

Abstract
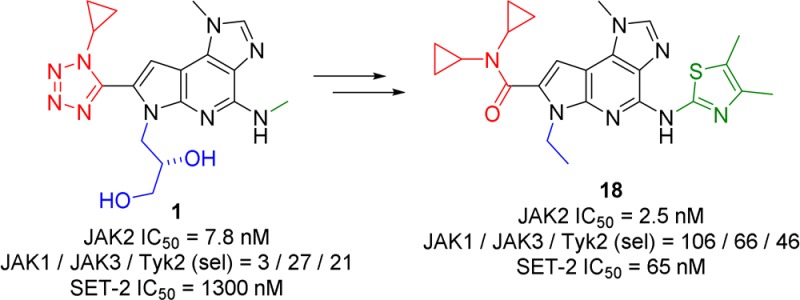
Early hit to lead work on a pyrrolopyridine chemotype provided access to compounds with biochemical and cellular potency against Janus kinase 2 (JAK2). Structure-based drug design along the extended hinge region of JAK2 led to the identification of an important H-bond interaction with the side chain of Tyr 931, which improved JAK family selectivity. The 4,5-dimethyl thiazole analogue 18 demonstrated high levels of JAK family selectivity and was identified as a promising lead for the program.
Keywords: Kinase, Janus kinase 2 (JAK2), thiazole, myeloproliferative neoplasms, structure-based drug design (SBDD)
Signal transduction cascades mediated by kinases have long been viewed as promising targets for therapeutic intervention. Janus kinase 2 (JAK2) is a nonreceptor cytoplasmic tyrosine kinase involved in signal transducer and activator of transcription (STAT) signaling. Imbalances in the JAK-STAT pathway have recently been associated in a variety of diseases, including inflammatory disease, various cancers, and myeloproliferative neoplasms (MPNs).1 MPNs are a class of myeloid malignancies that are represented by the uncontrolled growth of a multipotent hematopoietic progenitor stem cell. Identification of a mutation in the JH2 pseudokinase domain (JAK2 V617F) in MPN patients was found to render the JAK2 kinase constitutively active.2−5 This mutation was observed in high frequency in patients with MPNs (>95% in polycythemia vera, >50% in essential thrombocytopenia, >50% in primary myelofibrosis).
Success with kinase targeted therapies supported the feasibility of targeting JAK2 for therapeutic intervention in MPNs. Recent approval of the JAK1/JAK2 dual inhibitor ruxolitinib6 for the treatment of primary myelofibrosis reinforced the tractability of targeting the JAK family specifically. A key challenge associated with design of a selective JAK2 inhibitor is the high degree of homology in the kinase domains of the JAK family.
Herein we report on initial hit to lead (HTL) efforts leading to potent JAK2 inhibitors. Further structure-based drug design (SBDD) efforts to explore and capitalize on several single amino acid differences in the extended hinge region of the JAK kinases led to the identification of 4,5-disubstituted thiazoles (e.g., 18) as highly selective JAK2 inhibitors.
Screening of the Bristol-Myers Squibb compound collection using a homogeneous time-resolved fluorescence (HTRF) assay identified pyrrolopyridine 1 as a hit with good JAK2 potency but modest kinome selectivity (Figure 1). Replacement of the tetrazole with a secondary amide (2) resulted in significantly reduced potency, likely due to loss of the enforced s-cis amide conformation. Tertiary amides that were unable to position a cyclopropyl group in the optimal vector (4, 5), lost potency and JAK3 selectivity as compared to 1. Dicyclopropyl amide 3 regained the potency and selectivity of the initial hit, reinforcing the importance of the vector for the cyclopropyl group. Compound 3 demonstrated modest antiproliferative activity in a JAK2 pathway-dependent cell line (SET-2).
Figure 1.

Hit to lead SAR.
X-ray crystallographic analysis of compound 3(7) bound to the JAK2 kinase domain revealed that the tertiary amide is tucked under the P-loop. The dicyclopropyl amide is well suited to traverse the space between the floor of the binding pocket and the P-loop, likely contributing to enhanced selectivity among the JAK family. The diol side-chain on the azaindole nitrogen extends into the ribose pocket. Hydrogen bond contacts with the hinge occur from the imidazole nitrogen and 4-position −NHMe, thereby projecting the methyl group toward the extended hinge region of JAK2 (Figure 2).
Figure 2.
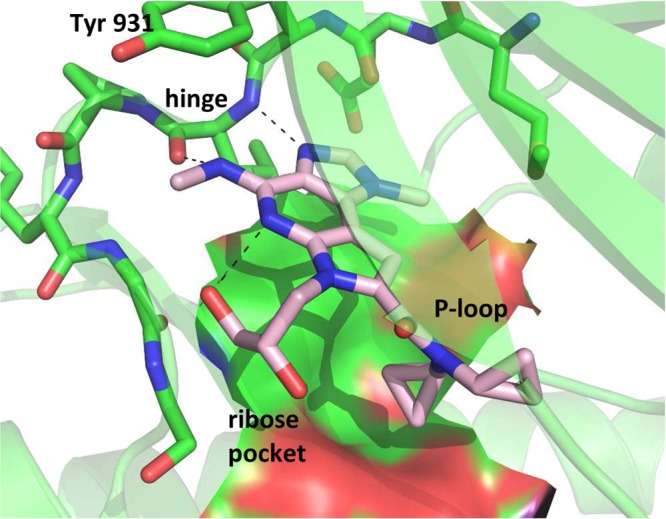
Crystal structure of compound 3 bound to the kinase catalytic domain of JAK2. The carbons of 3 are colored in pink and the carbons for JAK2 are colored in green. Oxygens are colored red, nitrogens blue, and sulfurs yellow. Hydrogen bonds are indicated with dashed lines.
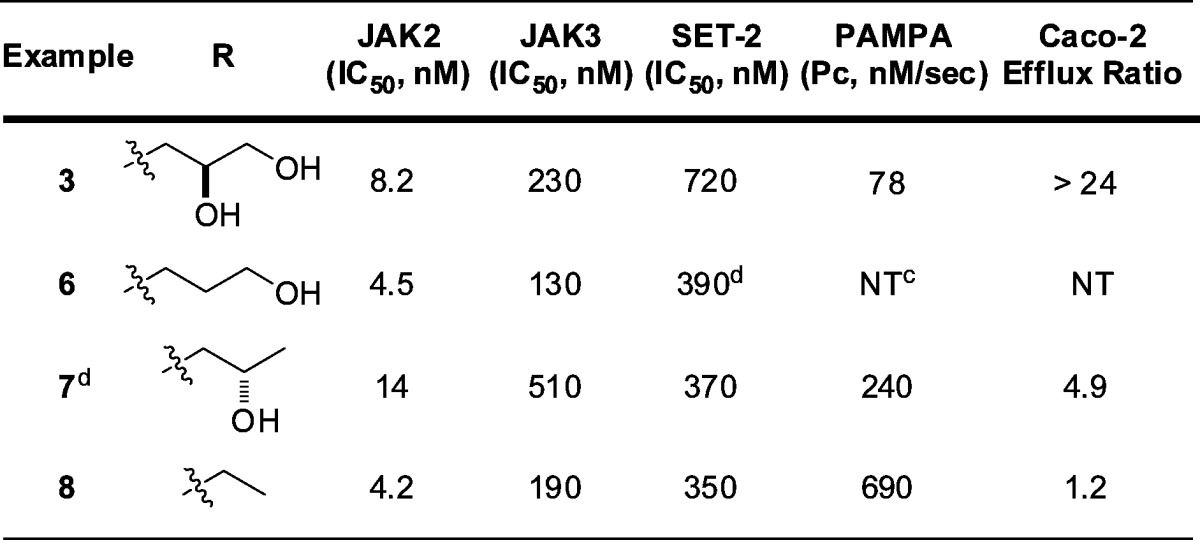
Compound 3 demonstrated poor intrinsic permeability (PAMPA) and high efflux (Caco-2). Sequential removal of the hydroxyl groups maintained or improved JAK2 activity, selectivity, and cellular activity (6, 7) (Table 1). The ethyl analogue (8) was optimal with respect to permeability and efflux and showed improved JAK3 selectivity (44×) and modest selectivity against the other JAK family members JAK1 and Tyk2 (15× and 16×, respectively).
Table 1. Azaindole SARa,b.
Assay protocol details are provided in the Supporting Information.
Assay results are the average of at least two replicates.
Not tested in this assay.
Assay results are N of 1.
Scheme 1 outlines the synthesis of compounds similar to 8 starting from amino imidazo[4,5-c]pyridine 9.8 The tricyclic core 10 was assembled in a two-step protocol wherein a Heck coupling-cyclization was preceded by iodination of 2-aminopyridine 9. Alkylation of the azaindole was followed by ester hydrolysis and amide coupling with the appropriate amine to yield the final compounds.
Scheme 1. Synthesis of Early Lead Compounds.

Reagents and conditions: (a) NIS, acetonitrile, RT, 75%; (b) ethyl pyruvate, Pd2(dba)3, Cy2NMe, DMA, 60 °C, 70%; (c) R-OTs or R-I, Cs2CO3, DMF, 60 °C, 65–75%; (d) NaOH, EtOH, 60 °C, 80–90%; (e) R1R2NH, EDCI, HOBt, i-Pr2NEt, MeCN, 50 °C, 70–75%; (f) TFA-CH2Cl2 (10:1), RT, 90%.
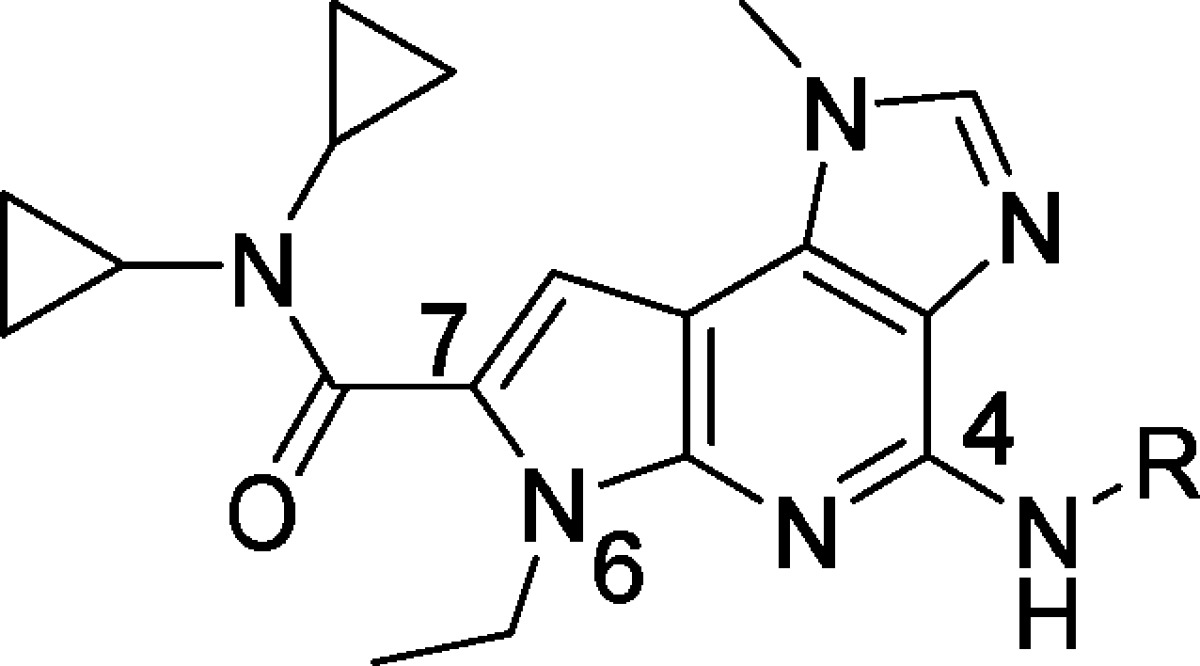
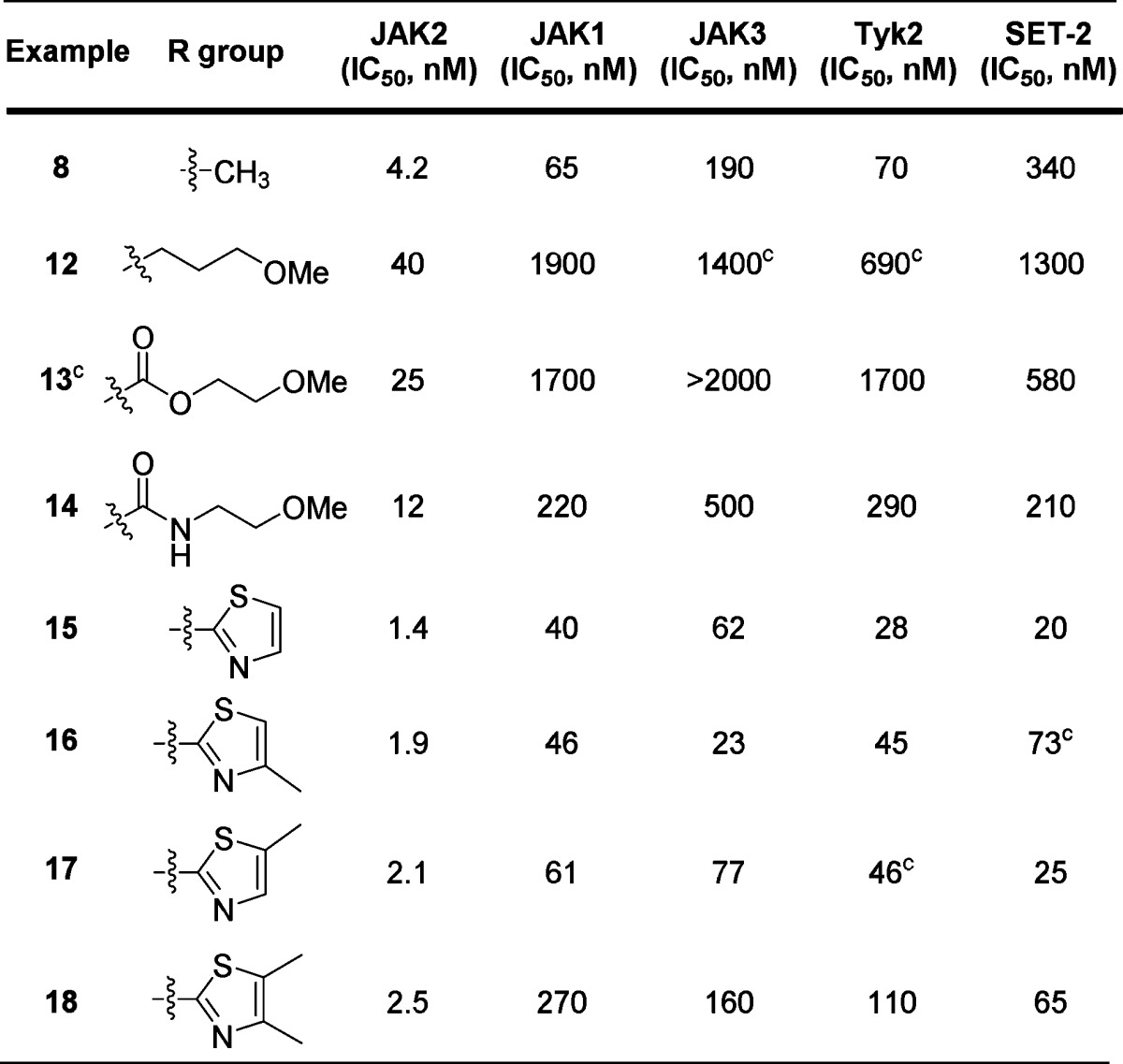
With a goal of improving the broader JAK family selectivity of 8, we began our SAR exploration around the C4 position while holding positions N6 and C7 as ethyl and dicyclopropylamide, respectively. Inspection of the X-ray cocrystal structure of 3 with the JAK2 kinase domain indicated that a hydrogen bond acceptor with an appropriate chain length appended at the C4 position may interact favorably with Tyr 931 in the hinge region. To probe this potential interaction, alkyl and acyl linkers were attached at the C4 position of the pyrrolopyridine core (Table 2).
Table 2. C4 SARa,b.
Assay protocol details are provided in the Supporting Information.
Assay results are the average of at least two replicates.
Assay results are N of 1.
Consistent with our prediction, the crystal structure of 12 bound to the JAK2 kinase domain showed a hydrogen bond interaction of the methoxypropyl group with the phenolic hydrogen of Tyr 931 (see Supporting Information). Molecular modeling suggested that a trajectory from a sp2 center would more effectively traverse the opening between the P-loop and extended hinge region toward solvent. Appending the 2-methoxyethyl side chain to the scaffold through a sp2 hybridized linker (13, 14) maintained planarity with the azaindole core and further improved potency against JAK2 while preserving JAK family selectivity. Despite the improved selectivity, these compounds (12, 13, 14) were less potent than the methyl analogue (8). An amide isostere, thiazole (15), improved potency and cellular activity, while retaining comparable levels of selectivity to the methoxyalkyl analogues.
In addition to Tyr 931, several amino acid differences among JAK family members along the extended hinge region drove us to postulate that substitution at the 4- or 5-position on the thiazole may further enhance JAK family selectivity. Directing hydrophobic groups toward Gln 853 may induce a repulsive interaction associated with JAK1 (Arg 868), JAK3 (Ser 826), and Tyk2 (Arg 901). Other nonconserved amino acids within the JAK family, namely, JAK2/Met 865 (near Gln 853), and along the extended hinge region (JAK2/Ser 936 and JAK2/Asp 939) may play a subtle role in influencing selectivity.
Selectivity of thiazole 15 was marginally enhanced through incorporation of methyl groups at the 4- or 5-position of the thiazole (16, 17) (Table 2). Incorporation of a methyl group at both the 4- and 5- positions of the thiazole (18) was found to significantly enhance JAK family selectivity while maintaining cellular potency in the JAK2-dependent cell line. Compound 18 displayed superior JAK family selectivity as compared to ruxolitinib9 and other JAK inhibitors in the clinic.10
The C4 analogues were prepared from the fully protected aminopyridine1119 (Scheme 2). Access to scaffold 19 was available via a similar synthetic sequence to that described for the C4-NHMe analogues. Deprotection provided the free amine 20, which could undergo reductive amination (12), chloroformate or isocyanate coupling (13, 14), or Buchwald–Hartwig coupling (15).
Scheme 2. Synthesis of C4 Analogues.

Reagents and conditions: (a) TFA, CH2Cl2 (10:1), RT, 95%; (b) 3-methoxypropanal, NaCNBH3, MeOH 32%; (c) 2-methoxyethyl chloroformate, pyridine, CH2Cl2, 20%; (d) 2-methoxyethanamine, DMF, phosgene, pyridine, CH2Cl2, 16%; (e) ethyl isocyanate, pyridine, 23%; (f) 2-bromothiazole, Pd2(dba)3, BINAP, NaOtBu, toluene, 110 °C, 11%; (g) benzoylisothiocyanate, acetone; then 1 N NaOH, ethanol, 60 °C, 72%; (h) α-bromoketone, ethanol, 65 °C.
The substituted thiazoles were also accessed from aminopyridine 20. Alkyl thiazoles (e.g., 16, 17, 18) were formed by reaction of thiourea 21 with an appropriately substituted α-bromoketone.
The crystal structure of 18(12) bound to the JAK2 kinase domain highlighted some of the extended hinge amino acid differences in the JAK family members, which may account for the high selectivity found for compound 18 (Figure 3). The thiazole ring nitrogen was observed to engage Tyr 931 in a hydrogen bonding interaction similar to that observed with methoxypropyl analogue 12. This interaction is not feasible in JAK1 (Phe). The thiazole sulfur is positioned to have a positive nonbonding interaction with the core pyridyl nitrogen, likely reinforcing the desired conformation of the inhibitor in the enzyme.13 Consistent with our prediction, the hydrophobic methyl groups of 18 are directed toward Gln 853, presumably contributing to the observed JAK family selectivity.
Figure 3.
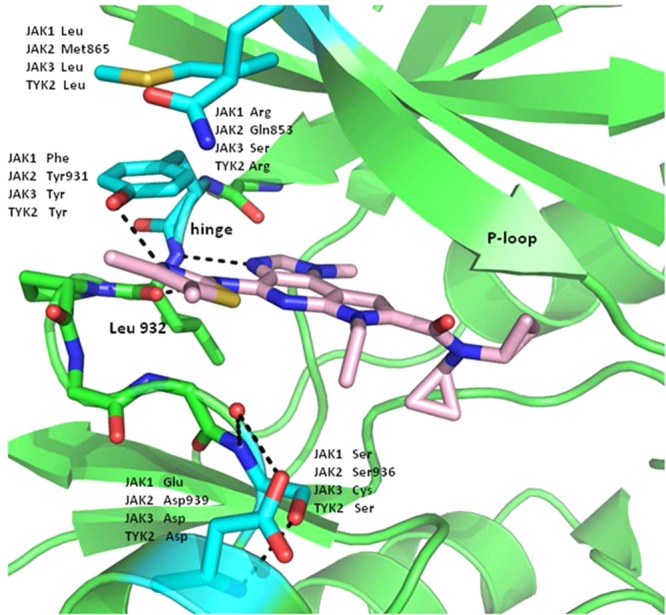
Crystal structure of 18 bound to the kinase catalytic domain of JAK2. The carbons of 18 are colored in pink and the carbons for JAK2 are colored in green except for the residues near the thiazole ring that differ in the JAK family (carbons are colored cyan). Oxygens are colored red and nitrogens blue. Hydrogen bonds are indicated with dashed lines.
Compound 18 was the first compound from the pyrrolopyridine series that demonstrated target potency (biochemical and cellular) and significant JAK family selectivity in our enzymatic assays (Table 3). The kinome selectivity of 18 was assessed in a single point format at 1 μM against a panel of 412 kinases at Ambit Biosciences (now DiscoverRx) and showed good selectivity across the Ambit profile, based on less than 10% control for 6 out of 412 kinases.14 Consistent with the biochemical assays, no other JAK family members were hits in Ambit profiling. Outside of the JAK family members, 18 also demonstrated excellent selectivity against a panel of 22 kinases tested in enzymatic assays. Although Src kinase was identified as a hit in the Ambit profile, a biochemical assay indicated Src was not significantly inhibited by 18.
Table 3. Compound 18 Selectivity.
| biochemical |
Ambit |
|||
|---|---|---|---|---|
| kinase | IC50, μM | x-fold | kinase | % control |
| JAK1 | 0.270 | 106 | JAK2 | 1.8 |
| JAK3 | 0.160 | 66 | Fyn | 4.4 |
| Tyk2 | 0.110 | 46 | Src | 6.5 |
| LCK | 1.80 | 700 | DAPK3 | 6.6 |
| DAPK1 | 1.40 | 550 | FLT3 N841I | 6.7 |
| Fyn | 0.010 | 4 | ACK | 9.4 |
| Src | >2 | 800 | ||
Compound 18 was rapidly metabolized across species but otherwise had an acceptable ADMET profile in in vitro assays (Table 4). The good permeability associated with earlier analogues (8) was maintained in 18. Initial assessments of in vitro safety (hERG flux, PXR, and HEPG2) for 18 were clean. In an in vivo setting, compound 18 exhibited excellent oral exposure (Rat, 5 mg/kg PO; Cmax 29 μM, AUC0–24h 131.4 μM·h).
Table 4. Compound 18 ADMET Profile.
| metabolic stability (T1/2 min) | 16 (H), 14 (R), 36 (D) |
| PAMPA (pH 7.4) | 532 nm/sec |
| HLM CYP IC50 | all >5 μM; except CYP3A4 (3 μM) |
| hERG (flux) | >80 μM |
| PXR EC50 | >16 μM |
| HEPG2 IC50 | >50 μM |
| plasma protein binding (% bound) | 99.1 (H), 99.0 (M), 99.0 (R) |
Early hit to lead efforts provided access to biochemical and cellular active compound 8. Structure-based design efforts around 8 identified extended hinge region modifications which enhanced JAK family selectivity. Incorporation of thiazoles along the extended hinge region led to the identification of a lead series, and compound 18 represented a significant advance in our efforts to identify potent and selective JAK2 inhibitors with promising pharmacokinetic properties. Additional SAR around this lead series will be the subject of an additional disclosure.15
Acknowledgments
The authors would like to acknowledge our Department of Discovery Synthesis and BBRC for scale-up synthetic work of intermediates and Bethanne Warrack for analytical work.
Glossary
ABBREVIATIONS
- JAK2
Janus kinase 2
- JAK1
Janus kinase 1
- JAK3
Janus kinase 3
- STAT
signal transducer and activator of transcription
- MPN
myeloproliferative neoplasm
- JH2
JAK homology domain 2
- HTL
hit to lead
- SBDD
structure-based drug design
- HTRF
homogeneous time-resolved fluorescence
- PAMPA
parallel artificial membrane permeability assay
- Tyk2
tyrosine kinase 2
- HLM
human liver microsome
- CYP
cytochrome P450
- BTK
Burton’s tyrosine kinase
- CDK2
cyclin-dependent kinase 2
- CK2
casein kinase
- GSK
glycogen synthase kinase
- IRAK4
interleukin-1 receptor-associated kinase 4
- AAK1
AP2-associated protein kinase 1
- Abl
Abelson leukemia oncogene cellular homologue
- DAPK1
death-associated protein kinase 1
- Fyn
a Src family tyrosine protein kinase
- IKK-ε
IκB kinase-ε
- JNK1
Jun N-terminal kinase 1
- LCK
lymphocyte-specific protein tyrosine kinase
- NEK2
NIMA-related kinase
- PKC
protein kinase C
- Src
Rous sarcoma oncogene cellular homologue
- IGF1R
insulin-like growth factor receptor 1
- FLT3
FMS-like tyrosine kinase 3
- ACK
activated CDC42 kinase
- ADMET
absorption, distribution, metabolism, excretion, and toxicity
Supporting Information Available
Full experimental details for key compounds and biological protocols. The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.5b00225.
Author Present Address
† Xenex, 121 Interpark, Suite 104, San Antonio, Texas 78216, United States.
Author Present Address
‡ Immunomedics, 300 The American Road, Morris Plains, New Jersey 07950, United States.
Author Present Address
§ Janssen Pharmaceutical Companies of Johnson and Johnson, 1400 McKean Road, Spring House, Pennsylvania 19002, United States.
Author Present Address
∥ BioMotiv, 20600 Chagrin Boulevard, Suite 210, Cleveland, Ohio 44122, United States.
Author Contributions
All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Morgan K. J.; Gilliland D. G. A role for JAK2 mutations in myeloproliferative diseases. Annu. Rev. Med. 2008, 59, 213.and references therein. 10.1146/annurev.med.59.061506.154159. [DOI] [PubMed] [Google Scholar]
- Levine R. L.; Wadleigh M.; Cools J.; Ebert B. L.; Wernig G.; Huntly B. J.; Boggon T. J.; Wlodarska I.; Clark J. J.; Moore S.; Adelsperger J.; Koo S.; Lee J. C.; Gabriel S.; Mercher T.; D’Andrea A.; Fröhling S.; Döhner K.; Marynen P.; Vandenberghe P.; Mesa R. A.; Tefferi A.; Griffin J. D.; Eck M. J.; Sellers W. R.; Meyerson M.; Golub T. R.; Lee S. J.; Gilliland D. G. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell 2005, 7, 387. 10.1016/j.ccr.2005.03.023. [DOI] [PubMed] [Google Scholar]
- Baxter E. J.; Scott L. M.; Campbell P. J.; East C.; Fourouclas N.; Swanton S.; Vassiliou G. S.; Bench A. J.; Boyd E. M.; Curtin N.; Scott M. A.; Erber W. N.; Green A. R. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet 2005, 365, 1054. 10.1016/S0140-6736(05)74230-6. [DOI] [PubMed] [Google Scholar]
- Kralovics R.; Passamonti F.; Buser A. S.; Teo S.-S.; Tiedt R.; Passweg J. R.; Tichelli A.; Cazzola M.; Skoda R. C. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N. Engl. J. Med. 2005, 352, 1779. 10.1056/NEJMoa051113. [DOI] [PubMed] [Google Scholar]
- James C.; Ugo V.; Le Couédic J.-P.; Staerk J.; Delhommeau F.; Lacout C.; Garçon L.; Raslova H.; Berger R.; Bennaceur-Griscelli A.; Villeval J. L.; Constantinescu S. N.; Casadevall N.; Vainchenker W. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature 2005, 434, 1144. 10.1038/nature03546. [DOI] [PubMed] [Google Scholar]
- Cervantes F.; Vannucchi A.; Kiladjian J.; Al-Ali H.; Sirulnik A.; Stalbovskaya V.; McQuitty M.; Hunter D.; Levy R.; Passamonti F.; Barbui T.; Barosi G.; Harrison C.; Knoops L.; Gisslinger H. Three-year efficacy, safety, and survival findings from COMFORT-II, a phase 3 study comparing ruxolitinib with best available therapy for myelofibrosis. Blood 2013, 122, 4047. 10.1182/blood-2013-02-485888. [DOI] [PubMed] [Google Scholar]
- PDB ID 5CF6.
- Kempson J.; Junqing Guo; Das J.; Moquin R.; Spergel S.; Watterson S.; Langevine C.; Dyckman A.; Pattoli M.; Burke J.; Yang X.; Gillooly K.; McIntyre K.; Chen L.; Dodd J.; McKinnon M.; Barrish J.; Pitts W. Synthesis, initial SAR and biological evaluation of 1,6-dihydroimidazo[4,5-d]pyrrolo[2,3-b]pyridin-4-amine derived inhibitos of IκB kinase. Bioorg. Med. Chem. Lett. 2009, 19, 2646. 10.1016/j.bmcl.2009.03.159. [DOI] [PubMed] [Google Scholar]
- Quintás-Cardama A.; Vaddi K.; Liu P.; Manshouri T.; Li J.; Scherle P. A.; Caulder E.; Wen X.; Li Y.; Waeltz P.; Rupar M.; Burn T.; Lo Y.; Kelley J.; Covington M.; Shepard S.; Rodgers J. D.; Haley P.; Kantarjian H.; Fridman J. S.; Verstovsek S. Preclinical characterization of the selective JAK1/2 inhibitor INCB018424: therapeutic implications for the treatment of myeloproliferative neoplasms. Blood 2010, 115, 3109. 10.1182/blood-2009-04-214957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tam C. S.; Verstovsek S. Investigational Janus kinase inhibitors. Expert Opin. Invest. Drugs 2013, 22, 687. 10.1517/13543784.2013.774373. [DOI] [PubMed] [Google Scholar]
- Purandare A.; Schroeder G.; Wan H.; Hart A.; Grebinski J.; Inghrim J.. Preparation of dicyclopropyldihydroimidazopyrrolopyridinecarboxamide derivatives for use as JAK2 inhibitors. PCT Int. Appl. WO 2011028864 A1 20110310, 2011.; Purandare A.; Grebinski J.; Hart A.; Inghrim J.; Schroeder G.; Wan H.. JAK2 inhibitors ans their use for the treatment of myeloproliferative diseases and cancer U.S.Patent 8202881, 2012.
- PDB ID 5CF5.
- Beno B. R.; Yeung K.-S.; Bartberger M. D.; Pennington L. D.; Meanwell N. A. A survey of the role of noncovalent sulfur interactions in drug design. J. Med. Chem. 2015, 58, 4383. 10.1021/jm501853m. [DOI] [PubMed] [Google Scholar]
- Fabian M. A.; Biggs W. H. III; Treiber D. K.; Atteridge C. E.; Azimioara M. D.; Benedetti M. G.; Carter T. A.; Ciceri P.; Edeen P. T.; Floyd M.; Ford J. M.; Galvin M.; Gerlach J. L.; Grotzfeld R. M.; Herrgard S.; Insko D. E.; Insko M. A.; Lai A. G.; Lélias J.-M.; Mehta S. A.; Milanov Z. V.; Velasco A. M.; Wodicka L. M.; Patel H. K.; Zarrinkar P. P.; Lockhart D. J. A small molecule-kinase interaction map for clinical kinase inhibitors. Nat. Biotechnol. 2005, 23, 329. 10.1038/nbt1068. [DOI] [PubMed] [Google Scholar]
- Wan H.; Schroeder G. M.; Hart A. C.; Inghrim J.; Grebinski J.; Lorenzi M. V.; You D.; McDevitt T.; Penhallow B.; Vuppugalla R.; Zhang Y.; Gu X.; Iyer R.; Lombardo L. J.; Trainor G. L.; Ruepp S.; Lippy J.; Tokarski J. S.; Blatt Y.; Sack J. S.; Khan J. A.; Stefanski K.; Sleckza B.; Arunachalam P.; Pregaglathan B.; Narayanan S.; Nanjundaswamy K. C.; Kuppusamy P.; Mathur A.; Sun J.-H.; Wong M. K.; Wu D.-R.; Li P.; Gupta A.; Purandare A. V. Discovery of a highly selective JAK2 inhibitor, BMS-911543 for the treatment of myeloproliferative neoplasms. ACS Med. Chem. Lett. 2015, 10.1021/acsmedchemlett.5b00226. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.