank the Fondazione Banco di Sardegna (grant to M.S), the KU Leuven (grant Geconcerteerde Onderzoeksacties, GOA/15/019/TBA) (to A.S. and L.N.), and the Italian Ministero dell’Istruzione, dell’Università e della Ricerca (PRIN 2010, grant 2010W2KM5L_003) (to M.S, D.R, and M.C.) for their financial support.
The authors declare no competing financial interest.
Supplementary Material
References
- Influenza (Seasonal)—Fact Sheet no. 211; World Health Organization: Geneva, 2014; http://www.who.int/mediacentre/factsheets/fs211/en/ (accessed 19 January 2015).
- Uyeki T. M. Preventing and controlling influenza with available interventions. N. Engl. J. Med. 2014, 370, 789–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanderlinden E.; Naesens L. Emerging antiviral strategies to interfere with influenza virus entry. Med. Res. Rev. 2014, 34, 301–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leang S. K.; Deng Y. M.; Shaw R.; Caldwell N.; Iannello P.; Komadina N.; Buchy P.; Chittaganpitch M.; Dwyer D. E.; Fagan P.; Gourinat A. C.; Hammill F.; Horwood P. F.; Huang Q. S.; Ip P. K.; Jennings L.; Kesson A.; Kok T.; Kool J. L.; Levy A.; Lin C.; Lindsay K.; Osman O.; Papadakis G.; Rahnamal F.; Rawlinson W.; Redden C.; Ridgway J.; Sam I. C.; Svobodova S.; Tandoc A.; Wickramasinghe G.; Williamson J.; Wilson N.; Yusof M. A.; Kelso A.; Barr I. G.; Hurt A. C. Influenza antiviral resistance in the Asia-Pacific region during 2011. Antiviral Res. 2013, 97, 206–210. [DOI] [PubMed] [Google Scholar]
- Ruigrok R. W.; Crépin T.; Hart D. J.; Cusack S. Towards an atomic resolution understanding of the influenza virus replication machinery. Curr. Opin. Struct. Biol. 2010, 20, 104–113. [DOI] [PubMed] [Google Scholar]
- Pflug A.; Guilligay D.; Reich S.; Cusack S. Structure of influenza A polymerase bound to the viral RNA promoter. Nature 2014, 516, 355–360. [DOI] [PubMed] [Google Scholar]
- Reich S.; Guilligay D.; Pflug A.; Malet H.; Berger I.; Crépin T.; Hart D.; Lunardi T.; Nanao M.; Ruigrok R. W.; Cusack S. Structural insight into cap-snatching and RNA synthesis by influenza polymerase. Nature 2014, 516, 361–366. [DOI] [PubMed] [Google Scholar]
- Plotch S. J.; Bouloy M.; Ulmanen I.; Krug R. M. A unique cap(m7GpppXm)-dependent influenza virion endonuclease cleaves capped RNAs to generate the primers that initiate viral RNA transcription. Cell 1981, 23, 847–858. [DOI] [PubMed] [Google Scholar]
- Dias A.; Bouvier D.; Crépin T.; McCarthy A. A.; Hart D. J.; Baudin F.; Cusack S.; Ruigrok R. W. The cap-snatching endonuclease of influenza virus polymerase resides in the PA subunit. Nature 2009, 458, 914–918. [DOI] [PubMed] [Google Scholar]
- Yuan P.; Bartlam M.; Lou Z.; Chen S.; Zhou J.; He X.; Lv Z.; Ge R.; Li X.; Deng T.; Fodor E.; Rao Z.; Liu Y. Crystal structure of an avian influenza polymerase PA(N) reveals an endonuclease active site. Nature 2009, 458, 909–913. [DOI] [PubMed] [Google Scholar]
- Das K.; Aramini J. M.; Ma L. C.; Krug R. M.; Arnold E. Structures of influenza A proteins and insights into antiviral drug targets. Nature Struct. Mol. Biol. 2010, 17, 530–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogolino D.; Carcelli M.; Sechi M.; Neamati N. Viral enzymes containing magnesium: metal binding as a successful strategy in drug design. Coord. Chem. Rev. 2012, 256, 3063–3086. [Google Scholar]
- Tomassini J. E.; Selnick H.; Davies M. E.; Armstrong M. E.; Baldwin J.; Bourgeois M.; Hastings J. C.; Hazuda D.; Lewis J.; McClements W.; Ponticello G.; Radzilowski E.; Smith G.; Tebben A.; Wolfe A. Inhibition of cap (m7GpppXm)-dependent endonuclease of influenza virus by 4-substituted 2,4-dioxobutanoic acid compounds. Antimicrob. Agents Chemother. 1994, 38, 2827–2837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hastings J. C.; Selnick H.; Wolanski B.; Tomassini J. E. Anti-influenza virus activities of 4-substituted 2,4-dioxobutanoic acid inhibitors. Antimicrob. Agents Chemother. 1996, 40, 1304–1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomassini J. E.; Davies M. E.; Hastings J. C.; Lingham R.; Mojena M.; Raghoobar S. L.; Singh S. B.; Tkacz J. S.; Goetz M. A. A novel antiviral agent which inhibits the endonuclease of influenza viruses. Antimicrob. Agents Chemother. 1996, 40, 1189–1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cianci C.; Chung T. D. Y.; Meanwell N.; Putz H.; Hagen M.; Colonno R. J.; Krystal M. Identification of N-hydroxamic acid and N-hydroxy-imide compounds that inhibit the influenza virus polymerase. Antiviral Chem. Chemother. 1996, 7, 353–360. [Google Scholar]
- Singh S. B.; Tomassini J. E. Synthesis of natural flutimide and analogous fully substituted pyrazine-2,6-diones, endonuclease inhibitors of influenza virus. J. Org. Chem. 2001, 66, 5504–5516. [DOI] [PubMed] [Google Scholar]
- Bauman J. D.; Patel D.; Baker S. F.; Vijayan R. S.; Xiang A.; Parhi A. K.; Martínez-Sobrido L.; Lavoie E. J.; Das K.; Arnold E. Crystallographic fragment screening and structure-based optimization yields a new class of influenza endonuclease inhibitors. ACS Chem. Biol. 2013, 8, 2501–2508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevaert A.; Dallocchio R.; Dessì A.; Pala N.; Rogolino D.; Sechi M.; Naesens L. Mutational analysis of the binding pockets of the diketo acid inhibitor L-742,001 in the influenza virus PA endonuclease. J. Virol. 2013, 87, 10524–10538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sagong H. Y.; Bauman J. D.; Patel D.; Das K.; Arnold E.; Lavoie E. J. Phenyl substituted 4-hydroxypyridazin-3(2H)-ones and 5-hydroxypyrimidin-4(3H)-ones: inhibitors of influenza A endonuclease. J. Med. Chem. 2014, 57, 8086–8098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuzuhara T.; Iwai Y.; Takahashi H.; Hatakeyama D.; Echigo N. Green tea catechins inhibit the endonuclease activity of influenza A virus RNA polymerase. PLoS Curr. 2009, 1, RRN1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao S.; Klein M. L.; LeBard D. N.; Levine B. G.; Liang H.; MacDermaid C. M.; Alfonso-Prieto M. Magnesium-dependent RNA binding to the PA endonuclease domain of the avian influenza polymerase. J. Phys. Chem. B 2014, 118, 873–889. [DOI] [PubMed] [Google Scholar]
- Berman H. M.; Westbrook J.; Feng Z.; Gilliland G.; Bhat T. N.; Weissig H.; Shindyalov I. N.; Bourne P. E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baughman B. M.; Jake Slavish P.; DuBois R. M.; Boyd V. A.; White S. W.; Webb T. R. Identification of influenza endonuclease inhibitors using a novel fluorescence polarization assay. ACS Chem. Biol. 2012, 7, 526–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carcelli M.; Rogolino D.; Bacchi A.; Rispoli G.; Fisicaro E.; Compari C.; Sechi M.; Stevaert A.; Naesens L. Metal-chelating 2-hydroxyphenyl amide pharmacophore for inhibition of influenza virus endonuclease. Mol. Pharmaceutics 2014, 11, 304–316. [DOI] [PubMed] [Google Scholar]
- Stevaert A.; Nurra S.; Pala N.; Carcelli M.; Rogolino D.; Shepard C.; Domaoal R. A.; Kim B.; Alfonso-Prieto M.; Marras S. A. E.; Sechi M.; Naesens L. An integrated biological approach to guide the development of metal-chelating inhibitors of influenza virus PA endonuclease. Mol. Pharmacol. 2015, 87, 323–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walters W. P.; Stahl M. T.; Murcko M. A. Virtual screening—an overview. Drug Discovery Today 1998, 3, 160–178. [Google Scholar]
- Jorgensen W. L. The many roles of computation in drug discovery. Science. 2004, 303, 1813–1818. [DOI] [PubMed] [Google Scholar]
- Pala N.; Dallocchio R.; Dessì A.; Brancale A.; Carta F.; Ihm S.; Maresca A.; Sechi M.; Supuran C. T. Virtual screening-driven identification of human carbonic anhydrase inhibitors incorporating an original, new pharmacophore. Bioorg. Med. Chem. Lett. 2011, 21, 2515–2520. [DOI] [PubMed] [Google Scholar]
- Ishikawa Y.; Fujii S. Binding mode prediction and inhibitor design of anti-influenza virus diketo acids targeting metalloenzyme RNA polymerase by molecular docking. Bioinformation 2011, 6, 221–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Z.; Zhang L.; Fu H.; Wang Z.; Lin J. Design of the influenza virus inhibitors targeting the PA endonuclease using 3D-QSAR modeling, side-chain hopping, and docking. Bioorg. Med. Chem. Lett. 2014, 24, 539–547. [DOI] [PubMed] [Google Scholar]
- Parkes K. E.; Ermert P.; Fässler J.; Ives J.; Martin J. A.; Merrett J. H.; Obrecht D.; Williams G.; Klumpp K. Use of a pharmacophore model to discover a new class of influenza endonuclease inhibitors. J. Med. Chem. 2003, 46, 1153–1164. [DOI] [PubMed] [Google Scholar]
- Kim J.; Lee C.; Chong Y. Identification of potential influenza virus endonuclease inhibitors through virtual screening based on the 3D-QSAR model. SAR QSAR Environ. Res. 2009, 20, 103–118. [DOI] [PubMed] [Google Scholar]
- Molecular Operating Environment, MOE 2009.10; Chemical Computing Group Inc.: 1010 Sherbooke St. West, Suite #910, Montreal, QC, Canada, H3A 2R7, 2009.
- Sechi M.; Angotzi G.; Dallocchio R.; Dessì A.; Carta F.; Sannia L.; Mariani A.; Fiori S.; Sanchez T.; Movsessian L.; Plasencia C.; Neamati N. Design and synthesis of novel dihydroxyindole-2-carboxylic acids as HIV-1 integrase inhibitors. Antiviral Chem. Chemother. 2004, 15, 67–81. [DOI] [PubMed] [Google Scholar]
- Sechi M.; Azzena U.; Delussu M. P.; Dallocchio R.; Dessì A.; Cosseddu A.; Pala N.; Neamati N. Design and synthesis of bis-amide and hydrazide-containing derivatives of malonic acid as potential HIV-1 integrase inhibitors. Molecules 2008, 13, 2442–2461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sechi M.; Rizzi G.; Bacchi A.; Carcelli M.; Rogolino D.; Pala N.; Sanchez T.; Taheri L.; Dayam R.; Neamati N. Design and synthesis of novel dihydroxyquinoline-3-carboxylic acids as HIV-1 integrase inhibitors. Bioorg. Med. Chem. Lett. 2009, 17, 2925–2935. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.