

Abstract
Novel, cellular, gain-of-signal, bioluminescent reporter assays for fatty acid synthesis type II (FASII) inhibitors were constructed in an efflux-deficient strain of Pseudomonas aeruginosa and based on the discovery that FASII genes in P. aeruginosa are coordinately upregulated in response to pathway disruption. A screen of 115,000 compounds identified a series of sulfonamidobenzamide (SABA) analogs, which generated strong luminescent signals in two FASII reporter strains but not in four control reporter strains designed to respond to inhibitors of pathways other than FASII. The SABA analogs selectively inhibited lipid biosynthesis in P. aeruginosa and exhibited minimal cytotoxicity to mammalian cells (50% cytotoxic concentration [CC50] ≥ 80 μM). The most potent SABA analogs had MICs of 0.5 to 7.0 μM (0.2 to 3.0 μg/ml) against an efflux-deficient Escherichia coli (ΔtolC) strain but had no detectable MIC against efflux-proficient E. coli or against P. aeruginosa (efflux deficient or proficient). Genetic, molecular genetic, and biochemical studies revealed that SABA analogs target the enzyme (AccC) catalyzing the biotin carboxylase half-reaction of the acetyl coenzyme A (acetyl-CoA) carboxylase step in the initiation phase of FASII in E. coli and P. aeruginosa. These results validate the capability and the sensitivity of this novel bioluminescent reporter screen to identify inhibitors of E. coli and P. aeruginosa FASII.
INTRODUCTION
Pseudomonas aeruginosa is a highly virulent, persistent human pathogen with both acquired and intrinsic drug resistances. It is the most common cause of nosocomial pneumonia, causing 15% to 20% of hospital-acquired pneumonias (1), and up to 75% of patients in intensive care units are colonized with this pathogen (2). P. aeruginosa is also becoming a major cause of community-acquired pneumonia in severely ill patients (3). An astounding 30% of clinical isolates from critically ill patients are resistant to three or more drugs, which leads to treatment failure (4). The discovery and development of new classes of antibiotics, which are not subject to existing target-based resistance mechanisms, is an important strategy in combating drug resistance, and targeting unexploited or underexploited essential bacterial pathways has been a successful strategy for discovering new compound classes (5, 6).
This study focused on the fatty acid synthesis type II (FASII) pathway in P. aeruginosa. While most bacteria utilize exogenous fatty acids for phospholipid synthesis (7), the FASII pathway is absolutely essential for bacterial membrane biogenesis in Gram-negative bacteria because the hydroxyl-fatty acid constituents of LPS cannot be obtained from an extracellular source (8). FASII inhibitors also block the production of Gram-negative signaling molecules, including homoserine lactones and hydroxyquinolines, which are important to establish and maintain P. aeruginosa virulence (9). This FASII macromolecular synthesis pathway is conserved and essential in Gram-negative bacteria but is absent from the mammalian cytoplasm, which utilizes a distantly related type I FAS enzyme complex (10). While mammalian mitochondria do contain a FASII system (11), its relevance in the treatment of acute infections by bacterial FASII inhibitors is unclear (12, 13). Significantly, a FabI inhibitor is about to enter phase III studies in humans for Staphylococcus aureus infections (14), and isoniazid, a FASII inhibitor, is currently used clinically to treat tuberculosis (15), indicating that these inhibitors are highly selective for bacterial FASII or that inhibition of mitochondrial FASII is not toxic during treatment of acute-phase infections. Clearly, the absence of significant cytotoxicity by a FASII inhibitor is an important criterion for further development of such a potential drug. Although there are no anti-pseudomonal agents in development or approved for clinical use that act on the FASII pathway, existing drugs targeting FASII in other bacterial species indicate the feasibility of targeting this pathway with antibiotics.
The high rate of failure in recent target-based biochemical screens for antibacterials (16, 17) prompted us to design novel cellular reporter screens with the potential to detect inhibitors of any rate-determining step in the entire FASII pathway. To construct such screening strains, we identified FASII promoters that were induced in response to decreased flux through the FAS pathway. These FASII depletion-responsive promoters were fused to the Photorhabdus luminescens luxCDABE operon to provide a gain-of-signal bioluminescent response upon reduction in the pathway flow. The resulting cellular screens of this study proved to be more sensitive to FASII inhibition than are growth assays, and they select for inhibitors that can penetrate the P. aeruginosa cell. In this study, we optimized and applied one of the FASII screens to over 100,000 diverse compounds and identified a novel series of compounds that generate highly significant luminescent responses in several FASII reporter strains but not in reporter strains designed to respond to inhibition of other targets (18, 19). To verify the ability of these new reporter screens to identify novel FASII inhibitors, we used molecular genetic tools and biochemical assays to identify the molecular target of the most potent hit compound series. These studies demonstrate that these screening hits target AccC, which carries out the biotin carboxylase half-reaction of the acetyl coenzyme A (acetyl-CoA) carboxylase step in the initiation phase of the FASII pathway.
MATERIALS AND METHODS
Strains, plasmids, and growth media.
Bacterial strains and plasmids used for assays are described in Table 1. All P. aeruginosa strains were derivatives of PAO1 (20). Escherichia coli TOP10 (Invitrogen), E. coli DB3.1 (host strain for Gateway vectors from Invitrogen, Inc.), E. coli SM10 (21), and E. coli S17-1 (ATCC 47055) were used as hosts for molecular cloning. Vogel-Bonner minimal medium (VBMM) was prepared as described previously (21). Luria-Bertani (LB) medium (Lennox version; liquid and agar) was purchased from Difco. LB was supplemented with gentamicin (LBG) and/or isopropyl-β-d-thiogalactopyranoside (IPTG), as noted for each experiment.
TABLE 1.
Strains and plasmids
| Straina or plasmid | Relevant genotypeb | Reference |
|---|---|---|
| P. aeruginosa strains | ||
| PAO1 | Wild-type laboratory strain | 20 |
| PAO1-LAC | PAO1 with lacIq lacZ′ inserted at attB in chromosome via mini-CTX-lacM15 | 37 |
| PAO397 | PAO1 Δ(mexAB-oprM) nfxB Δ(mexCD-oprJ) Δ(mexJKL) Δ(mexXY) Δ(mexEF-oprN) opmH+ | 28 |
| MDM1505 | PAO397 pGSV3-Spr-Paer-PA1365-luxCDABE (secA reporter) | 19 |
| MDM1519 | PAO397 pGSV3-Spr-Paer-PA0614-luxCDABE (gyrA reporter) | 19 |
| MDM1520 | PAO397 pGSV3-Spr-Paer-PA2030-luxCDABE (coaD reporter) | 19 |
| MDM1522 | PAO397 pGSV3-Spr-Paer-PA1494-luxCDABE (glmU reporter) | 19 |
| MDM1284 | PAO1-LAC ΔfabG::tet/pUCP24GW-lacPO-Paer-fabG | This study |
| MDM1325 | PAO1-LAC ΔaccD::tet/pUCP24GW-lacIqlacPO-Paer-accD | This study |
| MDM1460 | PAO397 mini-Tn7-P-PA2968(fabD)-luxCDABE | This study |
| MDM1471 | PAO397 mini-Tn7-P-PA3645(fabZ)-luxCDABE | This study |
| MDM1472 | PAO397 mini-Tn7-P-PA0500(bioB)-luxCDABE | This study |
| MDM1473 | PAO397 mini-Tn7-P-PA3112(accD)-luxCDABE | This study |
| MDM1480 | PAO397 pGSV3-Spr-P-PA4848(accC)-luxCDABE | This study |
| MDM1439 | PAO1-LAC mini-Tn7-P-PA3645(fabZ)-luxCDABE ΔaccD/pUCP24GW-lacIqlacPO-accD | This study |
| MDM1420 | PAO1-LAC mini-Tn7-P-PA3645(fabZ)-luxCDABE/pUCP24GW-lacIqlacPO (empty vector) | This study |
| MDM1440 | PAO1-LAC mini-Tn7-P-PA2968(fabD)-luxCDABE ΔaccD/pUCP24GW-lacIqlacPO-accD | This study |
| MDM1421 | PAO1-LAC mini-Tn7-P-PA2968(fabD)-luxCDABE/pUCP24-lacIqlacPO (empty vector) | This study |
| MDM1441 | PAO1-LAC mini-Tn7-P-PA0500(bioB)-luxCDABE ΔaccD/pUCP24GW-lacIqlacPO-accD | This study |
| MDM1422 | PAO1-LAC mini-Tn7-P-PA0500(bioB)-luxCDABE/pUCP24-lacIqlacPO (empty vector) | This study |
| MDM2363 | PAO397 mini-Tn7-P-PA2968(fabD)-luxCDABE ΔaccC/pUCP24GW-lacIqlacPO-E. coli-accC(WT) | This study |
| MDM2365 | PAO397 mini-Tn7-P-PA2968(fabD)-luxCDABE ΔaccC/pUCP24GW-lacIq PO-E. coli-accC(F193V) | This study |
| MDM2402 | PAO397 mini-Tn7-P-PA2968(fabD)-luxCDABE ΔaccC/pCUP24GW-lacIqlacPO-Pae-accC(WT) | This study |
| E. coli strains | ||
| K-12 | Wild type | 51 |
| KLE701 | K-12 tolC::tet | 51 |
| MDM2250 | K-12 tolC::tet accC(F193V) | This study |
| MDM2251 | K-12 tolC::tet accC(L61Q) | This study |
| MDM2252 | K-12 tolC::tet accC(L61R) | This study |
| MDM2254 | K-12 tolC::tet accC(R167H) | This study |
| MDM2298 | K-12 tolC::tet accC(F193V), derived by P1 transduction of the accC SABA-resistant allele from MDM2250 into E. coli K-12 tolC::tet | This study |
| MDM2295 | MDM2298/pUCP24GW-lacIqlacPO-E. coli accC(WT) | This study |
| MDM2296 | K-12 tolC::tet/pUCP24GW-lacIqlacPO-E. coli accC(F193V) | This study |
| MDM2430 | K-12 tolC::tet/pUCP24GW-lacIqlacPO-E. coli accC(L61R) | This study |
| MDM2431 | K-12 tolC::tet/pUCP24GW-lacIqlacPO-E. coli accC(R167H) | This study |
| Plasmids | ||
| pEX18ApGW | pEX18Ap modified with Invitrogen Gateway vector conversion kit | 52 |
| pUCP24GW | pUCP24 modified with Invitrogen Gateway vector conversion kit | 18 |
| pUCP24GW-lacIq | pUCP24GW modified by addition of lacIq at BspHI site | 18 |
| mini-Tn7-GW-lux | Mini-Tn7-GW modified to accept DraIII-cut DNA fragments proximal to the P. luminescens luxCDABE operon | 18 |
| pGSV3-lux-Spr | pGSV3-lux modified by replacement of Gmr with Spr element at SacI sites | 27 |
Commonly used E. coli hosts and cloning vectors are described and referenced in Materials and Methods.
Apr, Spr, Gmr, and Tcr indicate ampicillin, spectinomycin, gentamicin, and tetracycline resistance, respectively.
PCR and primers.
Synthetic oligonucleotide primers (Operon, Inc.) were designed using the published genome sequence for P. aeruginosa (22) or Escherichia coli (23) and web-based PRIMER3 (Whitehead Institute) (see Table S1 in the supplemental material). Primers were used at 10 μM in PCR amplifications with PhusionGC polymerase (New England BioLabs) for P. aeruginosa chromosomal DNA templates or with AccuPrime Taq DNA high-fidelity polymerase (Invitrogen, Inc.) with E. coli chromosomal DNA templates. Primers used for plasmid construction are noted below; those used for sequencing the E. coli accA, accB, accC, and accD genes following PCR amplification from sulfonamidobenzamide (SABA)-resistant mutants are shown as numbers 37 to 54 in Table S1 in the supplemental material.
Construction of P. aeruginosa complemented deletions.
Plasmid constructs to generate knockouts of accC, accD, and fabG in P aeruginosa were built in the vector pEX18ApGW by means of SOE (splicing by overlap extension) PCR and Gateway technology using primers 1 to 26 (see Table S1 in the supplemental material) as described previously (24). For deletion of the accD and fabG genes, the pEX18ApGW vectors carried a tetracycline resistance (Tetr) element in place of the genes and about 1 kb of homology on both sides of the locus to be deleted. For the accC gene, the pEX18ApGW vector carried a markerless, in-frame deletion that was flanked by about 1 kb of homologous DNA on each side. These constructs were used successfully to create merodiploids in P. aeruginosa strains PAO-LAC and PAO397, which were then resolved in the presence of a complementing copy of the appropriate wild-type or mutant gene on the extrachromosomally replicating plasmid pUCP24GW to create lac-regulated complemented deletions as described previously (18). The pUCP24 vector carried the lacIq gene for the accC and accD deletions but not for the fabG deletion, because complementation appeared to be inadequate to support growth in the presence of multiple copies of lacIq. Complemented fabG deletions were made in P. aeruginosa PAO-LAC carrying a single integrated copy of lacIq. Putative deletions were confirmed by PCR with flanking primers (primers out-F and out-R primers in Table S1 in the supplemental material) outside the regions cloned into pEX18ApGW.
Determination of mammalian-cell cytotoxicity.
The 50% cytotoxic concentration (CC50) of compounds for cultured mammalian cells (HeLa, ATCC CCL-2; American Type Culture Collection, Manassas, VA) was determined as the concentration of a compound that inhibits 50% of the conversion of MTS to formazan (25) using VP-SFM medium without serum, as previously described (26).
RNA preparation, labeling, and array hybridization.
Cultures of the lac-regulated strains MDM1284 (PAO-LAC ΔfabG/pUCP24-lacPO-fabG) and MDM1325 (PAO-LAC ΔaccD/pUCP24-lacIq-lacPO-accD) were grown in VBMM with either limiting IPTG (0.06 mM and 0.02 mM, respectively) or excess IPTG (1 mM) for about 120 min after detectable effects on growth were observed in the limiting IPTG cultures. The cultures were treated with a 2:1 mixture of RNA Protect bacterial reagent (Qiagen, Inc.) according to the manufacturer's instructions. Total RNA was prepared from each culture using an RNeasy kit (Qiagen, Inc.). Experimental replicates were generated by duplicating the entire growth, harvesting, and RNA preparation for each strain. The duplicate RNA preparations were sent to NimbleGen Systems, Inc. (Madison, WI), for labeling and hybridization to microarrays representing the P. aeruginosa PAO1 genome (22).
Construction of bioluminescent reporter strains.
The upstream regulatory regions of several target-responsive genes (i.e., fabD, fabZ, bioB, accC, and accD) were amplified by PCR (primers 27 to 36 in Table S1 in the supplemental material), cut with the restriction endonuclease DraIII or EcoRI (New England BioLabs, Inc.) as indicated by the primer name, gel purified, ligated to the Photorhabdus luminescens luxCDABE operon in the mini-Tn7 (18) or pGSV3 (27) vector, and integrated into an efflux-competent P. aeruginosa PAO1 strain (20) or into its efflux-deficient derivative PAO397 (28).
Compound screening libraries and analogs.
A high-throughput screen for FASII inhibitors (see below) was applied to 63,785 compounds in the Microbiotix, Inc., library and to 52,746 compounds in the collection at the National Screening Laboratory for the Regional Centers of Excellence in Biodefense and Emerging Infectious Disease (NSRB) at Harvard Medical School (Boston, MA). Analogs of the SABA scaffold were purchased from ChemBridge, Inc. (San Diego, CA). Liquid chromatography-mass spectrometry (LC-MS) analyses established that the analogs were ≥98% pure and of the correct molecular mass. Nuclear magnetic resonance (NMR) spectra were consistent with the assigned structures. Control compounds, including a benzoyl-amino-benzoate (BAB; compound 24c of reference 29) inhibitor of Enterococcus faecalis and Streptococcus pyogenes FabH, a pyridopyrimidine (PYP; compound 1 of reference 30) inhibitor of AccC, and a phenoxyacetamide (PhAA; compound MBX 2359 of reference 31) inhibitor of the P. aeruginosa type III secretion system (T3SS), were synthesized as described previously (29–31); thiolactomycin and ciprofloxacin were purchased (Sigma).
High-throughput screen for FASII inhibitors.
A cellular high-throughput screen (HTS) utilizing strain MDM1460 was carried out as follows. Overnight cultures of MDM1460 were inoculated from frozen stock into LB containing 10 μg/ml gentamicin. In the morning, MDM1460 was subcultured at an optical density at 600 nm (OD600) of 0.005 and grown to an OD600 of ∼0.1. Then, 100 μl of cells was added to each well of white, opaque 384-well plates containing 2 μl of screening compound per well (50 μM final concentration). The negative control was dimethyl sulfoxide (DMSO), and the positive control was the published acetyl-CoA carboxylase (ACC) inhibitor PYP (30) at 2 μM. Plates were sealed with a gas-permeable seal and incubated at 30°C overnight. In the morning, relative luminescence unit (RLU) values were measured in an Envision Multilabel multiplate reader (PerkinElmer) at an elapsed time of 18 to 25 h. Optimization studies demonstrated that the cellular assay RLU values were sensitive to DMSO, but the Z′ factor and signal/background ratio were suitable for HTS at 2% DMSO (see Fig. S1A in the supplemental material). A pilot screen of known bioactive compounds (Spectrum library; MicroSource Discovery Systems, Inc.) exhibited excellent screening parameters and identified a hit known to inhibit FASII (see Fig. S1B in the supplemental material). A Z score was calculated for each screened compound as the number of multiples of the DMSO negative-control standard deviation that the sample signal exhibited above the DMSO signal. A hit was defined as a compound that exhibited a Z score of ≥5.
MIC.
MICs were determined using the broth microdilution method described in the CLSI (formerly NCCLS) guidelines (32) and are expressed in micromolar units to facilitate convenient comparisons with the activities of the compounds in other assays. The upper limit of concentrations evaluated was 100 μM, since this is the aqueous solubility limit of the particular SABA analogs described here.
Selection for SABA-resistant mutants.
Approximately 109 cells from concentrated frozen stocks of E. coli tolC::tet in LB were spread on LB agar plates containing 12.5 μg/ml tetracycline and 2.6 μg/ml SABA-1 (about 8-fold MIC) and incubated overnight at 37°C. Colonies were picked and retested in MIC assays to identify clones which had resistance to SABA-1 and -2 but which exhibited unaltered sensitivity to three unrelated antibiotics. Genomic DNA was prepared from seven independent colonies with specific resistance to SABA-1 and -2. The accA, accB, accC, and accD genes were sequenced from all seven clones using the primers indicated in Table S1 in the supplemental material.
MMS evaluations.
Macromolecular synthesis (MMS) assays for lipid, protein, RNA, and DNA pathway labeling were performed essentially as described previously (33). Briefly, bacterial cells were grown to an OD600 of 0.5 before being split into 10-ml aliquots and incubated with a given inhibitor for 15 min. Then, either [14C]acetate (10 μCi) to determine fatty acid synthesis, a 3H-labeled amino acid mix (10 μCi) to determine protein synthesis, [3H]uracil (10 μCi) for stable RNA synthesis, or [3H]thymidine (10 μCi) for DNA synthesis was added to the cultures for 30 min. The labeled cells were then collected via vacuum filtration, and the radioactivity was counted and normalized to the OD600 of the culture. The amount of label incorporated by the untreated cell population was set at 100%. Averages and standard errors were obtained from triplicate samples derived from biological duplicate experiments.
E. coli ACC assay.
The radiochemical E. coli ACC assay used the reconstituted AccABCD multiprotein complex to determine the 50% inhibitory concentrations (IC50s) for SABA analogs. The assay was modeled after the work of Alves et al. (34); however, instead of using liquid chromatography-tandem mass spectrometry (LC-MS/MS) to detect malonyl-CoA, we monitored the rate of [14C]CO2 fixation onto acetyl-CoA. Briefly, AccAD were coexpressed and purified as a tetramer. AccC was expressed and purified as a dimer. Apo-AccB was purified and converted to AccB using purified BirA to biotinylate the carrier protein. Complete biotinylation was checked by intact protein mass spectrometry. The calculated mass of AccB is 18,850, and our observed mass was 18,719; the difference is accounted for by the presence of a histidine tag and the removal of the N-terminal methionine in the cells. The observed mass of the holo-AccB was 18,945, consistent with the addition of a biotin group with a mass of 226. The three protein preparations were mixed together (AccAD-AccC-AccB; 0.125/0.125/0.25 μM) and preincubated for 20 min at room temperature. The assay mixture contained 0.5 mM ATP, 5 mM MgCl2, 50 mM KCl, 1 mM dithiothreitol, and 0.1 M Tris-HCl (pH 8.0) in a total volume of 50 μl. NaH14CO2 (1 mM; 55 mCi/mmol) was added to start the reaction, and the mixture was incubated at 37°C for 5 min. The reaction was stopped by adding 12.5 μl of 6 N HCl, and the mixture was applied to a 2.3-cm Whatman filter disc and dried prior to scintillation counting. The assay was linear and reproducible, and unlike the spectrophotometric assays for the half-reactions, the fluorescent and UV absorbing properties of compounds did not interfere.
Array data accession number.
The gene expression profiles based on the NimbleGen, Inc., array data have been deposited in the NCBI Gene Expression Omnibus (GEO) database (www.ncbi.nlm.nih.gov/geo), which is a public functional genomics data repository supporting MIAME-compliant data submissions, under accession number GSE66045.
RESULTS
FASII genes are coordinately upregulated when fatty acid synthesis is impeded.
To detect FASII inhibitors that are active against P. aeruginosa cells, we undertook the construction of screening strains that would report inhibition of the FASII pathway as an increased luminescent signal. This strategy combined the simplicity and speed of a homogenous assay with the low cost of autobioluminescence and the enhanced specificity of a gain-of-signal assay. We used expression profiling by microarray hybridization to identify genes that are induced in response to reduced FASII pathway flow. Our strategy was to fuse FASII depletion-responsive promoters to the P. luminescens luxCDABE operon and create reporter strains for screening. To simulate reduced FASII pathway flow, we constructed P. aeruginosa strains in which the expression of FASII genes could be reduced to levels that inhibit normal growth. Since most genes encoding the FASII pathway are essential (35), these strains were constructed by deleting the essential gene in the presence of a Plac-regulated complementing copy carried on the replicating plasmid pUCP24GW (18, 19), enabling the expression of the FASII genes to be regulated by IPTG. Accordingly, we generated P. aeruginosa strains with regulated expression of accD, which encodes a subunit of the FASII initiation activity acetyl-CoA carboxylase, and fabG, which encodes β-ketoacyl-acyl carrier protein (ACP) reductase, the first reduction step in the FASII elongation cycle. As expected, both of these strains, MDM1284 (fabG) and MDM1325 (accD), failed to grow in the absence of IPTG. MDM1284 and MDM1325 were subjected to IPTG limitation during growth and harvested for RNA preparation when detectable effects on the growth rates were observed (∼1 to 2 h later). The resulting RNA was used to measure changes in gene expression by microarray hybridization (see Materials and Methods).
To identify target-responsive genes, we searched the resulting normalized gene expression levels (GEO accession number GSE66045) for genes that are upregulated in response to downregulation of each FASII gene. A total of 147 genes were upregulated ≥4-fold (≥2-log2 change) in response to downregulation of accD in the ΔaccD strain (MDM1325). These responsive genes fell into a variety of functional categories based on clusters of orthologous groups (COGs) (36); however, 12.6% and 7.5% of the upregulated genes are involved in translation and lipid metabolism, respectively (Fig. 1A). A total of 23 genes exhibit an upregulated expression response of a ≥8-fold change (≥3 log2), 13 of which encode proteins involved in lipid metabolism or biotin cofactor synthesis (Fig. 1A and B). Similarly, 28 genes were upregulated ≥4-fold (≥2-log2 change) in the complemented ΔfabG strain in response to downregulation of fabG, including 10 of the 13 genes involved in lipid metabolism or biotin cofactor synthesis in the ≥3-log2 set from the ΔaccD strain. The three missing genes in the lipid metabolism COG category are fabG itself, which was downregulated in this engineered strain, fabD, whose expression may have been affected by polarity due to its linkage to fabG, and fabB, which was upregulated but only 2.6-fold (1.4-log2 change). The lower levels of upregulation in general from the complemented ΔfabG strain were probably due to the accumulation of mutations in the single lacIq copy integrated into the chromosome of the PAO-LAC strain (37) (see Materials and Methods). All but two of the 13 highly upregulated lipid metabolism and biotin cofactor genes identified in the ΔaccD strain play known roles in the FASII pathway (Fig. 1B). The two exceptions are PA3644 (lpxA), which is likely upregulated because of its linkage in an apparent operon with fabZ, and PA3267, encoding a hypothetical protein of unknown function that clusters with several phospholipid/glycerol acyltransferases. Conversely, the only putative FASII genes that are not highly upregulated in the IPTG-limited growth of the ΔaccD strain are fabH1 (PA0999), fabH2 (PA3333), and fabF2 (PA1373), which have been shown previously to play no known role in FASII (38), and fabI (PA1806), whose encoded function is duplicated in P. aeruginosa by a triclosan-resistant enoyl-ACP reductase encoded by fabV (PA2950) (39). The accD gene was not highly upregulated because it was engineered in this strain; however, it exhibited a 5-fold (2.3-log2 change) during IPTG-limited growth of the ΔfabG strain (see GSE66045).
FIG 1.

Summary of expression profiling of P. aeruginosa ΔaccD cells complemented by lac-promoted accD in limited versus excess IPTG inducer levels. (A) Abundance of genes upregulated ≥4-fold (gray bars) and ≥8-fold (black bars) as a percentage of genes in each COG category. (B) Log2 fold upregulation of FASII-related genes from the lipid metabolism COG category. Error bars represent standard deviations (n = 5).
FASII-sensitive promoter-lux fusions respond to downregulated FASII genes.
To confirm the FASII depletion response of the genes identified in the expression profiling experiments, the upstream regulatory regions of several upregulated genes (accC, accD, fabD, fabZ, and bioB) were fused to the P. luminescens luxCDABE operon and transferred to the complemented ΔaccD deletion strains by using either mini-Tn7 integrations (18) or single-crossover insertions with pGSV3 (27). Cells with regulated complemented deletions or wild-type cells carrying the same reporter constructs were grown overnight in various inducer (i.e., IPTG) concentrations and were then subcultured in the morning without inducer. The resulting cultures were examined for their luminescent response to downregulation of accD. For example, reduction in accD expression in the PfabD-lux strain MDM1440 decreased the growth rate and induced luminescence (Fig. 2A). As expected, a wild-type strain carrying the same reporter construct, MDM1421, grew to a higher density than the IPTG-limited deletion strain but exhibited the lowest level of luminescence throughout its growth. The more rapid increase in RLU production by the complemented deletion versus the wild-type strain produced an increasingly larger separation between the luminescent responses of the two strains. The luminescence values (RLU) for three wild-type and complemented ΔaccD strain pairs containing three different FASII gene promoter fusions to luxCDABE (i.e., fabD, fabZ, and bioB), which were normalized to cell density (OD600), revealed a consistent luminescent response of cells to FASII pathway depletion by lowered IPTG at 270 min (Fig. 2B). These results were consistent with the expected responses of the FASII promoters to impedance of the FASII pathway, as determined by expression profiling.
FIG 2.

Luminescence reports from wild-type and regulated complemented ΔaccD strains. (A) Kinetics of growth (OD600 [open symbols, dotted lines]) and luminescence (RLU [closed symbols, solid lines]) for the ΔaccD/pUCP24-lacIqlacPO-accD strain MDM1440 and the wild-type PAO1 strain MDM1421, both carrying mini-Tn7-PfabD-luxCDABE reporter constructs (circles, WT; triangles, ΔaccD strain in 0.05 mM IPTG; diamonds, ΔaccD strain in 0.025 mM IPTG). (B) Luminescence normalized to cell number (RLU/OD600) at 270 min of growth in LB for the complemented ΔaccD strains (black) MDM1439, MDM1440, and MDM1441 in 0.025 mM IPTG and wild-type strains (gray) MDM1420, MDM1421, and MDM1422 carrying three different FASII depletion reporters as indicated. Error bars represent standard deviations (n = 3).
FASII-sensitive promoter-lux fusions respond to FASII inhibitors.
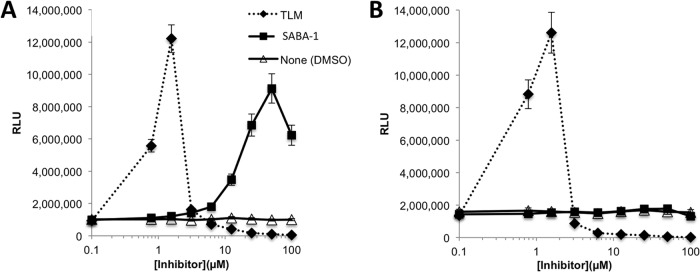
In preparation for high-throughput screening, we evaluated the responses of several reporter strains to known FASII inhibitors. Because most existing FASII inhibitors are subject to efflux by P. aeruginosa (35, 40), we integrated the PFASII-luxCDABE reporter constructs into the genome of PAO397, a derivative of PAO1 in which five RND-family efflux pump genes have been deleted (28), to generate FASII reporter strains with increased sensitivity to FASII inhibitors. We investigated the sensitivity, kinetics, and specificity of the luminescent response of the resulting strains to treatment with the FabB inhibitor thiolactomycin (TLM) (41) and a pyridopyrimidine (PYP) AccC inhibitor (30). As controls, the reporter strains were treated with inhibitors of targets unrelated to P. aeruginosa FASII, including the gyrase inhibitor ciprofloxacin (CIP), a phenoxyacetamide (PhAA) inhibitor of P. aeruginosa type III secretion (31), and a benzoylamino-benzoate (BAB) targeting E. faecalis and S. pyogenes FabH (29). The PfabD-lux reporter strain MDM1460 produced highly significant luminescence in response to doses of TLM and PYP 20- to 30-fold below their MICs (Fig. 3A and Table 2) but did not respond to CIP, PhAA, or BAB.
FIG 3.

Sensitivity, kinetics, and specificity of luminescent response of reporter strains to known FASII inhibitors. (A) Concentration dependence of RLU response of the efflux-deficient FASII reporter strain MDM1460 (PfabD-lux) at 20 h to two FASII inhibitors: a pyridopyrimidine AccC inhibitor (PYP) and a thiolactomycin FabB inhibitor (TLM). Also included are several non-FASII inhibitor negative controls: a phenoxyacetamide inhibitor of P. aeruginosa type III secretion (PhAA), the gyrase inhibitor ciprofloxacin (CIP), and a benzoyl aminobenzoate (BAB) inhibitor of E. faecalis and S. pyogenes FabH. Two active analogs of the SABA screening hit series, SABA-1 and SABA-2, and one inactive analog are shown. (B) RLU values versus time of incubation in the presence of sub-MICs of FabB inhibitor TLM (1.25 μM, closed symbols) or DMSO (open symbols) for efflux-deficient P. aeruginosa strains carrying luxCDABE driven by the accC (blue squares) (MDM1480) or fabD (red circles) (MDM1460) promoter. (C) Z score of RLU values after 20 h of incubation of efflux-deficient reporter strains in the presence of PYP (2 μM), TLM (1.25 μM), BAB (20 μM), a PhAA T3SS inhibitor (20 μM), and ciprofloxacin (CIP) (2 ng/ml) compared to DMSO. Promoter elements driving luxCDABE and their inhibitory response pathways are given in the key and were integrated into PAO397 to generate strains MDM1460, MDM1473, MDM1505, MDM1419, MDM1520, and MDM1522. Error bars represent standard deviations (n = 3).
TABLE 2.
MICs of FASII inhibitors for E. coli and P. aeruginosa wild-type, mutant, and engineered strains
| Strain | Description | MIC (μM) |
|||||
|---|---|---|---|---|---|---|---|
| CIP | TLM | PYP | SABA-1 | SABA-2 | SABA-3 | ||
| MDM2170 | E. coli | 0.06 | 300 | 40 | >100 | >100 | >100 |
| MDM2171 | E. coli tolC::tet | 0.03 | 30–60 | 0.5–1.0 | 0.45–0.9 | 3.5–7.0 | >100 |
| MDM2252 | E. coli tolC::tet accC(L61R) | 0.03 | 30–60 | 0.5–1.0 | >100 | >100 | >100 |
| MDM2254 | E. coli tolC::tet accC(R167H) | 0.03 | 30–60 | 0.5–1.0 | >100 | >100 | >100 |
| MDM2250 | E. coli tolC::tet accC(F193V) | 0.03 | 30–60 | 0.5–1.0 | >100 | >100 | >100 |
| MDM2295 | E. coli tolC::tet accC(F193V)/pUCP24-lacPO-E. coli accC(WT) | 0.03 | 30–60 | 0.5–1.0 | 25–50 | 25 | >100 |
| MDM2296 | E. coli tolC::tet/pUCP24-lacPO-E. coli accC(F193V) | 0.03 | 30–60 | 0.5–1.0 | 25 | 25 | >100 |
| MDM2430 | E. coli tolC::tet/pUCP24-lacPO-E. coli accC(L61R) | 0.03 | 30–60 | 0.5–1.0 | 25 | 25–50 | >100 |
| MDM2431 | E. coli tolC::tet/pUCP24-lacPO-E. coli accC(R167H) | 0.03 | 30–60 | 0.5–1.0 | 25 | 12–25 | >100 |
| MDM1233 | P. aeruginosa PAO1 | 0.18 | >100 | >160 | >100 | >100 | >100 |
| MDM1370 | P. aeruginosa PAO397 | 0.015 | 48 | 25–40 | >100 | >100 | >100 |
| MDM2263 | PAO397 ΔaccC/pUCP24-lacPO-E. coli accC(WT) | 0.015 | 14–24 | 0.8 | >100a | >100a | >100 |
| MDM2265 | PAO397 ΔaccC/pUCP24-lacPO-E. coli accC(F193V) | 0.015 | 14–24 | 0.8 | >100 | >100 | >100 |
| MDM2402 | PAO397 ΔaccC/pUCP24-lacPO- P.aeruginosa accC(WT) | 0.015 | 14–24 | 25 | >100b | >100b | >100 |
The time course of the luminescent response was examined with TLM at a concentration that was 8-fold below its MIC, using two different FASII reporter strains, PfabD-lux and PaccC-lux. The differences in the luminescent responses of the reporter cells to TLM versus the diluent DMSO at 270 min (Fig. 3B) were not as significant as those observed for reporter strains carrying wild-type and downregulated FASII genes (Fig. 2). However, cultures treated with TLM continued to produce luminescence after 21 h, and the luminescent responses were significantly elevated in the presence of the sub-MIC of TLM compared to the diluent DMSO. The reporter luminescent signal declined slightly from 21 h to 28 h but remained significantly selective for FASII inhibitor TLM throughout the observation period during the second day (>5-fold for PfabD-lux; >3-fold for PaccC-lux), providing a durable response to TLM and a choice of time points for an endpoint reading in a screen.
Finally, to demonstrate that the reporter strain responses are highly selective for FASII inhibitors and promoters, we evaluated two FASII inhibitor-sensitive reporter strains and four reporter strains designed to respond to inhibitors of other targets (18, 19, 27) for sensitivity to five inhibitors of FASII and non-FASII targets (Fig. 3C). The FASII inhibitor-sensitive reporter strains carrying PfabD-lux and PaccD-lux responded to the FASII inhibitors TLM and PYP but not to CIP, PhAA, or the BAB inhibitor of FASII, found in Gram-positive strains. The response of the PPA2030-lux reporter strain to incubation with BAB was unexpected and may indicate a degree of nonspecificity for BAB. Conversely, the non-FASII reporters did not respond to TLM or PYP, but the reporter for gyrase inhibitors (PPA0614-lux) did respond to ciprofloxacin as described previously (18). In summary, the luminescent signals generated by the FASII reporter strains were selectively induced in response to FASII inhibitors that target the initiation stage (AccC) or the elongation cycle (FabB) of the FASII pathway. In addition, the reporters were sensitive to inhibitors at levels as low as 1/30 the MIC, confirming the potential utility of these reporter strains for high-throughput screening to identify inhibitors of the FASII pathway.
Implementation of a high-throughput cellular bioluminescent reporter screen for inhibitors of the FASII pathway.
To develop a high-throughput screen (HTS) for FASII inhibitors, we selected the PfabD-luxCDABE reporter strain MDM1460 because it exhibited the strongest and most reproducible luminescent response to the known FASII inhibitors TLM and PYP. The cellular screening parameters, including culture volume, cell density, compound concentration, endpoint time, and DMSO concentration tolerance, were adjusted to optimize the luminescent response in 384-well microplates, as measured by Z′ factor (42), using 2 μM PYP and the diluent DMSO as positive and negative controls, respectively (see Materials and Methods). The optimized screen produced signal-to-background ratios (S/B) and Z′ factors in each 384-well screening plate typical of those shown in Fig. S1 in the supplemental material (S/B = 8.3; Z′ factor = 0.68). We applied the HTS to over 115,000 discrete small molecules, selected potential inhibitors with Z scores of ≥5, confirmed them by reassay in the primary screen, and validated them based on the following features: (i) specificity for FASII (i.e., highly significant Z scores of ≥5 versus PfabD-lux and PaccD-lux reporter strains but much reduced and mostly insignificant Z scores versus non-FASII reporter strains); (ii) minimal cytotoxicity versus HeLa cells (CC50 ≥ 80 μM), as measured in serum-free medium to ensure that serum binding did not mask cytotoxicity; and (iii) selectivity for inhibition of lipid synthesis versus inhibition of DNA synthesis in preliminary macromolecular synthesis assays (see Materials and Methods). Five compounds representing three distinct chemical scaffolds met these stringent criteria (data not shown). Here, we focus on two of these compounds, which share a core sulfonamidobenzamide (SABA) scaffold, SABA-1 and SABA-2, together with the structurally related but inactive SABA-3 (Table 3 and Fig. 3A and C).
TABLE 3.
Characterization of SABA screening hits and negative control in multiple assays

SABA compounds target the biotin carboxylase (AccC) of acetyl-CoA carboxylase.
To determine the molecular target of the SABA analogs, we first evaluated them for inhibition of the growth of efflux-competent and -deficient strains of P. aeruginosa and E. coli (Table 2). The two active SABA analogs in the P. aeruginosa reporter assay, SABA-1 and SABA-2, failed to exhibit antibacterial activity against wild-type E. coli or P. aeruginosa at concentrations up to 100 μM (45 μg/ml). However, SABA-1 and SABA-2 did exhibit MICs in the range of 0.45 to 9 μM (0.2 to 0.4 μg/ml) and 3.5 to 7 μM (1.5 to 3.0 μg/ml), respectively, when tested against an efflux-deficient strain of E. coli (ΔtolC) (Table 2), indicating that efflux likely rescued E. coli cells from growth inhibition. Furthermore, the compounds reduced the growth rate (increased the exponential-phase doubling time) of the efflux-deficient P. aeruginosa strain PAO397, but not the wild-type strain PAO1, by 30 to 40% when added at 50 μM (24 μg/ml) (Table 3; also, see Fig. S2 in the supplemental material), and they exhibited statistically significant increases in RLU values in the corresponding efflux-deficient reporter strain (MDM1460) at compound concentrations as low as 6 to 10 μM (3 to 5 μg/ml) (Fig. 3A). By comparison, the structurally related SABA-3 failed to generate a luminescent report in the screening strain and did not produce a significant effect in the growth or reporter assays (Fig. 3A and C and Table 2; also, see Fig. S2 in the supplemental material). These results demonstrated that SABA-1 and -2 are subject to efflux from E. coli and P. aeruginosa but are capable of inhibiting growth of E. coli and the growth rate of P. aeruginosa when efflux is reduced.
Next, we verified that SABA-1 and SABA-2 inhibit lipid synthesis selectively in the efflux-deficient P. aeruginosa strain PAO397 by examining their effect on cellular assays measuring the synthesis of several key macromolecules, including DNA, RNA, protein, and lipids. SABA-1 and SABA-2 inhibited acetate incorporation into lipids at least 2-4-fold more potently than they inhibited DNA, RNA, or protein synthesis, while SABA-3 did not inhibit any of the pathways significantly (Fig. 4A). This reduction in fatty acid synthesis is consistent with the growth rate inhibition observed for these compounds but is insufficient to generate detectable MICs in efflux-deficient P. aeruginosa.
FIG 4.
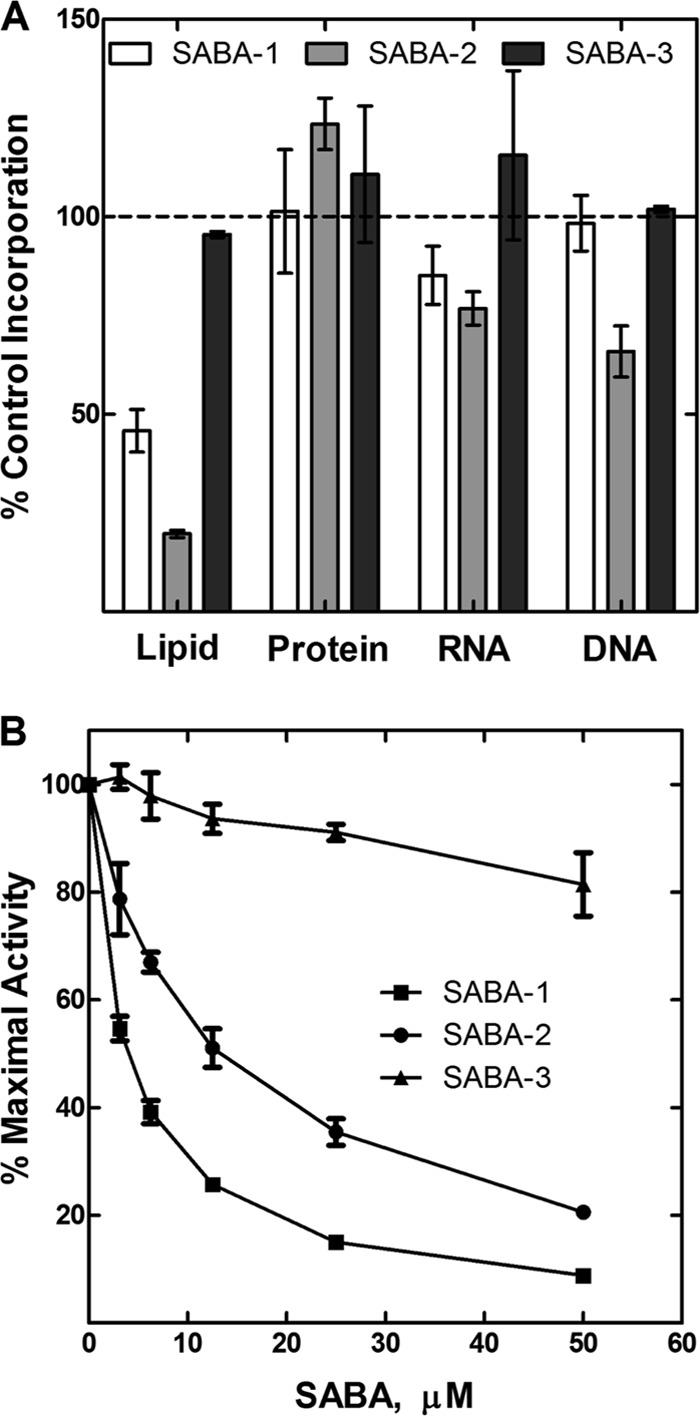
Identification of the SABA mechanism of action. (A) Effect of three SABA analogs at 50 μM on the synthesis of cellular macromolecules in P. aeruginosa PAO397 as measured by incorporation of radiolabeled precursors (see Materials and Methods). (B) Effect of three SABA analogs on the activity of purified E. coli acetyl-CoA carboxylase (ACC).
The antibacterial activity of SABA-1 and SABA-2 against the efflux-deficient strain of E. coli allowed a direct genetic approach to identifying the pathway target. Mutants resistant to SABA-1 at 8-fold its MIC were selected in the E. coli ΔtolC strain and arose at a frequency of about 10−9. Seven independent mutants exhibited resistance to SABA-1 and SABA-2 but not to PYP, TLM, or ciprofloxacin (e.g., strains MDM2250, MDM2252, and MDM2254 in Table 2). Since SABA analogs failed to inhibit the elongation cycle of FASII in a reconstituted E. coli fatty acid synthase assay (43) (data not shown), we reasoned that they do not target any of the enzymes comprising the two-carbon addition cycle (e.g., FabH, FabD, FabB, FabG, FabI, and FabZ). Therefore, we focused on acetyl-CoA carboxylase (ACC), which catalyzes the initial step in FASII and is not measured in the elongation cycle assay. Accordingly, we sequenced all four acc genes from all seven SABA-resistant mutants. Each of the strains contained a single codon-altering mutation only in accC. We found 5 distinct missense mutations that altered one of three amino acid codons: L61R, L61Q, R167G, R167H, and F193V. All three amino acid residues are conserved in E. coli and P. aeruginosa AccC (see Fig. S3 in the supplemental material), suggesting that the mutations may also affect P. aeruginosa susceptibility to SABA-1 and SABA-2. To verify the role of the mutations in accC in the resistance phenotype, we transferred the resistance from one of the original spontaneous resistant mutants [accC(F193V)] into the wild-type E. coli tolC::tet strain using P1 phage transduction and selected SABA-resistant transductants. DNA sequencing of SABA-resistant transductants revealed the presence of the F193V mutation in the accC locus, indicating that the single accC mutation is necessary and sufficient to confer resistance to SABA-1. Next, we expressed the WT, F193V, R167H, and L61R accC alleles in E. coli and P. aeruginosa using the expression vector pUCP24GW-lacIqlacPO (19). In MIC assays, expression of the plasmid-encoded copy of the accC(F193V), accC(R167H), and accC(L61R) alleles produced SABA resistance in E. coli ΔtolC carrying a wild-type chromosomal copy of the accC gene. In contrast, the plasmid-encoded wild-type E. coli accC allele had a minimal effect (≤2-fold) on the MIC when added to the SABA-resistant mutant E. coli strains (Table 2; also, see Fig. S4 in the supplemental material). As expected, addition of the SABA-resistant accC allele failed to alter the MICs for ciprofloxacin or PYP against E. coli ΔtolC cells. Taken together, these results indicate that the SABA-resistant allele is dominant in E. coli.
The ability of the SABA compounds to inhibit E. coli acetyl-CoA carboxylase (ACC) activity was determined using an in vitro biochemical assay (34). Acetyl-CoA carboxylase is a biotin-dependent, multifunctional enzyme that catalyzes the first regulated step in fatty acid synthesis. The enzyme complex is composed of a homodimeric biotin carboxylase (AccC2), biotinylated biotin carboxyl carrier protein (AccB, four copies), and an AccA2AccD2 heterotetrameric carboxyltransferase (44) and catalyzes two half-reactions to form malonyl-CoA. SABA-1 and SABA-2 inhibited ACC with IC50s of 4 and 12.5 μM, respectively (Fig. 4B). In contrast, SABA-3 failed to show any significant inhibition of E. coli ACC (Fig. 4B). Additional experiments will be required to determine the exact biochemical mechanism for SABA inhibition of ACC, but these results are consistent with the genetic locus of the resistance mutations and indicate that AccC is the target of SABA-1 and SABA-2.
The genetic experiments were expanded to confirm that the SABA compounds target AccC in P. aeruginosa. The wild-type P. aeruginosa accC gene was deleted from the FASII reporter strain MDM1460 in the presence of lac-regulated complementing copies of each of three different accC alleles—the P. aeruginosa wild-type allele and the E. coli wild-type and F193V resistant alleles. Growth of each of the three resulting P. aeruginosa strains with complemented deletions was directly proportional to the IPTG concentration, with little or no growth in the absence of IPTG, and the luminescent response was inversely proportional to the IPTG level except at very low IPTG levels, which inhibited growth very stringently (see Fig. S5 in the supplemental material). The presence of the E. coli accC(F193V) mutant allele, but not the wild-type E. coli accC allele, eliminated the bioluminescent response of P. aeruginosa PAO397 cells carrying the PfabD-lux reporter to SABA-1 but not to TLM (Fig. 5). Furthermore, the final cell density of the parental P. aeruginosa strain PAO397 in stationary phase is decreased by thiolactomycin and ciprofloxacin, but not by SABA-1 (Fig. 6A1 to A3). In contrast, the final cell densities of P. aeruginosa accC deletion strains complemented by a lac-regulated wild-type P. aeruginosa accC allele or by a lac-regulated E. coli accC allele were reduced by SABA-1 in an IPTG-dependent manner (Fig. 6B1 to B3 and C1 to C3), and complementation by the lac-regulated E. coli resistant accC(F193V) allele significantly alleviated the growth sensitivity to SABA-1 (Fig. 6D1 to D3) with no effect on the sensitivity of cells to CIP and PYP. These results confirmed that specific mutations in the accC gene confer resistance to SABA compounds in P. aeruginosa as well as in E. coli.
FIG 5.
Effect of a SABA-resistant accC allele on the bioluminescent response of a P. aeruginosa FASII reporter strain. PAO397 reporter (PfabD-luxCDABE) cells were engineered to carry the E. coli wild-type accC gene (A) or the E. coli SABA-resistant accC(F193V) allele (B) on the lac-regulated pUCP24GW-lacIqlacPO plasmid in place of the chromosomal copy of the accC gene and incubated with a concentration range of SABA-1, TLM, or diluent (DMSO) as shown. Luminescence (RLU) was measured after 20 h. Error bars represent standard deviations (n = 3).
FIG 6.

Effect of wild-type and SABA-resistant accC alleles on the growth of P. aeruginosa PAO397 in the presence of inhibitors. The extent of growth was measured at 24 h for PAO397 carrying the endogenous chromosomal copy of the wild-type accC gene (A) or for PAO397 engineered to carry the wild-type P. aeruginosa accC gene (MDM2402) (B), the E. coli wild-type accC allele (MDM2363) (C), or the E. coli SABA-resistant accC(F193V) allele (MDM2365) (D), all under lac regulation, on the pUCP24GW-lacIqlacPO plasmid in place of the chromosomal copy of the accC gene. Cells were incubated overnight in the presence of a concentration range of TLM (series 1), ciprofloxacin (series 2), or SABA-1 (series 3) in the presence of the indicated IPTG concentration (see the key in panel A2), and growth was measured as OD600.
DISCUSSION
This study describes a new cell-based reporter screen for inhibitors of the FASII pathway and its validation by the identification of a novel chemical series of inhibitors of ACC in the initiation stage of FASII. These new screening strains are based on the cellular transcriptional response to depletion of FASII. The observation that the genes induced strongly in response to FASII depletion in P. aeruginosa primarily encode FASII enzymes permits the construction of gain-of-signal reporter strains with promoters mechanistically linked to the targeted pathway. Application of the FASII cellular reporter screens in this study illustrates several important benefits of these types of screens, including the following: (i) simplicity and low cost of use, (ii) gain-of-signal output that reduces the frequency of false positives such as toxic compounds or luciferase inhibitors, (iii) sensitivity that is superior to that of cell growth-based outputs, (iv) the ability to detect inhibitors that cross the poorly permeable P. aeruginosa outer membrane, (v) the ability to detect inhibitors of several different steps in the pathway, and (vi) specificity for FASII. There are also limitations. For example, the high degree of sensitivity of the reporter screens to FASII inhibition is both beneficial and potentially problematic for screening. It enables the identification of weak inhibitors, providing an entry point for chemistry for further optimization as drugs. This sensitivity may prove useful in screening fragment libraries to identify small scaffolds but may preclude the identification of FASII inhibitors that are capable of killing cells or FASII inhibitors with cytotoxic off-target activities, since the reporter strain would be unable to produce a significant signal. In practice, such a scenario is unlikely, because few compounds in current small-molecule libraries are capable of arresting the growth of P. aeruginosa. This may be a more significant limitation for screens using efflux-deficient P. aeruginosa, as described in this report, but the efflux-deficient reporter strain improves the odds of detecting novel FASII inhibitors.
While the reporter screens described here respond to reduction in the flux through the FASII pathway, they may not detect inhibitors of each of the steps in the pathway for both genetic and biochemical reasons. First, there is redundancy of genes encoding enzymes for some pathway steps in P. aeruginosa. P. aeruginosa contains two AcpP proteins, two enoyl-ACP reductases (a triclosan-sensitive FabI and an insensitive FabV [39]), as well as multiple condensing enzymes (38). Three of the condensing enzymes appear to be critical in P. aeruginosa, controlling initiation condensation (FabY) and elongation condensation for saturated (FabF) and unsaturated (FabB) fatty acids (45, 46). Each of these three condensing enzymes is a potential drug target. The others are nonessential for P. aeruginosa growth in laboratory media and include a bypass condensation enzyme (FabH3) utilizing C8-CoA (47), an enzyme used for quinoline signal synthesis (FabH1; also called PqsD) not directly related to essential cell membrane synthesis (48), and two that are of unknown function and upregulated weakly in response to FASII inhibition (FabF2, FabH2). Second, some FASII steps appear to catalyze rapid equilibrium reactions, reducing the likelihood that inhibitors can be identified. These include the FabD and FabZ enzymes (49). Therefore, the most likely P. aeruginosa FASII steps to yield inhibitors are ACC, the elongation cycle-condensing enzymes FabB and FabF, the reductase FabG, and the initiation-condensing enzyme FabY (49).
The specificity of the screen is limited by the fact that other cellular stresses in addition to FASII depletion could lead to upregulation of the reporter. Because it is not feasible to examine every possible stressor, it is crucial to evaluate primary hits in secondary assays. In this case, we used additional luminescent reporter assays to rapidly identify compounds that affect only FASII-related reporters. Then, we applied another cellular assay, the relative inhibition of the incorporation of radiolabeled precursors into lipids and DNA. Inhibitors that demonstrated selectivity for inhibition of lipid synthesis were evaluated further using the genetic approach of resistance mutation selection. The selection and mapping of mutations conferring SABA resistance in E. coli indicates that the molecular target of SABA analogs is the biotin carboxylase AccC. Furthermore, our observation that the addition of any of the resistant accC alleles in E. coli ΔtolC strains on an extrachromosomal plasmid results in SABA resistance indicates that accC is the only SABA target in the cell necessary for growth inhibition. While this approach is very powerful, it is not applicable if the compound does not exhibit an MIC. In such cases, biochemical assays will be required to identify the targeted protein. In this case, we used biochemical assays to provide support for AccC as the target of the SABA series, and the results are quite consistent with this hypothesis. Further research is under way to fully characterize the mechanism of SABA inhibition of ACC.
At least two other AccC inhibitors, PYP and a benzimidazole, have been described and cocrystallized with AccC (PDB accession numbers 2V58 [30] and 3JZF [50], respectively). Both inhibitors bind in or near the ATP binding site, and four codons that can be mutated to give rise to PYP resistance have been identified (30). These residues line the ATP binding site and are distinct from the three residues that are altered in the SABA-resistant mutants (see Fig. S3 in the supplemental material). The sites of SABA-resistant mutations are located on the other side of the protein and appear to surround a cleft that may provide access for substrates (see Fig. S6 in the supplemental material). These mutations do not confer resistance to PYP (Table 2; also, see Fig. S3 in the supplemental material). Thus, we hypothesize that the SABA inhibitors bind AccC at a site distinct from those for PYP and the benzimidazole and likely do not interfere with ATP binding. The novelty of the SABA scaffold and its potentially unique binding site on AccC suggest the potential clinical utility for this series if the antibacterial activity can be optimized. Clearly, chemical optimization must include efforts to reduce efflux from Gram-negative bacteria, and application of the cellular screen configured in an efflux-competent host will provide an extremely sensitive cellular luminescent assay to identify analogs capable of escaping efflux.
In conclusion, the characterization of two SABA primary screening hits demonstrates that the novel screens described here provide sensitive detection of new chemical entities targeting the FASII pathway. As cellular pathway screens, they offer the potential to identify new cell-permeant inhibitors of a variety of FASII pathway steps. By using an efflux competent host strain, the reporter may also allow detection of new classes of FASII inhibitors that can penetrate the P. aeruginosa wall and membrane and also evade efflux; at least, such reporter strains will provide a rapid screening tool for identification of analogs that are less subject to efflux pumps in P. aeruginosa.
Supplementary Material
ACKNOWLEDGMENTS
We thank Herbert Schweizer (University of Florida) for advice and for providing plasmids. We acknowledge Lindsay Burt for help in making the P. aeruginosa complemented ΔaccC strain. We thank Su Chiang (NSRB, Harvard Medical School, Boston, MA) for assistance and coordination of the screening of the NSRB library.
This work was supported by National Institutes of Health grants AI079986 (D.T.M.) and GM034496 (C.O.R.), Cancer Center Support Grant CA21765, and the American Lebanese Syrian Associated Charities.
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.00686-15.
REFERENCES
- 1.Jarvis WR. 2003. Benchmarking for prevention: the Centers for Disease Control and Prevention's National Nosocomial Infections Surveillance (NNIS) system experience. Infection 31(Suppl 2):S44–S48. [PubMed] [Google Scholar]
- 2.Malangoni MA, Crafton R, Mocek FC. 1994. Pneumonia in the surgical intensive care unit: factors determining successful outcome. Am J Surg 167:250–255. doi: 10.1016/0002-9610(94)90086-8. [DOI] [PubMed] [Google Scholar]
- 3.Garau J, Gomez L. 2003. Pseudomonas aeruginosa pneumonia. Curr Opin Infect Dis 16:135–143. doi: 10.1097/00001432-200304000-00010. [DOI] [PubMed] [Google Scholar]
- 4.Flamm RK, Weaver MK, Thornsberry C, Jones ME, Karlowsky JA, Sahm DF. 2004. Factors associated with relative rates of antibiotic resistance in Pseudomonas aeruginosa isolates tested in clinical laboratories in the United States from 1999 to 2002. Antimicrob Agents Chemother 48:2431–2436. doi: 10.1128/AAC.48.7.2431-2436.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hernandez V, Crepin T, Palencia A, Cusack S, Akama T, Baker SJ, Bu W, Feng L, Freund YR, Liu L, Meewan M, Mohan M, Mao W, Rock FL, Sexton H, Sheoran A, Zhang Y, Zhang YK, Zhou Y, Nieman JA, Anugula MR, Keramane El, Savariraj MK, Reddy DS, Sharma R, Subedi R, Singh R, O'Leary A, Simon NL, De Marsh PL, Mushtaq S, Warner M, Livermore DM, Alley MR, Plattner JJ. 2013. Discovery of a novel class of boron-based antibacterials with activity against gram-negative bacteria. Antimicrob Agents Chemother 57:1394–1403. doi: 10.1128/AAC.02058-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Johnson KW, Lofland D, Moser HE. 2005. PDF inhibitors: an emerging class of antibacterial drugs. Curr Drug Targets Infect Disord 5:39–52. doi: 10.2174/1568005053174618. [DOI] [PubMed] [Google Scholar]
- 7.Yao J, Rock CO. 2015. How bacterial pathogens eat host lipids: implications for the development of fatty acid synthesis therapeutics. J Biol Chem 290:5940–5946. doi: 10.1074/jbc.R114.636241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Parsons JB, Frank MW, Subramanian C, Saenkham P, Rock CO. 2011. Metabolic basis for the differential susceptibility of Gram-positive pathogens to fatty acid synthesis inhibitors. Proc Natl Acad Sci U S A 108:15378–15383. doi: 10.1073/pnas.1109208108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pearson JP, Feldman M, Iglewski BH, Prince A. 2000. Pseudomonas aeruginosa cell-to-cell signaling is required for virulence in a model of acute pulmonary infection. Infect Immun 68:4331–4334. doi: 10.1128/IAI.68.7.4331-4334.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Heath RJ, White SW, Rock CO. 2001. Lipid biosynthesis as a target for antibacterial agents. Prog Lipid Res 40:467–497. doi: 10.1016/S0163-7827(01)00012-1. [DOI] [PubMed] [Google Scholar]
- 11.Witkowski A, Joshi AK, Smith S. 2007. Coupling of the de novo fatty acid biosynthesis and lipoylation pathways in mammalian mitochondria. J Biol Chem 282:14178–14185. doi: 10.1074/jbc.M701486200. [DOI] [PubMed] [Google Scholar]
- 12.Rock CO, Jackowski S. 2002. Forty years of bacterial fatty acid synthesis. Biochem Biophys Res Commun 292:1155–1166. doi: 10.1006/bbrc.2001.2022. [DOI] [PubMed] [Google Scholar]
- 13.Parl A, Mitchell SL, Clay HB, Reiss S, Li Z, Murdock DG. 2013. The mitochondrial fatty acid synthesis (mtFASII) pathway is capable of mediating nuclear-mitochondrial cross talk through the PPAR system of transcriptional activation. Biochem Biophys Res Commun 441:418–424. doi: 10.1016/j.bbrc.2013.10.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Butler MS, Blaskovich MA, Cooper MA. 2013. Antibiotics in the clinical pipeline in 2013. J Antibiot (Tokyo) 66:571–591. doi: 10.1038/ja.2013.86. [DOI] [PubMed] [Google Scholar]
- 15.Slayden RA, Lee RE, Barry CE III. 2000. Isoniazid affects multiple components of the type II fatty acid synthase system of Mycobacterium tuberculosis. Mol Microbiol 38:514–525. doi: 10.1046/j.1365-2958.2000.02145.x. [DOI] [PubMed] [Google Scholar]
- 16.Payne DJ, Gwynn MN, Holmes DJ, Pompliano DL. 2007. Drugs for bad bugs: confronting the challenges of antibacterial discovery. Nat Rev Drug Discov 6:29–40. doi: 10.1038/nrd2201. [DOI] [PubMed] [Google Scholar]
- 17.Silver LL. 2005. A retrospective on the failures and successes of antibacterial drug discovery. IDrugs 8:651–655. [PubMed] [Google Scholar]
- 18.Moir DT, Di M, Opperman T, Schweizer HP, Bowlin TL. 2007. A high-throughput, homogeneous, bioluminescent assay for Pseudomonas aeruginosa gyrase inhibitors and other DNA-damaging agents. J Biomol Screen 12:855–864. doi: 10.1177/1087057107304729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Moir DT, Di M, Wong E, Moore RA, Schweizer HP, Woods DE, Bowlin TL. 2011. Development and application of a cellular, gain-of-signal, bioluminescent reporter screen for inhibitors of type II secretion in Pseudomonas aeruginosa and Burkholderia pseudomallei. J Biomol Screen 16:694–705. doi: 10.1177/1087057111408605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Holloway BW, Krishnapillai V, Morgan AF. 1979. Chromosomal genetics of Pseudomonas. Microbiol Rev 43:73–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.de Lorenzo V, Timmis KN. 1994. Analysis and construction of stable phenotypes in gram-negative bacteria with Tn5- and Tn10-derived minitransposons. Methods Enzymol 235:386–405. doi: 10.1016/0076-6879(94)35157-0. [DOI] [PubMed] [Google Scholar]
- 22.Stover CK, Pham XQ, Erwin AL, Mizoguchi SD, Warrener P, Hickey MJ, Brinkman FS, Hufnagle WO, Kowalik DJ, Lagrou M, Garber RL, Goltry L, Tolentino E, Westbrock-Wadman S, Yuan Y, Brody LL, Coulter SN, Folger KR, Kas A, Larbig K, Lim R, Smith K, Spencer D, Wong GK, Wu Z, Paulsen IT, Reizer J, Saier MH, Hancock RE, Lory S, Olson MV. 2000. Complete genome sequence of Pseudomonas aeruginosa PAO1, an opportunistic pathogen. Nature 406:959–964. doi: 10.1038/35023079. [DOI] [PubMed] [Google Scholar]
- 23.Blattner FR, Plunkett G III, Bloch CA, Perna NT, Burland V, Riley M, Collado-Vides J, Glasner JD, Rode CK, Mayhew GF, Gregor J, Davis NW, Kirkpatrick HA, Goeden MA, Rose DJ, Mau B, Shao Y. 1997. The complete genome sequence of Escherichia coli K-12. Science 277:1453–1462. doi: 10.1126/science.277.5331.1453. [DOI] [PubMed] [Google Scholar]
- 24.Wolfgang MC, Lee VT, Gilmore ME, Lory S. 2003. Coordinate regulation of bacterial virulence genes by a novel adenylate cyclase-dependent signaling pathway. Dev Cell 4:253–263. doi: 10.1016/S1534-5807(03)00019-4. [DOI] [PubMed] [Google Scholar]
- 25.Marshall NJ, Goodwin CJ, Holt SJ. 1995. A critical assessment of the use of microculture tetrazolium assays to measure cell growth and function. Growth Regul 5:69–84. [PubMed] [Google Scholar]
- 26.Aiello D, Williams JD, Majgier-Baranowska H, Patel I, Peet NP, Huang J, Lory S, Bowlin TL, Moir DT. 2010. Discovery and characterization of inhibitors of Pseudomonas aeruginosa type III secretion. Antimicrob Agents Chemother 54:1988–1999. doi: 10.1128/AAC.01598-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Moir DT, Di M, Moore RA, Schweizer HP, Woods DE. 2008. Cellular reporter screens for inhibitors of Burkholderia pseudomallei targets in Pseudomonas aeruginosa. Trans R Soc Trop Med Hyg 102(Suppl 1):S152–S162. doi: 10.1016/S0035-9203(08)70033-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chuanchuen R, Murata T, Gotoh N, Schweizer HP. 2005. Substrate-dependent utilization of OprM or OpmH by the Pseudomonas aeruginosa MexJK efflux pump. Antimicrob Agents Chemother 49:2133–2136. doi: 10.1128/AAC.49.5.2133-2136.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nie Z, Perretta C, Lu J, Su Y, Margosiak S, Gajiwala KS, Cortez J, Nikulin V, Yager KM, Appelt K, Chu S. 2005. Structure-based design, synthesis, and study of potent inhibitors of beta-ketoacyl-acyl carrier protein synthase III as potential antimicrobial agents. J Med Chem 48:1596–1609. doi: 10.1021/jm049141s. [DOI] [PubMed] [Google Scholar]
- 30.Miller JR, Dunham S, Mochalkin I, Banotai C, Bowman M, Buist S, Dunkle B, Hanna D, Harwood HJ, Huband MD, Karnovsky A, Kuhn M, Limberakis C, Liu JY, Mehrens S, Mueller WT, Narasimhan L, Ogden A, Ohren J, Prasad JV, Shelly JA, Skerlos L, Sulavik M, Thomas VH, VanderRoest S, Wang L, Wang Z, Whitton A, Zhu T, Stover CK. 2009. A class of selective antibacterials derived from a protein kinase inhibitor pharmacophore. Proc Natl Acad Sci U S A 106:1737–1742. doi: 10.1073/pnas.0811275106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bowlin NO, Williams JD, Knoten CA, Torhan MC, Tashjian TF, Li B, Aiello D, Mecsas J, Hauser AR, Peet NP, Bowlin TL, Moir DT. 2014. Mutations in the Pseudomonas aeruginosa needle protein gene pscF confer resistance to phenoxyacetamide inhibitors of the type III secretion system. Antimicrob Agents Chemother 58:2211–2220. doi: 10.1128/AAC.02795-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.NCCLS. 1997. Approved standard M7-A4. Methods for dilution antimicrobial susceptibility tests for bacteria that grow aerobically, 4th ed National Committee for Clinical Laboratory Standards, Wayne, PA. [Google Scholar]
- 33.Cherian PT, Yao J, Leonardi R, Maddox MM, Luna VA, Rock CO, Lee RE. 2012. Acyl-sulfamates target the essential glycerol-phosphate acyltransferase (PlsY) in Gram-positive bacteria. Bioorg Med Chem 20:4985–4994. doi: 10.1016/j.bmc.2012.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Alves J, Westling L, Peters EC, Harris JL, Trauger JW. 2011. Cloning, expression, and enzymatic activity of Acinetobacter baumannii and Klebsiella pneumoniae acetyl-coenzyme A carboxylases. Anal Biochem 417:103–111. doi: 10.1016/j.ab.2011.05.041. [DOI] [PubMed] [Google Scholar]
- 35.Schweizer HP. 1998. Intrinsic resistance to inhibitors of fatty acid biosynthesis in Pseudomonas aeruginosa is due to efflux: application of a novel technique for generation of unmarked chromosomal mutations for the study of efflux systems. Antimicrob Agents Chemother 42:394–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tatusov RL, Koonin EV, Lipman DJ. 1997. A genomic perspective on protein families. Science 278:631–637. doi: 10.1126/science.278.5338.631. [DOI] [PubMed] [Google Scholar]
- 37.Hoang TT, Kutchma AJ, Becher A, Schweizer HP. 2000. Integration-proficient plasmids for Pseudomonas aeruginosa: site-specific integration and use for engineering of reporter and expression strains. Plasmid 43:59–72. doi: 10.1006/plas.1999.1441. [DOI] [PubMed] [Google Scholar]
- 38.Zhang YM, Rock CO. 2012. Will the initiator of fatty acid synthesis in Pseudomonas aeruginosa please stand up? J Bacteriol 194:5159–5161. doi: 10.1128/JB.01198-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhu L, Lin J, Ma J, Cronan JE, Wang H. 2010. Triclosan resistance of Pseudomonas aeruginosa PAO1 is due to FabV, a triclosan-resistant enoyl-acyl carrier protein reductase. Antimicrob Agents Chemother 54:689–698. doi: 10.1128/AAC.01152-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chuanchuen R, Beinlich K, Hoang TT, Becher A, Karkhoff-Schweizer RR, Schweizer HP. 2001. Cross-resistance between triclosan and antibiotics in Pseudomonas aeruginosa is mediated by multidrug efflux pumps: exposure of a susceptible mutant strain to triclosan selects nfxB mutants overexpressing MexCD-OprJ. Antimicrob Agents Chemother 45:428–432. doi: 10.1128/AAC.45.2.428-432.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Machutta CA, Bommineni GR, Luckner SR, Kapilashrami K, Ruzsicska B, Simmerling C, Kisker C, Tonge PJ. 2010. Slow onset inhibition of bacterial beta-ketoacyl-acyl carrier protein synthases by thiolactomycin. J Biol Chem 285:6161–6169. doi: 10.1074/jbc.M109.077909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang JH, Chung TD, Oldenburg KR. 1999. A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J Biomol Screen 4:67–73. doi: 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]
- 43.Heath RJ, Rock CO. 1995. Enoyl-acyl carrier protein reductase (fabI) plays a determinant role in completing cycles of fatty acid elongation in Escherichia coli. J Biol Chem 270:26538–26542. doi: 10.1074/jbc.270.44.26538. [DOI] [PubMed] [Google Scholar]
- 44.Parsons JB, Rock CO. 2013. Bacterial lipids: metabolism and membrane homeostasis. Prog Lipid Res 52:249–276. doi: 10.1016/j.plipres.2013.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jacobs MA, Alwood A, Thaipisuttikul I, Spencer D, Haugen E, Ernst S, Will O, Kaul R, Raymond C, Levy R, Chun-Rong L, Guenthner D, Bovee D, Olson MV, Manoil C. 2003. Comprehensive transposon mutant library of Pseudomonas aeruginosa. Proc Natl Acad Sci U S A 100:14339–14344. doi: 10.1073/pnas.2036282100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yuan Y, Sachdeva M, Leeds JA, Meredith TC. 2012. Fatty acid biosynthesis in Pseudomonas aeruginosa is initiated by the FabY class of beta-ketoacyl acyl carrier protein synthases. J Bacteriol 194:5171–5184. doi: 10.1128/JB.00792-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yuan Y, Leeds JA, Meredith TC. 2012. Pseudomonas aeruginosa directly shunts beta-oxidation degradation intermediates into de novo fatty acid biosynthesis. J Bacteriol 194:5185–5196. doi: 10.1128/JB.00860-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang YM, Frank MW, Zhu K, Mayasundari A, Rock CO. 2008. PqsD is responsible for the synthesis of 2,4-dihydroxyquinoline, an extracellular metabolite produced by Pseudomonas aeruginosa. J Biol Chem 283:28788–28794. doi: 10.1074/jbc.M804555200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Parsons JB, Rock CO. 2011. Is bacterial fatty acid synthesis a valid target for antibacterial drug discovery? Curr Opin Microbiol 14:544–549. doi: 10.1016/j.mib.2011.07.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cheng CC, Shipps GW Jr, Yang Z, Sun B, Kawahata N, Soucy KA, Soriano A, Orth P, Xiao L, Mann P, Black T. 2009. Discovery and optimization of antibacterial AccC inhibitors. Bioorg Med Chem Lett 19:6507–6514. doi: 10.1016/j.bmcl.2009.10.057. [DOI] [PubMed] [Google Scholar]
- 51.Tegos G, Stermitz FR, Lomovskaya O, Lewis K. 2002. Multidrug pump inhibitors uncover remarkable activity of plant antimicrobials. Antimicrob Agents Chemother 46:3133–3141. doi: 10.1128/AAC.46.10.3133-3141.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Choi KH, Schweizer HP. 2005. An improved method for rapid generation of unmarked Pseudomonas aeruginosa deletion mutants. BMC Microbiol 5:30. doi: 10.1186/1471-2180-5-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.