

Abstract
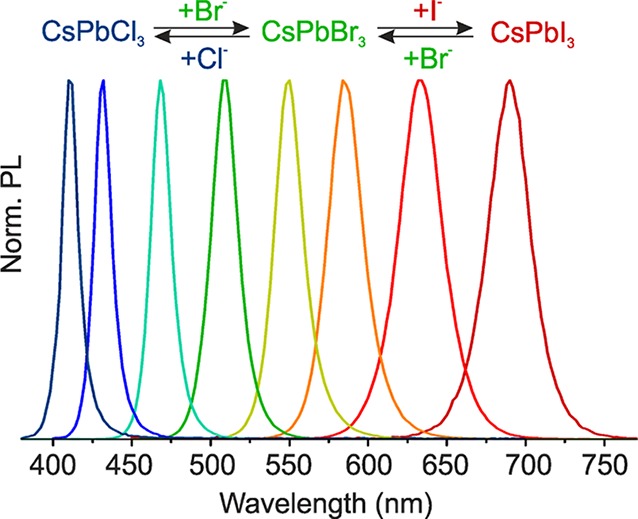
Postsynthetic chemical transformations of colloidal nanocrystals, such as ion-exchange reactions, provide an avenue to compositional fine-tuning or to otherwise inaccessible materials and morphologies. While cation-exchange is facile and commonplace, anion-exchange reactions have not received substantial deployment. Here we report fast, low-temperature, deliberately partial, or complete anion-exchange in highly luminescent semiconductor nanocrystals of cesium lead halide perovskites (CsPbX3, X = Cl, Br, I). By adjusting the halide ratios in the colloidal nanocrystal solution, the bright photoluminescence can be tuned over the entire visible spectral region (410–700 nm) while maintaining high quantum yields of 20–80% and narrow emission line widths of 10–40 nm (from blue to red). Furthermore, fast internanocrystal anion-exchange is demonstrated, leading to uniform CsPb(Cl/Br)3 or CsPb(Br/I)3 compositions simply by mixing CsPbCl3, CsPbBr3, and CsPbI3 nanocrystals in appropriate ratios.
Keywords: Nanocrystals, perovskites, metal halides, cation exchange, anion exchange, photoluminescence
Rational synthesis of colloidal nanorystals (NCs) is of paramaunt importance in NC research due to the growing demand for compositional diversity, shape engineering, and new optical, electronic, magnetic, or catalytic functionalities of NCs.1,2 In this regard, postsynthetic chemical transformations of metallic, semiconducting, and magnetic NCs are increasingly useful, such as by galvanic replacement,3,4 cation-exchange reactions,5−10 or the nanoscale Kirkendall effect.11−13 These transformation routes, particularly suited to NCs due to their high surface-to-volume ratios and short diffusion path lengths, give access to a myriad of structures that are difficult or impossible to synthesize directly. The initial (parent) NC serves as a template whose size, shape, and composition can be independently modified.
For semiconductor NCs, typically metal chalcogenides, cation-exchange reactions are particularly powerful, resulting in a partial or complete replacement of cations while maintaining an uninterrupted anionic sublattice and often preserving the pre-existing shape as well.5−7 A partial list of notable examples includes (with the parent NC in parentheses) the following: the first report on cation exchange leading to Ag2Se (from CdSe NCs),14 CdS-Ag2S nanorod superlattices (from CdS nanorods),15 core–shell PbTe/CdTe, PbSe/CdSe and PbS/CdS NCs (from PbTe, PbSe and PbS NCs),16,17 PbSe/PbS core–shell and dot-in-rod NCs (from respective CdSe/CdS nanomorphologies),18 PbS nanorods (from CdS nanorods),19 disk-shaped CdTe NCs (from Cu2Te nanodisks),20 CuInS2 NCs (from Cu2-xS NCs),21 InP nanoplatelets (from Cu3-xP nanoplatelets),22 and sequential multiple cation exchanges for obtaining metastable phases.5 In contrast to the facile extraction and replacement of cations, anion-exchange in NCs has remained elusive. Taking CdSe as an example, where Cd2+ has at most half the radius of Se2–, cations are much easier to manipulate within the voids of the anionic sublattice than vice versa. The scarcity of reported examples of successful anion-exchange post-treatments in NCs is reflective of both the typical difficulties encountered (e.g., substantial resturcturing or fracturing) and the demanding reaction conditions necessary (e.g., high reaction temperatures of 160–450 °C for ZnO to ZnS(Se) conversions23−25) or the incomplete nature of the process (e.g., partial or limited to only a few surface atomic layers26−29). In this work, we report a shift in the status quo; halide anions in metal halide semiconductor NCs can be easily extracted and replaced with another halide, owing to their single ionic charge, the rigid nature of the cationic sublattice, and an efficient vacancy-assisted diffusion mechanism.
We recently reported a simple one-step synthesis of cesium lead halide perovskites (CsPbX3, X = Cl, Br, I) in the form of monodisperse colloidal nanocubes (4–15 nm edge lengths).30 Through compositional modulations and quantum size-effects, the bandgap energies and photoluminescence (PL) spectra are readily tunable over the entire visible spectral region of 410–700 nm. A peculiar feature of CsPbX3 NCs is that, contrary to uncoated chalcogenide NCs, dangling bonds on the surface do not impart severe midgap trap states and the as-synthesized NCs exhibit bright emission with quantum efficiencies of up to 90% in green-to-red spectral region. The fact that metal halides are significantly different from metal chalcogenides (for example, from a structural standpoint, they consist of singly charged anions and exhibit highly ionic bonding) led us to explore postsynthetic chemical transformations of CsPbX3 NCs, the subject of this work. Cation- and anion-exchange reactions of ABX3 perovskite structures offer a very promising avenue to a plethora of optoelectronic materials.
With this motivation in mind, various attempts to exchange either Cs+ cations (with Rb+, Ag+, Cu+, or Ba2+) or Pb2+ cations (with Sn2+, Ge2+, or Bi3+) in CsPbX3 NCs were undertaken, though unfortunately leading to the decomposition of the parent CsPbX3 NCs in every case. To this end, most common outcome was the formation of a new halide such as AgX. In stark contrast, fast (for example, in several seconds), low-temperature, deliberately partial, or complete anion-exchange (Figure 1a) could easily be performed in the cases of Cl-to-Br, Br-to-Cl, Br-to-I, and I-to-Br anion-exchanges, via the formation of homogeneous solid solutions. Because of the large difference in ionic radii between Cl– and I– causing the instability of Cl/I solid solutions, no mixed-halide solid solutions could be obtained in CsPbCl3 + I– or CsPbI3 + Cl– systems, but rather slow and complete exchange occurred.
Figure 1.
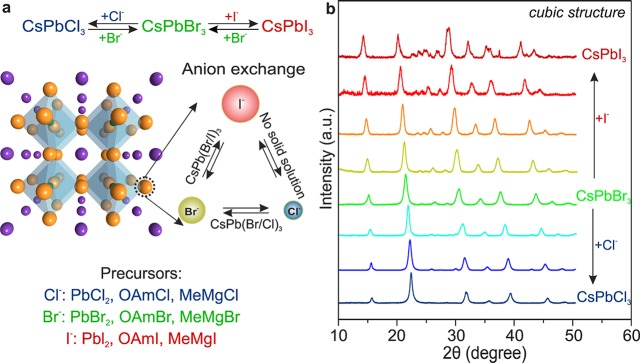
(a) Schematic of the anion-exchange within the cubic perovskite crystal structure of CsPbX3 along with a list of suitable reagents for each reaction when performed in organic media. A three-dimensional network is formed by corner-sharing PbX6 octahedra with Cs+ (purple spheres) occupying the interstitial voids. Ionic radii: Cs+, 1.88 Å; Pb2+, 1.16 Å; Cl–, 1.81 Å; Br–, 1.96 Å; and I–, 2.2 Å.35−37 (b) Powder X-ray diffraction (XRD) patterns of the parent CsPbBr3 NCs and anion-exchanged samples (using PbCl2 and PbI2 as halide sources), showing the retention of phase-pure cubic perovskite structure and an average (Scherrer) crystallite size of 8–10 nm. The shift of the XRD reflections is linearly dependent on the composition (Vegard’s law), indicating the formation of uniform solid solutions. Equivalent behaviors were also observed for CsPbCl3 + Br– and CsPbI3 + Br– systems. Formation of solid solutions has been also confirmed by energy dispersive X-ray spectroscopy (EDX) and Rutherford backscattering spectrometry (RBS).
The anion-exchange reactions reported herein were conducted in dry octadecene (ODE) as a solvent by mixing a specific ratio of the desired halide source and CsPbX3 NCs (see Supporting Information file SI2 and, for experimental details, Table S1). The concentrations of capping ligands (oleylamine and oleic acid) were adjusted to be similar to those used for the synthesis of the parent CsPbX3 NCs. All tested halide sources, from organometallic Grignard reagents (MeMgX) to oleylammonium halides (OAmX) and simple PbX2 salts, afforded fast anion-exchange at 40 °C; at this temperature, the solubility of all reagents and NCs could be maintained.
It is well-known that bulk CsPbX3 crystallize in orthorhombic, tetragonal, and cubic polymorphs of the perovskite lattice with the cubic phase being the high-temperature state for all compounds.31−33 By direct synthesis at 160–200 °C, CsPbX3 NCs are formed in the cubic phase.30 Interestingly, the subsequent anion-exchange manipulations of the halide ions do not seem to affect the cationic sublattice and the cubic perovskite crystal structure is maintained (Figure 1b) despite the low temperature of the anion-exchange reaction. The size and shape of the parent NCs are also preserved in the course of the anion-exchange (Figure 2). The direct synthesis of single- or mixed-halide CsPbX3 at such low temperatures yields either exclusively large and polydisperse crystallites of poorly or nonluminescent, wider-bandgap orthorhombic phases or simply no crystalline products at all. CsPbI3, for example, is highly luminescent and red in its three-dimensional cubic phase, yet yellow and nonluminescent upon conversion into its orthorhombic polymorph.31−34
Figure 2.

Transmission electron microscopy (TEM) images of ∼10 nm CsPbX3 NCs after treatment with various quantities of (a) chloride and (b) iodide anions. The insets show the evolution of emission colors (under a UV lamp, λ = 365 nm) upon forming mixed-halide CsPb(Br/Cl)3 and CsPb(Br/I)3 to fully exchanged CsPbCl3 and CsPbI3 NCs.
Along with the cubic crystal structure, bright PL (with quantum yields of 10–80%, the lowest values for CsPbCl3) is retained in anion-exchanged CsPbX3 NCs, with fwhm peak widths ranging from 12 nm for CsPbCl3 to 40 nm for CsPbI3 (Figure 3), comparable to directly synthesized CsPbX3 NCs.30 The PL spectra of such exchanged NCs are Stokes-shifted with respect to the optical absorption spectra. As in our previous report on the direct synthesis of mixed-halide NCs,30 the resulting composition of anion-exchanged NCs follows the overall halide ratio in the system, which is [X]parent/[X]incoming for anion-exchange or simply the precursor ratio in direct synthesis. The [X]parent/[X]incoming ratio in Cl-to-Br, Br-to-Cl, Br-to-I, and I-to-Br anion-exchanges was varied continuously from 3:1 to 1:3 for three tested halide sources to cover the entire visible spectral region (Table S1 and Figure S1). In general, the distribution of two halide ions between the solution and the crystal is governed by the balance of the crystal energies of the mixed solid-solution and single-halide perovskites on the one side and the solvation energies of halide ions in solution on the other side. The lack of a strong preference toward one of the halides either indicates that the crystal energies are similar for all halides, or that the preferred halide is also preferably solvated, thereby maintaining balance. Eventually, also an entropy of mixing should favor the formation of solid-solutions in the absence of strong enthalpic factors (crystal energy). A different picture is found for I-to-Cl or Cl-to-I exchanges. Treatment of CsPbCl3 NCs with a large excess of PbI2 (or OAmI and MeMgI) transforms the PL color from blue (410 nm) directly to red (690 nm) in ca. 30–60 s. The rate for backward transition is similar, also lacking any intermediate color. This can be explained by the larger difference in ionic radii between Cl– and I–, leading to the higher stability of the single-halide crystals as compared to the solid-solutions.
Figure 3.
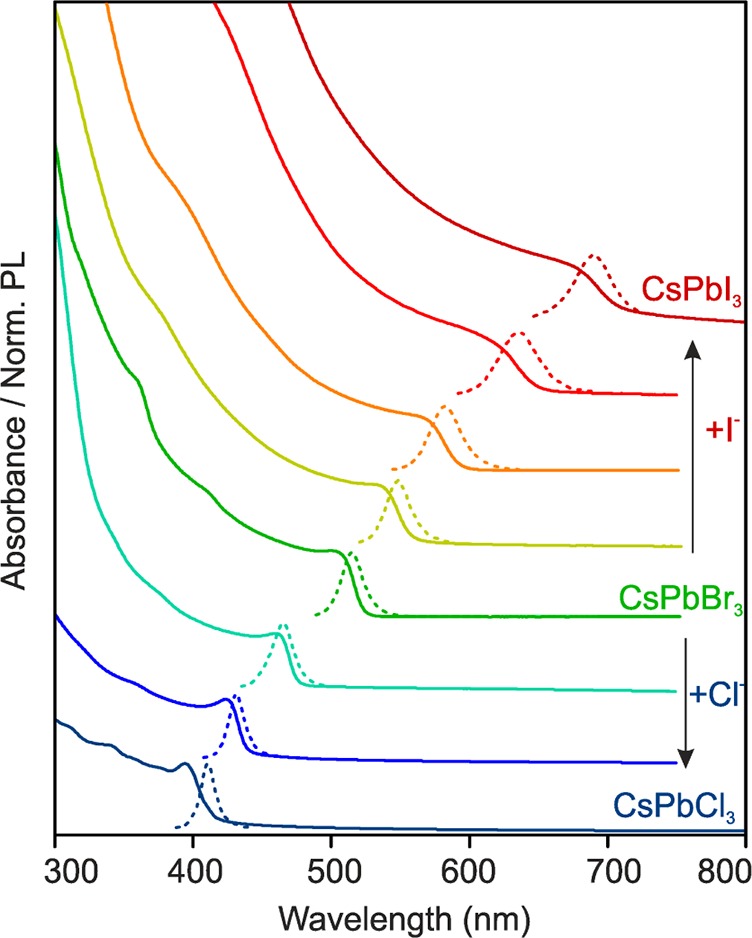
Evolution of the optical absorption (solid lines) and PL (dashed lines) spectra of CsPbBr3 NCs with increasing quantities of PbCl2 or PbI2, added as exchanging halide sources.
The high speed of anion-exchange in perovskite NCs is rooted in the ionic properties of perovskite metal halide crystals. The high ionic conductivity of halides in bulk CsPbX3 has been known for 30 years.38 The primary conduction mechanism is the diffusion of halide vacancies, VX* (X = Br, Cl), where the activation energy is 0.29 eV for CsPbCl3 and 0.25 eV for CsPbBr3. Recently, methylammonium hybrid organic–inorganic perovskite analogues (CH3NH3PbI3 and CH3NH3PbBr3) have been the subject of numerous studies due to their unprecedented (for solution-processed absorber materials) photovoltaic power conversion efficiencies of up to 20%.39−44 Also here understanding the ionics of perovskite halides holds a potential key to explaining a range of important observations such as the hysteresis of current–voltage characteristics45 or the self-compensating mechanism of electrical conductivity.46 Density functional theory calculations point to a prevalence of ionic versus electronic disorder with the charged vacancy concentration exceeding 0.4% at room temperature,46 and electron and hole traps being rather shallow with respect to hole and conduction bands.47−49 In an elegant study using specially designed ion-selective galvanic cells, Maier et al. have confirmed that of the three ions in the lattice (CH3NH3+, Pb2+, and I–), only the latest I– is responsible for ionic conductivity, via the vacancy diffusion mechanism.50
An in situ PL study of the Br-to-I exchange in CsPbBr3 is presented in Figure 4. Most of the anion-exchange occurs within the first several seconds. The gradual shift of the PL color is consistent with the continuous formation of homogeneous CsPbBrxI3–x solid solutions because compositional inhomogeneities or preferred compositions within the NC ensemble would lead to broad or multiple peaks. Supporting Information file SI1 presents video of the same reaction, again with clearly observed gradual change of PL colors. Importantly, the integrated intensities of the PL spectra of each exchanged sample remain comparable to that of the parent sample, indicating high PL quantum yields throughout the process. In the course of this work, rapid anion-exchange in thin films of hybrid perovskites of CH3NH3PbX3 was reported,51 along with in situ optical measurements. In such 2D extended films, CH3NH3PbBr3-to-CH3NH3PbI3 conversion was determined to progress via the formation of iodine-rich domains within the first seconds with the immediate appearance of red-emission closer to the PL of pure CH3NH3PbI3 followed by a slower homogenization of the composition over the entire sample. In accord with our study, CH3NH3PbCl3↔CH3NH3PbI3 conversions do not involve solid solutions due to the lattice mismatch between the parent and fully exchanged phases. Another study on the anion-exchange in the films of plate-type CH3NH3PbX3 NCs has appeared during the revision of our manuscript,52 where again Br–Cl and Br–I systems showed tunable solid solutions, while mixed Cl–I systems were not obtained.
Figure 4.
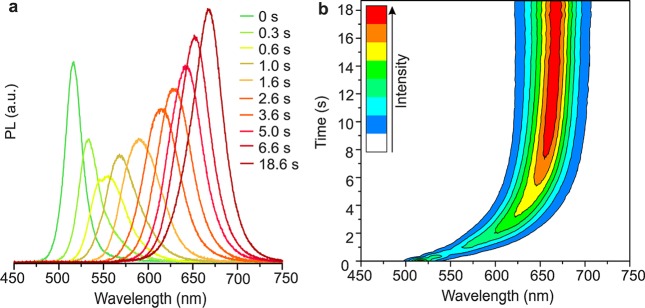
In-situ PL measurements during a CsPbBr3 to CsPbI3 NC conversion at 40 °C with [Br]parent/[I]incoming = 1:3, (a) plotted at specific times during conversion and (b) throughout the complete process (with three spectra acquired per second).
In addition to fast halide motion within the perovskite lattice, the ease of anion-exchange in CsPbX3 NCs is in part also due to fast exchange dynamics of the halide ions in solution. Even in the absence of added halide source, we find that the NCs themselves can serve as halide sources for each other (Figure 5a). For example, the mixing of CsPbBr3 and CsPbI3 in colloidal solution is followed by fast cross-exchange and homogenization of their compositions, forming CsPb(Br/I)3 solid solutions. Shuttling of halide ions between NCs is facilitated by the small concentration of the solvated halide ions in the colloidal dispersion, present as a residue of OAmX or similar species after the isolation of NCs or due to desorption from the NC surface. Only one PL peak is measured after the completion of ionic exchange. The time taken to reach full homogeneity in this case is 10–20 min, much longer than for direct anion-exchange. Investigations of intermediate stages of this process indicate that exchange occurs simultaneously in both kinds of particles; the CsPbBr3 PL peaks shift to longer wavelengths and the CsPbI3 PL moves to shorter wavelengths (Figure 5b and detailed in situ PL study in Figure S2). The tunability of the PL peaks, emission line widths, and quantum yields after full homogenization are equivalent to those obtained from direct synthesis or via direct ion-exchange as discussed above.
Figure 5.
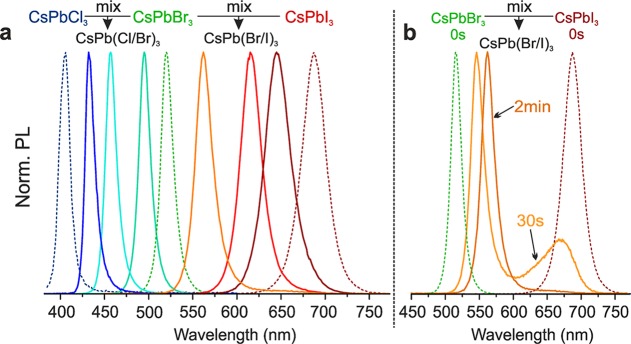
Inter-NC anion-exchange reactions in CsPbX3 NC systems. (a) An overview of the PL spectra of samples obtained by mixing CsPbBr3 NCs with either CsPbCl3 or CsPbI3 NCs in various ratios. (b) Time-dependent PL spectra showing an intermediate stage formed during inter-NC anion-exchange between CsPbBr3 and CsPbI3 in which two distinct NC species coexist with altered compositions.
In summary, remarkably fast anion-exchange was observed in perovskite CsPbX3 NCs. Overall, the behavior of perovskite halides with respect to anion-exchange is orthogonal to common metal chalcogenide NCs, namely since the cationic sublattice is substantially rigid and the singly charged halide ions are highly mobile. In metal chalcogenides, ion-exchange has been observed with such ease only for cations. Semiconducting properties of lead halide perovskites are highly defect-tolerant, maintaining bright excitonic emission throughout and upon completion of the anion-exchange. Of practical note, the herein demonstrated fine-tuning of the spectrally narrow and bright PL of anion-exchanged CsPbX3 NCs over the entire visible spectral region can be conveniently accomplished from numerous halide sources at low temperatures. In addition, fast anion-exchange between CsPbX3 NCs of different compositions can also be readily achieved. Future investigations of halide-exchange reactions in other nanoscale metal halide systems are clearly warranted, as high ionic conductivity may not be strictly necessary due to the short diffusion paths within the NCs.
Acknowledgments
This work was financially supported by the European Union via FP7 European Research Council Starting Grant (306733) and Horizon2020 MSCA-ETN phonsi (642656), by the Swiss National Science Foundation (Grant 200021_143638). M.B. is grateful to Swiss National Science Foundation for Ambizione Energy fellowship (Grant PZENP2_154287). All authors thank Nadia Schwitz for a help with producing the photographs and the video of anion-exchange reactions, Dr. M. Döbeli for RBS measurements (ETH Laboratory of Ion Beam Physics), Dr. F. Krumeich and M. Walter for EDX measurements, and Dr. N. Stadie for reading the manuscript. We also acknowledge the support of the Scientific Center for Optical and Electron Microscopy (ETH Zurich) and Empa Electron Microscopy Center.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.nanolett.5b02404.
Author Contributions
The manuscript was prepared through the contribution of all coauthors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Schaak R. E.; Williams M. E. ACS Nano 2012, 6, 8492–8497 10.1021/nn304375v. [DOI] [PubMed] [Google Scholar]
- Kovalenko M. V.; Manna L.; Cabot A.; Hens Z.; Talapin D. V.; Kagan C. R.; Klimov V. I.; Rogach A. L.; Reiss P.; Milliron D. J.; Guyot-Sionnnest P.; Konstantatos G.; Parak W. J.; Hyeon T.; Korgel B. A.; Murray C. B.; Heiss W. ACS Nano 2015, 9, 1012–1057 10.1021/nn506223h. [DOI] [PubMed] [Google Scholar]
- Oh M. H.; Yu T.; Yu S.-H.; Lim B.; Ko K.-T.; Willinger M.-G.; Seo D.-H.; Kim B. H.; Cho M. G.; Park J.-H.; Kang K.; Sung Y.-E.; Pinna N.; Hyeon T. Science 2013, 340, 964–968 10.1126/science.1234751. [DOI] [PubMed] [Google Scholar]
- Xia X.; Wang Y.; Ruditskiy A.; Xia Y. Adv. Mater. 2013, 25, 6313–6333 10.1002/adma.201302820. [DOI] [PubMed] [Google Scholar]
- Li H. B.; Zanella M.; Genovese A.; Povia M.; Falqui A.; Giannini C.; Manna L. Nano Lett. 2011, 11, 4964–4970 10.1021/nl202927a. [DOI] [PubMed] [Google Scholar]
- Gupta S.; Kershaw S. V.; Rogach A. L. Adv. Mater. 2013, 25, 6923–6944 10.1002/adma.201302400. [DOI] [PubMed] [Google Scholar]
- Rivest J. B.; Jain P. K. Chem. Soc. Rev. 2013, 42, 89–96 10.1039/C2CS35241A. [DOI] [PubMed] [Google Scholar]
- Sytnyk M.; Kirchschlager R.; Bodnarchuk M. I.; Primetzhofer D.; Kriegner D.; Enser H.; Stangl J.; Bauer P.; Voith M.; Hassel A. W.; Krumeich F.; Ludwig F.; Meingast A.; Kothleitner G.; Kovalenko M. V.; Heiss W. Nano Lett. 2013, 13, 586–593 10.1021/nl304115r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beberwyck B. J.; Surendranath Y.; Alivisatos A. P. J. Phys. Chem. C 2013, 117, 19759–19770 10.1021/jp405989z. [DOI] [Google Scholar]
- Ott F. D.; Spiegel L. L.; Norris D. J.; Erwin S. C. Phys. Rev. Lett. 2014, 113, 156803. 10.1103/PhysRevLett.113.156803. [DOI] [PubMed] [Google Scholar]
- Wang W. S.; Dahl M.; Yin Y. D. Chem. Mater. 2013, 25, 1179–1189 10.1021/cm3030928. [DOI] [Google Scholar]
- El Mel A. A.; Buffiere M.; Tessier P. Y.; Konstantinidis S.; Xu W.; Du K.; Wathuthanthri I.; Choi C. H.; Bittencourt C.; Snyders R. Small 2013, 9, 2838–2843 10.1002/smll.201202824. [DOI] [PubMed] [Google Scholar]
- Yin Y.; Rioux R. M.; Erdonmez C. K.; Hughes S.; Somorjai G. A.; Alivisatos A. P. Science 2004, 304, 711–714 10.1126/science.1096566. [DOI] [PubMed] [Google Scholar]
- Son D. H.; Hughes S. M.; Yin Y.; Alivisatos A. P. Science 2004, 306, 1009–1012 10.1126/science.1103755. [DOI] [PubMed] [Google Scholar]
- Robinson R. D.; Sadtler B.; Demchenko D. O.; Erdonmez C. K.; Wang L.-W.; Alivisatos A. P. Science 2007, 317, 355–358 10.1126/science.1142593. [DOI] [PubMed] [Google Scholar]
- Pietryga J. M.; Werder D. J.; Williams D. J.; Casson J. L.; Schaller R. D.; Klimov V. I.; Hollingsworth J. A. J. Am. Chem. Soc. 2008, 130, 4879–4885 10.1021/ja710437r. [DOI] [PubMed] [Google Scholar]
- Lambert K.; Geyter B. D.; Moreels I.; Hens Z. Chem. Mater. 2009, 21, 778–780 10.1021/cm8029399. [DOI] [Google Scholar]
- Jain P. K.; Amirav L.; Aloni S.; Alivisatos A. P. J. Am. Chem. Soc. 2010, 132, 9997–9999 10.1021/ja104126u. [DOI] [PubMed] [Google Scholar]
- Rivest J. B.; Swisher S. L.; Fong L.-K.; Zheng H.; Alivisatos A. P. ACS Nano 2011, 5, 3811–3816 10.1021/nn2001454. [DOI] [PubMed] [Google Scholar]
- Li H. B.; Brescia R.; Povia M.; Prato M.; Bertoni G.; Manna L.; Moreels I. J. Am. Chem. Soc. 2013, 135, 12270–12278 10.1021/ja404694k. [DOI] [PubMed] [Google Scholar]
- van der Stam W.; Berends A. C.; Rabouw F. T.; Willhammar T.; Ke X.; Meeldijk J. D.; Bals S.; de Mello Donega C. Chem. Mater. 2015, 27, 621–628 10.1021/cm504340h. [DOI] [Google Scholar]
- De Trizio L.; Gaspari R.; Bertoni G.; Kriegel I.; Moretti L.; Scotognella F.; Maserati L.; Zhang Y.; Messina G. C.; Prato M.; Marras S.; Cavalli A.; Manna L. Chem. Mater. 2015, 27, 1120–1128 10.1021/cm5044792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J.; Zheng H.; Jun Y.-w.; Alivisatos A. P. J. Am. Chem. Soc. 2009, 131, 13943–13945 10.1021/ja905732q. [DOI] [PubMed] [Google Scholar]
- Dloczik L.; Könenkamp R. Nano Lett. 2003, 3, 651–653 10.1021/nl0340879. [DOI] [Google Scholar]
- Dawood F.; Schaak R. E. J. Am. Chem. Soc. 2009, 131, 424–425 10.1021/ja808455u. [DOI] [PubMed] [Google Scholar]
- Brumer M.; Kigel A.; Amirav L.; Sashchiuk A.; Solomesch O.; Tessler N.; Lifshitz E. Adv. Funct. Mater. 2005, 15, 1111–1116 10.1002/adfm.200400620. [DOI] [Google Scholar]
- Saruyama M.; So Y.-G.; Kimoto K.; Taguchi S.; Kanemitsu Y.; Teranishi T. J. Am. Chem. Soc. 2011, 133, 17598–17601 10.1021/ja2078224. [DOI] [PubMed] [Google Scholar]
- Bailey R. E.; Nie S. J. Am. Chem. Soc. 2003, 125, 7100–7106 10.1021/ja035000o. [DOI] [PubMed] [Google Scholar]
- Choi D.; Lee S.; Lee J.; Cho K.-S.; Kim S.-W. Chem. Commun. 2015, 51, 899–902 10.1039/C4CC07438F. [DOI] [PubMed] [Google Scholar]
- Protesescu L.; Yakunin S.; Bodnarchuk M. I.; Krieg F.; Caputo R.; Hendon C. H.; Yang R. X.; Walsh A.; Kovalenko M. V. Nano Lett. 2015, 15, 3692–3696 10.1021/nl5048779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma S.; Weiden N.; Weiss A. Z. Phys. Chem. 1992, 175, 63–80 10.1524/zpch.1992.175.Part_1.063. [DOI] [Google Scholar]
- Trots D. M.; Myagkota S. V. J. Phys. Chem. Solids 2008, 69, 2520–2526 10.1016/j.jpcs.2008.05.007. [DOI] [Google Scholar]
- Stoumpos C. C.; Malliakas C. D.; Kanatzidis M. G. Inorg. Chem. 2013, 52, 9019–9038 10.1021/ic401215x. [DOI] [PubMed] [Google Scholar]
- Babin V.; Fabeni P.; Nikl M.; Nitsch K.; Pazzi G. P.; Zazubovich S. Phys. Status Solidi B 2001, 226, 419–428. [DOI] [Google Scholar]
- Verma A. S.; Jindal V. K. J. Alloys Compd. 2009, 485, 514–518 10.1016/j.jallcom.2009.06.001. [DOI] [Google Scholar]
- Verma A. S.; Kumar A.; Bhardwaj S. R. Phys. Status Solidi B 2008, 245, 1520–1526 10.1002/pssb.200844072. [DOI] [Google Scholar]
- Ida Y. Phys. Earth Planet. Inter. 1976, 13, 97–104 10.1016/0031-9201(76)90074-1. [DOI] [Google Scholar]
- Mizusaki J.; Arai K.; Fueki K. Solid State Ionics 1983, 11, 203–211 10.1016/0167-2738(83)90025-5. [DOI] [Google Scholar]
- Gratzel M. Nat. Mater. 2014, 13, 838–842 10.1038/nmat4065. [DOI] [PubMed] [Google Scholar]
- Green M. A.; Ho-Baillie A.; Snaith H. J. Nat. Photonics 2014, 8, 506–514 10.1038/nphoton.2014.134. [DOI] [Google Scholar]
- Park N.-G. J. Phys. Chem. Lett. 2013, 4, 2423–2429 10.1021/jz400892a. [DOI] [Google Scholar]
- Zhou H.; Chen Q.; Li G.; Luo S.; Song T.-b.; Duan H.-S.; Hong Z.; You J.; Liu Y.; Yang Y. Science 2014, 345, 542–546 10.1126/science.1254050. [DOI] [PubMed] [Google Scholar]
- Chung I.; Lee B.; He J.; Chang R. P. H.; Kanatzidis M. G. Nature 2012, 485, 486–489 10.1038/nature11067. [DOI] [PubMed] [Google Scholar]
- Stranks S. D.; Snaith H. J. Nat. Nanotechnol. 2015, 10, 391–402 10.1038/nnano.2015.90. [DOI] [PubMed] [Google Scholar]
- Xiao Z.; Yuan Y.; Shao Y.; Wang Q.; Dong Q.; Bi C.; Sharma P.; Gruverman A.; Huang J. Nat. Mater. 2014, 14, 193–198 10.1038/nmat4150. [DOI] [PubMed] [Google Scholar]
- Walsh A.; Scanlon D. O.; Chen S.; Gong X. G.; Wei S.-H. Angew. Chem., Int. Ed. 2015, 54, 1791–1794 10.1002/anie.201409740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buin A.; Pietsch P.; Xu J.; Voznyy O.; Ip A. H.; Comin R.; Sargent E. H. Nano Lett. 2014, 14, 6281–6286 10.1021/nl502612m. [DOI] [PubMed] [Google Scholar]
- Agiorgousis M. L.; Sun Y.-Y.; Zeng H.; Zhang S. J. Am. Chem. Soc. 2014, 136, 14570–14575 10.1021/ja5079305. [DOI] [PubMed] [Google Scholar]
- Yin W.-J.; Shi T.; Yan Y. Appl. Phys. Lett. 2014, 104, 063903. 10.1063/1.4864778. [DOI] [Google Scholar]
- Yang T.-Y.; Gregori G.; Pellet N.; Grätzel M.; Maier J. Angew. Chem., Int. Ed. 2015, 10.1002/anie.201500014. [DOI] [PubMed] [Google Scholar]
- Pellet N.; Teuscher J.; Maier J.; Grätzel M. Chem. Mater. 2015, 27, 2181–2188 10.1021/acs.chemmater.5b00281. [DOI] [Google Scholar]
- Jang D. M.; Park K.; Kim D. H.; Park J.; Shojaei F.; Kang H. S.; Ahn J.-P.; Lee J. W.; Song J. K. Nano Lett. 2015, 10.1021/acs.nanolett.5b01430. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.