

Abstract
We report the identification and synthesis of a series of aminopyrimidin-4-one IRAK4 inhibitors. Through high throughput screening, an aminopyrimidine hit was identified and modified via structure enabled design to generate a new, potent, and kinase selective pyrimidin-4-one chemotype. This chemotype is exemplified by compound 16, which has potent IRAK4 inhibition activity (IC50 = 27 nM) and excellent kinase selectivity (>100-fold against 99% of 111 tested kinases), and compound 31, which displays potent IRAK4 activity (IC50 = 93 nM) and good rat bioavailability (F = 42%).
Keywords: IRAK4, kinase inhibitor, inflammation, structure-based drug design
Interluekin-1 receptor associated kinase-4 (IRAK4) is a member of the IRAK4 family of serine-threonine kinases and is known to have biologically relevant kinase activity.1 In addition, all members of the family (including IRAK1, IRAK2, and IRAKM) are thought to have key protein scaffolding and regulatory functions in various signaling pathways.2 IRAK4 is a key mediator in the interluekin-1 receptor (IL-1R) and Toll-like receptor (TLR) signaling cascades.3 These signaling pathways play crucial roles in the innate immune response.4 IRAK4 functions as an upstream signal transducer that effects the activation of transcription factors kappa light chain enhancer of activated B cells (NF-κB) and activator protein-1 (AP-1) resulting in the generation of secondary inflammatory mediators.5 The importance of IRAK4 has been demonstrated with studies of IRAK4 kinase dead, knock-in mice. Mice possessing this genotype are resistant to joint inflammation in several rodent arthritis models.6 Additionally, a small IRAK4 deficient human population has been identified. Cells from these patients have impaired responses to ILR/TLR receptor stimulation.7 The data in rodents and humans implies that an IRAK4 inhibitor could modulate the production of key inflammatory cytokines and cytokine induced pathologies.
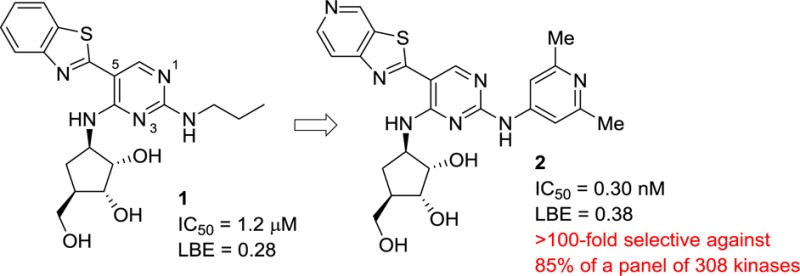
Recent reports have highlighted several novel and diverse series of IRAK4 inhibitors.8−10 We previously identified a unique series of aminopyrimidine compounds (1) from a high throughput screen.11 These hits were optimized to improve the IRAK4 potency and ligand binding efficiency (LBE)12 resulting in compounds such as pyrimidine 2 with subnanomolar IRAK4 inhibition and good LBE, but with modest kinase selectivity. Subsequent SAR work identified compounds with improved kinase selectivity (90–96%, cf., inhibitor 2 at 85%) and cellular potencies.13 These improvements in potency and selectivity were realized by altering the substituents around the periphery of the pyrimidine core, specifically at the C-2 and C-5 positions. An additional route that was investigated to improve the kinase selectivity of the series was to make changes to the pyrimidine core itself, specifically, changing the pyrimidine ring to a pyridine.13 While several pyridine analogues were examined, it was found that these compounds had similar kinase selectivity profiles to the parent pyrimidines.
Upon examination of the X-ray cocrystal structure of pyrimidine 1 (Figure 1), it was observed that there appeared to be a small pocket adjacent to the C-6 position of the pyrimidine ring of the inhibitor and in the vicinity of the Val263 backbone carbonyl. It had been previously shown that this space could be filled with a Cl, methyl, or amino group.11 Another substituent that was considered was a hydroxyl/carbonyl group. The presence of a hydroxyl group could potentially form an additional hydrogen bond interaction with the Val263 backbone carbonyl. However, studies of aminoisocytosine derivatives have shown that the oxo tautomeric form predominates in both solution and solid state.14 While it was believed that the pocket could spatially accommodate the carbonyl moiety, it was hypothesized that predominance of the oxo tautomer would lead to electrostatic repulsion from the Val263 carbonyl, which would force the inhibitor away from the hinge with a concomitant loss in potency. It was feasible that this altered binding mode could lead to improved kinase selectivity. If there was a loss in potency, it was assumed that it could be improved by modifications elsewhere in the molecule, and the perceived risk of loss of IRAK4 potency was acceptable in order to provide a greater overall kinase selectivity profile.
Figure 1.
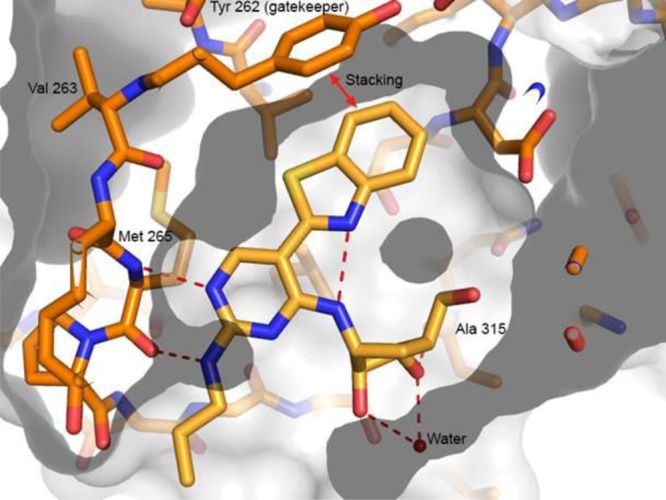
X-ray cocrystal structure of pyrimidine 1 bound to the IRAK4 hinge.
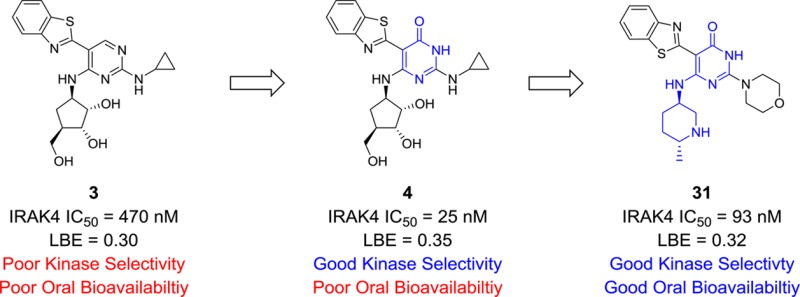
Pyrimidine 3 was chosen as the parent compound as it had reasonable IRAK4 potency and a smaller group (cyclopropyl) on the C-2 amino position, which would be more accommodating to shifts in binding mode, compared to the more potent pyrimidine compounds that had large substituted aryl rings at this position (cf., 2). When the corresponding pyrimidin-4-one 4 was synthesized, it was found to have a significant improvement in potency over the parent pyrimidine 3. Upon obtaining and solving the X-ray cocrystal structure, it was observed that the compound bound in a new binding mode as the pyrimidin-4-one tautomer (Figure 2A). The key change from the pyrimidine core was that the inhibitor had shifted down in the ATP binding site. This became readily apparent when the structures of pyrimidine 1 and pyrmidin-4-one 4 were overlaid (Figure 2B). While the pyrimidin-4-one was still forming two hydrogen bond interactions with the hinge motif (like the pyrimidine), these were now being made with a different donor–acceptor pair. Where the pyrimidine ring nitrogen was functioning as a hydrogen bond acceptor, now the pyrimidin-4-one carbonyl was forming this key bond (Figure 2C). Likewise, where the C-2 amino of the pyrimidine ring had functioned as the hydrogen bond donor, the pyrimidin-4-one ring NH was now filling this role.
Figure 2.
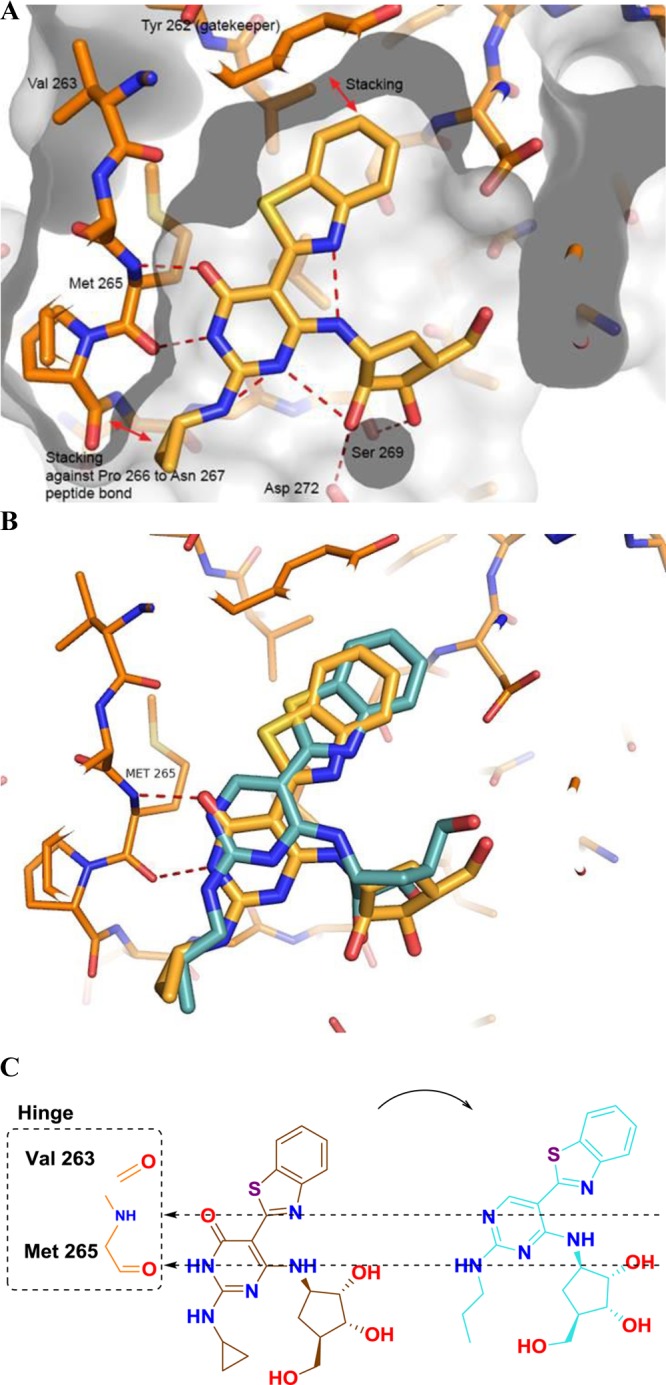
(A) X-ray cocrystal structure of pyrimidin-4-one 4. (B) Overlay of the X-ray structures of pyrimidine 1 (cyan) and pyrimidin-4-one 4 (orange). (C) Alignment of the hinge binding motifs between 1 (cyan) and 4 (orange).
Several key interactions were retained between the two chemotypes. It appeared that the π–π stacking interaction between the benzothiazole of the inhibitor and the tyrosine gatekeeper had been preserved. Additionally, an intramolecular hydrogen bond between the nitrogen of the benzothiazole ring and the C-4 amino group had also been maintained. Like for the case of the pyrimidine core,11 it is thought that this intramolecular hydrogen bond plays an important role in helping to preorganize the inhibitor into a coplanar orientation that facilitates binding.

It was hypothesized that the change in the binding mode would have SAR ramifications for the series, perhaps leading to a divergent set of active compounds. This became readily apparent when the most potent C-2 amino substituent, 2,6-dimethylpyridine, was appended to the pyrmidin-4-one core to generate compound 5. This substituent on the pyrimidine core provided compounds with single digit nanomolar or subnanomolar potency (cf., pyrimidine 2 above). In the case of pyrimidin-4-one 5, not only was there a loss of potency but there was also an associated drop in the ligand efficiency of the compound, particularly when compared to the cyclopropyl analogue 4. This result clearly indicated that the structure–activity relationships (SARs) between the pyrimidines and the pyrimidinones were divergent.
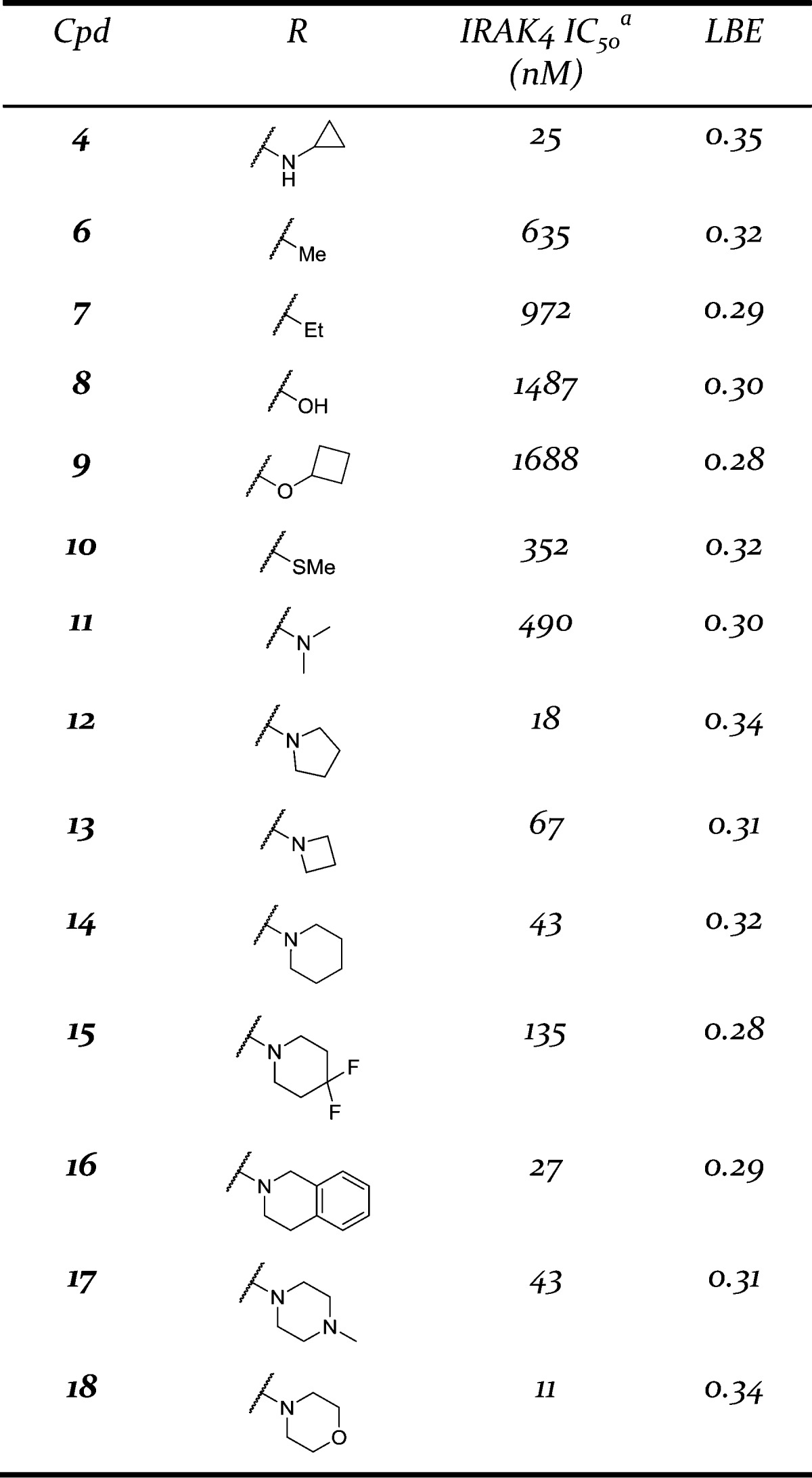
In another point of differentiation, the crystal structure of pyrimidin-4-one 4 indicated that the C-2 amino group was no longer participating in a hydrogen bond interaction with the IRAK4 hinge. This hinted at the possibility that it would be feasible to remove the C-2 amino group altogether. This modification would provide a step forward in improving the drug-like qualities of the compound by removing a hydrogen bond donor, and also provide additional SAR flexibility at this position. A survey of different substituents at the C-2 position was undertaken (Table 1). Methyl (compound 6) and ethyl (compound 7) substituents were tolerated, but with a loss in potency. Neither a hydroxyl/oxo group (compound 8) nor a cyclobutyl ether (compound 9) were able to generate potent inhibitors. For comparison, when these substituents were explored in the pyrimidine core they yielded compounds that were completely inactive (IRAK4 IC50 > 50 μM). In the case of the pyrimidin-4-one, although these modifications produced compounds that had a weaker potency than the parent (pyrimidin-4-one 4), it is in a range that warranted additional SAR optimization efforts. Switching to a thiomethyl ether (compound 10) or a dimethyl amine (compound 11) provided compounds of comparable potency to the alkyl substituents (6 and 7). This indicated that, while it was possible to remove the C-2 amino group, more electron rich groups appeared to be preferred at this position. In view of that, when electron rich cyclic amines (compounds 12–14) were examined, potencies below 100 nm could be achieved. When the more electron deficient difluoropiperidine 15 was tested, several fold loss of potency was observed compared to the parent piperidine (compound 14). However, increasing the size of the substituent to tetrahydroisoquinoline 16 maintained the potency.
Table 1. Modification of the C-2 Position.
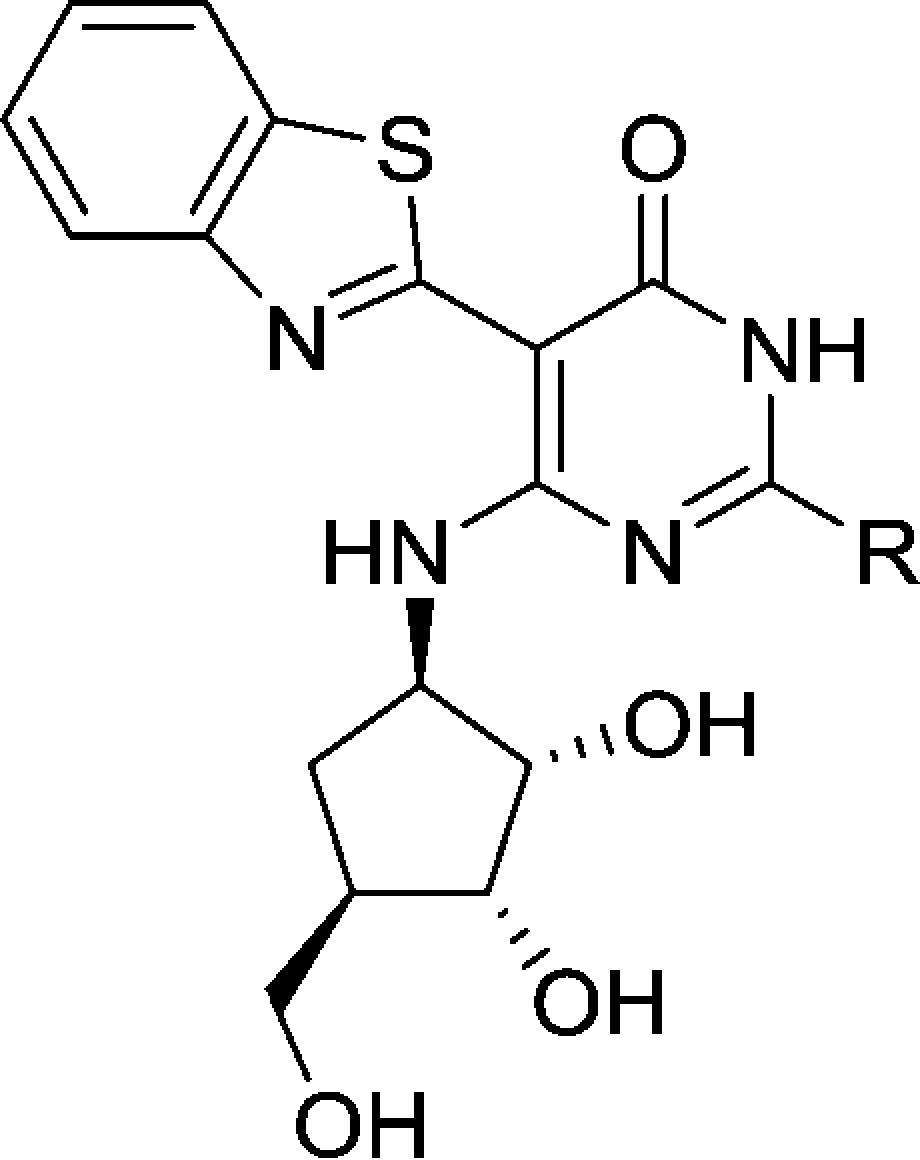
See Supporting Information for assay protocol.
The addition of a basic nitrogen in the form of an N-methyl piperizine ring (compound 17) was tolerated, as was an unsubstituted morpholine ring (compound 18). These last three changes were encouraging as they revealed that this position was amenable to changes that could improve the physiochemical properties or potentially the kinase selectivity of the compounds, but not have a drastic impact on the IRAK4 potency.
Having established promising SAR trends at the C-2 position, attention was focused on the carboribose ring. It was clear that in order to improve the drug-like properties of the series, it would be necessary to reduce the number of hydroxyl groups present on the carboribose ring. Metabolite identification studies indicated that oxidative as well as conjugative metabolism of the carboribose ring were major concerns. It was observed from the overlays of the pyrimidine and pyrimidin-4-one X-ray structures (Figure 2B) that, in addition to the core itself, the carboribose ring had shifted down in the binding site. This shift brought the carboribose ring in closer proximity to a region of an acidic residue, Asp272. In the structure of pyrimidin-4-one 4, a hydrogen bond interaction between a carboribose hydroxyl and the Asp272 was formed (Figure 2A).
This observation prompted the idea that perhaps it would be possible to remove all the hydroxyl groups and add a basic nitrogen into the ring (i.e., a pyrrolidine or a piperidine ring) that could still make the key hydrogen bond interaction with Asp272 (Figure 3). This would have the net benefit of reducing the overall number of hydrogen bond donors and acceptors, and also removing a metabolically labile primary hydroxyl group that did not appear to be involved in any interactions with the IRAK4 protein. In practice, when the carboribose ring of morpholine analogue 18 was replaced with a 3-piperidine ring to provide compound 19, it was found that the IRAK4 potency could be maintained with this modification and a slight increase in the binding efficiency was realized. Moreover, the properties of piperidine 19 were improved: the number of hydrogen bond acceptors was reduced (9 to 7), the number of hydrogen bond donors reduced (5 to 3), and the polar surface area was reduced as well (140 to 91). The X-ray cocrystal structure of compound 19 was solved confirming the hypothesis that a properly positioned basic group maintained a key interaction with the Asp272 residue (Figure 4).
Figure 3.
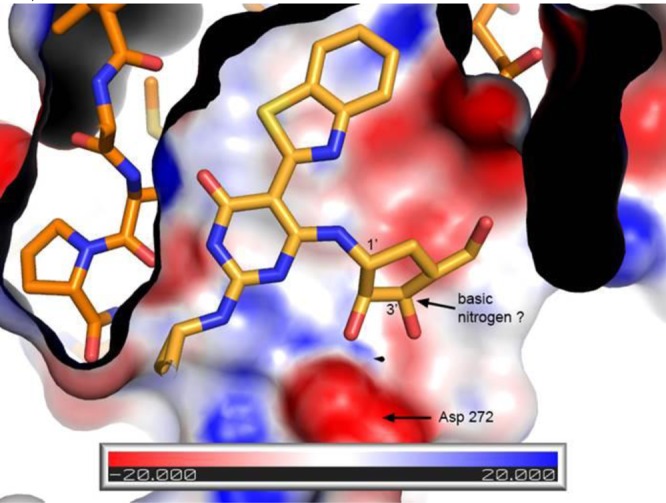
Charge surface map indicating potential interactions of the inhibitor with ASP272.
Figure 4.
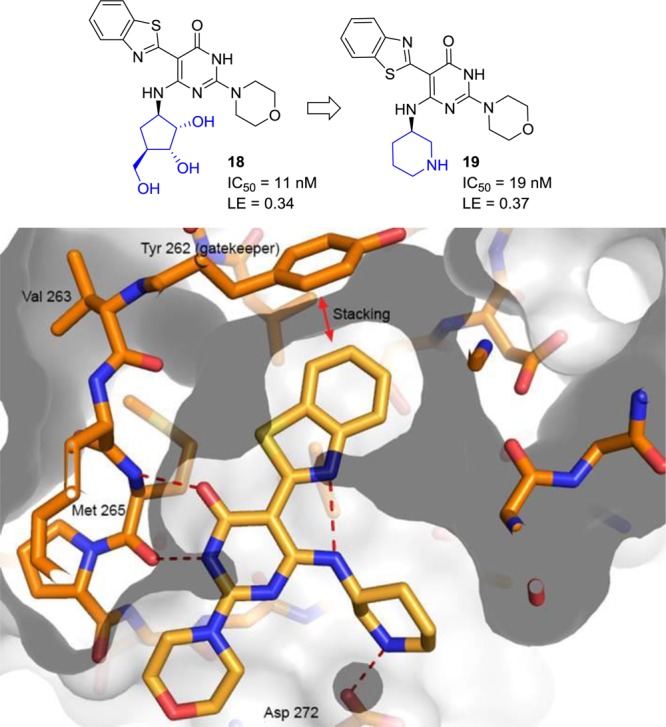
X-ray cocrystal structure of pyrimidine 19.
Capitalizing on the success with the piperidine ring, several additional carboribose replacement motifs were surveyed, and the results are presented in Table 2. In addition to piperidine, pyrrolidine (compounds 20 and 21) was tolerated although there was a clear stereochemical bias favoring the R configuration. This carried over into the piperidine modification as well as with the enantiomer of piperidine 19 having >13-fold loss in potency (data not shown). Methylation of the piperidine to give compound 22 resulted in a loss of potency compared to the parent piperidine 19. It was possible to move the basic nitrogen out of the ring, as in amino cyclohexane 23. The size of the ring could also be increased from 6 to 7 exemplified by azepane 24, having only a slight loss in potency. However, if the basicity of the nitrogen was reduced, a significant loss of potency was observed with lactam 25 having a potency over 2 μM. If the piperidine ring was substituted with a hydroxyl group (compound 26) a slight loss in potency was observed. Like the seven-membered ring example, when the basicity of the piperidine ring was reduced, in this case a piperidone ring (compound 27), a significant loss in potency was observed. Moving from a 3-piperidine to a 4-piperidine ring (compound 28) saw a slight drop in potency. A hydroxyl group could be reinstalled at the 3-position in place of the basic nitrogen (compound 29), but with a loss in potency. Finally, methyl substituents were tolerated vicinal to the piperidine nitrogen (compounds 30 and 31). Like the R vs S configuration bias for the parent piperidine, a preference for a single methyl isomer was observed (R, compound 31).
Table 2. Modification of the C-6 Position.
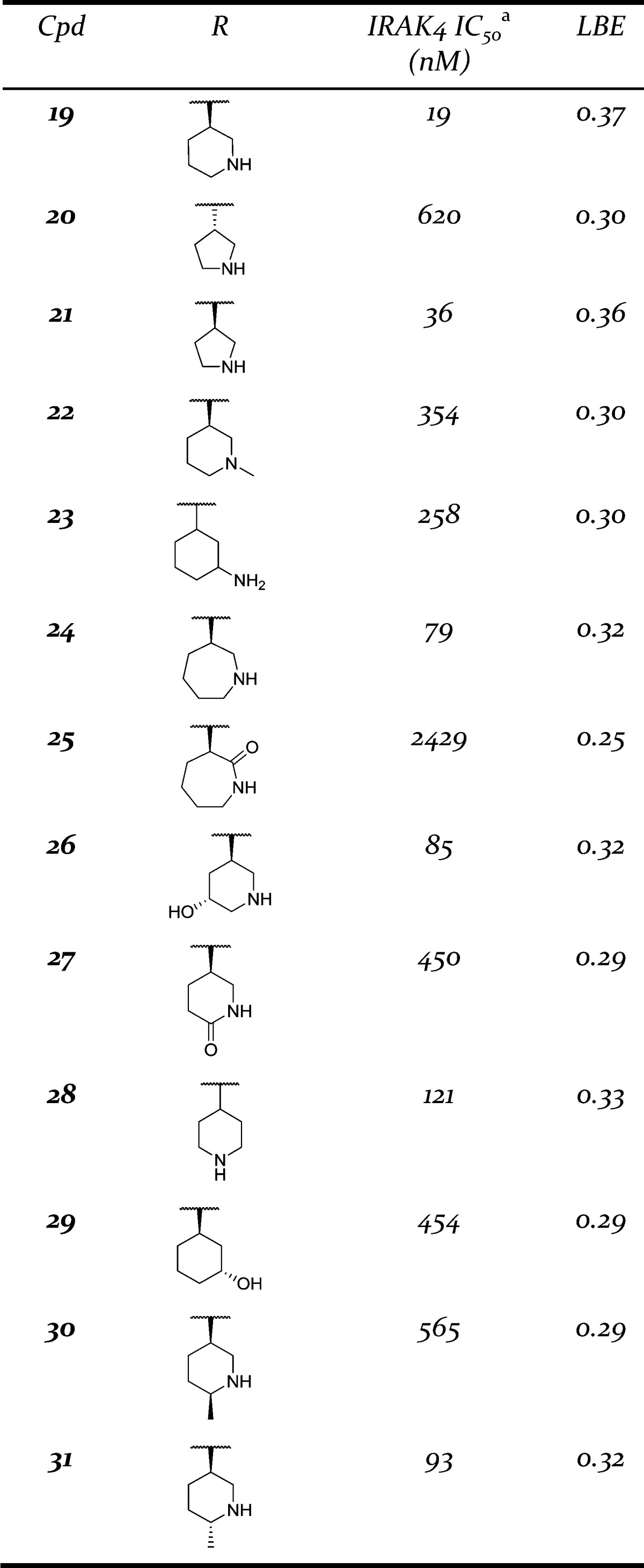
See Supporting Information for assay protocol.
Several potent pyrimidin-4-ones were selected for additional follow up (Table 3). These compounds were assessed for their potency in a cellular pathway engagement assay by measuring the effect of the IRAK4 inhibitors on NF-κB activation. A reporter cell line, THP1-XBlue, stably expressing NF-κB-inducible secreted embryonic alkaline phosphatase (SEAP), was stimulated with lipopolysaccharide (LPS) and the resulting SEAP activity assessed quantitatively.15 All of the selected compounds were active in this assay, although several compounds displayed a significant activity shift compared to the biochemical enzyme inhibition assay. In particular, pyrimidin-4-ones 12 and 19 displayed potency shifts of approximately 60-fold. This did not appear to be attributable to poor solubility (26 and 21 μM) or permeability (Papp = 12 and 29 × 10–6 cm/s) for these compounds. Pyrimidinone 19 was also active in human peripheral blood mononuclear cells (hPBMCs) stimulated with IL1β (IC50 = 1.9 μM) or the TLR 7/8 agonist R848 (IC50 = 4.2 μM), although the potency was right shifted analogous to the THP-1 data. Kinase selectivity was generally good across the class with most compounds displaying >100-fold selectivity against 95% of the kinases screened (n = 111). In particular compound 16 only had one off target kinase hit (FLT3). This kinase proved particularly problematic as all of the compounds had less than 100-fold selectivity against it. However, none of the compounds had any significant selectivity problems with inflammation-related kinases,16 a fact that makes them amenable to development for further in vivo studies. To this end, the pharmacokinetics of the selected compounds was also evaluated. Bioavailability was generally low or zero for the carboribose containing compounds (12, 13, 16, 18), but began to improve as the carboribose was replaced with a piperidine ring (19, 26, 31). Compound 19 possessed a reasonable 13% bioavailability, which could be improved to 42% in compound 31 after the addition of a methyl group adjacent to the piperidine nitrogen. Although further improvement of the in vivo clearance is warranted, replacement of the carboribose (13, 16, 18) with piperidine (19, 26, 31) and the concomitant improvement in clearance indicates this is a tractable issue with a potential path forward. This trend toward improving clearance could also be observed in hepatocyte stability studies (data not shown) providing a useful in vitro/in vivo correlation for further SAR efforts.
Table 3. PK, Cell Activity, and Kinase Selectivity of Selected Compounds.
| Cpd | IRAK4 IC50 (nM) | Cell IC50a (nM) | Kinase Selectivityb | Pappc (×10–6 cm/s) | Solubilityd (μM) | Rat Cle (mL/min/kg) | Rat %Ff |
|---|---|---|---|---|---|---|---|
| 12 | 18 | 78 | 96 | 12 | 26 | – | – |
| 13 | 67 | 292 | 96 | 18 | 23 | 75 | 4 |
| 16 | 27 | 141 | 99 | 5 | 12 | 88 | 0 |
| 18 | 11 | 375 | 94 | 7 | 63 | 98 | 0 |
| 19 | 19 | 187 | 90 | 29 | 21 | 45 | 13 |
| 26 | 85 | 333 | 96 | 9 | 8 | 43 | 2 |
| 31 | 93 | 580 | 97 | 15 | 13 | 51 | 42 |
See Supporting Information for assay protocol.
Percent kinases with >100-fold selectivity (n = 111) versus IRAK, IC50, see Supporting Information for additional details.
Measured in LLC-PK1 cells.
Measured kinetic solubility at pH 7.
IV dosed at 1 mg/kg as a solution of 1 mg/mL in 20% HPBCD.
PO dosed at 10 mg/kg as a suspension of 2 mg/mL in 0.4% methylcellulose. See Supporting Information for full PK parameters for compounds 19 and 31.
In summary, a structure enabled design strategy facilitated the development of a series of kinase selective IRAK4 aminopyrimidin-4-ones from an original aminopyrimidine lead series. Owing to a novel binding mode, these pyrimidin-4-ones displayed SAR that was divergent from the pyrimidines. This allowed for the incorporation of novel groups at the C-2 position, as well as replacement of the carboribose moiety. These modifications expedited the improvement of the physiochemical characteristics of the series, while maintaining excellent kinase selectivity, eventually leading to several compounds with moderate to good oral bioavailability in rat. These attributes make this series attractive for follow up optimization toward the development of a highly selective IRAK4 in vivo tool compound.
Supporting Information Available
Synthetic schemes, procedures, analytical data (including kinase selectivity) for compounds 4 and 19, conditions for the biological assays, kinase selectivity screen, and full PK data for 19 and 31. The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.5b00279.
Accession Codes
X-ray coordinates for compounds 1, 4, and 19 have been uploaded to the Protein Data Bank with the codes 4ZTL, 4ZTM, and 4ZTN, respectively.
The authors declare no competing financial interest.
Supplementary Material
References
- Staschke K. A.; Dong S.; Saba J.; Zhao J.; Brooks N. A.; Hepburn D. L.; Xia J.; Gulen M. F.; Kang Z.; Altuntas C. Z.; Tuohy V. K.; Gilmour R.; Li X.; Na S. IRAK4 Kinase Activity Is Required for Th17 Differentiation and Th17-Mediated Disease. J. Immunol. 2009, 183, 568–577. 10.4049/jimmunol.0802361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song K. W.; Talamas F. X.; Suttmann R. T.; Olson P. S.; Barnett J. W.; Lee S. W.; Thompson K. D.; Jin S.; Hekmat-Nejad M.; Cai T. Z.; Manning A. M.; Hill R. J.; Wong B. R. The kinase activities of interleukin-1 receptor associated kinase (IRAK)-1 and 4 are redundant in the control of inflammatory cytokine expression in human cells. Mol. Immunol. 2009, 46, 1458–1466. 10.1016/j.molimm.2008.12.012. [DOI] [PubMed] [Google Scholar]
- Brzezinska A. A.; Johnson J. L.; Munafo D. B.; Ellis B. A.; Catz S. D. Signaling mechanisms for Toll-like receptor-activated neutrophil exocytosis: key roles for interleukin-1-receptor-associated kinase-4 and phosphatidylinositol 3-kinase but not Toll/IL-1 receptor (TIR) domain-containing adaptor inducing IFN-beta (TRIF). Immunology 2009, 127, 386–397. 10.1111/j.1365-2567.2008.02980.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akira S.; Takeda K. Toll-like receptor signaling. Nat. Rev. Immunol. 2004, 4, 499–511. 10.1038/nri1391. [DOI] [PubMed] [Google Scholar]
- Suzuki N.; Saito T. IRAK-4 - a shared NF-kappa B activator in innate and acquired immunity. Trends Immunol. 2006, 27, 566–572. 10.1016/j.it.2006.10.003. [DOI] [PubMed] [Google Scholar]
- Koziczak-Holbro M.; Littlewood-Evans A.; Poellinger B.; Kovarik J.; Dawson J.; Zenke G.; Burkhart C.; Mueller M.; Gram H. The Critical Role of Kinase Activity of Interleukin-1 Receptor-Associated Kinase 4 in Animal Models of Joint Inflammation. Arthritis Rheum. 2009, 60, 1661–1671. 10.1002/art.24552. [DOI] [PubMed] [Google Scholar]
- Picard C.; Puel A.; Bonnet M.; Ku C. L.; Bustamante J.; Yang K.; Soudais C.; Dupuis S.; Feinberg J.; Fieschi C.; Elbim C.; Hitchcock R.; Lammas D.; Davies G.; Al-Ghonaium A.; Al-Rayes H.; Al-Jumaah S.; Al-Hajjar S.; Al-Mohsen I. Z.; Frayha H. H.; Rucker R.; Hawn T. R.; Aderem A.; Tufenkeji H.; Haraguchi S.; Day N. K.; Good R. A.; Gougerot-Pocidalo M. A.; Ozinsky A.; Casanova J. L. Pyogenic bacterial infections in humans with IRAK-4 deficiency. Science 2003, 299, 2076–2079. 10.1126/science.1081902. [DOI] [PubMed] [Google Scholar]
- Chaudhary D.; Robinson S.; Romero D. L. Recent Advances in the Discovery of Small Molecule Inhibitors of Interleukin-1 Receptor-Associated Kinase 4 (IRAK4) as a Therapeutic Target for Inflammation and Oncology Disorders. J. Med. Chem. 2015, 58, 96–110. 10.1021/jm5016044. [DOI] [PubMed] [Google Scholar]
- Hynes J.; Nair S. K. Advances in the Discovery of Small-Molecule IRAK4 Inhibitors. Annu. Rep. Med. Chem. 2014, 49, 117–133. 10.1016/B978-0-12-800167-7.00009-2. [DOI] [Google Scholar]
- Tumey L. N.; Boschelli D. H.; Bhagirath N.; Shim J.; Murphy E. A.; Goodwin D.; Bennett E. M.; Wang M.; Lin L.-L.; Press B.; Shen M.; Frisbie R. K.; Morgan P.; Mohan S.; Shin J.; Rao V. R. Identification and optimization of indolo[2,3-c]quinoline inhibitors of IRAK4. Bioorg. Med. Chem. Lett. 2014, 24, 2066–2072. 10.1016/j.bmcl.2014.03.056. [DOI] [PubMed] [Google Scholar]
- McElroy W. T.; Seganish W. M.; Herr R. J.; Harding J.; Yang J.; Yet L.; Komanduri V.; Prakash K. C.; Lavey B.; Tulshian D.; Greenlee W. J.; Sondey C.; Fischmann T.; Niu X. Discovery and hit to lead optimization of 2,6-diaminopyrimidine inhibitors of interleukin receptor-associated kinase 4. Bioorg. Med. Chem. Lett. 2015, 25, 1836–1841. 10.1016/j.bmcl.2015.03.043. [DOI] [PubMed] [Google Scholar]
- Hopkins A. L.; Groom C. R.; Alex A. Ligand efficiency: a useful metric for lead selection. Drug Discovery Today 2004, 9, 430–431. 10.1016/S1359-6446(04)03069-7. [DOI] [PubMed] [Google Scholar]
- Seganish W. M.; McElroy W. T.; Herr R. J.; Brumfield S.; Greenlee W. J.; Harding J.; Komanduri V.; Matasi J.; Prakash K. C.; Tulshian D.; Yang J.; Yet L.; Devito K.; Fossetta J.; Garlisi C. G.; Lundell D.; Niu X.; Sondey C. Initial optimization and series evolution of 2,6-diaminopyrimidine inhibitors of interleukin-1 receptor associated kinase 4. Bioorg. Med. Chem. Lett. 2015, 25, 3203–3207. 10.1016/j.bmcl.2015.05.097. [DOI] [PubMed] [Google Scholar]
- Craciun L.; Custelcean R.; Mager S. Molecular structure of an isocytosine analog: Combined X-ray structural and computational study of 2-Diethylalmino-6-methyl-5-n-propyl-4(3H)-pyrimidinone. Struct. Chem. 1999, 10, 303–310. 10.1023/A:1022051119505. [DOI] [Google Scholar]
- See Supporting Information for experimental protocol.
- Gaestel M.; Kotlyarov A.; Kracht M. Targeting innate immunity protein kinase signaling in inflammation. Nat. Rev. Drug Discovery 2009, 8, 480–499. 10.1038/nrd2829. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.