

Abstract
Novel mechanisms of action and new chemical scaffolds are needed to rejuvenate antibacterial drug discovery, and riboswitch regulators of bacterial gene expression are a promising class of targets for the discovery of new leads. Herein, we report the characterization of 5-(3-(4-fluorophenyl)butyl)-7,8-dimethylpyrido[3,4-b]quinoxaline-1,3(2H,5H)-dione (5FDQD)—an analog of riboflavin that was designed to bind riboswitches that naturally recognize the essential coenzyme flavin mononucleotide (FMN) and regulate FMN and riboflavin homeostasis. In vitro, 5FDQD and FMN bind to and trigger the function of an FMN riboswitch with equipotent activity. MIC and time-kill studies demonstrated that 5FDQD has potent and rapidly bactericidal activity against Clostridium difficile. In C57BL/6 mice, 5FDQD completely prevented the onset of lethal antibiotic-induced C. difficile infection (CDI). Against a panel of bacteria representative of healthy bowel flora, the antibacterial selectivity of 5FDQD was superior to currently marketed CDI therapeutics, with very little activity against representative strains from the Bacteroides, Lactobacillus, Bifidobacterium, Actinomyces, and Prevotella genera. Accordingly, a single oral dose of 5FDQD caused less alteration of culturable cecal flora in mice than the comparators. Collectively, these data suggest that 5FDQD or closely related analogs could potentially provide a high rate of CDI cure with a low likelihood of infection recurrence. Future studies will seek to assess the role of FMN riboswitch binding to the mechanism of 5FDQD antibacterial action. In aggregate, our results indicate that riboswitch-binding antibacterial compounds can be discovered and optimized to exhibit activity profiles that merit preclinical and clinical development as potential antibacterial therapeutic agents.
INTRODUCTION
Despite nearly 85 years of successful antibacterial chemotherapy, bacterial infections still pose a serious threat to human health. Continually emerging drug-resistant bacteria make existing agents less effective, and a paucity of new agents and decreased development effort from the pharmaceutical industry provide little hope of replenishing the arsenal (1, 2). Complications and mortality due to bacterial infections continue to increase, already reaching epidemic proportions in some areas of the world. If humans are to regain the upper hand in fighting bacterial infections, then innovation, investment, and new antibacterial agents will be needed. Although some of the barriers to the discovery and approval of new compounds are economic or policy related, there is also a desperate need for new targets and new mechanisms of action. A renewed effort to expand our knowledge of bacterial physiology and to translate discoveries into the clinic will be needed to address these challenges and to reinvigorate antibiotic development pipelines.
Recent advances in our understanding of how bacteria maintain physiological homeostasis revealed a promising class of potential antibiotic targets called riboswitches—noncoding mRNAs that form a structured receptor (or aptamer) which can directly bind to a specific small-molecule ligand or ion and thereby regulate gene expression (3–5). Ligand binding to a riboswitch aptamer stabilizes a conformationally distinct architecture in the mRNA that modulates the expression of the adjacent coding region(s) (4–8). To date, more than 35 riboswitch classes have been discovered and characterized (7). Three of these riboswitch classes have been revealed as important cellular targets of antibacterial small molecules whose mechanism of action had not been previously defined (9–13). More recently, several publications have demonstrated that novel small molecules that bind to selected riboswitch aptamers with affinities comparable to that of the cognate ligand can be rationally identified and optimized (14–21).
In some cases, synthetic or natural riboswitch ligand analogs have demonstrated potent antibacterial activity (12–15, 20, 22). For example, the phosphorylated form of roseoflavin (RoF) (Fig. 1A), a naturally produced analog of riboflavin that inhibits the growth of Gram-positive bacteria (23, 24), directly binds to riboswitches that recognize the essential coenzyme flavin mononucleotide (FMN) (12, 13, 22). Accordingly, the presence of RoF in growth media represses the expression of FMN riboswitch-regulated riboflavin biosynthesis and transport genes and inhibits bacterial growth (12, 22). The fact that RoF antibacterial action is partly due to FMN riboswitch binding underscores the potential for developing riboswitch-targeting antibiotics.
FIG 1.

Riboswitch binding and functional activity of 5FDQD [5-(3-(4-fluorophenyl)butyl)-7,8-dimethylpyrido[3,4-b]quinoxaline-1,3(2H,5H)-dione]. (A) Chemical structures of 5FDQD, flavin mononucleotide (FMN), roseoflavin (RoF), and roseoflavin phosphate (RoF-P). (B) Sequence and secondary structure model for B. subtilis 165 ribD, the 165-nucleotide aptamer region of the B. subtilis ribD FMN riboswitch. Nucleotides where spontaneous cleavage activity decreases upon addition of FMN or 5FDQD are identified with gray circles (regions 1 to 6, corresponding to the bands identified in panel C). The nucleotide position where spontaneous cleavage increases upon addition of 5FDQD is designated with an open circle (gray arrowhead in panel C). (C) PAGE analysis of an in-line probing assay with 5′ 32P-labeled 156 ribD RNA exposed to various concentrations of FMN or 5FDQD (18 pM to 300 nM in 4-fold increments). NR, −OH, and T1 designate no reaction (NR) and partial digestion with either hydroxide ions (cleaves after any nucleotide) or RNase T1 (cleaves after guanosine nucleotides), respectively. Precursor RNA (Pre) and certain RNase T1 cleavage product bands are identified. The locations of spontaneous RNA cleavage changes upon addition of FMN or 5FDQD (regions 1 through 6) are identified by numbered bars to the right of the gel. The location of an increase in spontaneous RNA cleavage upon addition of 5FDQD is identified by a gray arrowhead to the right of the gel. (D) Plot depicting the average of the normalized fraction of RNA cleaved at regions 1 to 6 versus the concentration of either FMN or 5FDQD. The curves indicate the best fit of the data to an equation for a two-state binding model (see Materials and Methods). (E) PAGE analysis of an in vitro transcription termination assay using the ribD FMN riboswitch from B. subtilis with the addition of various concentrations of 5FDQD (6 nM to 100 μM in 4-fold intervals). The positions of the riboswitch-terminated RNA transcript (T) and full-length transcript (FL) are indicated to the left of the gel. (F) Plot depicting the fraction of transcripts in the terminated form versus the concentration of 5FDQD or FMN. The curves indicate the best fit of the data to a two-state model for ligand-dependent termination activity.
Concurrent with these initial validation studies with RoF, we established a separate research and development program that identified and progressively improved a class of novel flavin analogs that bind to FMN riboswitches in vitro with potencies equal to that of FMN (P. D. G. Coish et al., 20 January 2011, patent application PCT/US2010/001876; P. D. G. Coish et al., 13 October 2011, patent application PCT/US2011/000617; P. A. Aristoff, P. D. G. Coish, and B. R. Dixon, 12 August 2012, patent application PCT/US2012/024507). A subset of these compounds were found to exhibit potent and highly selective antibacterial activity against Clostridium difficile. These findings suggest that a novel class of antibiotics that target FMN riboswitches could be developed to provide a useful alternative to currently approved therapies for C. difficile intestinal infections (CDI).
In a 2013 threat assessment report, the Centers for Disease Control and Prevention estimated that CDI cause 14,000 deaths and one billion dollars in excess medical costs each year in the United States (25). The current standard-of-care treatments for CDI, metronidazole or vancomycin, are usually effective, but the infection recurs in approximately 25% of patients, with subsequent recurrences even more likely (26–28). The broad-spectrum antibacterial activities of metronidazole and vancomycin are thought to contribute to recurrence by preventing the regrowth of healthy intestinal flora (29–33). A newer treatment, fidaxomicin, has a narrower spectrum of activity than vancomycin or metronidazole (34–36) and causes less alteration to the intestinal flora of CDI patients (32, 33). Consistent with this narrower spectrum of activity, multiple clinical trials have demonstrated a lower rate of CDI recurrence after treatment with fidaxomicin than after vancomycin or metronidazole treatment (37, 38). As a result, CDI therapies currently in development primarily include narrow- spectrum antibiotics or intestinal flora replacements (39–41).
Herein, we report the synthesis and characterization of 5-(3-(4-fluorophenyl)butyl)-7,8-dimethylpyrido[3,4-b]quinoxaline-1,3(2H,5H)-dione (5FDQD) (Fig. 1A), an analog of FMN that was identified via medicinal chemistry optimization of FMN riboswitch-binding ligands. 5FDQD is rapidly bactericidal against C. difficile in vitro and can cure mice of laboratory-induced CDI nearly as effectively as fidaxomicin. Among a collection of bacteria representative of natural intestinal microflora, 5FDQD has a narrower spectrum than those of fidaxomicin and vancomycin. Moreover, administration of a single dose of 5FDQD to healthy mice was less disruptive to the culturable cecal flora than fidaxomicin or vancomycin was.
MATERIALS AND METHODS
Chemicals, oligonucleotides, and bacterial strains.
5FDQD was resynthesized using a method similar to that published in patent literature (Coish et al., patent application PCT/US2010/001876; Coish et al., patent application PCT/US2011/000617; Aristoff et al., patent application PCT/US2012/024507). The detailed synthetic procedures and spectral characterization are described in the supplemental material. Except as specifically noted, all chemicals were purchased from Sigma-Aldrich (St. Louis, MO). Except as specifically noted, all growth media were purchased from BD Diagnostic Systems (Sparks, MD) and prepared in accordance with the manufacturer's instructions, and blood supplements were purchased from Cleveland Scientific (Bath, OH).
In-line probing assays.
The B. subtilis 165 ribD RNA used for in-line probing assays was prepared by in vitro transcription using a template generated from genomic DNA from Bacillus subtilis strain 168 (American Type Culture Collection, Manassas, VA). RNA transcripts were dephosphorylated, 5′ 32P labeled, and subsequently subjected to in-line probing using protocols similar to those described previously (42). The apparent dissociation constant (KD) for each ligand was derived by quantifying the amount of RNA cleaved at each nucleotide position over a range of ligand concentrations. For each region where modification was observed (regions 1 to 6 in Fig. 1B), the fraction of RNA cleaved at each ligand concentration was calculated by assuming that the maximal extent of cleavage is observed in the absence of ligand and the minimal cleavage is observed in the presence of the highest ligand concentration. The apparent KD was determined by using GraphPad Prism 6 software to fit the plot of the fraction cleaved, x versus the ligand concentration, [L], to the following equation: x = KD ([L] + KD)−1.
In vitro RNA transcription termination assays.
Single-round RNA transcription termination assays were conducted following protocols adapted from a previously described method (43). The DNA template covered the region from positions −382 to +13 (relative to the start of translation) of the B. subtilis ribD gene and was generated by PCR from genomic DNA from B. subtilis strain 168. To initiate transcription and form halted complexes, each sample was incubated at 37°C for 10 min and contained 1 pmol DNA template, 0.17 mM ApA dinucleotide (TriLink Biotechnologies, San Diego, CA), 2.5 μM ATP, 2.5 μM GTP, 2.5 μM UTP, 2 μCi 5′-[α-32P]UTP, and 0.4 U Escherichia coli RNA polymerase holoenzyme (Epicenter, Madison, WI) in 10 μl of 80 mM Tris-HCl (pH 8.0 at 23°C), 20 mM NaCl, 5 mM MgCl2, 0.1 mM EDTA, and 0.01 mg/ml bovine serum albumin (BSA) (New England BioLabs, Ipswich, MA). Halted complexes were restarted by the simultaneous addition of a mixture of all four nucleoside triphosphates (NTPs) that yield a final concentration of 50 μM, 0.2 mg/ml heparin to prevent reinitiation, and various concentrations of ligand as indicated to yield a final volume of 12.5 μl in a buffer containing 150 mM Tris-HCl (pH 8.0 at 23°C), 20 mM NaCl, 5 mM MgCl2, 0.1 mM EDTA, and 0.01 mg/ml BSA. Reaction mixtures were incubated for an additional 20 min at 37°C, and the products were separated by denaturing 10% polyacrylamide gel electrophoresis (PAGE) followed by quantitation of the fraction terminated at each ligand concentration by using a phosphorimager (Storm 860; GE Healthcare Life Sciences, Pittsburgh, PA).
In vitro antibacterial activity.
MIC analyses were conducted by Micromyx, LLC (Kalamazoo, MI) in accordance with standard microdilution methods for aerobic bacteria (44) and anaerobic bacteria (45). All procedures involving anaerobic bacteria were conducted within a Bactron 300 or Bactron II anaerobic chamber (Sheldon Manufacturing, Cornelius, OR) using prereduced media. MICs for anaerobic bacteria were determined in supplemented brucella broth, comprising BBL brucella broth, 5 μg/ml hemin, 10 μg/ml vitamin K, and 5% lysed horse blood. MICs for aerobic bacteria were determined in Mueller-Hinton II broth, and a supplement of 3% lysed horse blood was included for assays with Streptococcus pneumoniae. Stock solutions of fidaxomicin (Ontario Chemicals, Guelf, ON, Canada) and 5FDQD in dimethyl sulfoxide (DMSO) or vancomycin in water were used to prepare dilution plates such that the final concentration of DMSO in the assay did not exceed 5%. Time-kill assays for bactericidal activity were conducted at 35°C in supplemented brucella broth. A suspension of C. difficile strain VPI 10463 (ATCC 43255) was prepared from day-old colonies on supplemented brucella agar (BBL brucella agar, 5 μg/ml hemin, 10 μg/ml vitamin K, and 5% lysed sheep blood). Tenfold dilutions of the suspension spanning 102 to 105 were prepared and allowed to grow overnight in broth at 35°C. On the following morning, a culture with a measurable optical density at 600 nm (OD600) of less than 0.4 was diluted by 40-fold into 12 ml of fresh broth that had been prewarmed to 35°C. Two-milliliter aliquots of this culture were immediately mixed with 100 μl of DMSO or 100 μl of a solution of 5FDQD, fidaxomicin, or vancomycin sufficient to give the final concentrations indicated in Fig. 2. Serial dilutions of the starter culture into prereduced phosphate-buffered saline (PBS) were plated onto brain heart infusion agar supplemented with 0.5% yeast extract and 0.1% l-cysteine (BHIS) to measure the density at time zero and to confirm that the starting inoculum was greater than 5 × 105 CFU/ml (46). Serial dilutions of each test culture were prepared in PBS and plated onto BHIS agar at the indicated time points, and colonies were counted after 24 h and used to calculate the culture densities in CFU per milliliter. The lower limit of detection using this method was 200 CFU/ml.
FIG 2.

Time-kill analysis of 5FDQD, fidaxomicin, and vancomycin. Representative time-kill plot with DMSO, fidaxomicin (Fid), vancomycin (Van), or 5FDQD treatment of C. difficile VPI 10463. At 6 h, 4× and 8× MIC 5FDQD reduced the density of C. difficile VPI 10463 below 200 CFU/ml, the lower limit of detection (dashed black line).
Preparation of C. difficile spores for infecting mice.
C. difficile spores for the mouse infection model were prepared at Micromyx (Kalamazoo, MI). A frozen culture of C. difficile VPI 10463 was inoculated into 4 liters of prereduced sporulation medium comprising 80 g/liter Trypticase peptone, 5 g/liter proteose peptone no. 3, 1 g/liter ammonium sulfate, and 1.5 g/liter Tris adjusted to a final pH of 7.4. The culture was incubated in an anaerobic chamber at 35°C for 5 days, centrifuged, resuspended in 50% ethanol, and incubated at room temperature to eliminate any remaining vegetative bacterial cells. The spores were then washed twice in sterile PBS and enumerated by dilution plating onto supplemented brucella agar with 0.1% sodium taurocholate. Following quantitation, the spores were diluted to a concentration of 2 × 106 spores/ml and stored at −80°C.
Mouse model of CDI.
CDI efficacy studies were conducted by Aragen Biosciences (Morgan Hill, CA) and were reviewed and approved by Aragen's Institutional Animal Care and Use Committee (IACUC). Female C57BL/6 mice weighing 18 to 21 g were ordered from Harlan Laboratories (Indianapolis, IN), housed five per cage in a dedicated room, and allowed to acclimatize for at least 3 days. Animals were fed Teklad global rodent diet 2018-R (Harlan Laboratories, Indianapolis, IN) throughout the study. For the 7 days following acclimatization (days −10 through −4), mice were provided an antibiotic cocktail in their autoclaved drinking water that included kanamycin (0.5 mg/ml), gentamicin (0.044 mg/ml), colistin (1,062.5 U/ml), metronidazole (0.269 mg/ml), ciprofloxacin (0.156 mg/ml), ampicillin (0.1 mg/ml), and vancomycin (0.056 mg/ml). Mice were then provided normal autoclaved drinking water for 3 days (days −3 through −1), followed by a single oral dose of 10 mg of clindamycin per kg of body weight in a volume of 0.5 ml. Immediately following clindamycin dosing, animals were randomized and distributed into new cages of five animals such that the average weight across the groups of five animals was similar. On the following day (day 0), an inoculum of 2,000 spores of C. difficile VPI 10463 (ATCC 43255) in 0.5 ml of sterile PBS was orally administered to each mouse. Within 1 h of inoculation, infected mice were administered the treatments indicated in Fig. 3 by oral gavage twice a day (with approximately 10 h elapsing between morning and evening treatments) for 5 days, with 10 animals per treatment group. All test agents were dosed as a suspension in 0.5% aqueous methylcellulose. Body weights were measured three times a week prior to C. difficile infection and daily following infection. Animals were examined in the morning and evening for eight consecutive days following spore inoculation for clinical signs of morbidity, evidence of diarrhea, and mortality. At the end of the study, surviving animals were humanely euthanized.
FIG 3.

Efficacy of 5FDQD, fidaxomicin, and vancomycin in a mouse model of antibiotic-induced C. difficile infection. (A) Survival of C. difficile VPI 10463-infected C57BL/6 mice that were treated twice daily with 10 mg/kg 5FDQD, 3 mg/kg fidaxomicin (Fid), 0.5% methylcellulose vehicle control, or 50 mg/kg vancomycin (Van) once daily. All treatments were administered orally for 5 days beginning 1 h after inoculation, and each treatment group included 10 mice. One hundred percent survival was observed for 8 days with 5FDQD, fidaxomicin, and vancomycin as highlighted by an asterisk. (B) Average daily weight of surviving C. difficile VPI 10463-infected mice that received the treatments described above for panel A, normalized to the average weight of mice in each treatment group immediately prior to inoculation (set at a value of 1). Treatment groups are shown by the same symbols as shown in panel A. The weights of vehicle-treated mice that died during the study were omitted from the calculation of average weights after they died.
Pharmacokinetic analysis.
The Yale University IACUC reviewed and approved all animal experimental protocols for pharmacokinetic analysis (protocol number 20114-11652). Female C57BL/6 mice weighing 18 to 21 g were ordered from Harlan Laboratories, housed five per cage, and allowed to acclimatize for 14 days. Animals were fed Teklad global rodent diet 2018-R throughout the study. After acclimatization, 24 mice each received a single oral dose of 0.2 ml of a 1-mg/ml suspension of 5FDQD in aqueous 0.5% methylcellulose for a total dose of 10 mg/kg. At 1, 2, 4, 6, 8, and 24 h after dosing, three mice per time point were anesthetized with isoflurane and exsanguinated via cardiac puncture with a preheparinized syringe, and the collected blood was transferred to lithium heparinized tubes and held on ice until further processing. After exsanguination, the mice were humanely euthanized by cervical dislocation, the ceca were immediately removed, and the cecal contents were collected into preweighed tubes and frozen at −80°C until extraction. The previously collected blood samples were centrifuged at 4°C for 10 min at 6,000 rpm, and the plasma supernatant was collected into a separate tube and frozen at −80°C until extraction.
Sample extraction and bioanalyses were conducted by Northeast Bioanalytical (Hamden, CT). Cecal content samples were suspended in 2.0 ml of extraction/precipitation solution that contained 50% acetonitrile, 50% methanol, and 0.1% formic acid. The resulting suspensions were placed on a shaker table for 20 to 30 min, followed by centrifugation. Thawed plasma samples were combined with 3 volumes of extraction/precipitation solution, mixed, and allowed to warm to room temperature. A 100-μl aliquot of the cleared cecal extraction supernatant or plasma solution was transferred into a labeled autosampler vial and combined with 100 μl of an internal standard compound and 200 μl of extraction/precipitation solution. Samples were then capped, vortexed, and analyzed by liquid chromatography-tandem mass spectrometry (LC-MS/MS).
Test samples were injected onto a Phenomenex Synergi Polar-RP column (150 by 4.6 mm) with a corresponding guard column. The column temperature was maintained at 50°C. The analytes were separated using a variable composition of eluent A (acetonitrile) and eluent B (high-performance liquid chromatography [HPLC]-grade water with 0.1% formic acid). The initial composition of 40% eluent A and 60% eluent B was maintained at a flow rate of 1,000 μl/min for 1 min, at which point the composition of eluent A was increased to 95% over 0.5 min and held at 95% for 4 min at a flow rate of 1,300 μl/min. The composition of eluent A was then decreased to 40% at a flow rate of 1,100 μl/min and held at 40% for 0.5 min, at which point the flow rate was reduced to 1,000 μl/min and reequilibrated before injecting another sample. Analyte detection was performed by a Sciex API-4000 tandem mass spectrometer fitted with a Turbo Ion Spray source operating in positive-ion mode. Samples were quantitated against a calibration standard curve constructed from a stock solution of a known concentration of 5FDQD and using a linear regression of theoretical concentration of calibrators versus the response ratio (where the response ratio is the analyte peak area/labeled internal standard peak area). The regression used a 1/x2 weighting factor in the equation y = mx + b. An internal standard that is a structural analog of 5FDQD was included in all samples. The lower and upper limits of quantitation for 5FDQD in plasma and cecal contents were 12 ng/ml and 5,000 ng/ml, respectively.
Cecal flora analysis.
The Yale University IACUC reviewed and approved all animal experimental protocols for cecal flora analysis (protocol number 20114-11652). A total of 28 female C57BL/6 mice weighing 18 to 21 g were ordered from Harlan Laboratories, housed four per cage, and allowed to acclimatize for 7 days. Animals were fed Teklad global rodent diet 2018-R throughout the study. After acclimatization, mice were randomized into groups of four mice per cage for each treatment group. All four mice in a given treatment group received an oral dose of 0.2 ml of aqueous 0.5% methylcellulose (vehicle control) or 0.2 ml of an aqueous 0.5% methylcellulose suspension of 5FDQD, fidaxomicin, or vancomycin such that the total doses administered were 10 or 50 mg/kg 5FDQD, 3 or 50 mg/kg fidaxomicin, or 50 mg/kg vancomycin. Twenty-four hours after the doses were administered, the mice were humanely euthanized by cervical dislocation, the ceca were immediately removed, and the cecal contents were collected into preweighed tubes, weighed, and transferred immediately into an anaerobic chamber such that cecal contents were never exposed to the ambient atmosphere for greater than 10 min. Within the anaerobic chamber, the cecal contents were resuspended in 1 ml of prereduced 0.05% yeast extract per 100 mg of contents and homogenized by vortex mixing. A series of 10-fold serial dilutions ranging from 101 to 107 were prepared in 0.05% yeast extract, and 50-μl aliquots of each dilutions were plated in parallel onto agar media selective for each bacterial taxa, including Bacteroides bile esculin agar (47), Bifidobacterium-selective agar (48), Lactobacillus-selective agar (49), Enterococcosel, MacConkey agar, and mannitol salt agar. To enumerate Clostridium spores, an aliquot of the 101 and 102 dilutions for each animal was mixed with an equal volume of ethanol and incubated for 1 h to kill vegetative cells. Fifty microliters of each dilution was then plated onto egg yolk agar (Anaerobe Systems, Morgan Hill, CA). After growth at 35°C for the number of days appropriate to each medium and bacterial type, the colonies were counted, categorized according to visible morphology, and used to calculate the density of each taxa in the starting cecal contents in units of log CFU per milliliter. The reported densities represent an average of all countable dilution plates from four mice per treatment group. Each colony type was presumptively identified to the genus or species level based on visible appearance, Gram stain, and microscopic appearance. Where necessary, catalase, indole, and oxidase tests and antimicrobial disc susceptibility testing were used to confirm the presumptive identifications (47). Clostridium spiroforme was presumptively identified by colony morphology on egg yolk agar, Gram stain, ability to form spores, a unique helical shape viewable under a microscope, and antimicrobial susceptibility pattern including rifampin resistance (50, 51). The relative density of vegetative C. spiroforme was enumerated in parallel by dilution plating of selected cecal content samples onto brucella agar that contained 5% defibrinated sheep blood and 50 μg/ml rifampin.
RESULTS
The FMN analog 5FDQD is bound by FMN aptamers and modulates riboswitch activity.
An in-line probing assay (42) was used to assess the binding of 5FDQD to the ribD FMN riboswitch aptamer from B. subtilis (Fig. 1B) and to determine an apparent dissociation constant (KD) for the binding interaction. This assay exploits the fact that the internucleotide linkages in unstructured regions of an RNA will usually undergo phosphoester cleavage more rapidly than those in structured regions (52). The heightened flexibility of unstructured regions usually permits more frequent access to a conformation necessary for spontaneous cleavage by an internal RNA phosphoester transfer reaction. As the secondary and tertiary structures of the RNA change upon binding to a ligand, the reactivity of some of the internucleotide linkages will change, resulting in a different pattern of degradation products. The differences in RNA degradation products and the locations of structural changes can be detected and quantified via denaturing PAGE.
Using the in-line probing assay, 5FDQD and FMN induce nearly identical structural changes in the B. subtilis ribD FMN aptamer (Fig. 1C, regions 1 through 6). The only observable difference was that 5FDQD binding increases spontaneous cleavage of the internucleotide linkage between G33 and G34, whereas FMN decreases reactivity at this same position (Fig. 1C, gray arrowhead). The three-dimensional X-ray structure model of a Fusobacterium nucleatum FMN riboswitch aptamer complexed with FMN predicts an interaction between G34 and a hydroxyl group of the ribityl side chain of FMN (53). This same interaction cannot be formed in a complex with 5FDQD because the analog lacks hydroxyl groups, which likely explains this single difference in reactivity as revealed by in-line probing. Therefore, the natural FMN ligand and the FMN analog 5FDQD are most likely recognized by the same binding pocket and induce nearly identical structural changes in the aptamer.
RNA-ligand binding curves were established by evaluating the relative change in cleavage at regions 1 through 6 as a function of compound concentration (Fig. 1D). The concentrations of ligands needed to half-maximally modulate RNA aptamer structure, representing KD values, were 7.5 nM and 6.4 nM for 5FDQD and FMN, respectively. Thus, 5FDQD and FMN bind to the B. subtilis ribD FMN aptamer in vitro with equivalent potency. Moreover, the binding curves suggest that both compounds bind a single saturable site with 1-to-1 stoichiometry.
An in vitro transcription termination assay was performed to determine whether 5FDQD can induce FMN riboswitch function (Fig. 1E and F). Using a double-stranded DNA template that included the B. subtilis ribD endogenous promoter, FMN riboswitch region, and the first 13 nucleotides of the open reading frame, the concentration at which FMN or 5FDQD induce half-maximal transcription termination (T50) was determined. The T50 values for 5FDQD and FMN were 0.5 μM and 0.6 μM, respectively, indicating that 5FDQD can induce riboswitch function in vitro, again with the same potency as for FMN.
5FDQD exhibits robust and selective antibacterial activity.
Against a panel of 21 strains of C. difficile, including four hypervirulent North American pulsed-field gel electrophoresis type 1 (NAP1) ribotype 027 strains (54, 55) and 14 isolates from hospital patients, the MIC50 and MIC90 of 5FDQD were determined to be 1 μg/ml by broth microdilution methods (Table 1), compared to MIC50 and MIC90 values of 0.06 and 0.12 μg/ml for fidaxomicin. Although 5FDQD is less potent than fidaxomicin, it displays potencies equivalent to the published values for both vancomycin and metronidazole (56). Notably, there was less variability among the MIC values for 5FDQD than for fidaxomicin among the isolates tested.
TABLE 1.
Antibacterial activity of 5FDQD and fidaxomicin
| Organism (no. of isolates) | Strain | MIC (μg/ml) |
|
|---|---|---|---|
| 5FDQD | Fidaxomicin | ||
| Clostridium difficile (21)a | 0.5–1 | 0.03–0.5 | |
| Bacteroides fragilis group spp. (10)b | >32 | >32 | |
| Prevotella melaninogenica | 3437 | >32 | >32 |
| Prevotella bivia | 3447 | >32 | >32 |
| Porphyromonas asaccharolytica | 3553 | 4 | 0.12 |
| Porphyromonas levii | ATCC 29147 | 4 | 4 |
| Fusobacterium nucleatum | ATCC 25586 | >32 | >32 |
| Fusobacterium necrophorum | ATCC 25286 | >32 | >32 |
| Veillonella parvula | ATCC 17745 | >32 | 0.06 |
| Bifidobacterium bifidum | ATCC 15696 | >32 | 0.015 |
| Bifidobacterium breve | ATCC 15698 | >32 | 0.008 |
| Bifidobacterium infantis | ATCC 15702 | >32 | 0.008 |
| Bifidobacterium longum | ATCC 15707 | >32 | 0.03 |
| Lactobacillus plantarum | ATCC 39268 | >32 | 4 |
| Lactobacillus crispatus | ATCC 33820 | >32 | 4 |
| Lactobacillus acidophilus | ATCC 4356 | >32 | >32 |
| Lactobacillus lactis | ATCC 3711 | >32 | 2 |
| Lactobacillus acidophilus | ATCC 4356 | >32 | 1 |
| Lactobacillus gasseri | ATCC 33323 | >32 | 8 |
| Lactobacillus jensenii | ATCC 25258 | >32 | 32 |
| Lactobacillus casei | ATCC 393 | >32 | 2 |
| Lactobacillus rhamnosus | ATCC 7469 | >32 | 4 |
| Actinomyces naeslundii | ATCC 14699 | >32 | 1 |
| Actinomyces naeslundii | ATCC 12104 | >32 | 0.004 |
| Actinomyces viscosus | ATCC 43146 | >32 | 0.008 |
| Actinomyces israelii | ATCC 12102 | >32 | >32 |
| Escherichia coli (5) | >8c | NTd | |
| Pseudomonas aeruginosa (5) | >8 | NT | |
| Klebsiella pneumoniae (5) | >8 | NT | |
| Enterococcus faecalis (5) | 2–>8 | NT | |
| Streptococcus pneumoniae (5) | >8 | NT | |
| Staphylococcus aureus (6) | >8 | NT | |
ATCC 700057, ATCC 43255, BAA-1802 (NAP-1), BAA-1805 (NAP-1), BAA-1870 (NAP-1), and 16 recent clinical isolates.
B. fragilis (five isolates), B. thetaiotaomicron (three isolates), B. ovatus (one isolate), and B. vulgatus (one isolate).
Precipitation was observed at 16, 32, and 64 μg/ml in Mueller-Hinton II broth alone and Mueller-Hinton II broth plus 3% lysed horse blood.
NT, not tested.
The bactericidal activity of 5FDQD, fidaxomicin, and vancomycin were compared via a time-kill assay with C. difficile VPI 10463 (ATCC 43255) (Fig. 2). At a concentration 4- or 8-fold higher than the MIC, 5FDQD reduced the viable density of C. difficile by 99.9% to 1.7 × 103 and 8.0 × 103 CFU/ml, respectively, within 4 h. Within 6 h, incubated mixtures containing these concentrations of 5FDQD reduced viable cells below the 200 CFU/ml limit of quantitation. Similar bactericidal activity was observed for 5FDQD against C. difficile strain 630 (data not shown). In contrast, at a concentration 4-fold above their MIC values, fidaxomicin and vancomycin reduced the density of viable cells by only 6.3- and 64-fold, respectively, which is consistent with their previously reported activity (57). On the basis of these data, 5FDQD appears more rapidly bactericidal than these well-established CDI therapeutic compounds.
To assess the selectivity of 5FDQD antibacterial activity, we measured MIC values against a diverse panel of anaerobic and aerobic bacteria that included many common intestinal flora species (Table 1). Neither 5FDQD nor fidaxomicin showed appreciable activity against Bacteroides fragilis group species, whereas vancomycin and metronidazole are known to inhibit the B. fragilis group at pharmacologically relevant concentrations (35, 39, 58). Likewise, little to no activity was observed for 5FDQD and fidaxomicin against other anaerobic Gram-negative rods, which are usually susceptible to metronidazole but not vancomycin. Interestingly, 5FDQD showed no appreciable activity against the Bifidobacterium, Lactobacillus, or Actinomyces species tested, whereas fidaxomicin inhibited these strains as potently as it inhibits C. difficile. The activity of fidaxomicin, vancomycin, and metronidazole against these species has been well documented elsewhere (35, 58). On the basis of these data, the selectivity of 5FDQD with respect to anaerobic intestinal flora appears to be at least as good as, or superior to, currently marketed CDI therapeutic compounds. 5FDQD also exhibited very little activity against representative aerobic pathogens, with the exception of activity against some strains of Enterococcus faecalis.
In vivo efficacy.
An antibiotic-induced mouse CDI model was used to evaluate the in vivo efficacy of 5FDQD, fidaxomicin, and vancomycin. In accordance with published protocols (59), CDI was established in C57BL/6 mice by administering an antibiotic cocktail via their drinking water to disrupt intestinal flora and an oral dose of C. difficile VPI 10463 spores. If the infected mice are not subsequently treated with an effective antibiotic, they exhibit severe diarrhea, lethargy, weight loss, failure to groom, and 40 to 60% mortality within 48 h (59, 60).
Using this model, the efficacy of 5FDQD was compared to that of fidaxomicin and vancomycin, with all test agents suspended in aqueous 0.5% methylcellulose and administered orally twice each day for 5 days, except vancomycin, which was administered once a day for 5 days. Mice that received only the 0.5% methylcellulose vehicle orally developed signs of severe CDI within 3 days of inoculation, and 40% died by day 6 (Fig. 3A). The average weight of surviving vehicle-treated mice relative to their weight at the time of inoculation decreased by up to 17% during the study (Fig. 3B).
In contrast, treatment with 10 mg/kg 5FDQD prevented any mortality or weight loss, with the average weight increasing by 6% by day 8. Furthermore, although symptoms of mild diarrhea developed in one mouse, the symptoms resolved by day 3 of dosing. The activity of 10 mg/kg 5FDQD is comparable to the activity of both 3 mg/kg fidaxomicin and 50 mg/kg vancomycin. Symptoms of mild diarrhea were observed in two of the fidaxomicin-treated mice and one vancomycin-treated mouse, but the symptoms resolved by day 4. These data indicate that 5FDQD prevented the onset of severe CDI in mice as effectively as fidaxomicin and vancomycin did.
Pharmocokinetics of 5FDQD are advantageous for treating intestinal tract infections.
To assess the pharmacokinetic distribution of 5FDQD at an efficacious dose, mice were administered a single 10-mg/kg oral dose of 5FDQD, and the concentrations of the compound in the cecal contents and plasma were measured at specific time points after dosing. The levels of 5FDQD in the cecal contents were 192, 204, 174, 86, 82, and 0.3 μg/g at 1, 2, 4, 6, 8, and 24 h after dosing, respectively. This rapid transit into the lower intestinal tract followed by a sustained concentration over several hours is comparable to the distribution observed after a single dose of oral fidaxomcin (61). The levels of 5FDQD in the plasma were 23 ng/ml at 1 h and below the limit of quantitation (12 ng/ml) at the remaining times. These very low plasma 5FDQD levels would constitute a potential safety advantage for a drug intended to treat an intestinal tract infection such as that caused by C. difficile.
Cecal flora is only modestly affected by 5FDQD.
Since the disruption of endogenous intestinal flora is closely linked with both initial C. difficile infection and with recurrence rates (40, 62), we evaluated the extent to which 5FDQD, fidaxomicin, or vancomycin altered the culturable cecal flora in mice. A single oral dose of each compound or of the vehicle alone was administered to uninfected, healthy C57BL/6 mice. After 24 h, the mice were euthanized, the visible morphology of the ceca was recorded, and the densities of representative bacterial taxa within the cecal contents were determined by plating onto agar media selective for B. fragilis, bifidobacteria, lactobacilli, enterococci, Enterobacteriaceae, or staphylococci. In addition, the density of Clostridium spores in the cecal contents was determined by incubating an aliquot of the cecal contents with ethanol for 1 h followed by dilution plating onto egg yolk agar (63). The identity of each countable colony type on each type of medium subsequently was confirmed to the genus or species level (see Materials and Methods).
The ceca from mice treated with the aqueous 0.5% methylcellulose vehicle were visually indistinguishable from the ceca isolated from untreated mice. The cecal contents from vehicle-treated mice had a high density of culturable B. fragilis, bifidobacteria, and lactobacilli (2.1 × 107, 9.3 × 108, and 2.0 × 107 CFU/g, respectively) and a lower density of Clostridium spores, enterococci, Enterobacteriaceae, and staphylococci (6.8 × 104, 7 × 104, 4.6 × 103, and 3.0 × 104 CFU/g, respectively) (Fig. 4). This distribution was very similar to that measured for cecal contents from untreated mice (data not shown) and is consistent with previous characterizations of mouse cecal flora (64–66).
FIG 4.
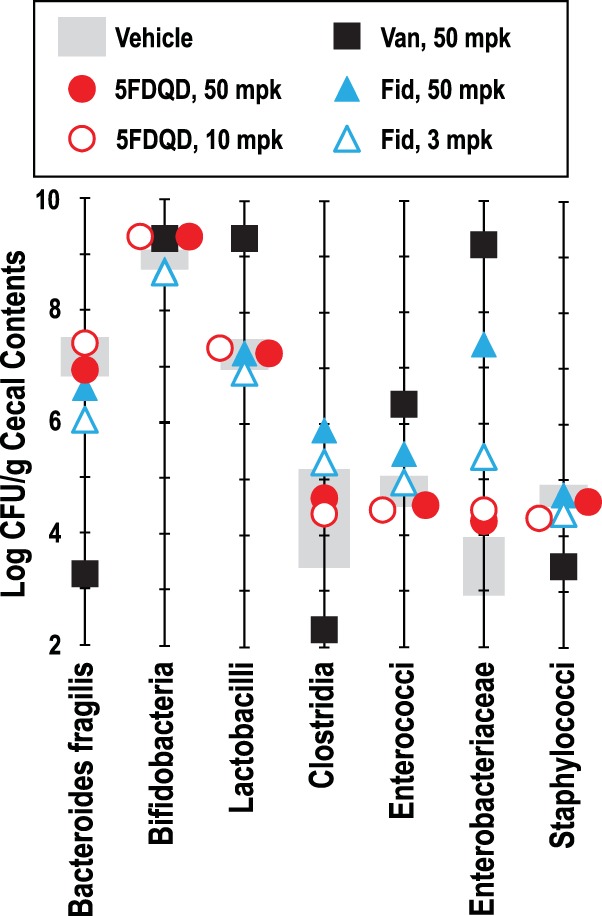
Effects of 5FDQD, fidaxomicin, and vancomycin on culturable cecal flora in mice. Ceca from healthy C57BL/6 mice were isolated 24 h after the mice were administered a single dose of each indicated treatment and dose (mpk, milligrams per kilogram), and cecal contents were dilution plated onto seven types of selective agar media to determine the densities of the indicated bacterial taxa in units of CFU/gram of cecal contents. All colony types counted were presumptively identified to the genus or species level as described in Materials and Methods. Data shown for clostridia are a measure of cecal spore density as determined by dilution plating an aliquot of ethanol-treated cecal contents. Data from vehicle-treated mice are shown as a range of the average ± 1 standard deviation (gray bars).
Treatment with a single 10-mg/kg or 5- mg/kg oral dose of 5FDQD did not affect the visible morphology of the ceca and had very little effect on the densities of the cultured cecal flora groups compared to the vehicle-treated mice. The only difference observed was a 5-fold increase in the density of members of the family Enterobacteriaceae. Thus, even at a dose 5-fold above the efficacious dose, 5FDQD did not dramatically alter the cecal morphology or flora. Speculation on why 5FDQD is highly selective is presented in Discussion (see below).
Ceca isolated from mice treated orally with 3 mg/kg fidaxomicin appeared normal, but those isolated from mice treated orally with 50 mg/kg fidaxomicin were visibly larger than those from vehicle- or 5FDQD-treated mice, and the mass of their cecal contents was increased by 40%. Treatment with a single 3-mg/kg oral dose of fidaxomicin also resulted in a 10-fold reduction of B. fragilis, a 10-fold increase of Enterobacteriaceae, and a 3-fold increase in Clostridium spores relative to vehicle-treated mice. These changes are consistent with alterations to bowel flora observed in human patients treated with fidaxomicin (32, 33). Treatment with 50 mg/kg fidaxomicin altered the flora to a greater extent, most notably a >1,000-fold increase in Enterobacteriaceae (Fig. 4). Although the density of Clostridium spores was not dramatically altered by either dose of fidaxomicin, the diversity of clostridial species was decreased as judged by visual appearance and microscopic examination of the colonies. One colony type presumptively identified as C. spiroforme (51) was increased 10-fold by fidaxomicin treatment to become the predominant culturable Clostridium species. Overall, fidaxomicin exhibited a moderate, dose-responsive effect on both cecal morphology and culturable cecal flora populations.
Ceca from mice treated orally with 50 mg/kg vancomycin were significantly enlarged, had visible gas bubbles, and contained 80% more contents by weight than ceca from vehicle- or 5FDQD-treated mice. Vancomycin also profoundly altered the culturable cecal flora (Fig. 4). B. fragilis, Clostridium spores, and staphylococci were decreased by 10,000-, 200-, and 10-fold, respectively, and lactobacilli, enterococci, and Enterobacteriaceae were increased by 100-, 32-, and >30,000-fold, respectively. These changes are consistent with the large alterations of bowel flora that vancomycin causes in human patients (30, 32). Overall, vancomycin treatment dramatically alters both cecal morphology and culturable cecal flora.
DISCUSSION
Given the urgent need for new antibiotic targets and lead compounds, we have established a research program aimed at identifying antibacterial compounds that target riboswitches. From a starting point of FMN and RoF, an intensive medicinal chemistry optimization effort yielded a series of flavin analogs that are potent FMN riboswitch ligands in vitro and that inhibit the growth of certain Gram-positive bacteria (Coish et al., patent application PCT/US2010/001876; Coish et al., patent application PCT/US2011/000617; Aristoff et al., patent application PCT/US2012/024507). Herein, we report the in vitro and in vivo characterization of one of these compounds, 5FDQD—a flavin analog that differs from FMN in that it has an N1-deaza flavin core and in that the ribityl phosphate moiety is replaced by an aryl-alkyl moiety.
The observation that in vitro 5FDQD binds to and triggers the function of an FMN riboswitch with the same potency as FMN is remarkable given the network of interactions that the aptamer appears to form with the ribose-phosphate moiety of FMN in crystallographic structure models (53, 67). Our in-line probing assay data indicate that 5FDQD binds to the riboswitch in the same region as FMN and causes nearly the same structural rearrangements, suggesting that the aptamer pocket is capable of forming equally favorable interactions with the aryl-alkyl side chain of 5FDQD. The transcription termination data confirm that these interactions are sufficient for 5FDQD to trigger riboswitch function in vitro with the same potency as for FMN.
Replacement of the ribityl-phosphate moiety of FMN with a hydrophobic moiety to yield compounds that retain riboswitch-binding function constitutes a key medicinal chemistry advance. This modification obviates the need to deliver a phosphorylated drug to the target cell or to deliver a prodrug that the target cell needs to phosphorylate in order to achieve full potency. These differences greatly expand the diversity of possible functional analogs. In contrast, the weak riboswitch-binding potency of the natural compound RoF in vitro suggests that it needs to be phosphorylated to roseoflavin phosphate (RoF-P) inside cells to be active (12), and this necessity likely diminishes the range of compound variations that could serve as effective antibiotics. Thus, 5FDQD and related analogs have two key advantages that are common characteristics of some classes of drugs (68). First, molecules in this series are uncharged and have reduced polarity compared to FMN or RoF, which might favor their uptake by target cells. Second, the compounds bind their RNA targets with high affinity without the need for modification by cellular enzymes. Moreover, our results suggest that 5FDQD is a representative of a much larger class of compounds that could be created based on this scaffold and that additional compounds could be generated and tested to further optimize the function of FMN riboswitch-binding antibiotic compounds.
In its current form, 5FDQD is a potent C. difficile antibacterial agent with an MIC90 of 1 μg/ml among strains tested herein, including four hypervirulent NAP-1 isolates. The C. difficile MIC90 values for 5FDQD are equivalent to those reported for metronidazole, vancomycin, and surotomycin, a phase III investigational agent for CDI therapy (56, 69). Although 5FDQD is less potent against C. difficile than fidaxomicin is, the available cecal concentration of 5FDQD after a single oral dose is well above the MIC and at a concentration that rapidly kills C. difficile in vitro. Accordingly, 5FDQD can completely prevent the onset of severe antibiotic-induced CDI in mice at an oral dose of 10 mg/kg twice daily for 5 days. In three repeated side-by-side comparisons, 10 mg/kg 5FDQD displayed potency equivalent to that of 3 mg/kg fidaxomicin. Given the high cecal bioavailability after dosing, it is likely that lower doses would also be effective. Future studies could be designed to determine a minimal effective dose of 5FDQD. However, it might also be beneficial to determine the maximal tolerated dose and whether higher concentrations of drug might clear an infection sooner and with fewer doses. Overall, the antibacterial activity and in vivo efficacy of 5FDQD against C. difficile are comparable to those of known CDI therapeutics or development candidates.
At present, the mechanism of 5FDQD antibacterial activity has not been definitively confirmed. Preliminary attempts to isolate 5FDQD-resistant C. difficile by single-step or serial passage methods have not yielded isolates with a stably increased MIC. These efforts indicate that the frequency of resistance development is <1 × 10−9 (data not shown). If confirmed, this low propensity to develop resistance could suggest either that mutations in the primary target are not well tolerated or that there are multiple targets. In support of a riboswitch-targeting component of the mechanism, 5FDQD binds to and triggers the function of an FMN riboswitch in vitro with a potency comparable to that of the active phosphorylated form of RoF, whose antibacterial activity is at least partly attributable to the repression of FMN riboswitch-regulated genes (12, 13, 22). Moreover, among a collection of 5FDQD analogs, a correlation exists between MIC and in vitro riboswitch-binding potency (data not shown). However, it is also conceivable that inhibition of flavoprotein-related processes could be a component of the 5FDQD antibacterial activity, as has been suggested for RoF (70, 71). Efforts are under way in R. R. Breaker's laboratory to isolate 5FDQD-resistant C. difficile and to definitively characterize the mode of action for 5FDQD, including elucidating the relative importance of FMN riboswitch binding.
It is well established that healthy, diverse intestinal flora protects against C. difficile colonization and reduces the likelihood of recurrence after an initial CDI episode (72–74). Although the specific taxa responsible for colonization resistance have not been unequivocally established, it is currently thought that bacteria of the Bacteroides, Bifidobacterium, and Prevotella genera and the Ruminococcaceae, Lachnospiraceae, and Porphyromonadaceae families are major contributors (75–80). Like fidaxomicin, 5FDQD is inactive against most of the aerobes and Gram-negative anaerobes tested herein, including the B. fragilis group and Prevotella species. Favorably, 5FDQD is less active than fidaxomicin against the Bifidobacterium, Lactobacillus, and Actinomyces strains tested herein. Likewise, the MICs of 5FDQD against representatives of these genera are higher than the published MICs of fidaxomicin, vancomycin, metronidazole, and two clinical development candidates for CDI treatment—LFF-571 and surotomycin (39, 41, 58, 81). Thus, the spectrum of 5FDQD appears to be narrower than those of established and developmental CDI therapeutics and should theoretically permit the regrowth of intestinal flora that protect against recurrence. At this time, the basis for the remarkable selectivity of 5FDQD is not entirely clear. Earlier analogs of 5FDQD were found to be modestly inhibitory toward an efflux-deficient ΔnorA strain of Staphylococcus aureus (82) or chemically permeabilized E. coli (83), suggesting that 5FDQD either does not readily enter or is effluxed from most of the bacterial species tested (data not shown). Future studies will seek to confirm the narrow spectrum of 5FDQD against a larger collection of representative species.
The narrower spectrum of 5FDQD compared to the spectra of fidaxomicin and vancomycin was evident from the effect of each agent on the spectrum of bacteria cultured from mouse cecal flora. At comparable doses in healthy mice, 5FDQD was significantly less disruptive to the cultured cecal flora than both fidaxomicin and vancomycin were. No changes in excess of 1 order of magnitude were observed after a single oral dose of up to 50 mg/kg 5FDQD, whereas a single fidaxomicin dose caused reduced cecal B. fragilis density and Clostridium diversity and increased Enterobacteriaceae density. Interestingly, overgrowth of Enterobacteriaceae is positively correlated with a number of intestinal disorders, including C. difficile colonization (74, 84–86). Although the taxa cultured herein represent a subset of the total flora, the measured changes are consistent with the known effects of fidaxomicin and vancomycin on bowel flora (32, 33). The lower impact of fidaxomicin on human bowel flora compared to vancomycin is widely believed to be the basis for a lower rate of CDI recurrence. Therefore, it is conceivable that treatment with a selective agent like 5FDQD would lead to fewer recurrences in humans. Efforts are now under way to evaluate the likelihood of CDI recurrence after treatment utilizing a hamster model (87) and a recently developed mouse recurrence model (88).
In summary, our efforts to optimize new FMN riboswitch-targeting antibiotics resulted in the identification of a novel series of flavin analogs that are potent and highly selective C. difficile antibacterial compounds. The most potent of these analogs examined to date, 5FDQD, can prevent lethal CDI in mice without disrupting the regrowth of beneficial flora. Further exploration of this compound series appears to be a promising route for the development of new CDI therapeutics. The success of this program also underscores the potential for the development of riboswitch-targeting compounds for treating other bacterial infections.
Supplementary Material
ACKNOWLEDGMENTS
The original design and synthesis of 5FDQD were supported by funds from BioRelix, Inc. (Foxborough, MA). Resynthesis of 5FDQD was funded by HHMI research support to R.R.B. and by the Yale Center for Molecular Discovery. Animal testing was supported by BioRelix, Yale University, and HHMI.
We thank Malavika Ghosh and Rashmi Munshi at Aragen for conducting in vivo studies. We thank Dean Shinabarger and Beverly Murray at Micromyx for conducting MIC studies. We thank Ron Mays and Vipin Agarwal at Northeast Bioanalytical for conducting pharmacokinetic bioanalyses.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.01282-15.
REFERENCES
- 1.Spellberg B, Guidos R, Gilbert D, Bradley J, Boucher HW, Scheld WM, Bartlett JG, Edwards J Jr, Infectious Diseases Society of America. 2008. The epidemic of antibiotic-resistant infections: a call to action for the medical community from the Infectious Diseases Society of America. Clin Infect Dis 46:155–164. doi: 10.1086/524891. [DOI] [PubMed] [Google Scholar]
- 2.Silver LL. 2011. Challenges of antibacterial discovery. Clin Microbiol Rev 24:71–109. doi: 10.1128/CMR.00030-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mandal M, Breaker RR. 2004. Gene regulation by riboswitches. Nat Rev Mol Cell Biol 5:451–463. doi: 10.1038/nrm1403. [DOI] [PubMed] [Google Scholar]
- 4.Serganov A. 2009. The long and the short of riboswitches. Curr Opin Struct Biol 19:251–259. doi: 10.1016/j.sbi.2009.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Roth A, Breaker RR. 2009. The structural and functional diversity of metabolite-binding riboswitches. Annu Rev Biochem 78:305–334. doi: 10.1146/annurev.biochem.78.070507.135656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Smith AM, Fuchs RT, Grundy FJ, Henkin TM. 2010. Riboswitch RNAs: regulation of gene expression by direct monitoring of a physiological signal. RNA Biol 7:104–110. doi: 10.4161/rna.7.1.10757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Breaker RR. 2011. Prospects for riboswitch discovery and analysis. Mol Cell 43:867–879. doi: 10.1016/j.molcel.2011.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Breaker RR. 2012. Riboswitches and the RNA world. Cold Spring Harb Perspect Biol 4(2):pii=a003566. doi: 10.1101/cshperspect.a003566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Koedam JC. 1958. The mode of action of pyrithiamine as an inducer of thiamine deficiency. Biochim Biophys Acta 29:333–344. doi: 10.1016/0006-3002(58)90192-6. [DOI] [PubMed] [Google Scholar]
- 10.Sudarsan N, Cohen-Chalamish S, Nakamura S, Emilsson GM, Breaker RR. 2005. Thiamine pyrophosphate riboswitches are targets for the antimicrobial compound pyrithiamine. Chem Biol 12:1325–1335. doi: 10.1016/j.chembiol.2005.10.007. [DOI] [PubMed] [Google Scholar]
- 11.Sudarsan N, Wickiser JK, Nakamura S, Ebert MS, Breaker RR. 2003. An mRNA structure in bacteria that controls gene expression by binding lysine. Genes Dev 17:2688–2697. doi: 10.1101/gad.1140003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lee ER, Blount KF, Breaker RR. 2009. Roseoflavin is a natural antibacterial compound that binds to FMN riboswitches and regulates gene expression. RNA Biol 6:187–194. doi: 10.4161/rna.6.2.7727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ott E, Stolz J, Lehmann M, Mack M. 2009. The RFN riboswitch of Bacillus subtilis is a target for the antibiotic roseoflavin produced by Streptomyces davawensis. RNA Biol 6:276–280. doi: 10.4161/rna.6.3.8342. [DOI] [PubMed] [Google Scholar]
- 14.Blount KF, Wang XJ, Lim J, Sudarsan N, Breaker RR. 2007. Antibacterial compounds that target lysine riboswitches. Nat Chem Biol 3:44–49. doi: 10.1038/nchembio842. [DOI] [PubMed] [Google Scholar]
- 15.Kim JN, Blount KF, Puskarz I, Lim J, Link KH, Breaker RR. 2009. Design and antimicrobial action of purine analogues that bind guanine riboswitches. ACS Chem Biol 4:915–927. doi: 10.1021/cb900146k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lünse CE, Schmidt MS, Wittmann V, Mayer G. 2011. Carba-sugars activate the glmS-riboswitch of Staphylococcus aureus. ACS Chem Biol 6:675–678. doi: 10.1021/cb200016d. [DOI] [PubMed] [Google Scholar]
- 17.Blount KF, Breaker RR. 2006. Riboswitches as antibacterial drug targets. Nat Biotechnol 24:1558–1564. doi: 10.1038/nbt1268. [DOI] [PubMed] [Google Scholar]
- 18.Blount KF, Puskarz I, Penchovsky R, Breaker RR. 2006. Development and application of a high-throughput assay for glmS riboswitch activators. RNA Biol 3:77–81. doi: 10.4161/rna.3.2.3102. [DOI] [PubMed] [Google Scholar]
- 19.Deigan KE, Ferre-D'Amare AR. 2011. Riboswitches: discovery of drugs that target bacterial gene-regulatory RNAs. Acc Chem Res 44:1329–1338. doi: 10.1021/ar200039b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ster C, Allard M, Boulanger S, Lamontagne Boulet M, Mulhbacher J, Lafontaine DA, Marsault E, Lacasse P, Malouin F. 2013. Experimental treatment of Staphylococcus aureus bovine intramammary infection using a guanine riboswitch ligand analog. J Dairy Sci 96:1000–1008. doi: 10.3168/jds.2012-5890. [DOI] [PubMed] [Google Scholar]
- 21.Gilbert SD, Reyes FE, Edwards AL, Batey RT. 2009. Adaptive ligand binding by the purine riboswitch in the recognition of guanine and adenine analogs. Structure 17:857–868. doi: 10.1016/j.str.2009.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mansjo M, Johansson J. 2011. The riboflavin analog roseoflavin targets an FMN-riboswitch and blocks Listeria monocytogenes growth, but also stimulates virulence gene-expression and infection. RNA Biol 8:674–680. doi: 10.4161/rna.8.4.15586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Matsui K, Wang HC, Hirota T, Matsukawa H, Kasai S, Shinagawa K. 1982. Riboflavin production by roseoflavin-resistant strains of some bacteria. Agric Biol Chem 46:2003–2008. doi: 10.1271/bbb1961.46.2003. [DOI] [Google Scholar]
- 24.Otani S, Takatsu M, Nakano M, Kasai S, Miura R. 1974. Roseoflavin, a new antimicrobial pigment from Streptomyces. J Antibiot (Tokyo) 27:86–87. (Letter.) 10.7164/antibiotics.27.86. [DOI] [PubMed] [Google Scholar]
- 25.Centers for Disease Control and Prevention. 2013. Antibiotic resistance threats in the United States, 2013. Centers for Disease Control and Prevention, Atlanta, GA. [Google Scholar]
- 26.Johnson S. 2009. Recurrent Clostridium difficile infection: causality and therapeutic approaches. Int J Antimicrob Agents 33(Suppl 1):S33–S36. doi: 10.1016/S0924-8579(09)70014-7. [DOI] [PubMed] [Google Scholar]
- 27.Johnson S. 2009. Recurrent Clostridium difficile infection: a review of risk factors, treatments, and outcomes. J Infect 58:403–410. doi: 10.1016/j.jinf.2009.03.010. [DOI] [PubMed] [Google Scholar]
- 28.Kelly CP. 2012. Can we identify patients at high risk of recurrent Clostridium difficile infection? Clin Microbiol Infect 18(Suppl 6):21–27. doi: 10.1111/1469-0691.12046. [DOI] [PubMed] [Google Scholar]
- 29.Krook A. 1981. Effect of metronidazole and sulfasalazine on the normal human faecal flora. Scand J Gastroenterol 16:587–592. doi: 10.3109/00365528109182016. [DOI] [PubMed] [Google Scholar]
- 30.Edlund C, Barkholt L, Olsson-Liljequist B, Nord CE. 1997. Effect of vancomycin on intestinal flora of patients who previously received antimicrobial therapy. Clin Infect Dis 25:729–732. doi: 10.1086/513755. [DOI] [PubMed] [Google Scholar]
- 31.Van der Auwera P, Pensart N, Korten V, Murray BE, Leclercq R. 1996. Influence of oral glycopeptides on the fecal flora of human volunteers: selection of highly glycopeptide-resistant enterococci. J Infect Dis 173:1129–1136. doi: 10.1093/infdis/173.5.1129. [DOI] [PubMed] [Google Scholar]
- 32.Tannock GW, Munro K, Taylor C, Lawley B, Young W, Byrne B, Emery J, Louie T. 2010. A new macrocyclic antibiotic, fidaxomicin (OPT-80), causes less alteration to the bowel microbiota of Clostridium difficile-infected patients than does vancomycin. Microbiology 156:3354–3359. doi: 10.1099/mic.0.042010-0. [DOI] [PubMed] [Google Scholar]
- 33.Louie TJ, Cannon K, Byrne B, Emery J, Ward L, Eyben M, Krulicki W. 2012. Fidaxomicin preserves the intestinal microbiome during and after treatment of Clostridium difficile infection (CDI) and reduces both toxin reexpression and recurrence of CDI. Clin Infect Dis 55:S132–S142. doi: 10.1093/cid/cis338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Finegold SA, Molitoris D, Vaisanen ML, Song YL, Liu CX, Bolanos M. 2004. In vitro activities of OPT-80 and comparator drugs against intestinal bacteria. Antimicrob Agents Chemother 48:4898–4902. doi: 10.1128/AAC.48.12.4898-4902.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Credito KL, Appelbaum PC. 2004. Activity of OPT-80, a novel macrocycle, compared with those of eight other agents against selected anaerobic species. Antimicrob Agents Chemother 48:4430–4434. doi: 10.1128/AAC.48.11.4430-4434.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gerber M, Ackermann G. 2008. OPT-80, a macrocyclic antimicrobial agent for the treatment of Clostridium difficile infections: a review. Expert Opin Invest Drugs 17:547–553. doi: 10.1517/13543784.17.4.547. [DOI] [PubMed] [Google Scholar]
- 37.Golan Y, Epstein L. 2012. Safety and efficacy of fidaxomicin in the treatment of Clostridium difficile-associated diarrhea. Therap Adv Gastroenterol 5:395–402. doi: 10.1177/1756283X12461294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Louie TJ, Miller MA, Mullane KM, Weiss K, Lentnek A, Golan Y, Gorbach S, Sears P, Shue YK, OPT-80-003 Clinical Study Group. 2011. Fidaxomicin versus vancomycin for Clostridium difficile infection. N Engl J Med 364:422–431. doi: 10.1056/NEJMoa0910812. [DOI] [PubMed] [Google Scholar]
- 39.Citron DM, Tyrrell KL, Merriam CV, Goldstein EJ. 2012. In vitro activities of CB-183,315, vancomycin, and metronidazole against 556 strains of Clostridium difficile, 445 other intestinal anaerobes, and 56 Enterobacteriaceae species. Antimicrob Agents Chemother 56:1613–1615. doi: 10.1128/AAC.05655-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bakken JS. 2009. Fecal bacteriotherapy for recurrent Clostridium difficile infection. Anaerobe 15:285–289. doi: 10.1016/j.anaerobe.2009.09.007. [DOI] [PubMed] [Google Scholar]
- 41.Debast SB, Bauer MP, Sanders IM, Wilcox MH, Kuijper EJ, ECDIS Study Group. 2013. Antimicrobial activity of LFF571 and three treatment agents against Clostridium difficile isolates collected for a pan-European survey in 2008: clinical and therapeutic implications. J Antimicrob Chemother 68:1305–1311. doi: 10.1093/jac/dkt013. [DOI] [PubMed] [Google Scholar]
- 42.Regulski EE, Breaker RR. 2008. In-line probing analysis of riboswitches. Methods Mol Biol 419:53–67. doi: 10.1007/978-1-59745-033-1_4. [DOI] [PubMed] [Google Scholar]
- 43.Landick R, Wang D, Chan CL. 1996. Quantitative analysis of transcriptional pausing by Escherichia coli RNA polymerase: His leader pause site as a paradigm. Methods Enzymol 274:334–353. doi: 10.1016/S0076-6879(96)74029-6. [DOI] [PubMed] [Google Scholar]
- 44.Clinical and Laboratory Standards Institute. 2012. Methods for dilution antimicrobial susceptibility tests for bacteria that grow aerobically—ninth edition. Approved standard M07-A9 Clinical and Laboratory Standards Institute, Wayne, PA. [Google Scholar]
- 45.Clinical and Laboratory Standards Institute. 2012. Methods for antimicrobial susceptibility testing of anaerobic bacteria—ninth edition. Approved standard M11-A8 Clinical and Laboratory Standards Institute, Wayne, PA. [Google Scholar]
- 46.Pearson RD, Steigbigel RT, Davis HT, Chapman SW. 1980. Method of reliable determination of minimal lethal antibiotic concentrations. Antimicrob Agents Chemother 18:699–708. doi: 10.1128/AAC.18.5.699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jousimies-Somer H, Summanen P, Citron DM, Baron EJ, Wexler HM, Finegold SM. 2002. Wadsworth-KTL anaerobic bacteriology manual, 6th ed Star Publishing Company, Belmont, CA. [Google Scholar]
- 48.Ferraris L, Aires J, Waligora-Dupriet AJ, Butel MJ. 2010. New selective medium for selection of bifidobacteria from human feces. Anaerobe 16:469–471. doi: 10.1016/j.anaerobe.2010.03.008. [DOI] [PubMed] [Google Scholar]
- 49.Hartemink R, Domenech VR, Rombouts FM. 1997. LAMVAB - a new selective medium for the isolation of lactobacilli from faeces. J Microbiol Methods 29:77–84. doi: 10.1016/S0167-7012(97)00025-0. [DOI] [Google Scholar]
- 50.Carman RJ, Wilkins TD. 1991. In vitro susceptibility of rabbit strains of Clostridium spiroforme to antimicrobial agents. Vet Microbiol 28:391–397. doi: 10.1016/0378-1135(91)90074-P. [DOI] [PubMed] [Google Scholar]
- 51.Borriello SP, Carman RJ. 1983. Association of iota-like toxin and Clostridium spiroforme with both spontaneous and antibiotic-associated diarrhea and colitis in rabbits. J Clin Microbiol 17:414–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Soukup GA, Breaker RR. 1999. Relationship between internucleotide linkage geometry and the stability of RNA. RNA 5:1308–1325. doi: 10.1017/S1355838299990891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Serganov A, Huang L, Patel DJ. 2009. Coenzyme recognition and gene regulation by a flavin mononucleotide riboswitch. Nature 458:233–237. doi: 10.1038/nature07642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Loo VG, Poirier L, Miller MA, Oughton M, Libman MD, Michaud S, Bourgault AM, Nguyen T, Frenette C, Kelly M, Vibien A, Brassard P, Fenn S, Dewar K, Hudson TJ, Horn R, Rene P, Monczak Y, Dascal A. 2005. A predominantly clonal multi-institutional outbreak of Clostridium difficile-associated diarrhea with high morbidity and mortality. N Engl J Med 353:2442–2449. doi: 10.1056/NEJMoa051639. [DOI] [PubMed] [Google Scholar]
- 55.O'Connor JR, Johnson S, Gerding DN. 2009. Clostridium difficile infection caused by the epidemic BI/NAP1/027 strain. Gastroenterology 136:1913–1924. doi: 10.1053/j.gastro.2009.02.073. [DOI] [PubMed] [Google Scholar]
- 56.Wong SS, Woo PC, Luk WK, Yuen KY. 1999. Susceptibility testing of Clostridium difficile against metronidazole and vancomycin by disk diffusion and Etest. Diagn Microbiol Infect Dis 34:1–6. doi: 10.1016/S0732-8893(98)00139-4. [DOI] [PubMed] [Google Scholar]
- 57.Babakhani F, Gomez A, Robert N, Sears P. 2011. Killing kinetics of fidaxomicin and its major metabolite, OP-1118, against Clostridium difficile. J Med Microbiol 60:1213–1217. doi: 10.1099/jmm.0.029470-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Snydman DR, Jacobus NV, McDermott LA. 2012. Activity of a novel cyclic lipopeptide, CB-183,315, against resistant Clostridium difficile and other Gram-positive aerobic and anaerobic intestinal pathogens. Antimicrob Agents Chemother 56:3448–3452. doi: 10.1128/AAC.06257-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chen X, Katchar K, Goldsmith JD, Nanthakumar N, Cheknis A, Gerding DN, Kelly CP. 2008. A mouse model of Clostridium difficile-associated disease. Gastroenterology 135:1984–1992. doi: 10.1053/j.gastro.2008.09.002. [DOI] [PubMed] [Google Scholar]
- 60.Collignon A. 2010. Methods for working with the mouse model. Methods Mol Biol 646:229–237. doi: 10.1007/978-1-60327-365-7_15. [DOI] [PubMed] [Google Scholar]
- 61.Swanson RN, Hardy DJ, Shipkowitz NL, Hanson CW, Ramer NC, Fernandes PB, Clement JJ. 1991. In vitro and in vivo evaluation of tiacumicins B and C against Clostridium difficile. Antimicrob Agents Chemother 35:1108–1111. doi: 10.1128/AAC.35.6.1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rolfe RD, Helebian S, Finegold SM. 1981. Bacterial interference between Clostridium difficile and normal fecal flora. J Infect Dis 143:470–475. doi: 10.1093/infdis/143.3.470. [DOI] [PubMed] [Google Scholar]
- 63.Chapin KC, Murray PR. 1999. Media, p 1687–1707. In Murray PR, Baron EJ, Pfaller MA, Tenover FC, Yolken RH (ed), Manual of clinical microbiology, 9th ed ASM Press, Washington, DC. [Google Scholar]
- 64.Savage DC, Dubos R. 1968. Alterations in the mouse cecum and its flora produced by antibacterial drugs. J Exp Med 128:97–110. doi: 10.1084/jem.128.1.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Antonopoulos DA, Huse SM, Morrison HG, Schmidt TM, Sogin ML, Young VB. 2009. Reproducible community dynamics of the gastrointestinal microbiota following antibiotic perturbation. Infect Immun 77:2367–2375. doi: 10.1128/IAI.01520-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Buffie CG, Jarchum I, Equinda M, Lipuma L, Gobourne A, Viale A, Ubeda C, Xavier J, Pamer EG. 2012. Profound alterations of intestinal microbiota following a single dose of clindamycin results in sustained susceptibility to Clostridium difficile-induced colitis. Infect Immun 80:62–73. doi: 10.1128/IAI.05496-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Vicens Q, Mondragon E, Batey RT. 2011. Molecular sensing by the aptamer domain of the FMN riboswitch: a general model for ligand binding by conformational selection. Nucleic Acids Res 39:8586–8598. doi: 10.1093/nar/gkr565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.O'Shea R, Moser HE. 2008. Physicochemical properties of antibacterial compounds: implications for drug discovery. J Med Chem 51:2871–2878. doi: 10.1021/jm700967e. [DOI] [PubMed] [Google Scholar]
- 69.Aspevall O, Lundberg A, Burman LG, Akerlund T, Svenungsson B. 2006. Antimicrobial susceptibility pattern of Clostridium difficile and its relation to PCR ribotypes in a Swedish university hospital. Antimicrob Agents Chemother 50:1890–1892. doi: 10.1128/AAC.50.5.1890-1892.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Langer S, Hashimoto M, Hobl B, Mathes T, Mack M. 2013. Flavoproteins are potential targets for the antibiotic roseoflavin in Escherichia coli. J Bacteriol 195:4037–4045. doi: 10.1128/JB.00646-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Langer S, Nakanishi S, Mathes T, Knaus T, Binter A, Macheroux P, Mase T, Miyakawa T, Tanokura M, Mack M. 2013. The flavoenzyme azobenzene reductase AzoR from Escherichia coli binds roseoflavin mononucleotide (RoFMN) with high affinity and is less active in its RoFMN form. Biochemistry 52:4288–4295. doi: 10.1021/bi400348d. [DOI] [PubMed] [Google Scholar]
- 72.Wilson KH, Silva J, Fekety FR. 1981. Suppression of Clostridium difficile by normal hamster cecal flora and prevention of antibiotic-associated cecitis. Infect Immun 34:626–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Britton RA, Young VB. 2012. Interaction between the intestinal microbiota and host in Clostridium difficile colonization resistance. Trends Microbiol 20:313–319. doi: 10.1016/j.tim.2012.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Reeves AE, Theriot CM, Bergin IL, Huffnagle GB, Schloss PD, Young VB. 2011. The interplay between microbiome dynamics and pathogen dynamics in a murine model of Clostridium difficile infection. Gut Microbes 2:145–158. doi: 10.4161/gmic.2.3.16333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Seekatz AM, Young VB. 2014. Clostridium difficile and the microbiota. J Clin Invest 124:4182–4189. doi: 10.1172/JCI72336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Manges AR, Labbe A, Loo VG, Atherton JK, Behr MA, Masson L, Tellis PA, Brousseau R. 2010. Comparative metagenomic study of alterations to the intestinal microbiota and risk of nosocomial Clostridium difficile-associated disease. J Infect Dis 202:1877–1884. doi: 10.1086/657319. [DOI] [PubMed] [Google Scholar]
- 77.Hopkins MJ, Macfarlane GT. 2002. Changes in predominant bacterial populations in human faeces with age and with Clostridium difficile infection. J Med Microbiol 51:448–454. [DOI] [PubMed] [Google Scholar]
- 78.Rea MC, O'Sullivan O, Shanahan F, O'Toole PW, Stanton C, Ross RP, Hill C. 2012. Clostridium difficile carriage in elderly subjects and associated changes in the intestinal microbiota. J Clin Microbiol 50:867–875. doi: 10.1128/JCM.05176-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Schubert AM, Rogers MA, Ring C, Mogle J, Petrosino JP, Young VB, Aronoff DM, Schloss PD. 2014. Microbiome data distinguish patients with Clostridium difficile infection and non-C. difficile-associated diarrhea from healthy controls. mBio 5(3):e01021-14. doi: 10.1128/mBio.01021-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Skraban J, Dzeroski S, Zenko B, Mongus D, Gangl S, Rupnik M. 2013. Gut microbiota patterns associated with colonization of different Clostridium difficile ribotypes. PLoS One 8:e58005. doi: 10.1371/journal.pone.0058005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Citron DM, Tyrrell KL, Merriam CV, Goldstein EJ. 2012. Comparative in vitro activities of LFF571 against Clostridium difficile and 630 other intestinal strains of aerobic and anaerobic bacteria. Antimicrob Agents Chemother 56:2493–2503. doi: 10.1128/AAC.06305-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kaatz GW, Seo SM. 1995. Inducible NorA-mediated multidrug resistance in Staphylococcus aureus. Antimicrob Agents Chemother 39:2650–2655. doi: 10.1128/AAC.39.12.2650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Leive L. 1968. Studies on the permeability change produced in coliform bacteria by ethylenediaminetetraacetate. J Biol Chem 243:2373–2380. [PubMed] [Google Scholar]
- 84.Hendrickson BA, Guo J, Laughlin R, Chen Y, Alverdy JC. 1999. Increased type 1 fimbrial expression among commensal Escherichia coli isolates in the murine cecum following catabolic stress. Infect Immun 67:745–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Berg RD. 1995. Bacterial translocation from the gastrointestinal tract. Trends Microbiol 3:149–154. doi: 10.1016/S0966-842X(00)88906-4. [DOI] [PubMed] [Google Scholar]
- 86.Gautreaux MD, Deitch EA, Berg RD. 1994. Bacterial translocation from the gastrointestinal tract to various segments of the mesenteric lymph node complex. Infect Immun 62:2132–2134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Douce G, Goulding D. 2010. Refinement of the hamster model of Clostridium difficile disease. Methods Mol Biol 646:215–227. doi: 10.1007/978-1-60327-365-7_14. [DOI] [PubMed] [Google Scholar]
- 88.Sun X, Wang H, Zhang Y, Chen K, Davis B, Feng H. 2011. Mouse relapse model of Clostridium difficile infection. Infect Immun 79:2856–2864. doi: 10.1128/IAI.01336-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.