

Abstract
The aim of this study was to improve the understanding of the pharmacokinetic-pharmacodynamic relationships of fosfomycin against extended-spectrum beta-lactamase (ESBL)-producing Escherichia coli strains that have different fosfomycin MICs. Our methods included the use of a hollow fiber infection model with three clinical ESBL-producing E. coli strains. Human fosfomycin pharmacokinetic profiles were simulated over 4 days. Preliminary studies conducted to determine the dose ranges, including the dose ranges that suppressed the development of drug-resistant mutants, were conducted with regimens from 12 g/day to 36 g/day. The combination of fosfomycin at 4 g every 8 h (q8h) and meropenem at 1 g/q8h was selected for further assessment. The total bacterial population and the resistant subpopulations were determined. No efficacy was observed against the Ec42444 strain (fosfomycin MIC, 64 mg/liter) at doses of 12, 24, or 36 g/day. All dosages induced at least initial bacterial killing against Ec46 (fosfomycin MIC, 1 mg/liter). High-level drug-resistant mutants appeared in this strain in response to 12, 15, and 18 g/day. In the study arms that included 24 g/day, once or in a divided dose, a complete extinction of the bacterial inoculum was observed. The combination of meropenem with fosfomycin was synergistic for bacterial killing and also suppressed all fosfomycin-resistant clones of Ec2974 (fosfomycin MIC, 1 mg/liter). We conclude that fosfomycin susceptibility breakpoints (≤64 mg/liter according to CLSI [for E. coli urinary tract infections only]) should be revised for the treatment of serious systemic infections. Fosfomycin can be used to treat infections caused by organisms that demonstrate lower MICs and lower bacterial densities, although relatively high daily dosages (i.e., 24 g/day) are required to prevent the emergence of bacterial resistance. The ratio of the area under the concentration-time curve for the free, unbound fraction of fosfomycin versus the MIC (fAUC/MIC) appears to be the dynamically linked index of suppression of bacterial resistance. Fosfomycin with meropenem can act synergistically against E. coli strains in preventing the emergence of fosfomycin resistance.
INTRODUCTION
In recent decades, there has been an increase in infections caused by Enterobacteriaceae that produce extended-spectrum beta-lactamases (ESBL). The most prevalent ESBL-producing species are Escherichia coli and Klebsiella pneumoniae (1–3). ESBL-producing Enterobacteriaceae are often resistant to other classes of antimicrobials, such as fluoroquinolones, sulfamaethoxazole/trimethoprim, tetracyclines, and aminoglycosides. Carbapenems are considered the drugs of choice for severe infections caused by ESBL producers (4). However, the extensive use of carbapenems may be contributing to the spread of carbapenemases (5). The repurposing of old antimicrobials with activity against drug-resistant Enterobacteriaceae is an important strategy to address the ever-present threat of antimicrobial resistance. However, due to the scarcity of information of the clinical value of these old drugs in well-designed studies, new clinical research trials are under way in order to reevaluate the efficacy of these older drugs against the current gold standard treatments (6).
Fosfomycin inhibits the formation of peptidoglycan via an interaction with the protein UDP-N-acetylglucosamine enolpyruvyl transferase (MurA). Inhibition of this enzyme results in decreased formation of N-acetylmuramic acid (the peptidoglycan precursor) from N-acetylglucosamine and phosphoenolpyruvate (7). According to the current susceptibility breakpoints, fosfomycin is active against the majority of ESBL-producing Enterobacteriaceae (8, 9). An oral formulation of fosfomycin is widely available for the treatment of acute uncomplicated urinary tract infection, and an intravenous formulation is also available in some countries (10). There is now renewed interest in using fosfomycin for the treatment of serious systemic infections caused by multidrug-resistant bacteria. However, there is a paucity of information related to the pharmacokinetics (PK) and pharmacodynamics (PD) of fosfomycin and considerable uncertainty regarding optimal regimens for systemic infections.
An improved understanding of the pharmacokinetic-pharmacodynamic relationships is a first critical step for the design of fosfomycin regimens that are safe, effective, and prevent the emergence of antimicrobial resistance (11). Here, we use a well-validated hollow fiber infection model (HFIM) to examine the PK and PD of fosfomycin against three isolates of ESBL-producing E. coli that demonstrate a range of resistance mechanisms and MIC values.
MATERIALS AND METHODS
Microorganisms.
Three clinical, non-clonally related, ESBL-producing E. coli isolates (Ec46 [CTX-M-15], Ec2974 [CTX-M-15], and Ec42444 [CTX-M-32]) were used. These strains were obtained from patients from the University Hospital Virgen de Macarena. The strains were identified to the species level by using a Bruker Biotyper matrix-assisted laser desorption ionization–time of flight mass spectrometry (MS) system (Bruker Daltonics, Billerica, MA) according to the manufacturer's instructions. The ESBL enzymes (CTX-M-15 and CTX-M-32) were identified by sequencing and clonality based on pulsed-field gel electrophoresis (data not shown).
Drugs.
Fosfomycin disodium salt (Sigma-Aldrich, Dorset, United Kingdom) and meropenem trihydrate (VWR, Leicestershire, United Kingdom) were used for the susceptibility testing, time-kill assays, preparation of drug-containing agar plates, and bioanalytical methods (ultra-high-performance liquid chromatography/tandem mass spectrometry [UHPLC/MS-MS] methods). Twenty-five milligrams per liter of glucose-6-phosphate (G6P; Sigma-Aldrich) was added to the media used for susceptibility testing, quantification of bacterial subpopulations in the pharmacodynamic experiments, and the HFIM. Intravenous clinical formulations of fosfomycin (Laboratorios ERN, Barcelona, Spain) and meropenem (Ranbaxy Limited, London, United Kingdom) were used for the HFIM experiments.
In vitro susceptibility testing.
The in vitro susceptibility to fosfomycin was measured using agar dilution according to CLSI methodology (9). MICs were performed in triplicate. Cation-adjusted Mueller-Hinton agar (Ca-MHA) plates (Becton-Dickinson, Sparks, MD) containing 25 mg/liter G6P and fosfomycin in concentrations ranging from 0.25 to 1,024 mg/liter was prepared. A replicator with 1-mm pins was used to place inocula of 1 × 104 CFU on the surface of the agar. Plates were incubated for 16 to 20 h in ambient air at 35°C. The meropenem susceptibility determination was performed using the standard broth microdilution methodology according to CLSI standards (9). E. coli ATCC 25922 was used as the quality control strain for these experiments.
Mutation frequency.
An initial inoculum of 102 CFU/ml was incubated overnight in Mueller-Hinton Broth and then plated on drug-free Ca-MHA plates (to estimate the total bacterial concentration) and drug-containing Ca-MHA plates (to estimate the subpopulation able to grow at a predetermined antimicrobial concentration). All Ca-MHA plates were supplemented with G6P. For meropenem, the bacterial suspension was plated on Ca-MHA plates that were drug free or contained 3 or 4 mg/liter of meropenem.
To investigate whether the mutants that grew on drug-containing plates had an elevated MIC, 10 colonies were selected and the fosfomycin and meropenem MICs were reestimated using agar dilution or the broth microdilution method, as previously described.
Hollow fiber infection model.
An HFIM was used to investigate the pharmacodynamics of fosfomycin against three ESBL-producing E. coli strains (12). Mueller-Hinton broth (MHB) was pumped from a central compartment through a hollow fiber cartridge (FiberCell Systems, Frederick, MD, USA) before being returned to the central compartment. A peristaltic pump (model 205U; Watson-Marlow, United Kingdom) was used. Fosfomycin and meropenem were administered to the central compartment by using a programmable syringe driver (Aladdin pump; World Precision Instruments, United Kingdom). Fresh Ca-MHB was pumped from a reservoir into the central compartment, and the same volume of drug-containing medium was removed as waste. The rate at which this occurred was controlled to simulate human pharmacokinetic profiles for both drugs (i.e., an elimination half-life of 4 h for fosfomycin and 1 h for meropenem [13, 14]).
The extracapillary space of each HFIM was inoculated with ∼40 ml of bacterial suspension. The desired inoculum was confirmed with quantitative cultures. The HFIM was incubated at 37°C in ambient air. Bacterial densities were determined by removing 1 ml from the extracapillary space via a sampling port. Serial dilutions in 100-μl volumes were then plated on both drug-free and drug-containing Ca-MH agar plates to enumerate total and resistant subpopulations, respectively.
Drug concentrations.
The fosfomycin and meropenem concentrations were determined using an UHPLC/MS-MS triple-quadrupole 6420 mass spectrometer (Agilent Technologies, Santa Clara, CA, USA) during the first dosing interval and at steady state. One milliliter was drawn from the central compartment at 1, 2, 4, 6, 8 and/or 12, and 24 h after drug administration, depending on the regimen being studied. Samples were immediately stored at −80°C until analysis.
A previously described method, with minor modification, was used to measure fosfomycin concentrations (15). Instrument parameters were optimized for fosfomycin (137.0 to 63 m/z) transitions. Fosfomycin was extracted from Ca-MHB by diluting the broth with LC/MS-grade water (Fisher Scientific, Loughborough, United Kingdom) with an internal standard (IS). HPLC separation was achieved using a Luna 3-μm CN 100-Å 50- by 2.0-mm column (Phenomenex, Torrance, CA, USA). The mobile phase consisted of 0.1% formic acid in water and 0.1% formic acid in acetonitrile at a flow rate of 0.35 ml/min. The column was maintained at room temperature (22°C). The internal standard was 6,7-dimethyl-2,3-di(2-pyridyl) quinoxaline (Sigma-Aldrich, Dorset, United Kingdom) at 0.125 ng/ml in water. The chromatographic run time for a sample was 2.5 min. The lower limit of quantitation of fosfomycin was 1 mg/liter. Intraday and interday coefficients of variation were <13.9% for concentrations ranging from 1 to 75 mg/liter.
For meropenem, the instrument parameters were optimized (384.2 to 68.1 m/z transitions). Meropenem was extracted from Ca-MHB as described above for fosfomycin. HPLC separation was achieved using a Zorbax Eclipse Plus 18 rapid-resolution 2.1- by 50-mm, 1.8-μm column (Agilent Technologies, Santa Clara, CA, USA). The mobile phases consisted of 0.1% formic acid in water and 0.1% formic acid in acetonitrile at a flow rate of 0.4 ml/min. The column was maintained at room temperature (22°C). The internal standard was 6,7-dimethyl-2,3-di(2-pyridyl) quinoxaline at 0.125 ng/ml in water. The chromatographic run time for one sample was 2.5 min. The lower limit of quantitation of meropenem was 0.005 mg/liter. Intraday and interday coefficients of variation were <14.2% for concentrations ranging from 0.005 to 8 mg/liter.
Fosfomycin pharmacokinetics and pharmacodynamics.
The fosfomycin concentration-time profiles were adjusted to mimic those observed in humans (13). As plasma protein binding of fosfomycin has been reported to be negligible (16), fosfomycin concentrations used in the HFIM were assumed to constitute unbound fosfomycin. A one-compartment PK model was fit to the in vitro fosfomycin concentration data. A population methodology was employed using the program Pmetrics (17). The structural model took the following form (equation 1):
| (1) |
where X1 is the total amount of fosfomycin (in milligrams) in the central compartment, R (in liters) is the infusion volume of fosfomycin into the central compartment, CL (in liters per hour) is the clearance of fosfomycin from the central compartment, and Vc (in liters) is the volume of the central compartment.
Dose range-finding studies.
The clinical strains Ec46 (MIC, 1 mg/liter) and Ec42444 (MIC, 64 mg/liter) were used for the initial dose range-finding studies in the HFIM. Human-like concentration-time profiles corresponding to regimens of 4 g every 8 hours (q8h; 12 g/day), 8 g/q8h (24 g/day), and 12 g/q8h (36 g/day) with an infusion time of 1 h were generated. An initial bacterial density of ∼1 × 106 CFU/ml was used for these experiments.
Determination of dose ranges that suppressed development of fosfomycin-resistant mutants.
To determine the drug exposure concentrations that suppressed the emergence of fosfomycin-resistant mutants, a dose range-finding study was conducted with strain Ec46 (MIC, 1 mg/liter). Human-like concentration-time profiles of fosfomycin corresponding to regimens of 4 g/q8h (12 g/day), 5 g/q8h (15 g/day), 6 g/q8h (18 g/day), and 8 g/q8h (24 g/day), with each dosage infused over 1 h, or 24 g/q24h (24 g/day), delivered in a single bolus, were used. The pharmacokinetic and pharmacodynamic data set was modeled via a population-based methodology. The structural model took the following form (equations 2 and 3):
| (2) |
| (3) |
where KgMAX-S and KgMAX-R represent the maximal growth rate constants for the susceptible and resistant populations (in CFU per millimeter per hour), respectively; CFUS and CFUR are the numbers of the susceptible and resistant organisms (in CFU per milliliter), respectively; POPMAX is the maximal bacterial population (in CFU per milliliter), KkMAX-S and KkMAX-R represent the maximal kill rate constants for the susceptible and resistant populations (in CFU per milliliter per hour), respectively; CFOS is the concentration of fosfomycin (in milligrams per liter); C50k-S and C50k-R denote the concentrations for which the effect is half-maximal for fosfomycin (in milligrams per liter) against the susceptible and resistant populations, respectively; and Hk-S and Hk-R represent the slope functions for killing of the sensitive and resistant populations, respectively.
Combination study of meropenem and fosfomycin.
The impact of fosfomycin-containing combination regimens on total bacterial killing and prevention of emergence of drug resistance were assessed. The effect of a clinically relevant regimen of fosfomycin at 4 g/q8h (1-h infusion) and meropenem at 1 g/q8h (0.5-h infusion) was assessed. Strain Ec2974 (MIC of 1 mg/liter for both fosfomycin and meropenem) was used for this experiment. An initial bacterial concentration of ∼1010 CFU/ml was used to promote the presence of resistant mutants (i.e., an inoculum in excess of the inverse of the mutation frequency).
Drug interaction modeling.
The interaction between fosfomycin and meropenem was quantified by using a mathematical model, as previously described (18). The interaction term was embedded in the bacterial kill portion of a series of five inhomogeneous differential equations. Two equations described the concentration-time profile of fosfomycin and meropenem. The other three dealt with growth/death of a subpopulation susceptible to both drugs, a population that was susceptible to fosfomycin but resistant to meropenem, and a population that was susceptible to meropenem but resistant to fosfomycin.
The system outputs were the following: (i) concentrations of drug 1; (ii) concentrations of drug 2; (iii) total bacterial density (i.e., the sum of all bacterial subpopulation densities); (iv) the bacterial density that was resistant to fosfomycin but susceptible to meropenem; (v) the bacterial density that was resistant to meropenem but susceptible to fosfomycin. A sixth differential equation (resistant to both drugs at baseline) was not required. The full description of the model is provided in reference (18).
In order to identify drug interactions (synergy, additivity, and antagonism) in a statistically robust way, the mathematical model has as its basis the definition of additivity (Loewe additivity) and identifies synergy or antagonism as a statistically significant excursion from additivity. This is accomplished by estimating the interaction parameter α. If α is positive and the lower end of the 95% confidence interval (CI) does not cross 0 (i.e., stays positive), then the interaction is a statistically significant synergy. If α is negative and the upper bound of the 95% confidence interval does not cross 0 (i.e., stays negative), then the interaction is antagonistic. Any time the 95% confidence bound around the point estimate of α crosses 0, the interaction is additive. There is an α value for each bacterial subpopulation. In this study, no meropenem-resistant clones were isolated. Hence, there were two rather than three estimates for α.
Population modeling was performed using the nonparametric adaptive grid (NPAG) program of Leary et al. (19). With respect to the model published in reference 18, there was one difference in our model. Because there were no resistant mutants identified until after 48 h in the fosfomycin-alone arm, a lag time for the production of a mutant was incorporated to allow for the substantial time from baseline for the emergence of a resistant isolate.
RESULTS
In vitro susceptibility.
The MIC for fosfomycin of strain Ec42444 was 64 mg/liter. For Ec46 and Ec2974, the MIC was 1 mg/liter. Strain Ec2974 had an MIC of meropenem of 1 mg/liter.
Mutation frequency.
Following 48 h of incubation, the frequencies of Ec46 mutants able to grow on plates that contained fosfomycin at 4× MIC, 16× MIC, 64× MIC, and 256× MIC (MIC, 1 mg/liter) were 1.07 × 10−5, 2.66 × 10−6, 1.9 × 10−8, and <7 × 10−8, respectively. The mutation frequencies for fosfomycin for strain Ec2974 were 2.79 × 10−6, 1.17 × 10−7, <1.22 × 10−9, and <1.22 × 10−9, respectively. The MICs of the recovered strains were ≤64 mg/liter. For meropenem, the mutation frequencies of Ec2974 at 3× MIC and 4× MIC (MIC, 1 mg/liter) were <1.75 ×10−9 and <1.84 × 10−9, respectively.
Fosfomycin pharmacokinetics.
A human-like profile was generated by Pfausler et al. (13) and was based on the plasma fosfomycin concentrations after intravenous administration of fosfomycin at 8 g/q8h. With this dosage, the area under the concentration-time curve for the free, unbound fraction of the drug from 0 to 24 h (fAUC0–24) at steady state of 3,105 mg · h/liter was observed in humans, which was similar to our result with the human simulation dose, 8 g/q8h, where an fAUC0–24 h of 3,136 mg · h/liter was obtained. The other dosages were simulated, and the pharmacodynamic parameters are shown in Table 1. For the strains with a MIC of 1 mg/liter, the time above the MIC (TMIC) covered the entire interval between doses. The observed and predicted concentrations are shown in Fig. 1.
TABLE 1.
Pharmacodynamic parameters for fosfomycin obtained from the hollow fiber infection model
| PD parameter | PD value for simulated human dosage (total dose/day) from: |
|||||
|---|---|---|---|---|---|---|
| Range-finding study |
Fractionation study |
|||||
| 4 g/q8h (12 g) | 8 g/q8h (24 g) | 12 g/q8h (36 g) | 6 g/q12h (12 g) | 12 g/q24h (12 g)a | 12 g/q24h (12 g)b | |
| fAUC0–24 (mean) | 1,744.94 | 3,136.03 | 4,287.82 | 2,121.83 | 2,366.83 | 2,404.61 |
| fAUC0–24/MIC (MIC = 1 mg/liter)c | 1,744.94 | 3,136.03 | 4,287.82 | 2,121.83 | 2,366.83 | 2,404.61 |
| fAUC0–24/MIC (MIC = 64 mg/liter)d | 27.26 | 49 | 67 | 33.15 | 36.98 | 37.57 |
Data are for fosfomycin delivered in 1-h infusion.
Data are for fosfomycin delivered via continuous infusion for 24 h.
Data are for strain Ec46, for which the MIC is 1 mg/liter.
Data are for strain Ec42444, for which the MIC is 64 mg/liter.
FIG 1.

Pharmacokinetic profiles of the fosfomycin dosages used in the hollow fiber infection model. Solid lines, predicted concentrations; crosses, observed concentrations.
Dose range-finding studies.
All regimens of fosfomycin led to rapid bactericidal activity against Ec46 (fosfomycin MIC, 1 mg/liter). However, there was growth in the 12-g/day arm of a highly resistant mutant that occurred after 24 h of treatment. Both 24 g/day and 36 g/day resulted in sterilization and complete suppression of drug-resistant mutants (Fig. 2A).
FIG 2.
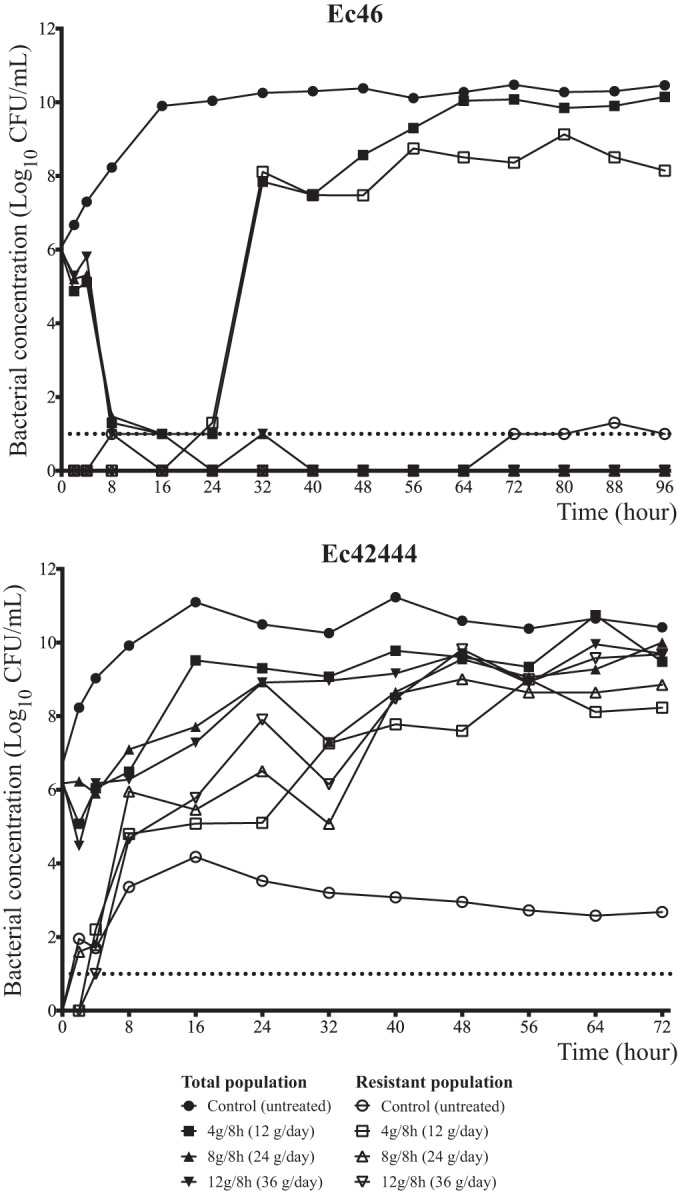
Fosfomycin dose range-finding results with two ESBL-producing E. coli strains. (A) Ec46 (MIC, 1 mg/liter); (B) Ec42444 (MIC, 64 mg/liter).
No efficacy was observed against the Ec42444 strain (fosfomycin MIC, 64 mg/liter) with the three dosages evaluated (4 g/q8h [12 g/day], 8 g/q8h [24 g/day], and 12 g/q8h [36 g/day]). With all the regimens, there was a small initial reduction in bacterial burden, but this was rapidly followed by growth of a resistant subpopulation (Fig. 2B).
Dose range-finding studies for suppression of development of resistant mutants.
To define the threshold of drug exposure that suppressed the emergence of drug-resistant mutants, the effects of progressively higher dosages of fosfomycin were examined. All dosages induced at least initial bacterial killing. In the arms receiving 12, 15, and 18 g of fosfomycin per day, there was growth of a high-level drug-resistant mutant which appeared after 30 to 40 h of exposure to fosfomycin. For the two study arms that received 24 g/day (i.e., 24 g once or in a divided dose), there was complete extinction of the bacterial inoculum and no subsequent emergence of drug resistance (Fig. 3). There was a clear delineation of an exposure (24 g/day) that suppressed resistance and eradicated the total population. Both 8 g every 8 h and 24 g once daily achieved this goal, indicating that the dynamic index that was best linked to resistance suppression was the fAUC/MIC ratio.
FIG 3.

Dose range-finding for suppression of development of fosfomycin-resistant mutants and fractionation studies with strain Ec46 (MIC, 1 mg/liter).
The results of the pharmacokinetic/pharmacodynamic model are shown in Table 2. The fit of the model to the data (regression analysis) included the following equations. For the observed versus predicted regression for all fosfomycin concentrations: Observed = (1.071 × Predicted) − 6.57; R2 = 0.967. For the observed versus predicted regression for all total colony counts: Observed = (1.067 × Predicted) − 0.892; R2 = 0.906. For the observed versus predicted regression for fosfomycin-resistant colony counts: Observed = (1.027 × Predicted) − 0.849; R2 = 0.903.
TABLE 2.
Parameter estimates from the mathematical model used for range-finding studies for suppression of emergence of fosfomycin-resistant mutants
| Parametera | Dosage (total dose/day) |
||||
|---|---|---|---|---|---|
| 4 g/q8h (12 g) | 5 g/q8h (15 g) | 6 g/q8h (18 g) | 8 g/q8h (24 g) | 24 g/q24h (24 g) | |
| V (liters) | 36.01 | 37.57 | 37.82 | 39.20 | 39.36 |
| SCL (liters/h) | 5.54 | 5.47 | 6.16 | 6.11 | 6.07 |
| KgMAX-S (CFU/ml/h) | 5 | 5 | 4.43 | 3.46 | 2.7 |
| KgMAX-R (CFU/ml/h) | 1.17 | 1.161 | 1.15 | 2.04 | 0.7 |
| KkMAX-S (CFU/ml/h) | 7.21 | 7.14 | 8.3 | 3.81 | 4.31 |
| KkMAX-R (CFU/ml/h) | 0.97 | 0.97 | 0.94 | 3.38 | 1.38 |
| C50k-S (mg/liter) | 41.09 | 51.02 | 56.1 | 1.01 | 46.01 |
| C50k-R (mg/liter) | 5 | 5 | 5.02 | 5.54 | 41.94 |
| Hk-S | 12.43 | 13.36 | 1.33 | 13.4 | 5.07 |
| Hk-R | 17.9 | 17.83 | 19.04 | 17.61 | 0.5 |
| POPMAX (CFU/ml) | 1.72 × 1010 | 7.94 ×109 | 1.43 ×1010 | 3 ×109 | 3 ×1010 |
| INITCOND_2 (CFU/ml) | 5.76 ×106 | 5.54 ×106 | 1 ×106 | 1 ×106 | 1.48 ×106 |
| INITCOND_3 (CFU/ml) | 1.69 ×102 | 1.69 ×102 | 1.69 ×102 | 1.73 ×102 | 4.71 ×102 |
SCL is the fosfomycin clearance. INITCOND_2 and INITCOND_3 are the initial conditions for the total and resistant bacterial populations, respectively. Other parameter abbreviations can be found in the text.
Combination of fosfomycin and meropenem.
In order to promote the emergence of drug resistant mutants, a high inoculum was used (Fig. 4). Fosfomycin at 4 g/q8h (12 g/day) administered as monotherapy reduced the bacterial concentrations by 3 log10 CFU/ml. However, mutants able to grow at 256 mg/liter appeared after 48 h of treatment and, 24 h later, the resistant mutants replaced the susceptible population.
FIG 4.
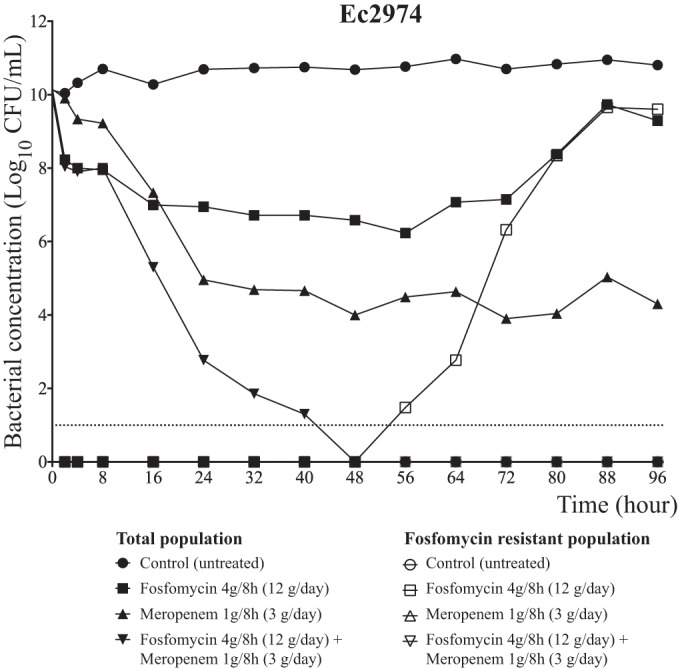
Efficacy of fosfomycin (at 4 g/q8h) and meropenem (at 1 g/q8h) in monotherapy and in combination against the Ec2974 strain in the HFIM.
On the other hand, meropenem at 1 g/q8h (3 g/day) alone diminished the bacterial burden by approximately 6 log10 CFU/ml. A reduction in the bactericidal activity was observed after 24 h. However, no mutants (able to grow at 4 mg/liter) appeared after 96 h of treatment. Finally, the combination of fosfomycin at 4 g/q8h and meropenem at 1 g/q8h produced a 10-log10 CFU/ml bacterial reduction, sterilizing the bacterial culture after 48 h. The combination not only added an extra 4-log10 CFU/ml decline compared to meropenem alone, but also completely suppressed all clones resistant to fosfomycin at a dose of this drug (4 g/q8h) that had previously allowed resistance emergence when employed as monotherapy. The results of the interaction model showed a synergistic effect for kill of the highly susceptible population (α = 4.78; 95% CI, 0.878 to 8.562) (Table 3). The fit of the model to the data was as follows. For the observed versus predicted regression for all meropenem concentrations: Observed = (1.142 × Predicted) − 0.874; R2 = 0.979. For the observed versus predicted regression for all fosfomycin concentrations: Observed = (0.985 × Predicted) + 1.089; R2 = 0.99. For the observed versus predicted regression for all total colony counts: Observed = (0.915 × Predicted) + 0.283; R2 = 0.903. For the observed versus predicted regression for all total colony counts (fosfomycin resistant): Observed = (1.142 × Predicted) − 0.874; R2 = 0.986.
TABLE 3.
Parameter estimates from the mathematical model from the drug combination study
| Parametera | Mean | Median | 95% CI |
|---|---|---|---|
| V1 (liters) | 11.71 | 12.56 | 10.0–12.60 |
| CL1 (liters/h) | 6.21 | 5.98 | 4.84–7.80 |
| V2 (liters) | 29.12 | 28.22 | 25.91–33.09 |
| CL2 (liters/h) | 6.59 | 6.32 | 4.48–8.94 |
| POPMAX (CFU/ml) | 1 × 1010 | 1 × 1010 | 1 × 1010–1.1 × 1010 |
| Kg-S (CFU/ml/h) | 1.59 | 1.50 | 1.27–2.00 |
| Kk-S (CFU/ml/h) | 2.43 | 2.21 | 2.08–3.00 |
| E50_1S (mg/liter) | 8.33 | 8.32 | 7.30–9.35 |
| E50_2S (mg/liter) | 13.51 | 14.84 | 10.00–15.75 |
| α_S | 4.78 | 4.88 | 0.878–8.56 |
| KgR-2 (CFU/ml/h) | 0.70 | 0.20 | 0.101–1.79 |
| KkR-2 (CFU/ml/h) | 2.52 | 2.97 | 1.23–3.38 |
| E50-2R2 (mg/liter) | 34.33 | 28.22 | 22.13–52.52 |
| α_R2 | 0.41 | 0.48 | −4.84–5.49 |
| INIT_3 (CFU/ml) | 1 × 1010 | 1 × 1010 | 1 × 1010–1.1 × 1010 |
| H1-S | 12.19 | 11.95 | 10.71–13.89 |
| H2-S | 2.25 | 1.58 | 1.12–4.00 |
| H2-R2 | 4.96 | 5.74 | 3.30–5.82 |
| TLAG3 (h) | 42.70 | 43.04 | 32.34–52.78 |
For parameters that include a subscript numeral (1 or 2), 1 denotes a meropenem parameter and 2 denotes a fosfomycin parameter. INIT_3 is the initial condition for the total bacterial population. TLAG3 is the lag time for the fosfomycin-resistant mutants' regrowth. E50 is the half-maximal effective concentration. α_S and α_R are the interaction parameters between the antimicrobials for the susceptible and resistant populations, respectively.
DISCUSSION
Fosfomycin is a phosphonic acid derivative with broad-spectrum antibacterial activity that often includes activity against organisms that are resistant to other first-line antibacterial agents (20). Fosfomycin was discovered in 1969 and has generally been used to treat uncomplicated urinary tract infections. Fosfomycin has a low molecular mass (138 Da), is highly hydrophilic, and has negligible protein binding. It is well tolerated and penetrates urine and most tissues, including the lung (21).
There is renewed interest in the use of fosfomycin for serious systemic infections caused by multidrug-resistant Enterobacteriaceae (11, 22). The intravenous formulation of fosfomycin is licensed in a limited number of countries for use in serious systemic infections (e.g., acute osteomyelitis, complicated urinary tract infections, nosocomial lower respiratory tract infections, bacterial meningitis, and bacteremia) that are caused by susceptible Gram-positive and Gram-negative bacteria (23–25).
Fosfomycin is a phosphoenolpyruvate analogue that covalently binds to amino acid residue Cys155 of MurA (UDP-GlcNAc enolpyruvyl transferase). Binding prevents the first steps of peptidoglycan biosynthesis, which ultimately leads to bacterial cell lysis and death (26). The glycerol-3-phosphate transporter (GlpT) and a hexose phosphate transporter (UhpT) have been described as the two main fosfomycin uptake transport systems in E. coli (26). The addition of G6P increases the expression of these transport proteins (UhpT), thus increasing the in vitro activity of fosfomycin (27). The supplementation of growth medium with 25 mg/liter G6P in the agar is part of the methodology for MIC determinations when using both the CLSI and the EUCAST methods (9, 28).
Our results with the E. coli isolate Ec2444, which had a fosfomycin MIC of 64 mg/liter, demonstrated that fosfomycin is ineffective as a monotherapy against this less-susceptible strain. In contrast, fosfomycin demonstrated a rapid and extensive bactericidal effect against the Ec46 strain, with an MIC of 1 mg/liter. However, all fosfomycin regimens with a total daily dose of <24 g per day resulted in the emergence of a resistant subpopulation after 30 to 40 h of drug exposure. There were no detectable high-level mutants (i.e., mutants able to grow in the presence of 256 mg/liter of fosfomycin) in the initial bacterial cultures, raising the question as to whether these were present in small numbers that were beneath the level of detection or whether they were truly absent. The latter case is more likely, simply because multiple samples were repeatedly negative. The appearance of highly resistant mutants may reflect progressive adaption of a low-level mutant or the development of sequential mutations that confer high-level resistance to fosfomycin. The molecular basis of such events requires further study: whether the appearance of mutants has any clinical implications in immunocompetent patients must be studied, but monotherapy would not seem to be a good option for E. coli infections in neutropenic and other immunosuppressed patients.
The pharmacodynamic index that best links drug exposure with antimicrobial efficacy is important for an understanding of the optimal use of fosfomycin (both in terms of bacterial killing and/or preventing the emergence of resistant subpopulations). Historically, fosfomycin has been considered an agent that exhibits time-dependent antibacterial activity (13, 29). In the current study, dose fractionation studies were conducted with two ESBL-producing strains with a fosfomycin MIC of 1 mg/liter. In both cases, the administration of fosfomycin resulted in the same rate and depth of bacterial killing, irrespective of the schedule of administration (Fig. 3, hours 8 to 16). This is consistent with the fAUC/MIC ratio being the pharmacodynamically linked index for resistance suppression. It remains possible, however, that a maximal effect was induced, because all regimens had TMIC values of 100% (baseline MIC, 1 mg/liter).
As resistance emergence was consistently observed, the pharmacodynamic index that is most closely linked to resistance suppression is of substantial importance. In Fig. 3, doses of 8 g/q8h (24 g/day) or 24 g q24h both completely suppressed resistance amplification. This is strong evidence that the fAUC/MIC ratio is linked to resistance suppression.
In serious infections, such as ventilator-acquired bacterial pneumonia (VABP), the bacterial burden is often substantial and can often exceed 9 log10 CFU/ml. We examined the utility of fosfomycin alone at the very dense inoculum of 1010 CFU/ml against Ec2974. In this experiment, the activity of fosfomycin alone was similar to that reported in the previous experiments, with a regrowth of resistant mutants. Due to the inability to prevent the appearance of mutants with monotherapy with fosfomycin for treatment of high-inoculum infections, we designed a study to test if the combination of fosfomycin (4 g/q8h) and meropenem (1 g/q8h) could improve bacterial killing and prevent the emergence of drug resistance. Meropenem alone resulted in increased killing relative to fosfomycin, but it did not result in extinction. We did not observe any meropenem-resistant mutants, although it is possible that a low level of mutants was present but not detectable (30).
The combination of fosfomycin and meropenem resulted in a >10-log10 CFU/ml kill and sterilization of the bacterial inoculum after 48 h of treatment. The mathematical model demonstrated a statistically significant synergistic interaction for killing of the fully susceptible population (α = 4.78; 95% CI, 0.878 to 8.562) (Table 3). The interaction was also additive for the subpopulation resistant to fosfomycin (α = 0.407; 95% CI, −4.84 to 5.49). This difference for the interaction being different for the fully susceptible and less-susceptible populations has been previously observed, although in this case α was positive, suggesting a favorable interaction of fosfomycin and meropenem (18). This highly promising result requires further validation in an in vivo setting, where immune effectors may contribute to the antimicrobial effect. The use of fosfomycin in vivo may result in the emergence of different and biologically distinct mutations to those observed in this study. Furthermore, the results may differ in the in vivo setting because of the lack of G6P supplementation.
In conclusion, we have observed different aspects for the use of fosfomycin against ESBL-producing E. coli strains. (i) The susceptibility breakpoints established for fosfomycin, ≤32 mg/liter (EUCAST) and ≤64 mg/liter (CLSI), for E. coli urinary tract infections only, appear to be too high for the treatment of serious systemic infections. (ii) Our results suggest that fosfomycin can be used to treat infections with lower MICs and lower bacterial densities, although relatively high daily dosages (i.e., 24 g/day) may be required to prevent the emergence of bacterial resistance. (iii) The fAUC/MIC appears to be the dynamically linked index for resistance suppression. (iv) Until more data are available, fosfomycin should not be used as monotherapy to treat systemic infections with either high MICs or with high bacterial densities. (v) The combination of fosfomycin and meropenem is synergistic and prevents the emergence of drug resistance. Such a strategy may be useful for the treatment of severe infections caused by ESBL-producing E. coli strains. This study provides the experimental basis for further clinical studies to identify optimal regimens of fosfomycin for the treatment of serious systemic infections.
ACKNOWLEDGMENTS
This work was supported by the Ministerio de Economía y Competitividad, Instituto de Salud Carlos III (PI 13/01885 and PI 13/01282) and the Consejería de Igualdad, Salud y Políticas Sociales, Junta de Andalucía (PI-0044-2013), Spain. The work was also partly supported by the Ministerio de Economía y Competitividad, Instituto de Salud Carlos III-FEDER, Spanish Network for Research in Infectious Diseases (REIPI RD12/0015). The work of Fernando Docobo-Pérez is supported by a Sara Borrell postdoctoral fellowship from the Instituto de Salud Carlos III. William Hope is supported by a National Institute of Health Research (NIHR) Clinician Scientist fellowship.
We declare the following potential conflicts of interest: J.R.B. acts as scientific advisor for AstraZeneca, Merck, and Achaogen and has been a speaker for AstraZeneca, Merck, Pfizer, and Astellas. He received funding for research from COMBACTE, COMBACTE-CARE, COMBACTE-MAGNET projects, and the Innovative Medicines Initiative (IMI), funded by the European Union and EFPIA companies. W.H. has received research funding and/or acted as a consultant for Pfizer, Gilead Sciences, Astellas, F2G, Basilea, and Pulmocide.
REFERENCES
- 1.Rodriguez-Baño J, Picon E, Gijon P, Hernandez JR, Ruiz M, Pena C, Almela M, Almirante B, Grill F, Colomina J, Gimenez M, Oliver A, Horcajada JP, Navarro G, Coloma A, Pascual A, Spanish Network for Research in Infectious Diseases (REIPI). 2010. Community-onset bacteremia due to extended-spectrum beta-lactamase-producing Escherichia coli: risk factors and prognosis. Clin Infect Dis 50:40–48. doi: 10.1086/649537. [DOI] [PubMed] [Google Scholar]
- 2.Rodriguez-Baño J, Navarro MD, Romero L, Muniain MA, de Cueto M, Rios MJ, Hernandez JR, Pascual A. 2006. Bacteremia due to extended-spectrum beta-lactamase-producing Escherichia coli in the CTX-M era: a new clinical challenge. Clin Infect Dis 43:1407–1414. doi: 10.1086/508877. [DOI] [PubMed] [Google Scholar]
- 3.Rodriguez-Bano J, Lopez-Prieto MD, Portillo MM, Retamar P, Natera C, Nuno E, Herrero M, del Arco A, Munoz A, Tellez F, Torres-Tortosa M, Martin-Aspas A, Arroyo A, Ruiz A, Moya R, Corzo JE, Leon L, Perez-Lopez JA, SAEI/SAMPAC Bacteraemia Group. 2010. Epidemiology and clinical features of community-acquired, healthcare-associated and nosocomial bloodstream infections in tertiary-care and community hospitals. Clin Microb Infect 16:1408–1413. doi: 10.1111/j.1469-0691.2010.03089.x. [DOI] [PubMed] [Google Scholar]
- 4.Pitout JD, Laupland KB. 2008. Extended-spectrum beta-lactamase-producing Enterobacteriaceae: an emerging public-health concern. Lancet Infect Dis 8:159–166. doi: 10.1016/S1473-3099(08)70041-0. [DOI] [PubMed] [Google Scholar]
- 5.Tzouvelekis LS, Markogiannakis A, Psichogiou M, Tassios PT, Daikos GL. 2012. Carbapenemases in Klebsiella pneumoniae and other Enterobacteriaceae: an evolving crisis of global dimensions. Clin Microbiol Rev 25:682–707. doi: 10.1128/CMR.05035-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rosso-Fernandez C, Sojo-Dorado J, Barriga A, Lavin-Alconero L, Palacios Z, Lopez-Hernandez I, Merino V, Camean M, Pascual A, Rodriguez-Bano J, FOREST Study Group . 2015. Fosfomycin versus meropenem in bacteraemic urinary tract infections caused by extended-spectrum beta-lactamase-producing Escherichia coli (FOREST): study protocol for an investigator-driven randomised controlled trial. BMJ Open 5:e007363. doi: 10.1136/bmjopen-2014-007363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Skarzynski T, Mistry A, Wonacott A, Hutchinson SE, Kelly VA, Duncan K. 1996. Structure of UDP-N-acetylglucosamine enolpyruvyl transferase, an enzyme essential for the synthesis of bacterial peptidoglycan, complexed with substrate UDP-N-acetylglucosamine and the drug fosfomycin. Structure 4:1465–1474. doi: 10.1016/S0969-2126(96)00153-0. [DOI] [PubMed] [Google Scholar]
- 8.de Cueto M, Lopez L, Hernandez JR, Morillo C, Pascual A. 2006. In vitro activity of fosfomycin against extended-spectrum beta-lactamase-producing Escherichia coli and Klebsiella pneumoniae: comparison of susceptibility testing procedures. Antimicrob Agents Chemother 50:368–370. doi: 10.1128/AAC.50.1.368-370.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Clinical and Laboratory Standards Institute. 2013. Performance standards for antimicrobial susceptibility testing, 23rd informational supplement. CLSI document M100-S23 Clinical and Laboratory Standards Institute, Wayne, PA. [Google Scholar]
- 10.Gupta K, Hooton TM, Naber KG, Wullt B, Colgan R, Miller LG, Moran GJ, Nicolle LE, Raz R, Schaeffer AJ, Soper DE, Infectious Diseases Society of America, European Society for Microbiology and Infectious Diseases. 2011. International clinical practice guidelines for the treatment of acute uncomplicated cystitis and pyelonephritis in women: a 2010 update by the Infectious Diseases Society of America and the European Society for Microbiology and Infectious Diseases. Clin Infect Dis 52:e103–e120. doi: 10.1093/cid/ciq257. [DOI] [PubMed] [Google Scholar]
- 11.Karageorgopoulos DE, Wang R, Yu XH, Falagas ME. 2012. Fosfomycin: evaluation of the published evidence on the emergence of antimicrobial resistance in Gram-negative pathogens. J Antimicrob Chemother 67:255–268. doi: 10.1093/jac/dkr466. [DOI] [PubMed] [Google Scholar]
- 12.Felton TW, Goodwin J, O'Connor L, Sharp A, Gregson L, Livermore J, Howard SJ, Neely MN, Hope WW. 2013. Impact of bolus dosing versus continuous infusion of piperacillin and tazobactam on the development of antimicrobial resistance in Pseudomonas aeruginosa. Antimicrob Agents Chemother 57:5811–5819. doi: 10.1128/AAC.00867-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pfausler B, Spiss H, Dittrich P, Zeitlinger M, Schmutzhard E, Joukhadar C. 2004. Concentrations of fosfomycin in the cerebrospinal fluid of neurointensive care patients with ventriculostomy-associated ventriculitis. J Antimicrob Chemother 53:848–852. doi: 10.1093/jac/dkh158. [DOI] [PubMed] [Google Scholar]
- 14.Mouton JW, Touzw DJ, Horrevorts AM, Vinks AA. 2000. Comparative pharmacokinetics of the carbapenems: clinical implications. Clin Pharmacokin 39:185–201. doi: 10.2165/00003088-200039030-00002. [DOI] [PubMed] [Google Scholar]
- 15.Li L, Chen X, Dai X, Chen H, Zhong D. 2007. Rapid and selective liquid chromatographic/tandem mass spectrometric method for the determination of fosfomycin in human plasma. J Chromatogr B 856:171–177. doi: 10.1016/j.jchromb.2007.05.037. [DOI] [PubMed] [Google Scholar]
- 16.Gonzalez D, Schmidt S, Derendorf H. 2013. Importance of relating efficacy measures to unbound drug concentrations for anti-infective agents. Clin Microbiol Rev 26:274–288. doi: 10.1128/CMR.00092-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Neely MN, van Guilder MG, Yamada WM, Schumitzky A, Jelliffe RW. 2012. Accurate detection of outliers and subpopulations with Pmetrics, a nonparametric and parametric pharmacometric modeling and simulation package for R. Ther Drug Monitor 34:467–476. doi: 10.1097/FTD.0b013e31825c4ba6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Drusano GL, Neely M, Van Guilder M, Schumitzky A, Brown D, Fikes S, Peloquin C, Louie A. 2014. Analysis of combination drug therapy to develop regimens with shortened duration of treatment for tuberculosis. PLoS One 9:e101311. doi: 10.1371/journal.pone.0101311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Leary RH, Jelliffe R, Schumitzky A, Van Guilder M. 2001. An adaptive grid non-parametric approach to population pharmacokinetic/dynamic (PK/PD) population models, p 389–394. In Proceedings 14th IEEE Symposium on Computer-Based Medical Systems. IEEE, New York, NY. [Google Scholar]
- 20.Hendlin D, Stapley EO, Jackson M, Wallick H, Miller AK, Wolf FJ, Miller TW, Chaiet L, Kahan FM, Foltz EL, Woodruff HB, Mata JM, Hernandez S, Mochales S. 1969. Phosphonomycin, a new antibiotic produced by strains of streptomyces. Science 166:122–123. doi: 10.1126/science.166.3901.122. [DOI] [PubMed] [Google Scholar]
- 21.Roussos N, Karageorgopoulos DE, Samonis G, Falagas ME. 2009. Clinical significance of the pharmacokinetic and pharmacodynamic characteristics of fosfomycin for the treatment of patients with systemic infections. Int J Antimicrob Agents 34:506–515. doi: 10.1016/j.ijantimicag.2009.08.013. [DOI] [PubMed] [Google Scholar]
- 22.Falagas ME, Kastoris AC, Kapaskelis AM, Karageorgopoulos DE. 2010. Fosfomycin for the treatment of multidrug-resistant, including extended-spectrum beta-lactamase producing, Enterobacteriaceae infections: a systematic review. Lancet Infect Dis 10:43–50. doi: 10.1016/S1473-3099(09)70325-1. [DOI] [PubMed] [Google Scholar]
- 23.Pulcini C, Bush K, Craig WA, Frimodt-Moller N, Grayson ML, Mouton JW, Turnidge J, Harbarth S, Gyssens IC, ESCMID Study Group for Antibiotic Policies. 2012. Forgotten antibiotics: an inventory in Europe, the United States, Canada, and Australia. Clin Infect Dis 54:268–274. doi: 10.1093/cid/cir838. [DOI] [PubMed] [Google Scholar]
- 24.Agencia Española de Medicamentos y Productos Sanitarios. 2013. Fosfomicina. Spanish Agency of Medicines and Medical Devices, Madrid, Spain: http://www.aemps.gob.es/cima/pdfs/es/ft/54165/FT_54165.pdf. [Google Scholar]
- 25.Medicines and Healthcare Products Regulatory Agency. 2015. Fosfomycin sodium. MHRA, London, England: http://www.mhra.gov.uk/home/groups/par/documents/websiteresources/con309596.pdf. [Google Scholar]
- 26.Kahan FM, Kahan JS, Cassidy PJ, Kropp H. 1974. The mechanism of action of fosfomycin (phosphonomycin). Ann N Y Acad Sci 235:364–386. doi: 10.1111/j.1749-6632.1974.tb43277.x. [DOI] [PubMed] [Google Scholar]
- 27.Grimm H. 1979. In vitro investigations with fosfomycin on Mueller-Hinton agar with and without glucose-6-phosphate. Infection 7:256–259. doi: 10.1007/BF01648937. [DOI] [PubMed] [Google Scholar]
- 28.European Committee on Antimicrobial Susceptibility Testing. 2015. Breakpoint tables for interpretation of MICs and zone diameters, version 5.0. EUCAST, Växjö, Sweden: http://www.eucast.org/ Accessed 30 April 2015. [Google Scholar]
- 29.Sauermann R, Karch R, Langenberger H, Kettenbach J, Mayer-Helm B, Petsch M, Wagner C, Sautner T, Gattringer R, Karanikas G, Joukhadar C. 2005. Antibiotic abscess penetration: fosfomycin levels measured in pus and simulated concentration-time profiles. Antimicrob Agents Chemother 49:4448–4454. doi: 10.1128/AAC.49.11.4448-4454.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tangden T, Adler M, Cars O, Sandegren L, Lowdin E. 2013. Frequent emergence of porin-deficient subpopulations with reduced carbapenem susceptibility in ESBL-producing Escherichia coli during exposure to ertapenem in an in vitro pharmacokinetic model. J Antimicrob Chemother 68:1319–1326. doi: 10.1093/jac/dkt044. [DOI] [PubMed] [Google Scholar]