

Abstract
Despite the availability of >30 effective drugs for managing HIV-1 infection, no current therapy is curative, and long-term management is challenging owing to the emergence and spread of drug-resistant mutants. Identification of drugs against novel HIV-1 targets would expand the current treatment options and help to control resistance. The highly conserved HIV-1 capsid protein represents an attractive target because of its multiple roles in replication of the virus. However, the low antiviral potencies of the reported HIV-1 capsid–targeting inhibitors render them unattractive for therapeutic development. To facilitate the identification of more-potent HIV-1 capsid inhibitors, we developed a scintillation proximity assay to screen for small molecules that target a biologically active and specific intersubunit interface in the HIV-1 capsid. The assay, which is based on competitive displacement of a known capsid-binding small-molecule inhibitor, exhibited a signal-to-noise ratio of >9 and a Z factor of >0.8. In a pilot screen of a chemical library containing 2,400 druglike compounds, we obtained a hit rate of 1.8%. This assay has properties that are suitable for screening large compound libraries to identify novel HIV-1 capsid ligands with antiviral activity.
INTRODUCTION
HIV/AIDS continues to be a major global health problem, with ∼1.5 million HIV-related deaths and >2 million new HIV infections reported annually. Currently, an estimated 35 million people live with HIV infection (1). However, the number of HIV-related deaths is declining, owing to the increased availability of and improvements in antiretroviral therapy (ART). The current arsenal of FDA-approved anti-HIV drugs used in ART is based on 28 molecular entities (2). ART has been fairly successful in containing the HIV/AIDS epidemic, but the side effects of long-term ART and the emergence of drug-resistant HIV-1 mutants warrant discovery and development of anti-HIV drugs. Because existing regimens predominantly target the three viral enzymes and are not curative, the development of drugs against novel targets is an important goal.
The HIV-1 capsid is a relatively unexploited therapeutic target that has gained attention as understanding of its structural and mechanistic roles in the HIV-1 replication cycle has improved. The capsid, which encases the viral genome and associated proteins, is critical for efficient reverse transcription, nuclear entry, and possibly integration. In addition, the capsid appears to cloak the reverse-transcribed viral genome, thus promoting innate immune evasion by limiting access to cytosolic DNA sensors (3). The capsid is composed of a lattice of capsid protein (CA) hexamers that are stabilized by three major intersubunit interfaces and connected through interactions between the carboxy-terminal domains of CA (4, 5). The conical shape of the HIV-1 capsid is a result of the asymmetric distribution of CA pentamers in the lattice, which promote the curvature necessary for closure of the ends (6–8).
Several HIV-1 capsid–targeting small-molecule antiviral compounds have been reported (9–13); however, their relatively low potencies and/or chemical properties have rendered them unsuitable for clinical development. Nonetheless, HIV-1 capsid–targeting inhibitors have been useful as probes in mechanistic and biochemical studies of HIV-1 capsid function (14, 15). The small-molecule PF-3450074 (PF74) inhibits HIV-1 infection at submicromolar concentrations and binds to a pocket in the amino-terminal domain of the CA at the intrahexamer interface formed by the carboxy-terminal domain of an adjacent subunit (10, 16, 17). The PF74 capsid binding pocket has been identified as an interaction site for several host proteins that play important roles in the HIV-1 infection cycle (16, 18). This region of the CA is highly conserved among HIV-1 isolates (19), and resistance of HIV-1 to PF74 appears to require multiple mutations in CA (10, 20). These observations suggest that the genetic barrier to the clinical resistance to inhibitors that bind to the PF74 binding pocket will be high in vivo, which is attractive from the perspective of therapeutic durability. Although the HIV-1 capsid is an attractive drug target, compounds with sufficient antiviral potency for clinical promise have yet to be identified.
It was recently shown that PF74 binds with higher affinity to the assembled CA hexamer in vitro than to monomeric CA, consistent with its functional target being the intersubunit interface (16, 17). Based on this observation, we developed a novel target-based assay for unbiased identification of compounds that interact with the PF74 binding pocket in the HIV-1 capsid. The radiometric assay detects the ability of compounds to competitively inhibit the binding of PF74 to the assembled CA hexamer in vitro. CA hexamers are immobilized on scintillation proximity assay (SPA) beads, which emit light upon binding of [3H]PF74. Unlabeled compounds that bind competitively to the PF74 binding pocket will compete for PF74 binding, thus reducing the signal. This mix-and-read endpoint assay is scalable to a 384-well format and sufficiently robust for high-throughput screening (HTS).
MATERIALS AND METHODS
Chemicals and reagents.
Streptavidin-coated polyvinyltoluene SPA beads were purchased from PerkinElmer and suspended in STE buffer (100 mM NaCl, 10 mM Tris-HCl, 1 mM EDTA, pH 7.4) at 25 mg/ml. Detergents were purchased from Thermo Scientific (Pierce Surfact-Amps detergent sampler). PF74, diiodo-PF74, PF74 analog compounds, and BI-2 were synthesized and purified in the Chemical Synthesis Core, Vanderbilt Institute of Chemical Biology. The structures of these compounds have been reported (10, 13). Working stocks of these chemicals were prepared by dissolution in dimethyl sulfoxide (DMSO) to 10 mM concentrations. [3H]PF74 was produced by 3H-I exchange reaction with diiodo-PF74 to a specific radioactivity of 52 Ci/mmol by Moravek Biochemicals and Radiochemicals (La Brea, CA). A compound library (Spectrum Collection) for pilot screening was obtained from Microsource Discovery, Inc., and accessed through the Vanderbilt High Throughput Screening Facility, Vanderbilt Institute of Chemical Biology.
Preparation of biotinylated HIV-1 CA hexamer.
HIV-1 hexameric CA (hCA) was produced by assembly of recombinant CA with cysteine and alanine substitutions to facilitate the formation of stable hexameric units, as previously reported (21). Biotinylated HIV-1 hexameric CA (bhCA) used in the assay was prepared by either (i) direct biotinylation of hexameric CA (bhCA-1) or (ii) coassembling of CA and biotinylated CA (bhCA-2). Biotinylation of CA was carried out using Pierce EZ-Link sulfo-NHS-biotin (Thermo Scientific) according to the manufacturer's recommendation, and the conditions were adjusted to limit the reaction to link 9 to 12 biotin molecules per hexamer, as quantified with a 4′-hydroxyazobenzene-2-carboxylic acid (HABA) assay (Pierce biotin quantitation kit). To produce bhCA-2, a 1:2 molar ratio mixture of bhCA-1 and hCA was disassembled and then coassembled into the hexamers as follows. First, the hexamer mixture was dialyzed into 50 mM Tris-HCl buffer (pH 8.0) in the presence of 100 mM β-mercaptoethanol for 16 h to achieve disassembly of the hexamers into individual CA subunits. Then the buffer was exchanged with 50 mM Tris-HCl buffer (pH 8.0) with 1.0 M NaCl for 24 h to promote coassembly of biotinylated and unbiotinylated CA and cross-linking of the subunits within hexamers in the lattice. The reaction mixture was then dialyzed into 50 mM Tris-HCl (pH 8.0) for 24 h to disassemble the tubes into disulfide cross-link–stabilized hexamers. All the dialysis steps were performed at 4°C. Final product was flash frozen in liquid nitrogen and stored in aliquots at −80°C.
Method for the SPA-based PF74 binding competition assay.
Various quantities of SPA beads and detergents were mixed in STE buffer and incubated for 10 min. Separately, various amounts of bhCA and [3H]PF74 were mixed in STE buffer and incubated for 10 min. The detergent reduced the nonspecific binding of [3H]PF74 to the beads, and it was necessary to mix beads with the detergent prior to adding bhCA to limit nonspecific binding. Following the incubation, the two mixtures were combined and dispensed into 96-well or 384-well white opaque scintillation counting plates (PerkinElmer OptiPlate). The total volume of the assay mix was 100 μl and 25 μl per well for the 96-well and 384-well plates, respectively. The assay plates were incubated at room temperature for 16 h, and scintillation was quantified on a PerkinElmer TopCount NXT instrument with at least 30 s counting time. To establish the background signal of the assay, the assay mixture was prepared without bhCA. Testing of the competing compounds was performed by dispensing the appropriate amounts of the compounds into the assay plate wells prior to the addition of the assay mixture.
The assay was optimized to achieve a satisfactory signal-to-background ratio (S/B) with a minimum reagent cost by adjusting the amounts of the beads, the detergents, the receptor protein, and [3H]PF74 in the assay mixture. The optimal assay mixture contained 1.25 mg/ml SPA beads, 0.2% (wt/vol) Brij 58, 0.3 μM bCA, and 0.1 μM [3H]PF74. The competence of the assay in 384-well plates was evaluated by preparing a checkerboard plate, in which the assay mixture with bhCA (positive samples) and without bhCA (for background binding) were distributed in alternating wells.
Testing of PF74 and analogs with the assay.
Unlabeled PF74 and its analog compounds were dissolved in DMSO and dispensed into 96-well plates to yield the desired concentrations (0 to 10 μM) in the final assay volume. The DMSO concentrations of all wells were kept at a constant value of <2%. The assay mixture was prepared and dispensed into the wells. Following 16 h of incubation at room temperature, the plate was read.
Testing of the assay for high-throughput screening.
Stock solutions of the compounds in the Spectrum Collection (2,400 compounds) were available at 10 mM concentrations in DMSO, and 25-nl aliquots of each compound and appropriate controls were dispensed into empty 384-well plates using a Labcyte Echo dispenser. The assay mixture containing SPA beads, protein, and [3H]PF74 was prepared and 25 μl dispensed into each well with a Multidrop Combi (Thermo Scientific) reagent dispenser. The assay contents were mixed by shaking the plates on an orbital shaker for 5 min and incubated at room temperature for 16 h, and scintillation was quantified with the TopCount instrument.
RESULTS AND DISCUSSION
Design of the assay.
To identify novel compounds that interact with the PF74 binding site in the HIV-1 capsid, we adopted an approach based on the scintillation proximity assay (SPA) (Fig. 1). Disulfide-stabilized HIV-1 CA hexamers were prepared by assembly of recombinant CA containing engineered Cys substitutions at positions 14 and 45 in the protein sequence. The CA hexamers were biotinylated in vitro, then immobilized onto streptavidin-coated SPA beads. Upon addition of [3H]PF74, binding of the compound to the hCA on the surface of the beads resulted in excitation of the scintillant within the beads by emitted β particles, resulting in emission of light. Emissions from unbound ligand molecules were quenched by the solution, owing to the low energy of the tritium emission. Thus, SPA allowed detection of bound radioligand in the presence of unbound ligand, obviating the need for a washing step. In principle, the assay may be used to screen compound libraries for compounds that compete with PF74 for binding to hCA.
FIG 1.

Diagram of the assay strategy. Streptavidin-coated SPA beads were first treated with a detergent to prevent nonspecific binding of the radioligand to the beads. Next, biotinylated hCA and [3H]PF74 were added to the mixture. Because of the limited penetration of the low-energy 3H-labeled β particles in aqueous media (<1.5 μm), β particles from unbound [3H]PF74 were unlikely to excite the scintillator in the SPA beads. Binding between hCA and [3H]PF74 retained a large fraction of [3H]PF74 molecules at the bead surface, thereby promoting the interaction between the β particles emitted from [3H]PF74 and the scintillators in the SPA beads, resulting in light emission (assay signal). The assay signal dropped in the presence of a molecule that competed with [3H]PF74 for binding to hCA.
Optimization of the biotinylated CA hexamer.
We employed polyvinyl toluene (PVT) beads coated with streptavidin, thus permitting the facile immobilization of the biotinylated CA hexamer. In initial studies, in which we employed equilibrium dialysis to quantify PF74 affinity for the CA hexamer, we observed that biotinylation of hCA reduced its affinity for PF74, and the reduction of the affinity correlated with the amount of biotin incorporated into the protein (data not shown). Therefore, to ensure adequate biotinylation for bead capture, while minimizing the loss of binding affinity, the biotinylation was controlled by changing the reaction time and biotin concentration in order to achieve 9 to 12 biotin molecules per hexamer.
We optimized the assay to achieve a high S/B and low reagent consumption. Although the assay was ultimately intended for 384-well plates, the initial steps of the assay development were performed in 96-well plates for the convenience of handling relatively higher reagent volumes compared to those of 384-well plates. Each assay condition was tested in duplicate and processed in parallel in the same plate.
The first objective of the assay development was to reduce the background due to binding of the relatively hydrophobic radioligand [3H]PF74 to hydrophobic PVT beads. The high background resulted in an S/B of <3 in all the assay conditions tested. To reduce the background binding, we tested several detergents, bovine serum albumin (BSA), and polyethylene glycols with the assay mixture. Among them, only the detergents Brij 35, Brij 58, Tween 80, and Igepal CA-630 showed a substantial reduction in the background signal, resulting in a higher S/B (Fig. 2A). Brij 58 at 0.2% (wt/vol) concentration was chosen for further development of the assay. In preparation of the assay mixture, we found it was important to mix the beads with the detergent prior to addition of bhCA in order to achieve a low background signal. Therefore, the order of addition was beads, detergent, bhCA, and [3H]PF74.
FIG 2.

Optimization of the SPA competition assay. (A) Detergent optimization of S/B. Shown are the results from Brij 58 titration in reaction mixtures containing 1.25 mg/ml beads, 0.125 μM bhCA-1, and 0.125 μM [3H]PF74 (assay signal) or the equivalent lacking bhCA (background). (B) Optimization of [3H]PF74 concentration. Two biotinylated CA hexamer preparations (bhCA-1 and bhCA-2) were tested at 0.125 μM concentration in reaction mixtures containing 1.25 mg/ml beads and 0.2% (wt/vol) Brij 58. (C) Optimization of the concentrations of the SPA beads. (D) Optimization of bhCA-2 concentration. Reaction mixtures in panels C and D contained 0.2% (wt/vol) Brij 58 and 0.1 μM [3H]PF74. (E) Specificity of the assay signal and stability of the assay with time. Background was established with assay mixture lacking bhCA (no bhCA). 5Mut–bhCA-1, which exhibits low affinity for PF74, was prepared by direct biotinylation of the 5Mut CA hexamer.
We next optimized the [3H]PF74 concentration. We observed that concentrations between 0.025 and 0.2 μM resulted in the highest S/Bs (Fig. 2B) for bhCA-1 and bhCA-2. To ensure a sufficient level of scintillation from an assay well while conserving the radioligand, 0.1 μM [3H]PF74 was chosen for the assay. Between the two bhCAs, bhCA-2 generated a higher S/B. This might be due to the reduction in the affinity of bhCA-1 for PF74 compared to that of bhCA-2 because of the differences in the amount of biotinylation (all subunits of bhCA-1 hexamer were biotinylated, whereas the assembly of bhCA-2 was controlled to have an average of two biotinylated subunits per hexamer). Because of its greater S/B, bhCA-2 was selected for further development of the assay. Concentrations of bhCA-2 and the beads were optimized in tandem, revealing that the bead concentration of 0.5 mg/ml and bhCA-2 concentration of 0.05 μM resulted in the highest S/B (Fig. 2C and D). Thus, the final optimized assay mixture contained 0.5 mg/ml streptavidin-coated PVT SPA beads, 0.2% (wt/vol) Brij 58, 0.3 μM bhCA-2 (quantified as a CA subunit), and 0.1 μM [3H]PF74. Upon mixing the assay ingredients, the assay signal reached equilibrium within 1 h and remained unchanged for 24 h (Fig. 2E).
Assay validation.
To test whether the SPA-based assay detects specific binding to the PF74 binding pocket in CA, we employed hCA derived from the PF74-resistant 5Mut virus. 5Mut hexamers, which contain 5 substitutions in CA, exhibit low PF74 affinity relative to wild-type CA hexamers (20, 22). Use of 5Mut–bhCA-1 in the assay resulted in a signal close to the background (Fig. 2E), confirming the specificity of the assay.
To determine whether the assay is capable of detecting differences in compound binding affinity, we performed a competition assay with various concentrations of unlabeled PF74 and BI-2 as competitors. BI-2 is another capsid-targeting HIV-1 inhibitor that interacts with the same PF74 binding pocket but with lower affinity (16). At 10 μM concentration, unlabeled PF74 reduced the signal to nearly background level (Fig. 3A). In contrast, BI-2 had a weak inhibitory effect (∼20%) at concentrations of up to 20 μM. These results confirm that the SPA-based competition assay is capable of detecting differences in the relative binding affinity between two compounds that interact with the PF74 binding site.
FIG 3.
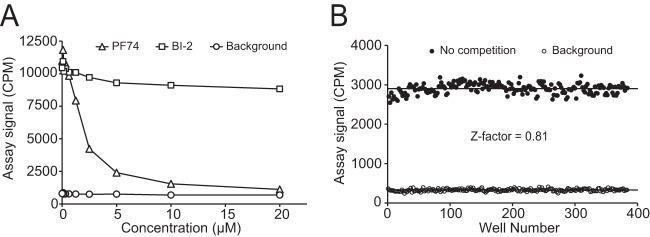
Validation of the assay with unlabeled competitor compounds and determination of Z factor. Assay conditions included 0.5 mg/ml beads, 0.2% (wt/vol) Brij 58, 0.3 μM bhCA-2, and 0.1 μM [3H]PF74. (A) Test of the response of the assay to unlabeled PF74 and BI-2 at various concentrations in 96-well plates. Reaction mixtures contained 1% (vol/vol) DMSO. (B) Results of the checkerboard evaluation of the assay in 384-well plates. Assay mixtures (25 μl) were manually pipetted into the wells.
To determine whether the assay can be miniaturized to 384-well plates for HTS, we performed a checkerboard assay in a 384-well plate. The assay yielded an S/B of 8.5 and a Z factor of 0.81 (Fig. 3B). Assays with a Z factor of >0.5 are considered excellent for HTS (23, 24). Therefore, the PF74 competition assay has statistical properties that are desirable for HTS.
To further validate the assay as a screen for compounds with antiviral activity, we tested a number of PF74 derivative compounds that were designed to have various minor modifications to the PF74 backbone structure (see Table S1 in the supplemental material). The synthesized compounds were dissolved in DMSO, so having some level of DMSO in the assay mixture was unavoidable. Therefore, a 1% DMSO concentration was maintained in all wells. In additional studies, we observed that DMSO concentrations up to 3% (vol/vol) did not significantly affect the assay signal (data not shown). At a 10 μM concentration of each compound, the assay showed a range of signal reduction. Evaluation of the antiviral potency of the PF74 derivatives against wild-type HIV-1 revealed that many were less potent inhibitors than PF74 (Fig. 4; see also Table S1). None of the compounds showed a 50% inhibitory concentration (IC50) higher than that of PF74, and several of them did not exhibit any detectable anti-HIV-1 activity. Interestingly, the IC50 values of the PF74 derivatives showed a significant correlation with the extent of inhibition observed in the SPA (Spearman 1-tailed r = −0.93; P < 0.0001) (Fig. 4). The results from compound BI-2, which is structurally different from PF74, also fell on the curve. Collectively, these data indicate that results from the SPA-based competition assay are likely to predict the antiviral potency of compounds identified during HTS.
FIG 4.
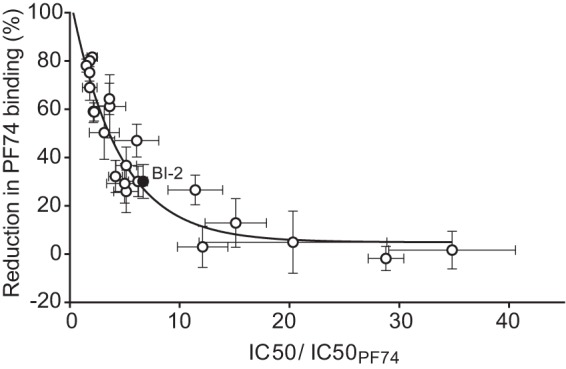
Correlation between the assay response (signal reduction at 10 μM concentration) and the IC50 of the PF74 derivatives relative to the IC50 of PF74. The two parameters were significantly correlated (Spearman r = −0.93; P < 0.0001).
Testing of the assay in HTS.
To determine the utility of the assay in HTS, we performed a small-scale pilot screen of the Spectrum Collection compound library at a compound concentration of 10 μM. The stock compounds were available at 10 mM concentrations in DMSO, thereby resulting in a 0.1% (vol/vol) DMSO concentration in the final assay mixture. Results of the screen showed that 43 compounds reduced the PF74 binding signal to a value below the threshold, set at 3σ below the mean signal value of the all compounds analyzed (Fig. 5). This represented a 1.8% hit rate, which seemed appropriate for HTS.
FIG 5.
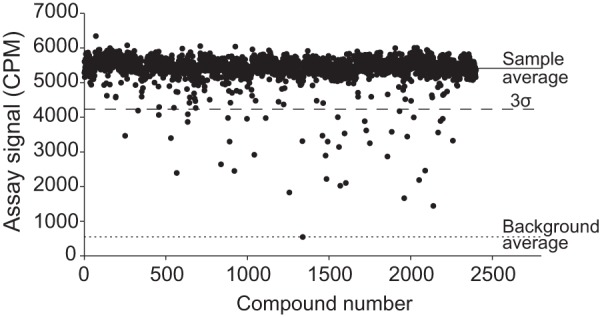
Results of the screening of the Spectrum Collection (2,400 compounds). Reactions (25-μl mixtures) were performed in 384-well plates and included 0.5 mg/ml beads, 0.2% (wt/vol) Brij 58, 0.3 μM bhCA-2, and 0.1 μM [3H]PF74. The screening scored 43 hits that produced an assay signal of <3 standard deviations from the average signal from all the screened compounds.
The screening assay we developed is a biochemical target-based assay based on the competition of binding of known antiviral compounds to the HIV-1 CA hexamer. The assay has properties desirable for HTS, including a simple mix-and-read endpoint format, a stable endpoint, relatively low reagent cost, and excellent statistical properties. It is radiometric, which may be undesirable to some laboratories because of the regulatory requirements for handling radioisotopes. The radiometric design of the assay also renders it susceptible to false-positive results due to the potential for quenching by colored compounds. This limitation may be overcome by using an instrument with an advanced quench correction method (e.g., Perkin-Elmer Microbeta Trilux) or by screening with an imaging-based instrument using SPA beads that are less susceptible to color quenching. Additionally, nonradiometric direct binding approaches, such as surface plasmon resonance and biolayer interferometry, may be used as orthogonal assays to test hits.
For convenience, our competition-binding assay employs a CA hexamer that is stabilized by an engineered disulfide bond to ensure the preservation of the PF74 binding interface within the hexamer. We and others have shown that PF74 binds specifically and with higher affinity to the disulfide-stabilized CA hexamer than to the isolated CA subunit (16, 17). While the cross-linking ensures efficient and specific PF74 binding, the substitutions could alter some structural or biological properties of the viral capsid. Specifically, the A14C and E45C substitutions could affect the intrinsic stability of the capsid and interactions of the viral capsid with host factors. Additionally, biotinylation of the CA hexamer may reduce its affinity for PF74. Notwithstanding these issues, the SPA-based assay that we developed should be useful in screening large compound libraries to identify novel chemical scaffolds that interact with the specific binding site for PF74 in the HIV-1 capsid, thus expanding the chemical space for development of capsid-targeting antiviral drugs.
Supplementary Material
ACKNOWLEDGMENTS
We thank David Weaver, Paige Vinson, and Rey Redha of the Vanderbilt High Throughput Screening Core Facility for advice and technical support. The Vanderbilt High Throughput Screening Core Facility is an institutionally supported core.
This work was supported by grants R01-AI089401, R01-AI114339, and 1U54MH084659-01 from the National Institutes of Health.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.00646-15.
REFERENCES
- 1.World Health Organization. 2014. Global update on the health sector response to HIV. World Health Organization, Geneva, Switzerland. [Google Scholar]
- 2.Kinch MS, Patridge E. 2014. An analysis of FDA-approved drugs for infectious disease: HIV/AIDS drugs. Drug Discov Today 19:1510–1513. doi: 10.1016/j.drudis.2014.05.012. [DOI] [PubMed] [Google Scholar]
- 3.Rasaiyaah J, Tan CP, Fletcher AJ, Price AJ, Blondeau C, Hilditch L, Jacques DA, Selwood DL, James LC, Noursadeghi M, Towers GJ. 2013. HIV-1 evades innate immune recognition through specific cofactor recruitment. Nature 503:402–405. doi: 10.1038/nature12769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Byeon IJ, Meng X, Jung J, Zhao G, Yang R, Ahn J, Shi J, Concel J, Aiken C, Zhang P, Gronenborn AM. 2009. Structural convergence between cryo-EM and NMR reveals intersubunit interactions critical for HIV-1 capsid function. Cell 139:780–790. doi: 10.1016/j.cell.2009.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pornillos O, Ganser-Pornillos BK, Kelly BN, Hua Y, Whitby FG, Stout CD, Sundquist WI, Hill CP, Yeager M. 2009. X-ray structures of the hexameric building block of the HIV capsid. Cell 137:1282–1292. doi: 10.1016/j.cell.2009.04.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhao G, Perilla JR, Yufenyuy EL, Meng X, Chen B, Ning J, Ahn J, Gronenborn AM, Schulten K, Aiken C, Zhang P. 2013. Mature HIV-1 capsid structure by cryo-electron microscopy and all-atom molecular dynamics. Nature 497:643–646. doi: 10.1038/nature12162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ganser BK, Li S, Klishko VY, Finch JT, Sundquist WI. 1999. Assembly and analysis of conical models for the HIV-1 core. Science 283:80–83. doi: 10.1126/science.283.5398.80. [DOI] [PubMed] [Google Scholar]
- 8.Pornillos O, Ganser-Pornillos BK, Yeager M. 2011. Atomic-level modelling of the HIV capsid. Nature 469:424–427. doi: 10.1038/nature09640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tang C, Loeliger E, Kinde I, Kyere S, Mayo K, Barklis E, Sun Y, Huang M, Summers MF. 2003. Antiviral inhibition of the HIV-1 capsid protein. J Mol Biol 327:1013–1020. doi: 10.1016/S0022-2836(03)00289-4. [DOI] [PubMed] [Google Scholar]
- 10.Blair WS, Pickford C, Irving SL, Brown DG, Anderson M, Bazin R, Cao J, Ciaramella G, Isaacson J, Jackson L, Hunt R, Kjerrstrom A, Nieman JA, Patick AK, Perros M, Scott AD, Whitby K, Wu H, Butler SL. 2010. HIV capsid is a tractable target for small molecule therapeutic intervention. PLoS Pathog 6:e1001220. doi: 10.1371/journal.ppat.1001220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kortagere S, Madani N, Mankowski MK, Schon A, Zentner I, Swaminathan G, Princiotto A, Anthony K, Oza A, Sierra LJ, Passic SR, Wang X, Jones DM, Stavale E, Krebs FC, Martin-Garcia J, Freire E, Ptak RG, Sodroski J, Cocklin S, Smith AB III. 2012. Inhibiting early-stage events in HIV-1 replication by small-molecule targeting of the HIV-1 capsid. J Virol 86:8472–8481. doi: 10.1128/JVI.05006-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lemke CT, Titolo S, von Schwedler U, Goudreau N, Mercier JF, Wardrop E, Faucher AM, Coulombe R, Banik SS, Fader L, Gagnon A, Kawai SH, Rancourt J, Tremblay M, Yoakim C, Simoneau B, Archambault J, Sundquist WI, Mason SW. 2012. Distinct effects of two HIV-1 capsid assembly inhibitor families that bind the same site within the N-terminal domain of the viral CA protein. J Virol 86:6643–6655. doi: 10.1128/JVI.00493-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lamorte L, Titolo S, Lemke CT, Goudreau N, Mercier JF, Wardrop E, Shah VB, von Schwedler UK, Langelier C, Banik SS, Aiken C, Sundquist WI, Mason SW. 2013. Discovery of novel small-molecule HIV-1 replication inhibitors that stabilize capsid complexes. Antimicrob Agents Chemother 57:4622–4631. doi: 10.1128/AAC.00985-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Matreyek KA, Yucel SS, Li X, Engelman A. 2013. Nucleoporin NUP153 phenylalanine-glycine motifs engage a common binding pocket within the HIV-1 capsid protein to mediate lentiviral infectivity. PLoS Pathog 9:e1003693. doi: 10.1371/journal.ppat.1003693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shah VB, Shi J, Hout DR, Oztop I, Krishnan L, Ahn J, Shotwell MS, Engelman A, Aiken C. 2013. The host proteins transportin SR2/TNPO3 and cyclophilin A exert opposing effects on HIV-1 uncoating. J Virol 87:422–432. doi: 10.1128/JVI.07177-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Price AJ, Jacques DA, McEwan WA, Fletcher AJ, Essig S, Chin JW, Halambage UD, Aiken C, James LC. 2014. Host cofactors and pharmacologic ligands share an essential interface in HIV-1 capsid that is lost upon disassembly. PLoS Pathog 10:e1004459. doi: 10.1371/journal.ppat.1004459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bhattacharya A, Alam SL, Fricke T, Zadrozny K, Sedzicki J, Taylor AB, Demeler B, Pornillos O, Ganser-Pornillos BK, Diaz-Griffero F, Ivanov DN, Yeager M. 2014. Structural basis of HIV-1 capsid recognition by PF74 and CPSF6. Proc Natl Acad Sci U S A 111:18625–18630. doi: 10.1073/pnas.1419945112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee K, Ambrose Z, Martin TD, Oztop I, Mulky A, Julias JG, Vandegraaff N, Baumann JG, Wang R, Yuen W, Takemura T, Shelton K, Taniuchi I, Li Y, Sodroski J, Littman DR, Coffin JM, Hughes SH, Unutmaz D, Engelman A, KewalRamani VN. 2010. Flexible use of nuclear import pathways by HIV-1. Cell Host Microbe 7:221–233. doi: 10.1016/j.chom.2010.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li G, Verheyen J, Rhee SY, Voet A, Vandamme AM, Theys K. 2013. Functional conservation of HIV-1 Gag: implications for rational drug design. Retrovirology 10:126. doi: 10.1186/1742-4690-10-126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shi J, Zhou J, Halambage UD, Shah VB, Burse MJ, Wu H, Blair WS, Butler SL, Aiken C. 2015. Compensatory substitutions in the HIV-1 capsid reduce the fitness cost associated with resistance to a capsid-targeting small-molecule inhibitor. J Virol 89:208–219. doi: 10.1128/JVI.01411-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pornillos O, Ganser-Pornillos BK, Banumathi S, Hua Y, Yeager M. 2010. Disulfide bond stabilization of the hexameric capsomer of human immunodeficiency virus. J Mol Biol 401:985–995. doi: 10.1016/j.jmb.2010.06.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shi J, Zhou J, Shah VB, Aiken C, Whitby K. 2011. Small-molecule inhibition of human immunodeficiency virus type 1 infection by virus capsid destabilization. J Virol 85:542–549. doi: 10.1128/JVI.01406-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Iversen PW, Eastwood BJ, Sittampalam GS, Cox KL. 2006. A comparison of assay performance measures in screening assays: signal window, Z′ factor, and assay variability ratio. J Biomol Screen 11:247–252. doi: 10.1177/1087057105285610. [DOI] [PubMed] [Google Scholar]
- 24.Zhang JH, Chung TD, Oldenburg KR. 1999. A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J Biomol Screen 4:67–73. doi: 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.