

Abstract
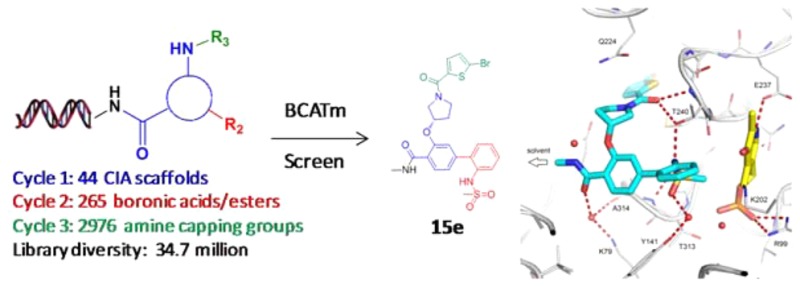
As a potential target for obesity, human BCATm was screened against more than 14 billion DNA encoded compounds of distinct scaffolds followed by off-DNA synthesis and activity confirmation. As a consequence, several series of BCATm inhibitors were discovered. One representative compound (R)-3-((1-(5-bromothiophene-2-carbonyl)pyrrolidin-3-yl)oxy)-N-methyl-2′-(methylsulfonamido)-[1,1′-biphenyl]-4-carboxamide (15e) from a novel compound library synthesized via on-DNA Suzuki–Miyaura cross-coupling showed BCATm inhibitory activity with IC50 = 2.0 μM. A protein crystal structure of 15e revealed that it binds to BCATm within the catalytic site adjacent to the PLP cofactor. The identification of this novel inhibitor series plus the establishment of a BCATm protein structure provided a good starting point for future structure-based discovery of BCATm inhibitors.
Keywords: BCATm, obesity, DNA Encoded Library, ELT, on-DNA Suzuki−Miyaura cross-coupling reaction
Branched chain aminotransferases (BCAT) catalyze transamination of the branched chain amino acids (BCAAs) leucine, isoleucine, and valine to their respective α-keto acids, which are further converted to succinyl-CoA and acetyl-CoA, and participate in tricarboxylic acid cycle and glycolysis.1 There are two isozymes, mitochondrial (BCATm) and cytosolic (BCATc), and the human BCAT isozymes are 58% identical in amino acid sequence.2 While BCATm is in most tissues, BCATc is largely restricted to the central nervous system.1 BCAAs are essential amino acids that must be consumed in the diet of mammals since they do not possess the necessary enzymes to synthesize these molecules de novo. An increasing body of evidence supports the notion that BCAAs stimulate protein synthesis in skeletal muscle, with leucine being primarily responsible for the stimulation of protein synthesis after intake of a mixed meal.3 Conversely, it is also known that the product of leucine transamination, α-keto isocaproic acid, inhibits protein degradation resulting in an overall accumulation of protein.4 A recent mouse BCATm knockout study demonstrated that lack of this enzyme increased the mice’s energy expenditure, associated with a futile protein turnover cycle, and protected them from obesity when subjected to a high fat diet.5 Furthermore, metabolomic profiling of obese versus lean humans reveals a BCAA-related metabolite signature that is suggestive of increased catabolism of BCAAs and correlated with insulin resistance.6 To further validate BCATm as a therapeutic target for the intervention of metabolic diseases, a suitable small molecular inhibitor is highly desirable. To date, no human BCATm inhibitors have ever been reported. Herein we report our effort in identifying small molecule inhibitors of human BCATm via DNA encoded library technology. More than 14 billion compounds from a superpool of various DNA encoded libraries of distinct scaffolds were screened against BCATm followed by off-DNA hit synthesis and activity confirmation. As a consequence, several potent BCATm chemotypes were discovered. In this report we describe the design and synthesis of a 34.7-million-member DNA-encoded library and BCATm hits identified from this library.
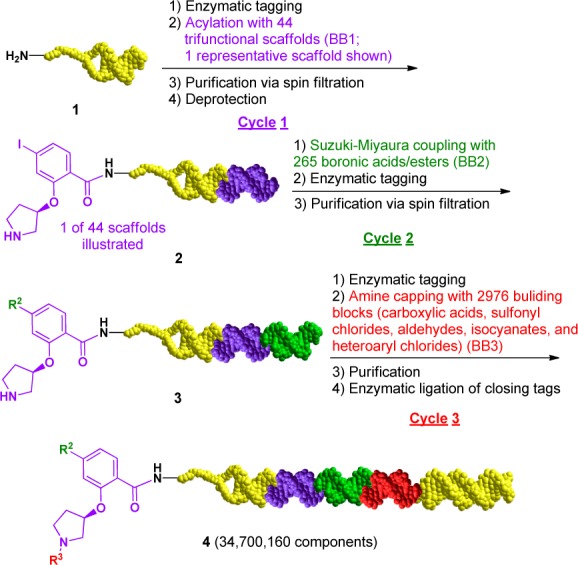
Encoded Library Technology (ELT) has proven to be an important lead discovery platform for various therapeutic targets.7−16 To increase library diversity and expand drug-like chemical space, we recently developed a trifunctional scaffold based novel DNA encoded library dubbed the “CIA” (carboxylic acid, iodide, and amine) library. Each of the trifunctional scaffolds contains a carboxylic acid, an aryl iodide group, and an Fmoc-protected amine group. Similar to our previously reported libraries, this library was synthesized using the split-and-pool strategy, beginning from a short sequence of duplex DNA stabilized by a synthetic hairpin (the “headpiece”).10,11 The design of the headpiece allows synthesis of the small molecule library starting from the free amine group, while DNA tags encoding the building blocks (BBs) are enzymatically ligated at the nonlinked end of the DNA duplex. In this instance, 44 CIA scaffolds (referred to as cycle 1 building blocks or BB1) were first installed onto the headpiece DNA starting material (1, Scheme 1) through amide bond formation followed by pooling and Fmoc-deprotection.11 After splitting into separate reaction wells, 265 cycle 2 boronic acids/ester building blocks (BB2) were introduced through on-DNA Suzuki–Miyaura cross-coupling reactions.17 This is our first reported application of on-DNA cross-coupling reactions in a library synthesis setting. After standard pooling, purification via spin filtration, and splitting into different reaction wells, 2976 unique cycle 3 building blocks (BB3) including carboxylic acids, sulfonyl chlorides, aldehydes, isocyanates, and heteroaryl chlorides were reacted with the amine group thus providing the final library with a size of 34.7 million. After pooling and purification, the DNA sequence was then “closed” with a final tag that encodes a library identifier, a priming region for PCR amplification, and a degenerate sequence to control for PCR amplification biases.
Scheme 1. Synthesis of the Three Cycle DNA Encoded CIA Library.
The affinity selections were performed on a chemically biotinylated human BCATm protein construct. The target protein was immobilized on a streptavidin agarose resin packed column. After immobilization, the column was washed with buffer containing 1 mM biotin to ensure occupation of all biotin binding sites. The immobilized protein was then exposed to the on-DNA library prior to extensive washing to remove nonbinders. Bound library molecules were eluted by heat denaturation of the protein at 80 °C. The eluant was then incubated with fresh immobilized protein to start a new round of affinity selection. A parallel selection against a column lacking protein was done to allow for the identification of molecules enriched by nontarget affinity. After three rounds of selection, the DNA tags of the eluted population were PCR amplified and sequenced, and the sequences were translated to identify structures of putative BCATm binders.
Further analysis of the affinity selection output could be conveniently performed in a Spotfire cube view. In Figure 1, encoded small molecule compounds enriched at the level of two independent DNA sequences or greater are shown with the cube axes representing building blocks used in cycle 1, cycle 2, and cycle 3 chemistry, respectively. As discussed in earlier reports, families of related compounds are of particular interest because they contain one or more building blocks in common and are easily recognized in these views as lines or planes.10,11 Here, several such features were evident. First of all, there is a strong selection for BB1 building block (R)-2-((1-(((9H-fluoren-9-yl)methoxy)carbonyl)pyrrolidin-3-yl)oxy)-4-iodobenzoic acid (5) as most of the data points shown in the cube are on the plane defined by this particular building block (i.e., all of the corresponding compounds to each of the data points have the same building block at cycle 1). Second, on this plane there are several lines: two major horizontal ones and three major perpendicular ones. The horizontal lines are defined by BB2 borates N-(2-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl)methanesulfonamide (6) and 3-boronobenzoic acid (7), correspondingly, and the three perpendicular lines are defined by BB3 carboxylates 5-bromo-2-((tert-butoxycarbonyl)amino)benzoic acid (8), 5-bromothiophene-2-carboxylic acid (9), and 2-amino-5-bromobenzoic acid (10). The structures corresponding to these lines are enumerated as general structures I, II, III, IV, and V.
Figure 1.

Spotfire cube data view of BCATm selection features. BB1, cycle 1 building blocks; BB2, cycle 2 building blocks; and BB3, cycle 3 building blocks. Color of the dots represents different levels of enrichment (pink is 50-fold, blue is 20-fold, green is 10-fold, and yellow is 5-fold). To simplify the visualization, only selection features of building block 5 with copy counts ≥2 were shown in the cube.
The above analysis suggested that affinity selection of the 34.7 million member CIA library over BCATm provides several series of molecules with a common cycle 1 trifunctional moiety. To confirm these hits, we synthesized representative off-DNA compounds from each of these series by standard synthetic methods as outlined in Scheme 2. Starting from the trifunctional scaffold 12, the methyl carboxylate was first converted to methylamide (13) followed by Suzuki–Miyaura cross-coupling to install the second substituted phenyl ring (14a–d). Then Boc-deprotection of the pyrrolidine nitrogen followed by acylation affords the desired compounds 15a–j. Meanwhile palladium mediated reduction of 13 removed the iodo group, which was followed by pyrrolidine nitrogen Boc-deprotection and subsequent acylation to afford compound 16.
Scheme 2. Off-DNA Synthesis of Representative Compounds Selected by BCATm.

Reagents and conditions: (a) diisopropyl azodicarboxylate, PPh3, tert-butyl 3-hydroxypyrrolidine-1-carboxylate; (b) (i) NaOH, THF-MeOH, H2O; (ii) HATU, DIEA, MeNH2·HCl, AcCN; (c) boronic acids, Pd(PPh3)4, Cs2CO3, THF/H2O, N2, 80 °C, 2 h; (d) (i) 10% TFA/DCM; (ii) HATU, DIEA, R2COOH, AcCN; (e) H2 balloon, 10% Pd/C, MeOH; (f) (i) 10% TFA/DCM; (ii) HATU, DIEA, 5-bromothiophene-2-carboxylic acid, AcCN.
Representative compounds 15a–c (Figure 2) from combination of lines 1–3 and 4 were first synthesized and tested in a BCATm enzymatic assay. As indicated in Figure 2, while both compounds 15a and 15b showed IC50 greater than 100 μM, compound 15c seems better than 15a and 15b with a measurable IC50 = 74.1 μM. We then switched the 3-carboxylphenyl group in 15c to the 2-methylsulfonamidophenyl group featured in line 5 thus leading to compound 15d, which displayed significant BCATm enzyme inhibition with IC50 = 3.9 μM.
Figure 2.

First group of off-DNA compounds and activities against BCATm.
Since compound 15d was initially prepared as a racemic mixture, its (R)-enantiomer (15e, Figure 3), which incorporates the actually selected BB1 (5), was subsequently synthesized. Compound 15e showed about 2-fold increase in BCATm enzyme inhibition potency (IC50 = 2.0 μM). Further structure–activity relationship exploration around compound 15e indicated that the 5-bromo group on the thiophene ring can be replaced by a chloro group (15f) with mostly maintained potency (IC50 = 2.8 μM). However, cyano substitution at the same position (15g) led to more than 5-fold decrease of activity (IC50 = 15.1 μM) suggesting either size or electron negativity of the cyano group is not tolerated at this position. It was further discovered that the 2-methylsulfonamidophenyl group is also critical for the observed activity. As shown earlier, the corresponding 3-carboxylphenyl analogue 15c was much less active than 15d. Replacement of the 2-methylsufonamidophenyl group with either 2-methylphenyl (15h, IC50 = 64.6 μM) or a simple unsubstituted phenyl group (15i, IC50 > 100 μM) caused more than 10-fold decrease in enzymatic activity, and removal of the entire phenyl group (16) rendered the compound almost inactive.
Figure 3.

Structures and activities for analogues of compound 15d.
To better understand its binding mode and mechanism of action, a high resolution (1.6 Å) X-ray crystal structure of BCATm in complex with 15e was obtained through cocrystallization in the presence of aldimine cofactor pyridoxal 5′-phosphate (PLP). As shown in Figure 4a, compound 15e sits deep in the BCATm active site with the methylamide moiety pointing into a solvent exposed area accounting for the tolerance of DNA linkage at this position. Compound 15e adopts a T-shaped conformation within the BCATm active site (Figure 4a). The phenyl 2-methylsulfonamido is directed toward the covalently bound PLP cofactor, such that one of the sulfone oxygens makes a direct H-bond with the backbone amide of A314, while the other oxygen engages in a water bridged interaction with the hydroxyl of Y141 (Figure 4b). The bromothiophene of 15e sits snugly in a hydrophobic pocket surrounded by residues including C318, V182, M241, V238, and V320 (Figure 4c). This results in a decrease in activity when the bromine is replaced by the longer cyano group of 15g but tolerates its substitution by chlorine (15f). We also briefly considered the importance of the chirality of the ether linkage. The T-shape binding mode of 15e is helped by a quasi-planar pyrrolidine with an axially branching methoxy moiety. Modeling suggests that the alternative enantiomer could adopt a similar binding mode with only small changes to the solvent-exposed pyrrolidinyl ring pucker and equatorial branching of the methoxy moiety. The key interactions offered by the phenyl 2-methylsulfonamido and the bromothiophene would still be in place, potentially explaining the relatively small difference between the enzyme inhibition for 15e and the racemate 15d.
Figure 4.
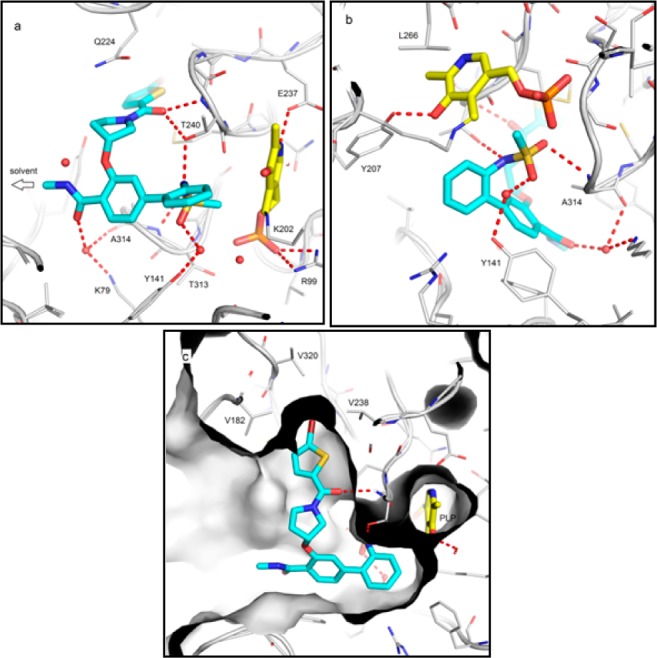
Views of X-ray crystal structure of BCATm in complex with 15e (in cyan) and cofactor PLP (in yellow).
In summary, a novel 34.7 million member trifunctional scaffold-based DNA encoded library was synthesized utilizing the recently reported on-DNA Suzuki–Miyaura reaction. Affinity based screening of this library against BCATm led to the discovery of a novel and attractive biphenyl pyrrolidine ether hit series that showed potent BCATm inhibition. SAR exploration indicated that both the methylsulfonamido and the bromothiophene moieties are crucial for the activity. A binding mode study revealed that representative compound 15e binds to BCATm at its catalytic site through multiple H-bond and van der Waals interactions. While this work exemplifies another successful application of DNA encoded libraries to hit/lead identification for novel therapeutic targets, the identification of the novel biphenyl pyrrolidine ether BCATm inhibitor series and the establishment of the liganded BCATm crystal structure also lays a good foundation for future structure-based BCATm inhibitor design.
Acknowledgments
We thank Dr. Argyrides Argyrou for a critical review of the manuscript and helpful discussions. We’d also like to thank Julia Christie for optical purity analysis of chiral compounds.
Glossary
ABBREVIATIONS
- BCAAs
branched chain amino acids
- BCATm
mitochondrial branched-chain aminotransferase
- BCATc
cytosolic branched-chain aminotransferase
- ELT
encoded library technology
- CIA
carboxylic acid, iodide, and amine
- BB
building block
- NTC
no-target control
- PLP
pyridoxal 5′-phosphate
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.5b00179.
Experimental details for the synthesis of the DNA encoded library, the synthesis of off-DNA compounds, affinity screening of encoded library, BCATm fluorescent assay, BCATm/PLP protein production and crystallization, X-ray crystal structure of BCATm/PLP complexed with inhibitor 15e, and the X-ray data summary table for 15e (PDF)
Accession Codes
The PDB code for 15e is 5CR5
Author Present Address
⊥ Lilly China Research and Development Center (LCRDC), Eli Lilly and Company, 338 Jia Li Lue Road, Shanghai 201203, P. R. China.
Author Present Address
# Roche Innovation Center Basel, Grenzacherstrasse 124, 4070 Basel, CH, Switzerland.
Author Present Address
∇ X-Chem Inc., 100 Beaver Street, Waltham, Massachusetts 02453, United States.
Author Present Address
○ University of Massachusetts Medical School, Worcester, Massachusetts 01655, United States.
Author Present Address
◊ AstraZeneca, Shanghai, China.
The authors declare no competing financial interest.
Supplementary Material
References
- Hutson S. Structure and Function of Branched Chain Aminotransferases. Prog. Nucleic Acids Res. Mol. Biol. 2001, 70, 175–206. 10.1016/S0079-6603(01)70017-7. [DOI] [PubMed] [Google Scholar]
- Goto M.; Miyahara K.; Hirotsu K.; Conway M.; Yennawar N.; Islam M. M.; Hutson S. M. Structural Determinants for Branched-Chain Aminotransferase Isozyme-Specific Inhibition by the Anticonvulsant Drug Gabapentin. J. Biol. Chem. 2005, 280, 37246–37256. 10.1074/jbc.M506486200. [DOI] [PubMed] [Google Scholar]
- Kimball S. R.; Jefferson L. S. Signaling Pathways and Molecular Mechanisms through which BCAA Mediate Translational Control of Protein Synthesis. J. Nutr. 2006, 136, 227S–231S. [DOI] [PubMed] [Google Scholar]
- Tischler M. E.; Desautels M.; Goldberg A. L. Does Leucine, Leucyl-tRNA, or Some Metabolite of Leucine Regulate Protein Synthesis and Degradation in Skeletal and Cardiac Muscle?. J. Biol. Chem. 1982, 257, 1613–1621. [PubMed] [Google Scholar]
- She P.; Reid T. M.; Bronson S. K.; Vary T. C.; Hajnal A.; Lynch C. J.; Hutson S. M. Disruption of BCATm in Mice Leads to Increased Energy Expenditure Associated with the Activation of a Futile Protein Turnover Cycle. Cell Metab. 2007, 6, 181–194. 10.1016/j.cmet.2007.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newgard C. B.; An J.; Bain J. R.; Muehlbauer M. J.; Stevens R. D.; Lien L. F.; Haqq A. M.; Shah S. H.; Arlotto M.; Slentz C. A.; Rochon J.; Gallup D.; Ilkayeva O.; Wenner B. R.; Yancy W. S. Jr.; Eisenson H.; Musante G.; Surwit R. S.; Millington D. S.; Butler M. D.; Svetkey L. P. A Branched-Chain Amino Acid-Related Metabolic Signature that Differentiates Obese and Lean Humans and Contributes to Insulin Resistance. Cell Metab. 2009, 9, 311–26. 10.1016/j.cmet.2009.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franzini R. M.; Neri D.; Scheuermann J. DNA-Encoded chemical libraries: Advancing beyond conventional small-molecule libraries. Acc. Chem. Res. 2014, 47, 1247–1255. 10.1021/ar400284t. [DOI] [PubMed] [Google Scholar]
- Kleiner R. E.; Dumelin C. E.; Liu D. R. Small-molecule discovery from DNA-encoded chemical libraies. Chem. Soc. Rev. 2011, 40, 5707–5717. 10.1039/c1cs15076f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodnow R. A., Jr.Handbook for DNA-Encoded Chemistry; John Wiley & Sons Press: Hoboken, NJ, 2014. [Google Scholar]
- Clark A. M.; Acharya R. A.; Arico-Muendel C. C.; Belyanskaya S. L.; Benjamin D. R.; Carlson N. R.; Centrella P. A.; Chiu C. H.; Creaser S. P.; Cuozzo J. W.; Davie C. P.; Ding Y.; Franklin G. J.; Franzen K. D.; Gefter M. L.; Hale S. P.; Hansen N. JV.; Israel D. I.; Jiang J.; Kavarana M. J.; Kelley M. S.; Kollmann C. S.; Li F.; Lind K.; Mataruse S.; Medeiros P. F.; Messer J. A.; Myers P.; O’Keefe H.; Oliff M. C.; Rise C. E.; Satz A. L.; Skinner S. R.; Svendsen J. L.; Tang L.; Vloten K. V.; Wagner R. W.; Yao G.; Zhao B.; Morgan B. A. Design, Synthesis and Selection of DNA-Encoded Small-Molecule Libraries. Nat. Chem. Biol. 2009, 5, 647–654. 10.1038/nchembio.211. [DOI] [PubMed] [Google Scholar]
- Deng H.; O’Keefe H.; Davie C. P.; Lind K. E.; Acharya R. A.; Franklin G. J.; Larkin J.; Lohr T.; Matico R.; Thompson M.; Gross J. W.; Neeb M.; O’Donovan G. K.; Centrella P. A.; Mataruse S.; Bedard K. L.; Vloten K. V.; Skinner S. R.; Belyanskaya S. L.; Cuozzo J. W.; Clark M. A.; Arico-Muendel C. C.; Morgan B. A. Discovery of Highly Potent and Selective Small Molecule ADAMTS-5 Inhibitors that Inhibit Human Cartilage Degradation via Encoded Library Technology (ELT). J. Med. Chem. 2012, 55, 7061–7079. 10.1021/jm300449x. [DOI] [PubMed] [Google Scholar]
- Gentile G.; Merlo G.; Pozzan A.; Bernasconi G.; Bax B.; Bamborough P.; Bridges A.; Carter P.; Neu M.; Yao G.; Brough C.; Cutler G.; Coffin A.; Belyanskaya S. 5-Aryl-4-carboxamide-1,3-oxazoles: Potent and selective GSK-3 inhibitors. Bioorg. Med. Chem. Lett. 2012, 22, 1989–1994. 10.1016/j.bmcl.2012.01.034. [DOI] [PubMed] [Google Scholar]
- Disch J. S.; Evindar G.; Chiu C. H.; Blum C. A.; Dai H.; Jin L.; Schuman E.; Lind K. E.; Belyanskaya S. L.; Deng J.; Coppo F.; Aquilani L.; Graybill T. L.; Cuozzo J. W.; Lavu S.; Mao C.; Vlasuk G. P.; Perni R. B. Discovery of thieno[3,2-d]pyrimidine-6-carboxamides as potent inhibitors of SIRT1, SIRT2, and SIRT3. J. Med. Chem. 2013, 56, 3666–3679. 10.1021/jm400204k. [DOI] [PubMed] [Google Scholar]
- Thalji R. K.; McAtee J. J.; Belyanskaya S.; Brandt M.; Brown G. D.; Costell M. H.; Ding Y.; Dodson J. W.; Eisennagel S. H.; Fries R. E.; Gross J. W.; Harpel M. R.; Holt D. A.; Israel D. I.; Jolivette L. J.; Krosky D.; Li H.; Lu Q.; Mandichak T.; Roethke T.; Schnackenberg C. G.; Schwartz B.; Shewchuk L. M.; Xie W.; Behm D. J.; Douglas S. A.; Shaw A. L.; Marino J. P. Jr. Discovery of 1-(1,3,5-triazin-2-yl)piperidine-4-carboxamides as inhibitors of soluble epoxide hydrolase. Bioorg. Med. Chem. Lett. 2013, 23, 3584–3588. 10.1016/j.bmcl.2013.04.019. [DOI] [PubMed] [Google Scholar]
- Encinas L.; O’Keefe H.; Neu M.; Remuiñán M. J.; Patel A. M.; Guardia A.; Davie C. P.; Pérez-Macías N.; Yang H.; Convery M. A.; Messer J. A.; Pérez-Herrán E.; Centrella P. A.; Álvarez-Gómez D.; Clark M. A.; Huss S.; O’Donovan G. K.; Ortega-Muro F.; McDowell W.; Castañeda P.; Arico-Muendel C. C.; Pajk S.; Rullás J.; Angulo-Barturen I.; Álvarez-Ruíz E.; Mendoza-Losana A.; Pages L. B.; Castro-Pichel J.; Evindar G. Encoded Library Technology as a Source of Hits for the Discovery and Lead Optimization of a Potent and Selective Class of Bactericidal Direct Inhibitors of Mycobacterium tuberculosis InhA. J. Med. Chem. 2014, 57, 1276–1288. 10.1021/jm401326j. [DOI] [PubMed] [Google Scholar]
- Kollmann C. S.; Bai X.; Tsai C.-H.; Yang H.; Lind K. E.; Skinner S. R.; Zhu Z.; Israel D. I.; Cuozzo J. W.; Morgan B. A.; Yuki K.; Xie C.; Springer T. A.; Shimaoka M.; Evindar G. Application of encoded library technology (ELT) to a protein–protein interaction target: Discovery of a potent class of integrin lymphocyte function-associated antigen 1 (LFA-1) antagonists. Bioorg. Med. Chem. 2014, 22, 2353–2365. 10.1016/j.bmc.2014.01.050. [DOI] [PubMed] [Google Scholar]
- Ding Y.; Clark M. A. Robust Suzuki–Miyaura Cross-Coupling on DNA-Linked Substrates. ACS Comb. Sci. 2015, 17, 1–4. 10.1021/co5001037. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.