

Abstract
We previously demonstrated that ginsenosides Rg1 and Re enhanced the immune response in C3H/HeB mice but not in C3H/HeJ mice carrying a mutation in the Tlr4 gene. The results of the present study showed that both Rg1 and Re inhibited mRNA expression and production of proinflammatory mediators that included tumor necrosis factor α, interleukin-1β, interleukin-6, cyclooxygenase-2, and inducible nitric oxide synthase from lipopolysaccharide (LPS)-stimulated macrophages. Rg1 was found to be distributed both extracellularly and intracellularly but Re was located only extracellularly to compete with LPS for binding to Toll-like receptor 4. Preinjection of Rg1 and Re into rats suppressed LPS-induced increases in body temperature, white blood cell counts, and levels of serum proinflammatory mediators. Preinjection of Rg1 and Re into mice prevented the LPS-induced decreases in total white blood cell counts and neutrophil counts, inhibited excessive expression of multiple proinflammatory mediators, and successfully rescued 100% of the mice from sepsis-associated death. More significantly, when administered after lethal LPS inoculation, Rg1, but not Re, still showed a potent antisepsis effect and protected 90% of the mice from death. The better protection efficacy of Rg1 could result from its intracellular distribution, suggesting that Rg1 may be an ideal antisepsis agent.
INTRODUCTION
Despite significant advances in the development of antimicrobial chemotherapy and supportive strategies, sepsis remains a significant cause of morbidity and mortality in humans (1). In North America, more than 750,000 patients develop sepsis annually and about 215,000 of the cases of sepsis result in death. The incidence is gradually increasing by about 1.5% per year (2). Evidence indicates that sepsis-induced lethality is often accompanied by a failure to develop appropriate immune responses to invading pathogens, particularly to their components (3, 4). Lipopolysaccharide (LPS) is a main constituent of Gram-negative bacterial cell walls (5) and is considered a leading cause of sepsis (6). Toll-like receptor 4 (TLR4) and its coreceptor, myeloid differentiation factor 2 (MD-2), form a heterodimer to recognize LPS because mice lacking either molecule are hyporesponsive to LPS (7, 8). The interaction between LPS and TLR4–MD-2 activates the LPS signaling pathway, resulting in phosphorylation of nuclear factor κB (NF-κB) (9). NF-κB activation induces high levels of proinflammatory cytokines, enzymes, and other mediators, including tumor necrosis factor alpha (TNF-α), interleukin-1β (IL-1β), IL-6, inducible nitric oxide synthase (iNOS), and cyclooxygenase-2 (COX-2) (10). By prompting the release of these inflammatory mediators, LPS not only activates both innate and adaptive immune responses at distal sites from the infection but also, in many cases, causes shock and death (11). Consequently, strategies that include prevention of ligand binding to TLR4 (12), blocking the interactions of TLR4 and adaptors in signaling pathways (13), and suppressing NF-κB signaling pathways (14) have been reported to be effective in intervening in the development of experimental sepsis.
Ginseng, the root of Panax ginseng C.A. Meyer (Araliaceae), has been used as a tonic in traditional Chinese medicine for more than 2,000 years. Ginseng saponins (ginsenosides) have been found to have adjuvant effects on the immune responses in cattle, pigs, mice, and rats (15–18). To date, more than 40 ginsenosides have been identified in the plant (19). Our investigation has demonstrated that the adjuvant activities of ginsenosides Rg1 and Re are closely related to the TLR4 signaling pathway, as the saponins exhibit adjuvant activity in normal animals but not in mice carrying a mutation in the Tlr4 gene (20). In addition, reinduced expression of NF-κB in macrophages has been suppressed by blocking TLR4 with specific antibodies. We therefore hypothesized that Rg1 and Re may compete with LPS to bind to TLR4 and could be useful in the treatment of LPS-induced sepsis. In the present study, we have investigated Rg1 and Re for their inhibition of proinflammatory mediators induced by LPS in vitro and in vivo, interference with LPS binding to TLR4, and protection against LPS-induced lethality.
MATERIALS AND METHODS
TAK-242 and ginsenosides Rg1 and Re.
Ginsenosides Rg1 and Re, members of a class of steroid glycosides and triterpene saponins found exclusively in Panax ginseng, were purchased from Hongjiu Biotech Co. Ltd. (Dalian, China). The purity of each ginsenoside was >98% as determined by the high-performance liquid chromatography (HPLC) method. Ethyl (6R)-6-[N-(2-chloro-4-fluorophenyl) sulfamoyl] cyclohex-1-ene-1-carboxylate (TAK-242) was purchased from Shanghai Biochembest Co. Ltd. The chemicals were dissolved in dimethyl sulfoxide (DMSO) for the preparation of the stock solutions, which were further diluted in culture medium to make the working solutions. The working solutions were passed through a gel endotoxin-removing column (Pierce), and the endotoxin level was tested using a Limulus reagent kit. Endotoxin levels were <0.5 U/ml.
Cells.
RAW264.7 and RAW-Blue cells were purchased from InvivoGen (San Diego, CA, USA). RAW-Blue cells were derived from RAW264.7 macrophages with chromosomal integration of pNiFty-SEAP, a plasmid expressing the secreted embryonic alkaline phosphatase (SEAP) gene under the control of an NF-κB-inducible ELAM-1 (E-selectin) promoter (21).
Animals.
Male Sprague Dawley rats and BALB/c mice were purchased from the Shanghai Laboratory Animal Center and Charles River Laboratories (St. Constant, Quebec, Canada). Animals were housed under specific-pathogen-free conditions and used in accordance with the recommendations of the National Institutes of Health Guide for the Care and Use of Laboratory Animals. Animal care/use protocols were approved by the Zhejiang University Institutional Animal Care and Use Committee and the Institute for Biological Sciences (National Research Council Canada) Animal Care Committee.
Rabbit anti-Rg1 and anti-Re polyclonal antibodies.
Rg1- and Re-bovine serum albumin (BSA) conjugates were synthesized by a modified procedure (22). Rabbit anti-Rg1 and ant-Re polyclonal antibodies (1:40,000) were prepared as described previously (22).
SEAP reporter assay.
RAW-Blue cells (2 × 106 cells/ml) were cultured with Rg1 or Re at 10, 30, 50, or 70 μg/ml in macrophage–serum-free medium (macrophage-SFM) (Invitrogen, Grand Island, NY, USA) for 1 h and then stimulated with LPS (Escherichia coli O111:B4) (InvivoGen) at 1 μg/ml for 20 h. Supernatants were collected and added to QuantiQuanta-blue substrate (InvivoGen). The SEAP activity was measured at 630 nm by the use of an enzyme-linked immunosorbent assay (ELISA) plate reader.
Quantitative RT-PCR.
RAW264.7 cells (1 × 107/ml) were cultured with Rg1 or Re at 50 μg/ml in macrophage-SFM for 1 h and then stimulated with LPS at 1 μg/ml for 20 h. After that, cells were lysed in 1 ml of RNAiso Plus (TaKaRa, Dalian, China) for RNA extraction. The extracted total RNA was mixed with iScript reagent (Bio-Rad) for reverse transcription (RT). Relative quantification of target cDNA genes to β-actin was conducted on an ABI 7500 system (PE/Applied Biosystems). The primers for target genes and β-actin were designed using Primer Express 5.0 (PE Applied Biosystems). Results are reported as n-fold differences relative to target gene mRNA expression in the calibrator group (23).
The following primers were used: for β-actin, 5′-AGCGGTTCCGATGCCCT-3′ and 5′-AGAGGTCTTTACGGATGTCAACG-3′; for TNF-α, 5′-CCTATGTCTCAGCCTCTTCT-3′ and 5′-CCTGGTATGAGATAGCAAAT-3′; for IL-1β, 5′-GGCAACTGTTCCTGAACTCAACTG-3′ and 5′-CCATTGAGGTGGAGAGCTTTCAGC-3′; for iNOS, 5′-CCCTTCCGAAGTTTCTGGCAGCAGC-3′ and 5′-GCCTGTCAGAGCCTCGTGGCTTTGG-3′; and for COX-2, 5′-TTCAAAAGAAGTGCTGGAAAAGGT-3′ and 5′-GATCATCTCTACCTGAGTGTCTTT-3′.
Western blotting.
The lysed cells in radioimmunoprecipitation assay (RIPA) lysis buffer and sera were adjusted to the same protein concentration by the use of bicinchoninic acid (BCA) reagent (Sangon Biotech, Shanghai, China) and then diluted with 2× Laemmli sample buffer (1:1) and boiled for 5 min. After centrifugation (12,000 × g) at 4°C for 5 min, 20 μl of diluted samples was loaded onto a 12% (wt/vol) separating gel and 5% stacking gel (SDS-polyacrylamide gel electrophoresis). The protein was transferred onto an Immobilon-p transfer membrane. The membrane was washed and then blocked in 5% blocking buffer (skim milk–Tris-buffered saline [TBS]) for 1 h at 37°C in the incubator shaker. After three washes in TBST (TBS containing 0.1% Tween 20), the membrane was incubated with target protein antibody at 4°C overnight. The membrane was washed three times in TBST, followed by incubation with the secondary antibody for 1.5 h at 4°C. After the final wash, the immunoblot was developed with BeyoECL Plus (Beyotime) according to the manufacturer's instructions and the images were captured by the use of a ChemiScope Mini imaging system (Clinx Science Instruments Co., Beijing, China).
Inhibition of LPS binding to TLR4.
RAW264.7 cells (1 × 105 cells/ml) were incubated with Rg1 or Re at 10, 30, 50, or 70 μg/ml and then stimulated with Alexa Fluor 594-LPS (Molecular Probes, Grand Island, NY, USA) at 10 μg/ml. After removing the supernatant, cells were fixed with Immunol staining fix solution. TLR4 in the cells was labeled with rat anti-mouse TLR4 monoclonal antibody (1:300), and rabbit anti-rat IgG conjugated with DyLight 488 (Cell Signaling Technology) (1:200) was used as a secondary antibody. The cells were observed under an inverse confocal laser scanning microscope (FV1000; Olympus). The images were acquired using Olympus confocal software (FV10-ASW 3.1).
Distribution of Rg1 and Re in cells.
RAW264.7 cells (1 × 105 cells/ml) were incubated with Rg1 or Re at 50 μg/ml, and the cells were then fixed with Immunol staining fix solution (Beyotime Biotechnology, Haimen, China). The fixed cells were incubated with rabbit anti-Rg1 or anti-Re antibody (1:300), and goat anti-rabbit IgG (Alexa Fluor 594 conjugate) (Cell Signaling Technology, Shanghai, China) (1:500) was used as a secondary antibody. The cell nucleus was stained with 2-(4-amidinophenyl)-6-indolecarbamidine dihydrochloride (DAPI) (Roche, Shanghai, China) (1 μg/ml), and the cell was stained with 3,3′-dioctadecyloxacarbo-cyanine perchlorate (Dio) (Biotium, Hayward, CA, USA) (10 μg/ml). The stained cells were observed under an inverse confocal laser scanning microscope (FV1000; Olympus). Images were acquired using Olympus confocal software (FV10-ASW 3.1).
Log P of Rg1 and Re.
Log octanol/water partition coefficient (log P) values were calculated in MOE according to the Wildman and Crippen model (24–26).
LPS-induced inflammation in rats.
Sprague Dawley rats were intravenously administered saline solution, 1 mg of Rg1/kg body weight (1 mg/kg Rg1), 1 mg/kg Re, or 1 mg/kg TAK-242, a novel TLR4 inhibitor (27). Fifteen minutes later, rats were challenged with 2.5 μg/kg LPS. Body temperature was measured before and after drug administration. Anticoagulated blood samples with EDTA were collected for white blood cell (WBC) counts at indicated time points using a ProCyte Dx automatic blood cell analyzer (IDEXX Laboratories, Westbrook, ME). Additional blood samples were collected at 4 h post-drug injection and used for preparation of serum to analyze proinflammatory mediators. The proinflammatory cytokine responses were detected by Western blot assay. The levels of nitric oxide (NO) and prostaglandin E2 (PGE2) in samples were determined by the use of a NO detection kit (Beyotime) and a PGE2 assay kit (R&D Systems, Shanghai, China), respectively, according to the manufacturer's protocols.
LPS-induced lethality in mice.
To examine the prophylactic effect of Rg1 and Re on LPS-induced lethality, 6- to 8-week-old BALB/c mice were randomly assigned to 7 groups with 10 mice in each group. The mice were either left untreated or subcutaneously injected with 2.5, 5, 10, or 20 mg/kg Rg1, 20 mg/kg Re, or 5 mg/kg TAK-242 3 times at 30-min intervals and then challenged with LPS (20 mg/kg) 15 min later.
To test the therapeutic effect of Rg1 and Re on LPS-induced lethality, BALB/c mice were assigned to 5 groups with 10 mice in each group. The mice were injected intraperitoneally (i.p.) with 20 mg/kg LPS. Fifteen minutes later, mice were subcutaneously injected with 10 mg/kg Rg1, 20 mg/kg Re, or 5 mg/kg TAK-242 3 times at 30-min intervals or left untreated. Survival rates were recorded for 60 h.
The effect of Rg1 and Re treatment on LPS-induced sepsis in mice.
Six- to 8-week-old BALB/c mice were randomly assigned to 5 groups (n = 9 per group). The mice in the first 4 groups were subcutaneously injected with 10 mg/kg Rg1, 20 mg/kg Re, or 5 mg/kg TAK-242 3 times at 30-min intervals or left untreated and were then challenged with 20 mg/kg LPS 15 min later. The mice in the fifth group were injected with LPS first and then subcutaneously administered 10 mg/kg Rg1 3 times at 30-min intervals (referred to here as the posttreatment mice). Three mice in each group were euthanized at 4, 8, or 12 h post-LPS administration for blood collection. Another 5 naive mice were euthanized and sampled at h 0. Samples of blood treated with EDTA were collected for WBC counts, and those without anticoagulant were harvested for serum separation to analyze proinflammatory mediators.
Total WBCs were enumerated using a hemocytometer after removal of red blood cells by the use of ammonium-chloride-potassium (ACK) lysis buffer, and differential cell counts were determined on blood smears stained with Hema-3 (Fisher Scientific, Middletown, VA, USA). Concentrations of proinflammatory cytokines and chemokines were measured by the use of a Milliplex mouse cytokine/chemokine kit (Millipore, Billerica, MA) and a Luminex 100IS system (Luminex Corporation, Austin, TX, USA). Levels of PGE2 and NO were determined by the use of a PGE2 assay kit and a total nitric oxide assay kit (R&D Systems), respectively, according to the manufacturer's protocol.
Statistics.
Data were expressed as means ± standard deviations (SD) and evaluated by the two-tailed Student's t test. Survival data were analyzed using the log-rank test. Differences between groups were considered significant at the level of P <0.05.
RESULTS
Rg1 or Re inhibits proinflammatory responses in LPS-stimulated macrophages.
Previous investigation showed that Rg1, Re, and LPS enhanced the immune responses to ovalbumin in C3H/HeB but not C3H/HeJ mice with a mutation in the Tlr4 gene (20). Based on that result, we hypothesized that Rg1 or Re might share the binding site of TLR4 in mammalian cells with LPS. To determine whether Rg1 and Re affect LPS-induced NF-κB activation, we first treated RAW-Blue cells with Rg1 or Re at various concentrations and then stimulated them with LPS. Figure 1A shows that LPS stimulated NF-κB phosphorylation, resulting in the high SEAP activity. However, pretreatment with Rg1 at 30, 50, and 70 μg/ml or Re at 50 and 70 μg/ml significantly inhibited SEAP activity. Furthermore, Rg1 exhibited significantly higher inhibitory activity than Re (Fig. 1A).
FIG 1.

Inhibitory effect of Rg1 and Re on LPS-induced inflammatory responses in macrophages. (A) Effect of Rg1 and Re on SEAP secretion by RAW-Blue cells stimulated with LPS. RAW-Blue cells were pretreated with Rg1 or Re at 10, 30, 50, or 70 μg/ml for 1 h and then incubated with LPS for 20 h. After incubation, supernatants were collected for a SEAP secretion assay using QUANTI-Blue substrate. Absorbance was measured at 630 nm by an ELISA plate reader. OD, optical density. (B to E) Effects of Rg1 and Re treatment on the expression and production of inflammatory mediators in LPS-stimulated macrophages. RAW264.7 cells were pretreated with Rg1 or Re at 50 μg/ml for 1 h and were then incubated with LPS for 20 h. After that, real-time PCR was used to analyze the mRNA expression of TNF-α, IL-1β, IL-6, COX-2, and iNOS of the cells (B); Western blot analysis was performed to measure TNF-α, IL-1β, IL-6, COX-2, and iNOS levels of the cell lysates (C); and relevant reagent kits were used to analyze NO (D) and PGE2 (E) in the supernatants. Values are means ± SD (n = 3). PBS, phosphate-buffered saline. *, P < 0.05; **, P < 0.01 (two-tailed Student's t test).
To analyze Rg1 and Re for their effects on proinflammatory mediators released from LPS-stimulated cells, we incubated RAW264.7 cells with 50 μg/ml Rg1 or Re and then stimulated the cells with LPS. Levels of mRNA and protein expression of these mediators were measured by real-time PCR or Western blot analysis. Results showed that both Rg1 and Re significantly suppressed mRNA expression of TNF-α, IL-1β, IL-6, COX-2, and iNOS (Fig. 1B) and inhibited the upregulation of the corresponding proteins (Fig. 1C). In addition, Rg1 or Re treatment also significantly decreased the production of NO (Fig. 1D) and PGE2 (Fig. 1E).
Rg1 or Re pretreatment prevents LPS from binding to TLR4.
To test the effect of Rg1 and Re on LPS binding to TLR4, we first incubated RAW264.7 cells with Rg1 or Re at 10, 30, 50, or 70 μg/ml and then stimulated the cells with LPS. Figure 2 shows that the staining intensity of LPS colocated with TLR4 decreased with the increased concentrations of Rg1 (Fig. 2A) and Re (Fig. 2B) pretreatment, indicating that Rg1 or Re prevented LPS from binding to TLR4.
FIG 2.

Blocking of LPS binding to TLR4 by Rg1 and Re. (A) LPS binding to macrophages pretreated with Rg1. RAW264.7 cells were pretreated with Rg1 for 1 h and then stimulated by Alexa Fluor 594-conjugated LPS (red). TLR4 was labeled with a specific antibody and a secondary antibody conjugated with DyLight 488 (green). Representative images from three independent experiments are shown. Bar, 10 μm. (B) LPS binding to macrophages pretreated with Re. RAW264.7 cells were treated in the same way as described for panel A except that Re was used in the pretreatment.
Distribution of ginsenosides Rg1 and Re in cells.
We previously found that incubation of mouse macrophages with Rg1 or Re in vitro activated NF-κB expression whereas pretreatment of the cells with TLR4 neutralizing antibody suppressed Re- but not Rg1-induced NF-κB expression, suggesting that Rg1, but not Re, penetrates the cell membrane and activates intracellular TLR4 signaling (20). To confirm this hypothesis, we incubated RAW264.7 cells with 50 μg/ml Rg1 or Re and then stained the cells with Rg1- or Re-specific antibody. As shown in Fig. 3, red staining was detected in both extracellular and intracellular areas of the cells incubated with Rg1 (Fig. 3A), while the red staining was found only on the cell surface of the cells treated with Re (Fig. 3B), suggesting that Rg1 can go inside the cells but Re cannot.
FIG 3.

Localization of Rg1 and Re in macrophages. (A) Rg1 was localized both inside and outside cells. RAW264.7 cells were incubated with Rg1 (50 μg/ml) for 2 or 6 h and then stained with primary antibody and Alexa Fluor 594-labeled secondary antibody (red). Cells without Rg1 treatment were set as the control (Ctrl). Images are representative of three independent experiments. Bars, 10 μm. (B) Re was localized only on the cell membrane. RAW264.7 cells were treated in the same way as described for panel A except that Re and anti-Re antibody were used instead of Rg1 and anti-Rg1 antibody. Bars, 10 μm.
The log octanol/water partition coefficient (log P) of a molecule reflects its ability to pass through the lipid bilayer (28). According to the Wildman and Crippen model (25, 26), log P of Rg1 is 1.12 and log P of Re is −0.03.
Rg1 or Re treatment suppresses the inflammation induced by LPS in rats.
To investigate the in vivo anti-inflammatory potential of Rg1 and Re, we first injected 1 mg/kg Rg1 or Re into rats and then challenged the animals with LPS. Changes in body temperature in response to LPS with or without pretreatment are shown in Fig. 4A. The injection procedure itself caused a transient stress-induced increase in body temperature of ∼1.2°C in each group. Thereafter, LPS-challenged rats without pretreatment developed a robust biphasic fever, with the first peak reaching ∼1.5°C at 2 h and the second peak reaching 1.8°C at 4 h. In contrast, the temperature changes for the Rg1-, Re-, and TAK-242-treated groups were only 0.9, 1.2, and 0.8°C at 2 h and 1.3, 1.4, and 1.0°C at 4 h, respectively. These results indicate that pretreatment with Rg1, Re, or TAK-242 significantly attenuated LPS-induced alterations in body temperature.
FIG 4.

Anti-inflammatory effect of Rg1 and Re in rats. (A and B) Effect of Rg1 and Re on body temperature and white blood cell counts in rats injected with LPS. Groups of rats (n = 6 per group) were intravenously injected with saline solution, Rg1, Re, or TAK-242 (1 mg/kg) 15 min before challenge with LPS (2.5 μg/kg). An additional group of rats were injected with saline solution only as a control. Body temperature was measured (A) and WBCs were counted (B) at the indicated time points. (C to E) Effect of Rg1 and Re on inflammatory mediators. Rats were treated in the same way as described for panel A. Blood samples were collected 4 h after LPS challenge for determination of TNF-α, IL-1β, IL-6, COX-2, and iNOS levels by Western blot analysis (C), and analyses of NO (D) and PGE2 (E) levels were performed using NO and prostaglandin E2 assay kits. Values are means ± SD (n = 6).*, P < 0.05; ** P < 0.01; ***, P < 0.001 (two-tailed Student's t test).
Figure 4B shows that LPS challenge increased the WBC counts whereas pretreatment with 1 mg/kg Rg1, Re, or TAK-242 significantly decreased this change. Besides, LPS elevated the levels of serum inflammatory mediators TNF-α, IL-1β, IL-6, COX-2, and iNOS (Fig. 4C) as well as the levels of NO and PGE2 (Fig. 4D and E). Pretreatment with Rg1, Re, or TAK-242 suppressed the increases seen with all the mediators.
Rg1 or Re treatment increases the survival of mice inoculated with LPS.
Intraperitoneal injection of LPS at a dose of 20 mg/kg was lethal to mice, and 70% to 80% of the mice died within 60 h (Fig. 5). However, pretreatment of the mice with Rg1 (Fig. 5A) or Re (Fig. 5B) increased their survival rates in a dose-dependent manner. With the doses of Rg1 or Re increased from 2.5 to 5 mg/kg, the survival rate was elevated from 60% to 90% (Rg1) or from 30% to 40% (Re). All the mice administered Rg1 at a minimal dose of 10 mg/kg were protected from death compared to 80% survival of mice treated with an equal dose of Re. To protect all the mice, 20 mg/kg Re was needed.
FIG 5.
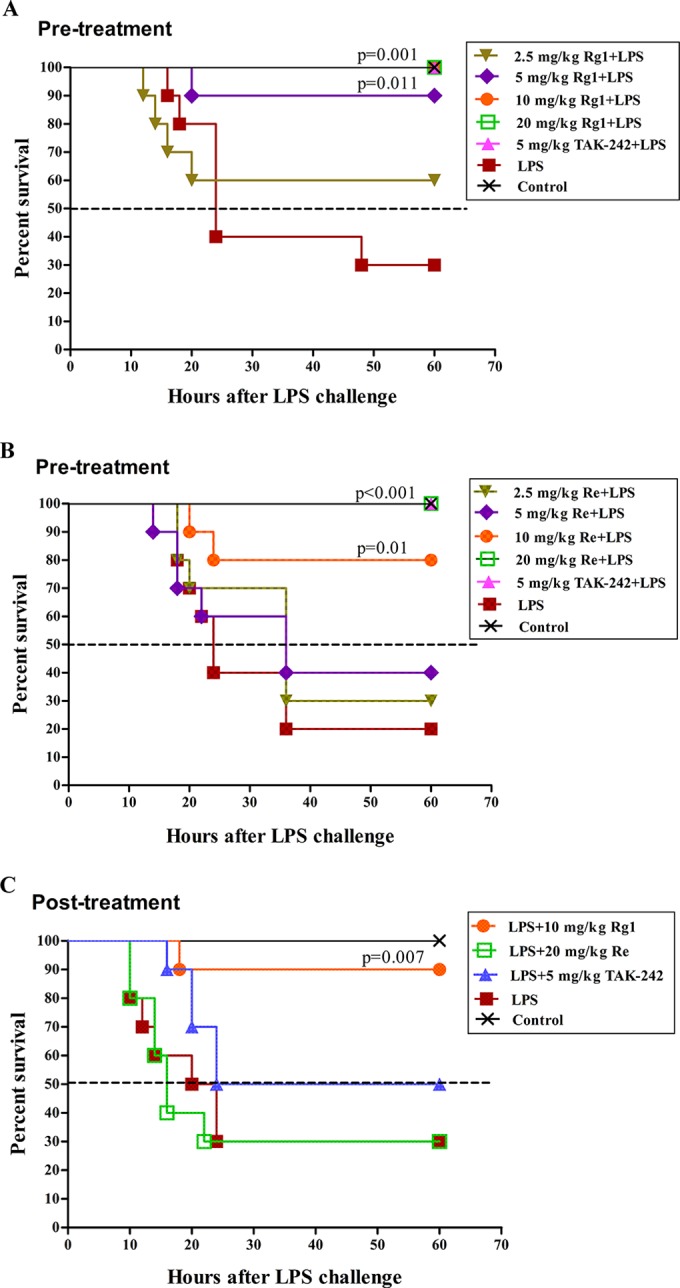
Protective effect of Rg1 and Re on LPS-induced lethality in mice. (A and B) Preventive effect of Rg1 (A) and Re (B). Mice were randomly allocated into 7 groups (n = 10) and were subcutaneously injected with various doses of Rg1 (A) or Re (B), TAK-242, or saline solution 3 times at 30-min intervals as indicated in the figure legends. Fifteen minutes after the last injection, mice were challenged with LPS (i.p.), except for the mice in the control group, which received saline solution only. (C) Therapeutic effects of Rg1 and Re. Mice were randomly allocated into 5 groups (n = 10) and left untreated or injected with LPS (20 mg/kg, i.p.). After 15 min, mice were subcutaneously administered Rg1 (10 mg/kg), Re (20 mg/kg), TAK-242 (5 mg/kg), or saline solution three times at 30-min intervals. Survival data were analyzed using the log-rank test.
On the basis of the results described above, administration of 10 mg/kg Rg1 and 20 mg/kg Re after LPS challenge was performed to evaluate the therapeutic effect of Rg1 and Re on LPS-induced sepsis in mice. As shown in Fig. 5C, 10 mg/kg Rg1 significantly raised the survival rate to 90% compared to 30% in the control group. However, no increased survival was observed in posttreatment mice administered 20 mg/kg Re.
Rg1 or Re treatment attenuates the sepsis induced by LPS in mice.
As shown in Fig. 6, the lethal dose of LPS administration led to a wave-shaped change in the populations of total WBCs and lymphocytes, in which the numbers decreased at 4 h postadministration and slightly rebounded at 8 h but dropped again at 12 h, while the number of neutrophils decreased at 4 h and gradually recovered between 8 and 12 h compared to the numbers seen with naive mice (0 h). In contrast, the number of total WBCs in the pre- or post-Rg1 (10 mg/kg Rg1 plus LPS or LPS plus 10 mg/kg Rg1)-treated mice increased remarkably and became significantly higher than in the mice in the untreated LPS group at 12 h (P < 0.05). Similar although not statistically significant changes were also observed with the number of neutrophils at 12 h. The number of lymphocytes from Rg1 posttreatment mice was significantly higher than in the LPS group (P < 0.05) at 12 h, while the levels of the lymphocytes from Rg1-pretreated mice stayed low after LPS administration and were significantly lower at 4 and 8 h (P < 0.05 at 4 h; P < 0.01 at 8 h) than the levels of those from mice in the LPS group. Moreover, Re pretreatment resulted in strong to significant increases in the number of total WBCs at 12 h (P < 0.05) and the numbers of neutrophils at 4, 8, and 12 h (P < 0.01 at 8 h) and in significant decreases in the number of lymphocytes at 8 h (P < 0.001), compared to those in the group of mice administered LPS alone. Treatment with TAK-242, as a positive control in this study, significantly increased the number of neutrophils at 4 h (P < 0.01) and reduced the numbers of total WBCs and lymphocytes at 8 h (P < 0.05 for total WBCs; P < 0.01 for lymphocytes).
FIG 6.

Effect of Rg1 and Re treatment on WBC populations in mice. BALB/c mice were randomly assigned to 5 groups (9 mice per group). The mice were subcutaneously injected with 10 mg of Rg1/kg body weight (mg/kg Rg1), 20 mg/kg Re, or 5 mg/kg TAK-242 3 times at 30-min intervals or left untreated and were then challenged with 20 mg/kg LPS 15 min later. An additional group of mice were injected with LPS first and then subcutaneously administered 10 mg/kg Rg1 3 times. Three mice in each group were euthanized for blood collection at the indicated time points. Another 5 naive mice were euthanized and sampled at h 0. Total and differential WBC counts in the EDTA-treated blood samples were determined. Values are means ± SD. *, P < 0.05; **, P < 0.01; ***, P < 0.001 (compared to the group administered LPS alone by the two-tailed Student's t test).
After lethal LPS administration, the serum levels of proinflammatory cytokines and chemokines increased significantly and stayed high during the following 12 h (Fig. 7A). Rg1 pretreatment significantly reduced the production of IL-1β (P < 0.01 at 8 h; P < 0.001 at 12 h), TNF-α (P < 0.05 at 4 h; P < 0.01 at 8 h; P < 0.05 at 12 h), IL-6 (P < 0.01 at 12 h), KC (P < 0.05 at 4 h), and MIP-2 (P < 0.01 at 8 h; P < 0.001 at 12 h). Similarly, Re pretreatment strongly decreased the amounts of IL-1β (P < 0.001 at 8 and 12 h), TNF-α (P < 0.01 at 4 h; P < 0.001 at 8 h; P < 0.05 at 12 h), and MIP-2 (P < 0.01 at 4 h; P < 0.001 at 8 and 12 h). Moreover, obvious declines in production of IL-1β (P < 0.001 at 8 h; P < 0.05 at 12 h), TNF-α (P < 0.05 at 4 h; P < 0.001 at 8 h), and MIP-2 (P < 0.05 at 4 h; P < 0.001 at 8 h; P < 0.05 at 12 h) were also observed in the Rg1 posttreatment group after LPS administration.
FIG 7.

Antisepsis effect of Rg1 and Re on the release of proinflammatory mediators in mice. BALB/c mice were randomly assigned to 5 groups (9 mice per group). The mice were subcutaneously injected with 10 mg/kg Rg1, 20 mg/kg Re, or 5 mg/kg TAK-242 3 times at 30-min intervals or left untreated and then challenged with 20 mg/kg LPS 15 min later. An additional group of mice were injected with LPS first and then subcutaneously administered 10 mg/kg Rg1 3 times. Three mice in each group were euthanized for blood collection at the indicated time points. Another 5 naive mice were euthanized and sampled at h 0. Sera were prepared and used for the measurement of levels of proinflammatory cytokines and chemokines (A), PGE2 (B), and NO (C). Data are presented as means ± SD (A and B) or means (C). *, P < 0.05; **, P < 0.01; ***, P < 0.001 (compared to LPS alone group by the two-tailed Student's t test).
Consistently, the serum PGE2 level also strongly increased after LPS administration (Fig. 7B). As expected, pretreatment with Rg1or Re significantly reduced the amount of PGE2 at 4 h (P < 0.05 for Re), 8 h (P < 0.05 for both Rg1 and Re), and 12 h (P < 0.05 for Rg1; P < 0.01 for Re). Also, Rg1 posttreatment significantly decreased the amount of PGE2 at 8 h (P < 0.05). For the NO analyses, due to the availability of only limited amounts of serum samples from each group at each time point, we pooled all the samples from each group at each time point. Still, we found that the serum NO level increased strongly after LPS inoculation, especially at 12 h, but was obviously reduced by the pretreatment with Rg1, Re, and TAK-242 and by the Rg1 posttreatment as well (Fig. 7C).
DISCUSSION
TLR4 selectively recognizes bacterial LPS (7, 11), rapidly triggering proinflammatory processes. However, hyperinflammatory host responses can cause life-threatening syndromes such as acute sepsis and septic shock. Infection by Gram-negative bacteria has become the major cause of deaths in intensive care units and is associated with a mortality rate of about 45% (29). Molecules with LPS antagonistic activity that can inhibit TLR4 activation are potential compounds for clinical management of sepsis. In this study, we demonstrated that Rg1 and Re suppressed production of multiple inflammatory mediators such as TNF-α, IL-1β, IL-6, COX-2, and iNOS in LPS-stimulated cells (Fig. 1), inhibited LPS-induced increases in the levels of body temperature, WBCs, serum TNF-α, IL-1β, IL-6, COX-2, iNOS, NO, and PGE2 in rats (Fig. 4), attenuated lethal sepsis (Fig. 6 and 7), and protected mice from death (Fig. 5) in a mouse model of endotoxin shock.
Excessive production of multiple inflammatory mediators has been shown to be closely connected with the symptomatology and pathogenesis of sepsis in clinical studies of both animals and humans (30). Consequently, regulation of multiple mediators could be more important than suppression of a single mediator. In fact, agents targeting a single mediator such as anti-TNF-α monoclonal antibody and IL-1 receptor antagonist are usually ineffective in the treatment of sepsis, since other inflammatory signaling pathways are not controlled (31, 32). In contrast, Rg1 and Re suppressed the production of multiple inflammatory mediators both in vitro (Fig. 1B, C, D, and E) and in vivo (Fig. 4C, D, and E and 7).
The induction of inflammatory responses by endotoxin is achieved by the orchestrated action of LPS-binding protein (LBP), CD14, MD-2, and TLR4 (33). LBP interacts with LPS and transfers it to CD14 (34, 35). CD14 is expressed on the myelomonocytic cells as a glycosylphosphatidylinositol-linked glycoprotein or in soluble form (sCD14) in serum (36). Monomeric LPS-CD14 and LPS–MD-2 are the complexes for activation of TLR4/MD-2 and TLR4 (37). After bonding to TLR4, LPS initiates cascade signaling involving interactions between a series of adapter proteins, which ultimately leads to phosphorylation of NF-κB, an important transcription factor in regulating expression of various proinflammatory cytokines (38). In this study, we demonstrated that pretreatment of Raw-Blue cells with Rg1 (30, 50, and 70 μg/ml) or Re (50 and 70 μg/ml) significantly downregulated LPS-induced phosphorylation of NF-κB (Fig. 1A). This is consistent with the suppressive effect of Rg1 and Re on LPS binding to TLR4 (Fig. 2).
Figure 5 shows that intraperitoneal injection of 20 mg/kg LPS killed 70% to 80% of the mice. Although the pathogenesis of LPS-induced lethality is not completely clear, systemic microcirculatory injury is considered to be a fundamental cause (39). Excessive circulating levels of NO and proinflammatory cytokines increase endothelial permeability, which in turn provokes internal-organ edema and induces widespread tissue injury and multiple-organ dysfunction (40, 41). In rats, 2.5 μg/kg LPS caused acute inflammatory responses, including increases in body temperature and total WBC populations and in production of various proinflammatory mediators (Fig. 4), while in the mouse shock model, besides high levels of multiple proinflammatory mediators, the lethal dose of LPS (20 mg/kg) led to decreases in the populations of total WBCs and lymphocytes and only negligible increases in the numbers of neutrophils (Fig. 6 and 7), indicating that the host immune systems were severely damaged by the lethal LPS inoculation. However, in both cases, Rg1 or Re pretreatment effectively downregulated those responses and successfully protected 100% of the mice from septic death (Fig. 5A and B). Such an antisepsis effect of Rg1 and Re may be due to their inhibition of LPS binding to TLR4. More importantly, the antisepsis effect of Rg1 at a dose of 10 mg/kg was even evident when it was given as a posttreatment, which rescued 90% of LPS-administered mice, suggesting that Rg1 has both preventative and therapeutic potential effects on LPS-induced sepsis. However, no protective effect was found in the posttreatment with 20 mg/kg Re.
The differences between Rg1 and Re in their antisepsis activities may be attributed to their differences in distribution in cells. Rg1 was able to pass through the membrane and interfere with LPS binding to both extracellular and intracellular TLR4 (Fig. 2A and 3A), while Re located on the cell membrane and blocked LPS binding only to extracellular TLR4 (Fig. 2B and 3B). In addition to the hyperinflammatory response in sepsis, immunosuppression also occurs and can be lethal due to substantial impairment of the innate immune system during the early stage of the sepsis development (42). Treatment strategies aiming to suppress the excessive inflammation have been proven ineffective. Clinical trials performed with monoclonal antibodies targeting LPS, monoclonal antibodies targeting TNF-α, and soluble receptors of TNF-α or recombinant IL-1rα failed to show a clinical benefit (43). Therefore, regulation of immunosuppression as well as hyperinflammation could be more beneficial than suppressing hyperinflammation alone. Rg1 has dual roles in regulation of the immune responses: upregulation of the immune response as found in our previous studies (18, 20) and downregulation of the proinflammatory response as presented in this study. The bidirectional activities of Rg1 may be the reason for the higher rate of survival of Rg1-treated mice than of TAK-242-treated mice shown in Fig. 5C.
In conclusion, Rg1 and Re can prevent LPS from binding to TLR4 and block the LPS-triggered signaling pathway. Injection of Rg1 and Re into rats before LPS challenge suppressed LPS-induced increases in body temperature and in levels of WBCs and serum proinflammatory mediators. Preinjection of Rg1 at 10 mg/kg and Re at 20 mg/kg into mice increased the population levels of total and differential WBCs, downregulated the expression and production of multiple proinflammatory mediators, and rescued 100% of the mice from LPS-induced lethality. Even administered after LPS challenge, Rg1 still effectively suppressed inflammatory responses and protected 90% of the mice from death, while Re administration had no beneficial effect, with the treated mice showing the same survival rate as the control mice (30%). Higher anti-LPS activity of Rg1 than Re could be attributed to its ability to pass through the host cell membrane and prevent LPS from triggering both extracellular and intracellular TLR4. More importantly, Rg1 exerts up- and downregulatory actions on the immune system, which leads to higher survival in Rg1-treated mice than in TAK-242-treated mice, suggesting that Rg1 might be an ideal therapeutic agent for sepsis.
ACKNOWLEDGMENTS
This study was supported by the National Natural Scientific Foundation of China (31372471) and Special Fund for Agro-Scientific Research in the Public Interest (201303040-03).
We thank Cenrong Zhang, Jia Yu, Lijia Zhang, Mengling Zhou, Xiaoyan Su, Yan Huang, Yisong Lu, and Yuhong Zhang for technical assistance in rat experiments, Xiaoling Gao for assistance in mouse sepsis experiment, Greg Harris for the Luminex and NO assays, and Feng Xie and Lin Yuan for valuable discussion.
REFERENCES
- 1.Song R, Kim J, Yu D, Park C, Park J. 2012. Kinetics of IL-6 and TNF-alpha changes in a canine model of sepsis induced by endotoxin. Vet Immunol Immunopathol 146:143–149. doi: 10.1016/j.vetimm.2012.02.008. [DOI] [PubMed] [Google Scholar]
- 2.Angus DC, Linde-Zwirble WT, Lidicker J, Clermont G, Carcillo J, Pinsky MR. 2001. Epidemiology of severe sepsis in the United States: analysis of incidence, outcome, and associated costs of care. Crit Care Med 29:1303–1310. doi: 10.1097/00003246-200107000-00002. [DOI] [PubMed] [Google Scholar]
- 3.Calandra T, Echtenacher B, Roy DL, Pugin J, Metz CN, Hultner L, Heumann D, Mannel D, Bucala R, Glauser MP. 2000. Protection from septic shock by neutralization of macrophage migration inhibitory factor. Nat Med 6:164–170. doi: 10.1038/72262. [DOI] [PubMed] [Google Scholar]
- 4.Lee SK, Kim SD, Lee HY, Baek SH, Ko MJ, Son BG, Park S, Choi YW, Bae YS. 2012. α-Iso-cubebene, a natural compound isolated from Schisandra chinensis fruit, has therapeutic benefit against polymicrobial sepsis. Biochem Biophys Res Commun 426:226–231. doi: 10.1016/j.bbrc.2012.08.070. [DOI] [PubMed] [Google Scholar]
- 5.Ohto U, Fukase K, Miyake K, Shimizu T. 2012. Structural basis of species-specific endotoxin sensing by innate immune receptor TLR4/MD-2. Proc Natl Acad Sci U S A 109:7421–7426. doi: 10.1073/pnas.1201193109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lynn M, Rossignol DP, Wheeler JL, Kao RJ, Perdomo CA, Noveck R, Vargas R, D'Angelo T, Gotzkowsky S, McMahon FG. 2003. Blocking of responses to endotoxin by E5564 in healthy volunteers with experimental endotoxemia. J Infect Dis 187:631–639. doi: 10.1086/367990. [DOI] [PubMed] [Google Scholar]
- 7.Poltorak A, He X, Smirnova I, Liu MY, Van Huffel C, Du X, Birdwell D, Alejos E, Silva M, Galanos C, Freudenberg M, Ricciardi-Castagnoli P, Layton B, Beutler B. 1998. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science 282:2085–2088. doi: 10.1126/science.282.5396.2085. [DOI] [PubMed] [Google Scholar]
- 8.Nagai Y, Akashi S, Nagafuku M, Ogata M, Iwakura Y, Akira S, Kitamura T, Kosugi A, Kimoto M, Miyake K. 2002. Essential role of MD-2 in LPS responsiveness and TLR4 distribution. Nat Immunol 3:667–672. [DOI] [PubMed] [Google Scholar]
- 9.Farrow B, Evers BM. 2002. Inflammation and the development of pancreatic cancer. Surg Oncol 10:153–169. doi: 10.1016/S0960-7404(02)00015-4. [DOI] [PubMed] [Google Scholar]
- 10.Surh YJ, Chun KS, Cha HH, Han SS, Keum YS, Park KK, Lee SS. 2001. Molecular mechanisms underlying chemopreventive activities of anti-inflammatory phytochemicals: down-regulation of COX-2 and iNOS through suppression of NF-kappa B activation. Mutat Res 480–481:243–268. [DOI] [PubMed] [Google Scholar]
- 11.Beutler B. 2000. Tlr4: central component of the sole mammalian LPS sensor. Curr Opin Immunol 12:20–26. doi: 10.1016/S0952-7915(99)00046-1. [DOI] [PubMed] [Google Scholar]
- 12.Hawkins LD, Christ WJ, Rossignol DP. 2004. Inhibition of endotoxin response by synthetic TLR4 antagonists. Curr Top Med Chem 4:1147–1171. doi: 10.2174/1568026043388123. [DOI] [PubMed] [Google Scholar]
- 13.Takashima K, Matsunaga N, Yoshimatsu M, Hazeki K, Kaisho T, Uekata M, Hazeki O, Akira S, Iizawa Y, Ii M. 2009. Analysis of binding site for the novel small-molecule TLR4 signal transduction inhibitor TAK-242 and its therapeutic effect on mouse sepsis model. Br J Pharmacol 157:1250–1262. doi: 10.1111/j.1476-5381.2009.00297.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lian LH, Jin Q, Song SZ, Wu YL, Bai T, Jiang S, Li Q, Yang N, Nan JX. 2013. Ginsenoside Rh2 downregulates LPS-induced NF-kappa B activation through inhibition of TAK1 phosphorylation in RAW 264.7 murine macrophage. Evid Based Complement Alternat Med 2013:646728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hu S, Concha C, Lin F, Persson WK. 2003. Adjuvant effect of ginseng extracts on the immune responses to immunisation against Staphylococcus aureus in dairy cattle. Vet Immunol Immunopathol 91:29–37. doi: 10.1016/S0165-2427(02)00264-7. [DOI] [PubMed] [Google Scholar]
- 16.Rivera E, Daggfeldt A, Hu S. 2003. Ginseng extract in aluminium hydroxide adjuvanted vaccines improves the antibody response of pigs to porcine parvovirus and Erysipelothrix rhusiopathiae. Vet Immunol Immunopathol 91:19–27. doi: 10.1016/S0165-2427(02)00269-6. [DOI] [PubMed] [Google Scholar]
- 17.Song X, Chen J, Sakwiwatkul K, Li R, Hu S. 2010. Enhancement of immune responses to influenza vaccine (H3N2) by ginsenoside Re. Int Immunopharmacol 10:351–356. doi: 10.1016/j.intimp.2009.12.009. [DOI] [PubMed] [Google Scholar]
- 18.Sun J, Song X, Hu S. 2008. Ginsenoside Rg1 and aluminum hydroxide synergistically promote immune responses to ovalbumin in BALB/c mice. Clin Vaccine Immunol 15:303–307. doi: 10.1128/CVI.00448-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xiang YZ, Shang HC, Gao XM, Zhang BL. 2008. A comparison of the ancient use of ginseng in traditional Chinese medicine with modern pharmacological experiments and clinical trials. Phytother Res 22:851–858. doi: 10.1002/ptr.2384. [DOI] [PubMed] [Google Scholar]
- 20.Su F, Yuan L, Zhang L, Hu S. 2012. Ginsenosides Rg1 and Re act as adjuvant via TLR4 signaling pathway. Vaccine 30:4106–4112. doi: 10.1016/j.vaccine.2012.03.052. [DOI] [PubMed] [Google Scholar]
- 21.Fitzgerald KA, Rowe DC, Barnes BJ, Caffrey DR, Visintin A, Latz E, Monks B, Pitha PM, Golenbock DT. 2003. LPS-TLR4 signaling to IRF-3/7 and NF-kappaB involves the toll adapters TRAM and TRIF. J Exp Med 198:1043–1055. doi: 10.1084/jem.20031023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fukuda N, Tanaka H, Shoyama Y. 2000. Formation of monoclonal antibody against a major ginseng component, ginsenoside Rg1 and its characterization. Monoclonal antibody for a ginseng saponin. Cytotechnology 34:197–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Livak KJ, Schmittgen TD. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods 25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 24.Katritzky AR, Kuanar M, Slavov S, Hall CD, Karelson M, Kahn I, Dobchev DA. 2010. Quantitative correlation of physical and chemical properties with chemical structure: utility for prediction. Chem Rev 110:5714–5789. doi: 10.1021/cr900238d. [DOI] [PubMed] [Google Scholar]
- 25.Mannhold R, Poda GI, Ostermann C, Tetko IV. 2009. Calculation of molecular lipophilicity: state-of-the-art and comparison of log P methods on more than 96,000 compounds. J Pharm Sci 98:861–893. doi: 10.1002/jps.21494. [DOI] [PubMed] [Google Scholar]
- 26.Wildman SA, Crippen GM. 1999. Prediction of physicochemical parameters by atomic contributions. J Chem Inf Comput Sci 39:868–873. doi: 10.1021/ci990307l. [DOI] [Google Scholar]
- 27.Hussey SE, Liang H, Costford SR, Klip A, DeFronzo RA, Sanchez-Avila A, Ely B, Musi N. 2013. TAK-242, a small-molecule inhibitor of Toll-like receptor 4 signalling, unveils similarities and differences in lipopolysaccharide- and lipid-induced inflammation and insulin resistance in muscle cells. Biosci Rep 33:37–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Walter A, Gutknecht J. 1986. Permeability of small nonelectrolytes through lipid bilayer membranes. J Membr Biol 90:207–217. doi: 10.1007/BF01870127. [DOI] [PubMed] [Google Scholar]
- 29.Martin GS, Mannino DM, Eaton S, Moss M. 2003. The epidemiology of sepsis in the United States from 1979 through 2000. N Engl J Med 348:1546–1554. doi: 10.1056/NEJMoa022139. [DOI] [PubMed] [Google Scholar]
- 30.Blackwell TS, Christman JW. 1996. Sepsis and cytokines: current status. Br J Anaesth 77:110–117. doi: 10.1093/bja/77.1.110. [DOI] [PubMed] [Google Scholar]
- 31.Fisher CJ Jr, Dhainaut JF, Opal SM, Pribble JP, Balk RA, Slotman GJ, Iberti TJ, Rackow EC, Shapiro MJ, Greenman RL, et al. 1994. Recombinant human interleukin 1 receptor antagonist in the treatment of patients with sepsis syndrome. Results from a randomized, double-blind, placebo-controlled trial. Phase III rhIL-1ra Sepsis Syndrome Study Group. JAMA 271:1836–1843. [PubMed] [Google Scholar]
- 32.Abraham J, Lewis G. 1998. Secrecy and transparency of medicines licensing in the EU. Lancet 352:480–482. doi: 10.1016/S0140-6736(97)11282-X. [DOI] [PubMed] [Google Scholar]
- 33.Jerala R. 2007. Structural biology of the LPS recognition. Int J Med Microbiol 297:353–363. doi: 10.1016/j.ijmm.2007.04.001. [DOI] [PubMed] [Google Scholar]
- 34.Wright SD, Ramos RA, Tobias PS, Ulevitch RJ, Mathison JC. 1990. CD14, a receptor for complexes of lipopolysaccharide (LPS) and LPS binding protein. Science 249:1431–1433. doi: 10.1126/science.1698311. [DOI] [PubMed] [Google Scholar]
- 35.Schumann RR, Leong SR, Flaggs GW, Gray PW, Wright SD, Mathison JC, Tobias PS, Ulevitch RJ. 1990. Structure and function of lipopolysaccharide binding protein. Science 249:1429–1431. doi: 10.1126/science.2402637. [DOI] [PubMed] [Google Scholar]
- 36.Gegner JA, Ulevitch RJ, Tobias PS. 1995. Lipopolysaccharide (LPS) signal transduction and clearance. Dual roles for LPS binding protein and membrane CD14. J Biol Chem 270:5320–5325. [DOI] [PubMed] [Google Scholar]
- 37.Shimazu R, Akashi S, Ogata H, Nagai Y, Fukudome K, Miyake K, Kimoto M. 1999. MD-2, a molecule that confers lipopolysaccharide responsiveness on Toll-like receptor 4. J Exp Med 189:1777–1782. doi: 10.1084/jem.189.11.1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bremner P, Heinrich M. 2002. Natural products as targeted modulators of the nuclear factor-kappaB pathway. J Pharm Pharmacol 54:453–472. doi: 10.1211/0022357021778637. [DOI] [PubMed] [Google Scholar]
- 39.Lehr HA, Bittinger F, Kirkpatrick CJ. 2000. Microcirculatory dysfunction in sepsis: a pathogenetic basis for therapy? J Pathol 190:373–386. doi:. [DOI] [PubMed] [Google Scholar]
- 40.Cohen J. 2002. The immunopathogenesis of sepsis. Nature 420:885–891. doi: 10.1038/nature01326. [DOI] [PubMed] [Google Scholar]
- 41.Netea MG, van der Meer JW, van Deuren M, Kullberg BJ. 2003. Proinflammatory cytokines and sepsis syndrome: not enough, or too much of a good thing? Trends Immunol 24:254–258. doi: 10.1016/S1471-4906(03)00079-6. [DOI] [PubMed] [Google Scholar]
- 42.Riedemann NC, Guo RF, Ward PA. 2003. Novel strategies for the treatment of sepsis. Nat Med 9:517–524. doi: 10.1038/nm0503-517. [DOI] [PubMed] [Google Scholar]
- 43.Kimura A, Kishimoto T. 2010. IL-6: regulator of Treg/Th17 balance. Eur J Immunol 40:1830–1835. doi: 10.1002/eji.201040391. [DOI] [PubMed] [Google Scholar]