

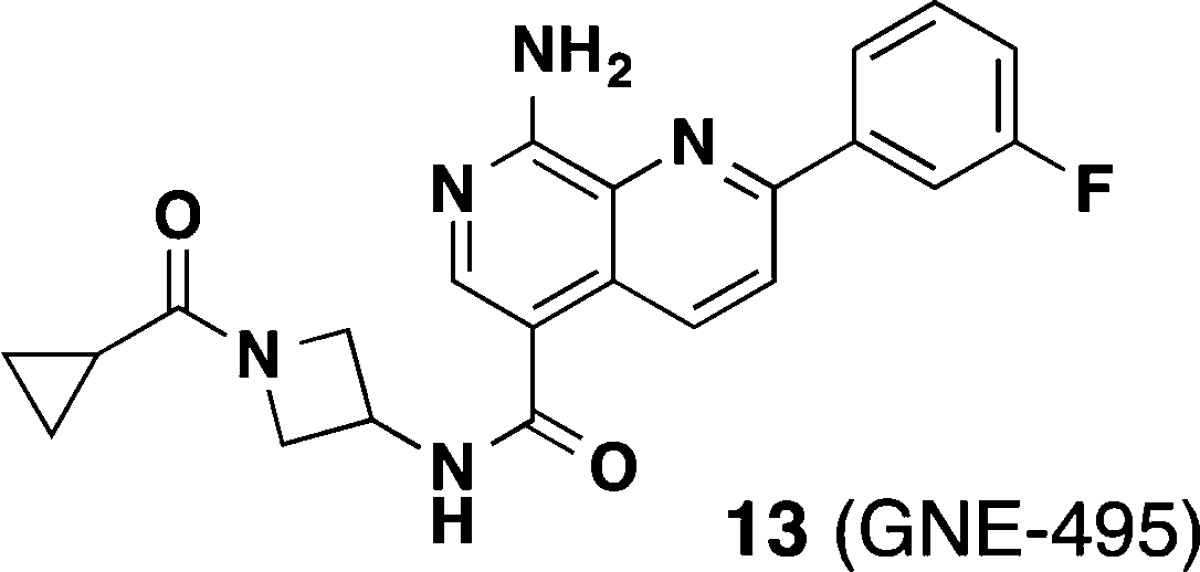
Abstract
Diverse biological roles for mitogen-activated protein kinase kinase kinase kinase 4 (MAP4K4) have necessitated the identification of potent inhibitors in order to study its function in various disease contexts. In particular, compounds that can be used to carry out such studies in vivo would be critical for elucidating the potential for therapeutic intervention. A structure-based design effort coupled with property-guided optimization directed at minimizing the ability of the inhibitors to cross into the CNS led to an advanced compound 13 (GNE-495) that showed excellent potency and good PK and was used to demonstrate in vivo efficacy in a retinal angiogenesis model recapitulating effects that were observed in the inducible Map4k4 knockout mice.
Keywords: Angiogenesis, MAP4K4, naphthyridine, GNE-495, P loop
Mitogen-activated protein kinase kinase kinase kinase 4 (MAP4K4, or HGK), and its homologues Misshapen and MIG-15 have been shown to regulate a multitude of biological processes, including embryonic development,1,2 metabolism,3,4 inflammation,5 neural regeneration,6 angiogenesis,7 and cancer.8,9 Given its diverse roles in these processes, we became interested in the therapeutic potential of MAP4K4 inhibition and began an endeavor to identify an inhibitor that would function in vivo.
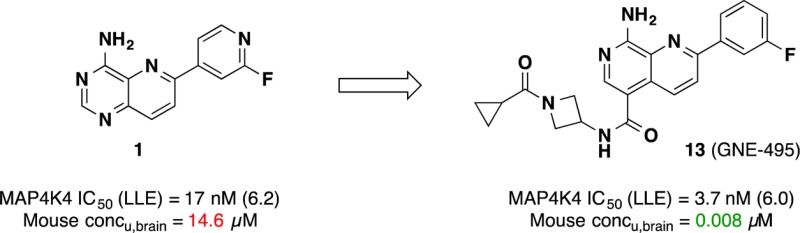
Our group recently reported two types of MAP4K4 inhibitors that were derived from separate fragment-based lead discovery efforts.10,11 Between the two chemical series, we were particularly attracted to the series bearing a pyridopyrimidine scaffold because of the potency and broad kinase selectivity that it conferred.10 An optimized compound 1 showed low nanomolar biochemical activity, excellent kinase selectivity, high exposure in vivo and also demonstrated a pharmacodynamic (PD) response in an HT-1080 human fibrosarcoma xenograft mouse model (Figure 1).
Figure 1.
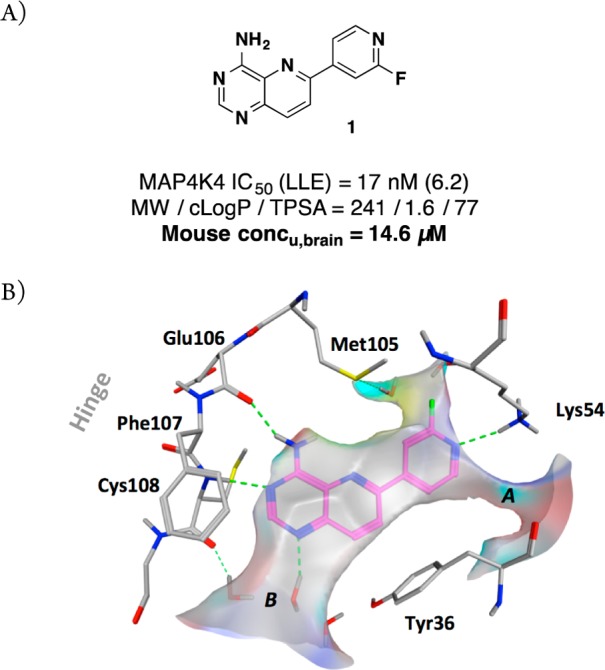
(A) Profile of pyridopyrimidine MAP4K4 inhibitor 1;10 (B) X-ray structure of 1 in MAP4K4 (PDB: 4OBP).
While this compound represented a valuable tool for our early in vivo experiments, a significant shortcoming was that 1 was poorly tolerated when dosed repeatedly in a multiday study. We hypothesized that this issue can be attributed to the ability of 1 to permeate into the CNS as analysis of the mouse in vivo PK showed that the brain-to-plasma ratio was substantial (unbound brain concentration = 14.6 μM, Figure 1). The exact mechanism of toxicity is not well understood at present, but there is the possibility of off-target toxicity as 1 also inhibits the closely related kinases MINK and TNIK to an equal extent. These kinases have been previously shown to impact neuronal integrity.12 Due to the fact that even an extremely selective MAP4K4 inhibitor would still harbor significant inhibitory activity against these two highly homologous kinases, we proceeded with a target profile lacking brain penetration.
We focused further medicinal chemistry efforts to decrease CNS penetration.13−15 It is well accepted that parameters such as MW, logP, TPSA, and number of hydrogen bond donors have a strong impact on the ability of compounds to cross the blood–brain barrier (BBB). Since the properties of 1 fell within the favorable range for brain penetration (MW = 241; logP = 1.6; TPSA = 77; #HBD = 1),16 we decided to redesign the series in order to limit CNS penetration. At the same time, we recognized that the 2-fluoropyridyl back pocket group was a potential safety liability due to its electrophilic nature. We desired to install a more stable group here in order to remove this liability. We previously showed that the 3-fluorophenyl was well tolerated in this region. In the optimization process, our intent was to maintain or improve all of the other favorable attributes including potency, kinase selectivity, and pharmacokinetics.
Our previously reported cocrystal structure of 1 (PDB: 4OBP) served as a guide for further optimization (Figure 1B). Compound 1 binds to a folded P loop conformation of MAP4K4 that has been observed for only a small subset of kinases.17 This conformation was therefore deemed advantageous for maintaining adequate kinase selectivity. In the cocrystal structure of 1 in this constricted ATP pocket, Tyr36 (contained within the P loop) rotates inward to make an edge-to-face interaction with the pyridopyrimidine core, thus producing a form-fitting enclosure for the ligand. Two potential vectors for further expansion from the scaffold were evident. We previously described ligand augmentation into the open channel near the catalytic Lys54 (A) with groups that were designed to favorably interact with protein residues surrounding those cavities.10 However, this pursuit led to losses in ligand efficiency without favorable improvements to other features including stability and permeability.
We next evaluated the area near the hinge residues (B). A cavity was observed between the hinge Cys108, Phe107, and P-loop Tyr36, which contained three crystallographically observed waters toward the opening to bulk solvent.18 WaterMap calculations19,20 predicted relatively high free energy of hydration values (ΔGhyd = 1.77–3.8 kcal/mol) at these locations suggesting the potential to displace them concomitant with displacement of these waters. This strategy included the opportunity to replace a water-mediated hydrogen bond to the hinge residue Cys108 backbone carbonyl. In particular, we recognized that the N1 position of the pyridopyrimidine presented an appropriate vector to extend into the available pocket. Importantly, modeling suggested that expansion off of this vector would not impinge on the induced Tyr36 orientation and therefore would maintain the desired folded P loop conformation.
To quickly test the tractability of this new design direction, we selected targets that utilized a 1-aminoisoquinoline core due to synthetic accessibility. This core allowed for rapid establishment of the SAR before transferring to the more synthetically challenging naphthyridine core. From previous studies, we knew that going from the quinazoline to pyridopyrimidine (by insertion of a “5-aza” nitrogen) typically resulted in a 5- to 10-fold improvement in potency.10 If this new vector was productive, we believed that the corresponding naphthyridine scaffold would show a similar potency increase. In that case, a campaign to identify a suitable route to the corresponding 1,7-naphthyridine would then be warranted.
An outline of the synthetic route for the isoquinolines is shown in Scheme 1. The starting 7-bromo-1-hydroxyisoquinoline (15) was readily converted to the 1-amino-4-bromoisoquinoline (17) intermediate. Further derivatization provided a carboxylic acid intermediate that was then parlayed into a variety of isoquinoline-4-amido analogues (3–9).
Scheme 1. Synthesis of Isoquinoline MAP4K4 Inhibitors (3–9).

Reagents and conditions: (a) POCl3, 100 °C, 3 h, 80%; (b) aq. NH3, NMP, 148 °C, 89%; (c) 3-fluorophenylboronic acid, Pd(dppf)Cl2, Na2CO3, 1,4-dioxane/H2O, 99%; (d) NBS, DMF, 0 °C, 49%; (e) CO (50 psi), Pd(dppf)Cl2, DMAP, MeOH, 70 °C; NaOH, H2O, MeOH, THF, 44%, 2 steps; (f) RNH2, HATU, DIPEA, DMF or THF; (g) (i) HCl, EtOAc/DCM; (ii) for 8, CH3CO2H, HATU, DIPEA, THF; (iii) for 9, CDI, DIPEA, CH3NH2.
As seen in Table 1, we were highly encouraged that even the simple 1° amide extension resulted in isoquinoline 3 that had improved potency (IC50 = 18 nM) relative to the comparative quinazoline compound 2 (IC50 = 58 nM). This result is consistent with earlier WaterMap predictions suggesting that potency gains could be achieved by substitutions in that area. However, compound 3 showed equally high metabolic clearance in rat liver microsomes despite being less lipophilic. We explored additional substituents that reach deeper into this pocket and span a wide range of MW, cLogP, and TPSA with the goal of limiting penetration into the CNS. We evaluated brain penetration using the rat IV cassette brain-to-plasma assay.21 Gratifyingly, we observed that further substitution in this pocket with a cyanomethyl amide (4) led to decreased total B/P ratio while retaining good potency. Introducing conformationally constrained substituents in the pocket including cyclopropyl (5), 3-hydroxycylobutyl (6), and several azetidines (7–9) led to improved potency and stability (except for the unsubstituted azetidine 7, which reduced potency). We also saw markedly reduced brain penetration for 6 and 7 presumably due to the presence of an additional H-bond donor. From this survey, we discovered that we could indeed expand the molecular size of our inhibitors, reduce the lipophilicity, and increase TPSA without sacrificing the potency or otherwise favorable properties of our compounds. We also saw improved metabolic stability that showed that the 2-fluoropyridine right-hand side substituent in compound 1 was not required for stability. These promising findings led us to explore further increasing potency by reinsertion of the “5-aza” moiety in the form of the 1,7-naphthyridine scaffold.
Table 1. Structure–Activity Relationships of Isoquinoline MAP4K4 Inhibitors (2–9).
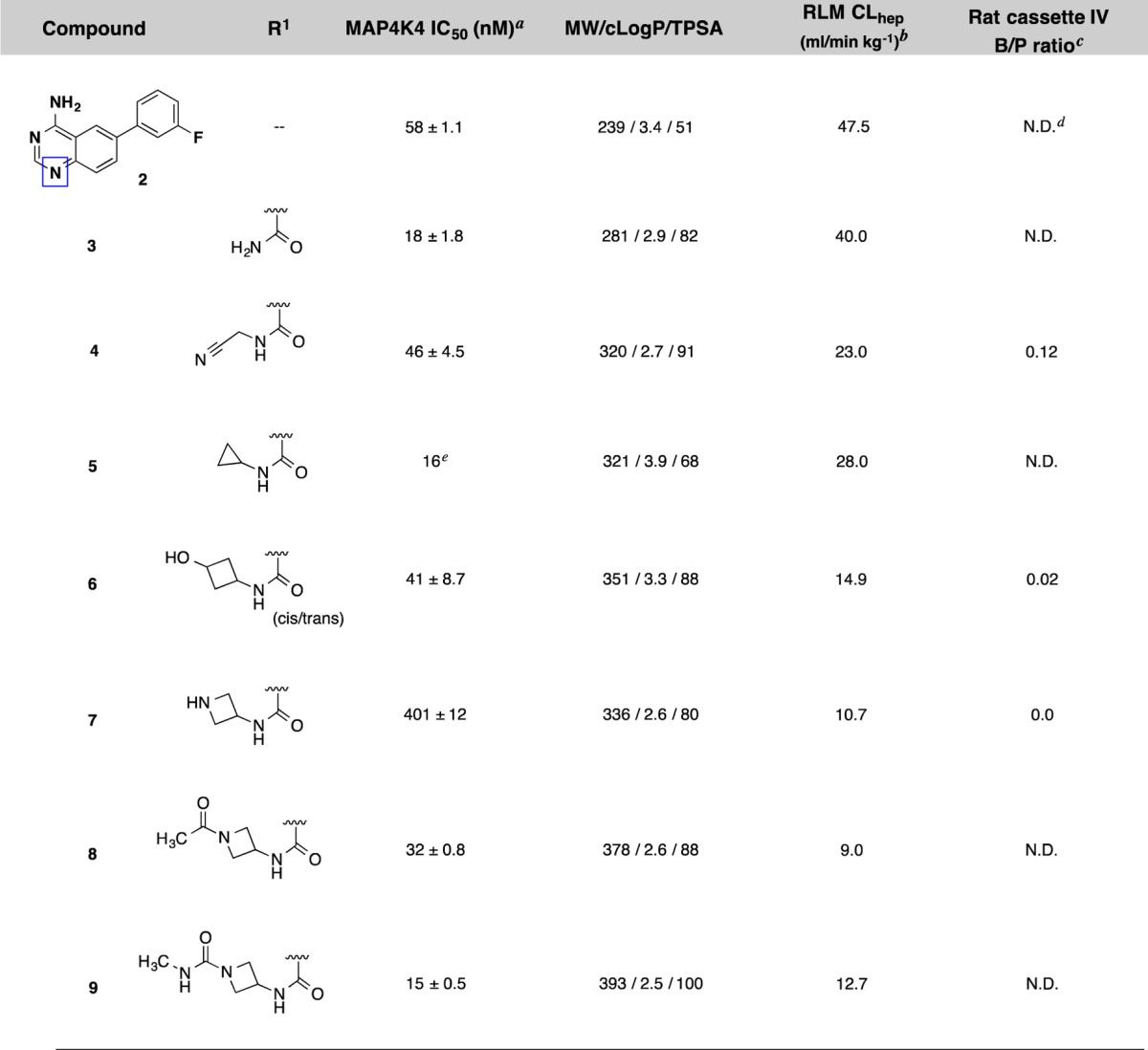
IC50 data are an average of at least 3 independent experiments (Z’LYTE, see Supporting Information). Values are given as the mean ± standard deviation.
Compounds were incubated with rat liver microsomes for 1 h, and percent remaining was determined by LC–MS/MS analysis.
Compounds were formulated in 10%DMSO/60%PEG400/30%SQ-H2O.
Not determined.
IC50 was determined using Caliper LabChip 3000 (LC3K) technology (see Supporting Information). Only one data point was obtained.
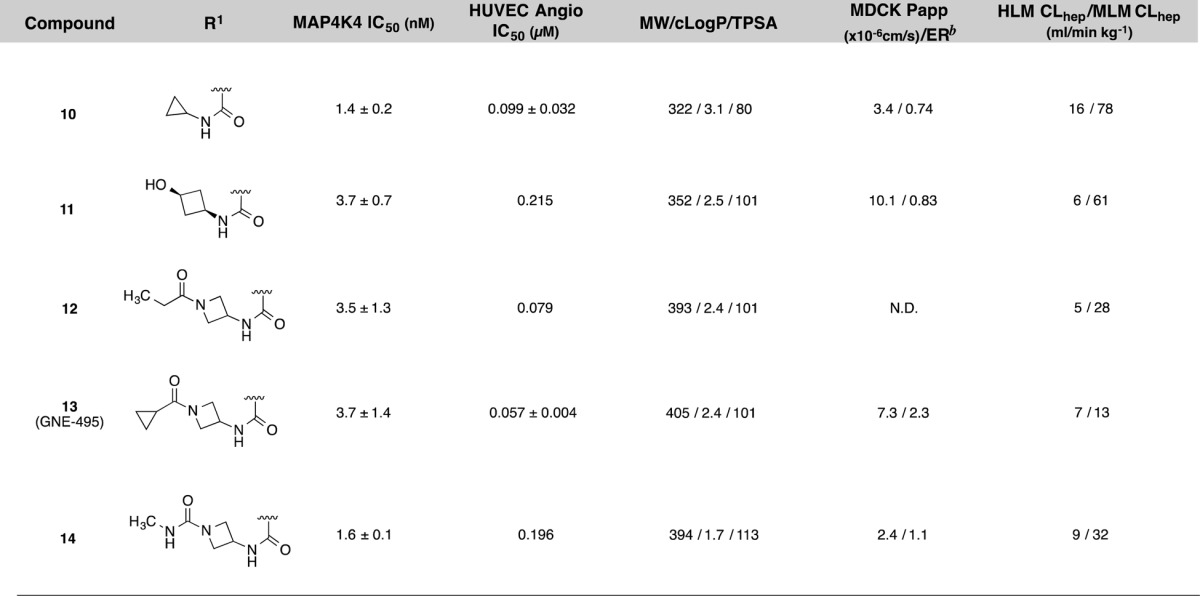
As a result of the successful SAR exploration with the isoquinoline scaffold, we were eventually able to invent a robust chemical route to 1,7-naphthyridines.22 This route enabled the preparation of compounds contained in Table 2. We readily applied lessons from the SAR survey conducted with the isoquinoline compounds described in Table 1, and the resulting compounds (10–14) showed even greater increases in biochemical potency than the expected 5–10-fold shifts that had been observed in earlier series (Table 2). These optimized compounds were within desirable molecular property space with TPSA values that are expected to prevent entry into the CNS but still had sufficient MDCK permeability (Papp) to allow for distribution into peripheral tissues. Consistent with the good permeability properties, these compounds exhibited adequate potency preventing migration of HUVEC cells in vitro, an assay that is used to evaluate antiangiogenic behavior.23 Notably, compound 13 showed the best balance of MAP4K4 inhibition, permeability, microsomal stability, and cellular potency.
Table 2. Structure–Activity Relationships of Naphthyridine MAP4K4 Inhibitors (10–14)a.
The X-ray cocrystal structure of compound 13 was solved and maintained all of the expected interactions that were present in the original pyridopyrimidine scaffold including multiple hinge binding interactions and the folded P-loop conformation (Figure 2). Additionally, the amide linker extending from the naphthyridine scaffold effectively replaces two of the water molecules and engages in a hydrogen-bonding interaction to the hinge Cys108 carbonyl. Overall, it was evident that the binding pocket was completely occupied by the optimized ligand.
Figure 2.
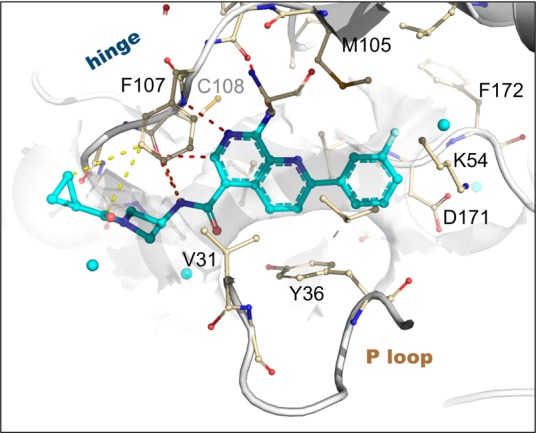
X-ray structure of 13 bound to MAP4K4 at 2.89 Å (PDB: 4ZK5). Select hydrogen bond (red), pi stacking (orange), and van der Waals (yellow) interactions are highlighted. Proximal ordered waters are shown in cyan spheres. The solvent-accessible protein surface in the vicinity of the ligand is shown in white, demonstrating the extensive contribution to the constricted enclosure created by the approach of Tyr36 enabled by a buckled P loop conformation. The ligand emerges toward solvent at the left, where the azetidine moiety creates a bend around the Phe107 side chain to maintain close contacts to the protein surface.
Given the favorable overall profile of 13 we proceeded to evaluate its in vivo PK behavior in three preclinical species: mouse, rat, and dog (Table 3). The compound was administered by intravenous (IV) and oral (PO) routes at 1 and 5 mg/kg, respectively, in each of the species (total of three animals per group). Compound 13 showed good in vivo profile in all species tested, with low clearances, moderate terminal half-lives, and reasonable oral exposure levels (F = 37–47%). Importantly, very minimal brain exposure was observed in mice (measured unbound brain concentration at 1 h time point = 0.008 μM),24 consistent with our design hypothesis around mitigating adverse effects. Compound 13 was also administered intraperitoneally to neonatal mouse pups at high doses: 25 and 50 mg/kg (Supporting Information, Figure S2). In these experiments, we also observed sustained exposures over the 24 h postdose. In order to determine if compound 13 (referred to as GNE-495 herein) can inhibit MAP4K4 function in vivo, we utilized the neonatal retinal vascular development model because inducible knockout of Map4k4 inhibited retinal vascular outgrowth and altered retinal vascular morphology.7 We found that IP injection of GNE-495 into newborn mice dose-dependently delayed retinal vascular outgrowth (Figures 3A,B and S3) and induced abnormal retinal vascular morphology (Figure 3C,D). These phenotypes recapitulated the retinal vascular defects observed in the inducible Map4k4 knockout mice,7 indicating that GNE-495 is indeed active in vivo. It is important to note that though GNE-495 also inhibits the related kinases MINK and TNIK, the observed in vivo effects were attributable solely to MAP4K4 inhibition as has been shown previously.7
Table 3. Cross-Species Pharmacokinetics of 13 (GNE-495) Following Dosing via IV and PO Routesa.
| IV bolus
(1 mg/kg); solutionb |
PO
(5 mg/kg); suspensionc |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| species | %PPB | CL (mL/min kg–1) | T1/2 (h) | Cmax (μM) | V (L/kg) | AUCinf (h·μM) | Cmax (μM) | Tmax (h) | F% | Cu,brain (μM) |
| mouse | 94.4 | 19 | 1.5 | 2.7 | 1.6 | 5.3 | 1.4 | 1.0 | 47 | 0.008 |
| rat | 97.8 | 7.5 | 3.4 | 4.0 | 1.2 | 12.0 | 1.5 | 4.0 | 40 | N.D. |
| dog | N.D. | 8.9 | 1.8 | 3.9 | 1.1 | 21.0 | 1.2 | 1.7 | 37 | N.D. |
Mean values are shown. Further details provided in the Supporting Information.
Vehicle: 10% EtOH, 30% PEG400, 60% 50 mM citrate, pH 3.0.
Vehicle: 0.5% (w/v) methylcellulose/0.2% (w/v) Tween 80 in sterile water (MCT).
Figure 3.

(A) Representative images of Isolectin-B4 (vascular marker) stained flat-mounted retinas at postnatal day 6 (P6) from mice treated with vehicle or 100 mg/kg GNE-495 daily from P1–P5. Areas in the retina without blood vessels (Avascular) are marked with dashed lines. Scale bar represents 500 μm. (B) Quantification of avascular area normalized to total retina from similar images shown in A. Each dot represents one retina. P value was calculated using Mann–Whitney unpaired test. (C) Representative images of isolectin-B4 stained P7 retinas at the vascular edges from mice treated with vehicle or 100 mg/kg GNE-495 daily from P1–P6. Arrows indicate long membrane protrusions in vascular endothelial cells. Scale bar represents 50 μm. (D) Numbers of long membrane protrusions (longer than 40 μm) along the vascular front per centimeter of vascular perimeter. Each dot represents one retina. P value was calculated using Mann–Whitney unpaired test. Nonspecific fluorescence dusts in the images shown in A and C were manually eliminated.
We previously reported the discovery of a potent and highly selective MAP4K4 tool compound 1. However, due to the possibility that high brain penetration precluded our ability to achieve long-term administration of the compound, we successfully optimized the molecular properties to limit the level of brain exposure in subsequent molecules. We were able to identify a new class of isoquinoline and naphthyridine-based MAP4K4 inhibitors that reduced brain exposures but maintained potent activity and good kinase selectivity. GNE-495 demonstrates high exposure in peripheral tissues but minimal brain penetration validating our design strategy. The fact that IP injection of GNE-495 recapitulated the phenotypes in Map4k4 inducible knockout mice confirms the notion that GNE-495 is active in vivo. This compound provides an opportunity to investigate the multitude of MAP4K4 functions in animal models and ultimately in patient diseases.
Acknowledgments
We thank Mengling Wong, Chris Hamman, Michael Hayes, and Amber Guillen for compound purification. We also thank Baiwei Lin, Deven Wang, and Yutao Jian for analytical support.
Glossary
ABBREVIATIONS
- MAP4K4
mitogen-activated protein kinase kinase kinase kinase 4
- CNS
central nervous system
- TPSA
topological polar surface area
- HUVEC
human umbilical vein endothelial cells
Supporting Information Available
Experimental procedures, kinase selectivity, details of in vitro and in vivo assays, and characterization of compounds. The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.5b00174.
Author Present Address
§ WIL Research, Ashland, Ohio 44805, United States.
Author Present Address
∥ Gilead Sciences, Foster City, California 94404, United States
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Diffraction data were collected at beamline 08ID-1 at the Canadian Light Source, which was supported by the NSERC, the NRC, the Canadian Institutes of Health Research, the Province of Saskatchewan, Western Economic Diversification Canada, and the University of Saskatchewan at beamline 5.0.2 of the Advanced Light Source. The Berkeley Center for Structural Biology was supported in part by the NIH, the NIGMS, and the Howard Hughes Medical Institute. The Advanced Light Source was supported by the Director, Office of Basic Energy Sciences, of the U.S. Department of Energy under Contract No. DE-AC02–05CH11231.
The authors declare no competing financial interest.
Supplementary Material
References
- Köppen M.; Fernández B. G.; Carvalho L.; Jacinto A.; Heisenberg C.-P. Coordinated Cell-Shape Changes Control Epithelial Movement in Zebrafish and Drosophila. Development 2006, 133, 2671–2681. 10.1242/dev.02439. [DOI] [PubMed] [Google Scholar]
- Poinat P.; De Arcangelis A.; Sookhareea S.; Zhu X.; Hedgecock E. M.; Labouesse M.; Georges-Labouesse E. A Conserved Interaction Between Beta1 Integrin/PAT-3 and Nck-Interacting Kinase/MIG-15 That Mediates Commissural Axon Navigation in C. Elegans. Curr. Biol. 2002, 12, 622–631. 10.1016/S0960-9822(02)00764-9. [DOI] [PubMed] [Google Scholar]
- Bouzakri K.; Ribaux P.; Halban P. A. Silencing Mitogen-Activated Protein 4 Kinase 4 (MAP4K4) Protects Beta Cells From Tumor Necrosis Factor-Alpha-Induced Decrease of IRS-2 and Inhibition of Glucose-Stimulated Insulin Secretion. J. Biol. Chem. 2009, 284, 27892–27898. 10.1074/jbc.M109.048058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang X.; Guilherme A.; Chakladar A.; Powelka A. M.; Konda S.; Virbasius J. V.; Nicoloro S. M. C.; Straubhaar J.; Czech M. P. An RNA Interference-Based Screen Identifies MAP4K4/NIK as a Negative Regulator of PPAR, Adipogenesis, and Insulin-Responsive Hexose Transport. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 2087–2092. 10.1073/pnas.0507660103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aouadi M.; Tesz G. J.; Nicoloro S. M.; Wang M.; Chouinard M.; Soto E.; Ostroff G. R.; Czech M. P. Orally Delivered siRNA Targeting Macrophage Map4k4 Suppresses Systemic Inflammation. Nature 2009, 458, 1180–1184. 10.1038/nature07774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loh S. H. Y.; Francescut L.; Lingor P.; Bähr M.; Nicotera P. Identification of New Kinase Clusters Required for Neurite Outgrowth and Retraction by a Loss-of-Function RNA Interference Screen. Cell Death Differ. 2008, 15, 283–298. 10.1038/sj.cdd.4402258. [DOI] [PubMed] [Google Scholar]
- Vitorino P.; Yeung S.; Crow A.; Bakke J.; Smyczek T.; West K.; McNamara E.; Eastham-Anderson J.; Gould S.; Harris S. F.; Ndubaku C.; Ye W. MAP4K4 Regulates Integrin-FERM Binding to Control Endothelial Cell Motility. Nature 2015, 519, 425–430. 10.1038/nature14323. [DOI] [PubMed] [Google Scholar]
- Liang J. J.; Wang H.; Rashid A.; Tan T.-H.; Hwang R. F.; Hamilton S. R.; Abbruzzese J. L.; Evans D. B.; Wang H. Expression of MAP4K4 Is Associated with Worse Prognosis in Patients with Stage II Pancreatic Ductal Adenocarcinoma. Clin. Cancer Res. 2008, 14, 7043–7049. 10.1158/1078-0432.CCR-08-0381. [DOI] [PubMed] [Google Scholar]
- Wright J. H.; Wang X.; Manning G.; LaMere B. J.; Le P.; Zhu S.; Khatry D.; Flanagan P. M.; Buckley S. D.; Whyte D. B.; Howlett A. R.; Bischoff J. R.; Lipson K. E.; Jallal B. The STE20 Kinase HGK Is Broadly Expressed in Human Tumor Cells and Can Modulate Cellular Transformation, Invasion, and Adhesion. Mol. Cell. Biol. 2003, 23, 2068–2082. 10.1128/MCB.23.6.2068-2082.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawford T. D.; Ndubaku C. O.; Chen H.; Boggs J. W.; Bravo B. J.; Delatorre K.; Giannetti A. M.; Gould S. E.; Harris S. F.; Magnuson S. R.; McNamara E.; Murray L. J.; Nonomiya J.; Sambrone A.; Schmidt S.; Smyczek T.; Stanley M.; Vitorino P.; Wang L.; West K.; Wu P.; Ye W. Discovery of Selective 4-Amino-Pyridopyrimidine Inhibitors of MAP4K4 Using Fragment-Based Lead Identification and Optimization. J. Med. Chem. 2014, 57, 3484–3493. 10.1021/jm500155b. [DOI] [PubMed] [Google Scholar]
- Wang L.; Stanley M.; Boggs J. W.; Crawford T. D.; Bravo B. J.; Giannetti A. M.; Harris S. F.; Magnuson S. R.; Nonomiya J.; Schmidt S.; Wu P.; Ye W.; Gould S. E.; Murray L. J.; Ndubaku C. O.; Chen H. Fragment-Based Identification and Optimization of a Class of Potent Pyrrolo[2,1-F][1,2,4]Triazine MAP4K4 Inhibitors. Bioorg. Med. Chem. Lett. 2014, 24, 4546–4552. 10.1016/j.bmcl.2014.07.071. [DOI] [PubMed] [Google Scholar]
- Hussain N. K.; Hsin H.; Huganir R. L.; Sheng M. MINK and TNIK Differentially Act on Rap2-Mediated Signal Transduction to Regulate Neuronal Structure and AMPA Receptor Function. J. Neurosci. 2010, 30, 14786–14794. 10.1523/JNEUROSCI.4124-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wager T. T.; Villalobos A.; Verhoest P. R.; Hou X.; Shaffer C. L. Strategies to Optimize the Brain Availability of Central Nervous System Drug Candidates. Expert Opin. Drug Discovery 2011, 6, 371–381. 10.1517/17460441.2011.564158. [DOI] [PubMed] [Google Scholar]
- Young R. C.; Mitchell R. C.; Brown T. H.; Ganellin C. R.; Griffiths R.; Jones M.; Rana K. K.; Saunders D.; Smith I. R.; Sore N. E. Development of a New Physicochemical Model for Brain Penetration and Its Application to the Design of Centrally Acting H2 Receptor Histamine Antagonists. J. Med. Chem. 1988, 31, 656–671. 10.1021/jm00398a028. [DOI] [PubMed] [Google Scholar]
- Doan K. M. M.; Humphreys J. E.; Webster L. O.; Wring S. A.; Shampine L. J.; Serabjit-Singh C. J.; Adkison K. K.; Polli J. W. Passive Permeability and P-Glycoprotein-Mediated Efflux Differentiate Central Nervous System (CNS) and Non-CNS Marketed Drugs. J. Pharmacol. Exp. Ther. 2002, 303, 1029–1037. 10.1124/jpet.102.039255. [DOI] [PubMed] [Google Scholar]
- Due to intramolecular H-bond only one HBD is available (observable by 1H NMR).
- Guimarães C. R. W.; Rai B. K.; Munchhof M. J.; Liu S.; Wang J.; Bhattacharya S. K.; Buckbinder L. Understanding the Impact of the P-Loop Conformation on Kinase Selectivity. J. Chem. Inf. Model. 2011, 51, 1199–1204. 10.1021/ci200153c. [DOI] [PubMed] [Google Scholar]
- These waters are not always present in various crystal structure complexes obtained suggesting that extensions off the quinazoline scaffold with groups with the ability to make electrostatic interactions within this region could lead to enhanced potency.
- Abel R.; Young T.; Farid R.; Berne B. J.; Friesner R. A. Role of the Active-Site Solvent in the Thermodynamics of Factor Xa Ligand Binding. J. Am. Chem. Soc. 2008, 130, 2817–2831. 10.1021/ja0771033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young T.; Abel R.; Kim B.; Berne B. J.; Friesner R. A. Motifs for Molecular Recognition Exploiting Hydrophobic Enclosure in Protein–Ligand Binding. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 808–813. 10.1073/pnas.0610202104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X.; Ding X.; Deshmukh G.; Liederer B. M.; Hop C. E. C. A. Use of the Cassette-Dosing Approach to Assess Brain Penetration in Drug Discovery. Drug Metab. Dispos. 2012, 40, 963–969. 10.1124/dmd.111.044420. [DOI] [PubMed] [Google Scholar]
- Chen J.; Xu Z.; Wang T.; Lyssikatos J.; Ndubaku C. A Versatile Annulation Route to Primary-Amino-Substituted Naphthyridine Esters. Synlett 2014, 25, 89–92. 10.1055/s-0033-1340111. [DOI] [Google Scholar]
- Wolfe A.; O’Clair B.; Groppi V. E.; McEwen D. P. Pharmacologic Characterization of a Kinetic in Vitro Human Co-Culture Angiogenesis Model Using Clinically Relevant Compounds. J. Biomol. Screening 2013, 18, 1234–1245. 10.1177/1087057113502085. [DOI] [PubMed] [Google Scholar]
- Rankovic Z. CNS Drug Design: Balancing Physicochemical Properties for Optimal Brain Exposure. J. Med. Chem. 2015, 58, 2584–2608. 10.1021/jm501535r. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.