

Abstract
Plasmid-encoded protein QnrB1 protects DNA gyrase from ciprofloxacin inhibition. Using a bacterial two-hybrid system, we evaluated the physical interactions between wild-type and mutant QnrB1, the GyrA and GyrB gyrase subunits, and a GyrBA fusion protein. The interaction of QnrB1 with GyrB and GyrBA was approximately 10-fold higher than that with GyrA, suggesting that domains of GyrB are important for stabilizing QnrB1 interaction with the holoenzyme. Sub-MICs of ciprofloxacin or nalidixic acid reduced the interactions between QnrB1 and GyrA or GyrBA but produced no reduction in the interaction with GyrB or a quinolone-resistant GyrA:S83L (GyrA with S83L substitution) mutant, suggesting that quinolones and QnrB1 compete for binding to gyrase. Of QnrB1 mutants that reduced quinolone resistance, deletions in the C or N terminus of QnrB1 resulted in a marked decrease in interactions with GyrA but limited or no effect on interactions with GyrB and an intermediate effect on interactions with GyrBA. While deletion of loop B and both loops moderately reduced the interaction signal with GyrA, deletion of loop A resulted in only a small reduction in the interaction with GyrB. The loop A deletion also caused a substantial reduction in interaction with GyrBA, with little effect of loop B and dual-loop deletions. Single-amino-acid loop mutations had little effect on physical interactions except for a Δ105I mutant. Therefore, loops A and B may play key roles in the proper positioning of QnrB1 rather than as determinants of the physical interaction of QnrB1 with gyrase.
INTRODUCTION
Quinolones are antimicrobials that target the essential bacterial enzymes DNA gyrase and topoisomerase IV and form drug-enzyme-DNA complexes that block movement of the DNA replication complex (1, 2). Bacterial resistance mutations have been selected with wide clinical use of quinolones. These mutations reduce drug binding by altering the target enzymes or eliminate quinolones by increased expression of efflux pumps.
Unexpectedly, transferable resistance has also emerged to compromise the clinical utility of quinolones further. First recognized in an isolate of Klebsiella pneumoniae in 1998 (3), transferable quinolone resistance is now widespread in enteric Gram-negative bacteria. Such resistance is transmissible by plasmids which often contain additional genes conferring resistance to other classes of antimicrobial agents (4–7). Plasmid-mediated quinolone resistance (PMQR) is caused by genes encoding either components of quinolone efflux pumps, a quinolone-modifying enzyme, or Qnr proteins that can protect gyrase and topoisomerase IV from quinolone inhibition. Although low-level resistance results from the presence of PMQR genes alone, their presence facilitates selection of higher level mutational resistance (8), and these genes have been documented to have spread worldwide (9).
Qnr proteins are members of the pentapeptide repeat family of proteins. Six types of plasmid-encoded proteins (QnrA, -B, -C, -D, -S, and -VC), are known, some with multiple variants, as well as others encoded on the chromosome especially of aquatic bacteria (3, 10–14). QnrB variants appear to have the highest prevalence (14). QnrB1 dimerizes via its C-terminal domain to form a rod-like structure that mimics B-form DNA (15). Each monomer has two external loops, a smaller loop, loop A (8 amino acids, Y46-Q53) and a larger loop, loop B (12 amino acids, M102-S113), which are required for Qnr-mediated protection of gyrase from ciprofloxacin action.
Alanine mutagenesis and deletion analysis of QnrB1 have provided information on structural elements that are important for the ability of the protein to confer resistance (16). By convention, the central amino acid in the pentapeptide unit is termed i, with neighboring amino acids i+1 and i−1 and i+2 and i−2 (17). Essential amino acids for protection of gyrase were found at the central amino acid positions i and i−2 in the pentapeptide repeat module and in the larger loop, loop B, where deletion of only a single amino acid compromised activity. For the single-alanine substitutions in the two loops, there was little effect on protective activity except for F111A in loop B. While alanine substitution of T106, T107, and R108 showed little effect on protection, their single or multiple deletions strongly reduced protection. Furthermore, deletion of 15 to 20 N-terminal amino acids and as few as 3 C-terminal amino acids also abolished resistance (16). Therefore, loop B seems to be a key for QnrB1's protective positioning on gyrase (18). Purified QnrA has been shown to bind to both the GyrA and GyrB subunits as well as the ParC and ParE subunits of topoisomerase IV (19, 20). QnrA also affects gyrase binding of DNA, consistent with a model in which the DNA-like structure of Qnr proteins and the related MfpA pentapeptide repeat protein position themselves in the gyrase DNA binding site (21). There are, however, no data on the effects of specific Qnr structural modifications on its physical interactions with gyrase proteins or on the effects of gyrase resistance mutations on this interaction. It also remains unclear how Qnr proteins are able to protect from quinolone action without impairing gyrase catalytic function.
In this study, we evaluated the physical interactions between QnrB1 and gyrase subunits, GyrA and GyrB, and a GyrBA fusion protein using a bacterial two-hybrid (B2H) system to analyze protein-protein interactions in vivo (22) and to assess the effects of QnrB and gyrase mutations on these interactions.
MATERIALS AND METHODS
Bacterial strains, plasmids, and media.
Escherichia coli DH5α or One Shot TOP10 (Invitrogen, Life Technology, CA, USA) was used for electrotransformation. BacterioMatch II validation reporter competent cells [Δ(mcrA)183 Δ(mcrCB-hsdSMR-mrr)173 endA1 hisB supE44 thi-1 recA1 gyrA96 (Nalr) relA1 lac containing F′ lacIq HIS3 aadA Kanr] (Agilent Technology, CA, USA) were used for chemical transformation and as the reporter strain in the bacterial two-hybrid (B2H) assay. The B2H system used 3.2-kb bait plasmid pBT (Cmr) and 4.4-kb target plasmid pTRG (Tcr) (Agilent Technology). Superoptimal broth with catabolic repressor (SOC) was used for bacterial growth and transformation (23).
Cloning of QnrB1 and gyrase.
Primers used for cloning are listed in Table S1 in the supplemental material. Potentially significant amino acid positions for QnrB1-DNA gyrase interactions were selected, based on their effect on the protective activity of QnrB1 (Fig. 1) (16). Site-directed mutagenesis was performed by using a Phusion site-directed mutagenesis kit (Thermo Scientific, MA, USA) to delete or substitute alanines for amino acids Q51, I105, T106, and F111. This method was also used to make a GyrA:S83L (GyrA with the S83L substitution) mutant. Amino acids to be deleted in total loop deletion mutants were determined in a previous study: amino acids 46 to 51 for loop A deletion mutant, amino acids 104 to 113 for loop B deletion mutant, and both for dual-loop deletion mutant (15). Wild-type qnrB1 and its N- or C-terminal deletion mutants were cloned into plasmid pBT using EcoRI and XhoI; gyrA or gyrB was similarly cloned into plasmid pTRG using BamHI and EcoRI or BamHI and XhoI, respectively. A GyrB-GyrA fusion protein with a 3-amino-acid (Gly-Ala-Pro) linker (GyrBA) was constructed, and its catalytic functions and interaction with quinolone were confirmed (C. Chen, R. Villet, G. A. Jacoby, and D. C. Hooper, unpublished data). These findings are consistent with those previously reported in which the catalytic function of the same construct was shown (24) and is consistent with the similarity of the crystal structure of a GyrBA fusion and native gyrase (25). This GyrBA construct gyrBA was cloned into pTRG using BamHI and XhoI. After sequencing to confirm mutagenesis, pBT-QnrB1, pTRG-GyrA, pTRG-GyrB, and pTRG-GyrBA were transformed into the E. coli reporter strain by chemical transformation. Tetracycline (12.5 μg/ml) was used to select pTRG derivatives, chloramphenicol (50 μg/ml) for pBT constructs, and both antibiotics for pBT-pTRG cotransformants. Colony PCR was performed to confirm successful cotransformation of both plasmids.
FIG 1.
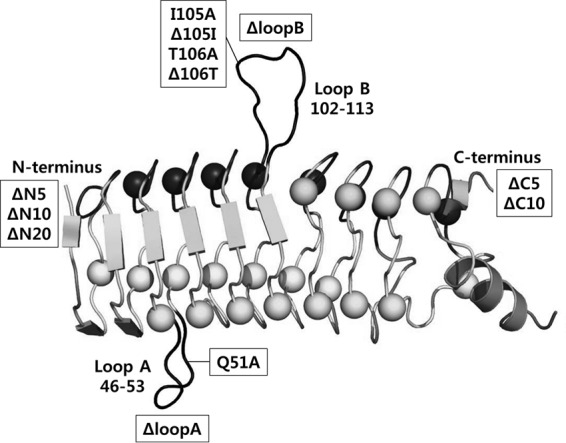
Illustration of QnrB1 and positions of the various mutations that were constructed in this study (modified from reference 15). Loop B is made up of the amino acids from positions 102 to 113, and loop A is made up of the amino acids from positions 46 to 53. Five, ten, or twenty amino acids were deleted from the N or C terminus to construct the ΔN and ΔC mutations.
Bacterial two-hybrid assay.
The B2H assay was performed using the BacterioMatch II two-hybrid system vector system (Agilent Technology) according to the manufacturer's instructions (26). In brief, this system uses a HIS3-aadA reporter cassette, and detection of protein-protein interaction is based on transcriptional activation of the HIS3 reporter gene, which allows growth in the presence of 3-amino-1,2,4-triazole (3-AT), a competitive inhibitor of HIS3 protein. When both proteins (bait and target) in the pBT and pTRG plasmids interact, they recruit and stabilize the binding of RNA polymerase at the promoter and activate the transcription of the HIS3 gene. The reporter strain is able to grow on media lacking histidine and containing 5 mM 3-AT, as transcriptional activation increases expression of the HIS3 gene product to levels that are sufficient to overcome the competitive inhibition by 3-AT.
After cotransformation was confirmed, 100-μl aliquots of serially diluted cotransformants were spread on agar containing nonselective screening medium (NS medium), selective screening medium containing 5 mM 3-AT (3-AT medium), and 3-AT medium with sub-MIC of ciprofloxacin (CIP medium) or nalidixic acid (NAL medium). The NS medium was made using 1.5% Bacto agar with 1× M9 minimal salts (Sigma-Aldrich, MO, USA), 0.4% glucose, 0.2 mM adenine HCl, 1× His dropout amino acid supplement (Clontech, CA, USA), 1 mM MgSO4, 1 mM thiamine HCl, 10 μM ZnSO4, 100 μM CaCl2, 50 μM isopropyl-β-d-thiogalactopyranoside (IPTG), 25 μg/ml of chloramphenicol, and 12.5 μg/ml of tetracycline. The plates were incubated at 37°C for 24 h and at 30°C for an additional 2, 4, and 6 days for NS, 3-AT, and CIP/NAL medium, respectively. The numbers of CFU were then counted.
The strength of the interaction between two constructs was calculated as the ratio of colonies obtained on 3-AT medium compared to those on NS medium. Self-activation was evaluated by performing the B2H assay with an empty vector, e.g., empty pBT vector with pTRG-gyrase subunits or wild-type or mutant pBT-QnrB1 with an empty pTRG vector. The cutoff for noninteraction for the empty vectors was defined as a ratio of ≤0.1%, a value more stringent than that recommended by the manufacturer (≤1%). The fold changes were calculated by comparing the ratio for each cotransformant construct with the ratio in the corresponding control with an empty pBT and pTRG-gyrase vector. In the case of CIP medium or NAL medium, the strength of the interaction was compared by ratios of the colony count on CIP or NAL medium to the count on NS medium instead of the fold change because only a small number of colonies of control strains having an empty vector grew in the presence of ciprofloxacin or nalidixic acid. Independent assays were performed at least 5 times, and data for fold changes are presented as median and interquartile range (IQR). MICs of ciprofloxacin and nalidixic acid were determined by broth microdilution using Mueller-Hinton II broth (Becton Dickinson, Franklin Lakes, NJ) with an additional 50 μM IPTG, 25 μg/ml of chloramphenicol, and 12.5 μg/ml of tetracycline in the serially diluted quinolone-containing media.
Fold change of QnrB1 mutant interactions with gyrase was compared with that of wild-type QnrB1. Wilcoxon signed-rank test was performed for statistical analyses using IBM SPSS Statistics for Windows, version 20.0. (Armonk, NY, USA). P values of <0.05 were considered statistically significant.
RESULTS
The number of colonies on plates with empty vector controls was always well within the acceptable limits for the procedure (i.e., ≤0.1%). QnrB1 interacted with individual gyrase subunits (Table 1); QnrB1 interacted most strongly with GyrB (median fold change, 2,865) and GyrBA (median fold change, 4,219) and less strongly with GyrA (median fold change, 383). In individual experiments, which were performed 9 times, including 3 wild-type cotransformants simultaneously, the GyrB interaction was consistently higher (5.5-fold median [IQR, 3.7- to 8.1-fold]) than that with GyrA and similar to the interaction with GyrBA (1.0-fold [IQR, 0.6- to 1.6-fold]).
TABLE 1.
Effects of sub-MIC amounts of ciprofloxacin or nalidixic acid on interaction of wild-type QnrB1 with gyrasea
| Gyrase gene on pTRG | MIC (μg/ml) |
Strength of interactionb (%) |
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CIP | NAL | No drug (control,c fold changed) | CIP (μg/ml) |
NAL (μg/ml) |
||||||||||
| 0.0156 | 0.0313 | 0.0625 | 0.125 | 0.25 | 1 | 2 | 4 | 8 | 16 | 32 | ||||
| No gyrase gene (empty) | 0.5 | 64 | ||||||||||||
| GyrA | 0.125 | 16 | 3.83 (0.01, 383) | 0.008 | 0.001 | |||||||||
| GyrA:S83L | 0.5 | 32 | 2.74 (0.01, 274) | 19.3 | 18.3 | 0.529 | 0.176 | 0.030 | ||||||
| GyrB | 0. 5 | 64 | 12.5 (0.00434, 2,865) | 71.9 | 41.9 | 1.59 | 17.7 | 4.36 | 0.685 | |||||
| GyrBA | 0.125 | 8 | 0.679 (0.000161, 4,219) | 0.016 | 0.001 | <0.001 | 0.040 | 0.003 | <0.001 | |||||
The effects of sub-MIC amounts of ciprofloxacin and nalidixic acid in E. coli BacterioMatch II validation reporter competent cells with plasmids pBT-QnrB1 (bait plasmid) and pTRG (target plasmid). Abbreviations: CIP, ciprofloxacin; NAL, nalidixic acid.
The strength of interaction represents the ratio (as a percentage) of numbers of colonies in selective screening medium containing 3-amino-1,2,4-triazole to those on nonselective screening medium.
The first value in the parentheses, the control value, represents the strength of interaction in contransformants having empty pBT and pTRG-gyrase.
The second value in the parentheses, fold change, was calculated by comparing the ratio in each cotransformant with the ratio in the control strain.
The magnitude of QnrB protection of gyrase supercoiling varies inversely with quinolone concentration, suggesting a potential competitive interaction in the binding of the two components (27). We thus evaluated the effects of quinolones on the interaction of QnrB with the gyrase subunits and GyrBA. The interactions of QnrB1 with gyrase components were measured after adding sub-MICs of ciprofloxacin (0.0156 to 0.25 μg/ml) or nalidixic acid (1 to 32 μg/ml) to evaluate the effect on the interaction of the two proteins. In comparison to the MIC of ciprofloxacin or nalidixic acid in the presence of plasmids with wild-type QnrB1 and empty pTRG, cotransformants with QnrB1 and either pTRG-GyrA, pTRG-GyrB, or pTRG-GyrBA had MICs that were 4-fold lower, the same, or 4- to 8-fold lower, respectively (Table 1). Since the reporter strain has a chromosomal quinolone resistance gyrA96 mutation, the lowered MIC is in keeping with the poison mechanism of quinolone action in which increases in quinolone-susceptible target expression increase the likelihood of lethal enzyme-DNA complex formation and consequently lower the MIC (28). In order to adjust for the different MICs of the various cotransformant constructs, interaction signals were measured in the presence of a range of sub-MICs of ciprofloxacin. There were substantial reductions (<0.1%) in the interaction signals between QnrB1 and GyrA and GyrBA after adding sub-MICs of ciprofloxacin or nalidixic acid (Table 1). In contrast, quinolones somewhat stimulated the interaction of QnrB1 with GyrB. Notably, the interaction of QnrB1 with quinolone-resistant GyrA:S83L was also substantially less affected than with wild-type GyrA. GyrA resistance mutations reduce the binding of quinolones to the gyrase-DNA complex (29). The quinolone-mediated reduced binding of QnrB1 to GyrA was lost in the presence of such a resistance mutation in our study. This finding is consistent with the concept that quinolones and QnrB each competitively affect the binding of the other to gyrase.
The structural features of QnrB1 that are important for its ability to confer resistance to quinolones have been evaluated (15, 16), but mutant QnrB proteins have not been studied specifically for their physical interactions with gyrase. Deletion mutations at the C or N terminus in QnrB1 resulted in a marked decrease in interactions with GyrA, a limited or no effect on GyrB interactions, and an intermediate effect on GyrBA interactions compared with wild-type QnrB1 (Table 2). Each of these mutations, with the exception of the ΔN5 (five N-terminal amino acids deleted) mutation, was previously shown to eliminate the resistance phenotype of QnrB1 (16). These findings together suggest that N-terminal and C-terminal amino acids of QnrB1 are more important for binding to domains of GyrA rather than GyrB. In addition, the reduced but residual binding to GyrBA, possibly representing an overall domain structure most closely related to that of gyrase holoenzyme, may reflect binding of QnrB incorrectly positioned to effect resistance.
TABLE 2.
Interaction of DNA gyrases with QnrB1 and QnrB1 mutants having C- or N-terminal deletions
| QnrB1 mutant (ciprofloxacin MIC [μg/ml])a | No. of expts | Fold changeb |
P valuee | |
|---|---|---|---|---|
| Median (ratioc) | Interquartile ranged | |||
| Interaction with GyrA | ||||
| Wild type (0.25) | 14 | 404 (1) | 317–602 | |
| ΔC5 (0.032) | 5 | 3 (<0.01) | 1–47 | 0.043 |
| ΔC10 (0.032) | 5 | 3 (<0.01) | 2–32 | 0.043 |
| ΔN5 (0.25) | 5 | 22 (0.05) | 14–159 | 0.043 |
| ΔN10 (0.064) | 5 | 4 (0.01) | 1–116 | 0.043 |
| ΔN20 (0.032) | 6 | 86 (0.21) | 61–201 | 0.046 |
| Interaction with GyrB | ||||
| Wild-type QnrB1 | 14 | 3,587 (1) | 1,582–10,826 | |
| ΔC5 | 5 | 3,482 (0.97) | 2,930–7,482 | 0.893 |
| ΔC10 | 5 | 2,689 (0.75) | 1,610–4,333 | 0.043 |
| ΔN5 | 10 | 1,451 (0.40) | 1,058–2,839 | 0.241 |
| ΔN10 | 5 | 1,958 (0.55) | 1,526–5,684 | 0.893 |
| ΔN20 | 5 | 4,998 (1.39) | 2,180–8,920 | 0.345 |
| Interaction with GyrBA | ||||
| Wild type | 9 | 3,777 (1) | 2,368–5,436 | |
| ΔC5 | 5 | 831 (0.22) | 788–2,843 | 0.043 |
| ΔC10 | 5 | 485 (0.13) | 320–1,441 | 0.043 |
| ΔN5 | 5 | 444 (0.12) | 149–1,352 | 0.043 |
| ΔN10 | 5 | 239 (0.06) | 20–1,598 | 0.043 |
| ΔN20 | 9 | 2,547 (0.67) | 584–3,340 | 0.051 |
The interaction of DNA gyrases with wild-type QnrB1 and the QnrB1 mutants with C- or N-terminal deletions is shown. The ciprofloxacin MICs of E. coli M15(pREP4) carrying pQE-60-QnrB1 derivatives in the presence of 100 μM IPTG in a previous study (16) are shown in the parentheses.
Fold changes for the interaction of wild-type QnrB1 in this table and subsequent tables differ slightly from the values in Table 1 since they include data from additional repetitions.
The ratio represents the median fold change in the mutant compared to that in the wild type.
The interquartile range is the 25th percentile to the 75th percentile.
The P value compares the value for the mutant to the value for the wild type. A boldface P value indicates a statistically significant decrease in interaction.
The QnrB1 β-helix structure is interrupted by two external loops, loops A and B, that extend from the helix (15). Deletion of these loops also results in reduced resistance conferred by QnrB1, with the deletion of larger loop B causing the greater reduction in resistance (15). Deletion of loop B and both loops, but not deletion of loop A, significantly reduced the interaction signal with GyrA by three- to fourfold (Table 3). For interaction with GyrB, the QnrB1 loop A deletion had a significant but small reduction (30%), and the loop B and dual-loop deletions were associated with an increased interaction. Surprisingly, the loop A deletion had a substantial reduction (17-fold) in interaction with GyrBA, with little effect of the loop B and dual-loop deletions. Thus, reductions in the overall interaction signal of QnrB1 loop mutants with GyrA exhibited the best correlation with reductions in resistance phenotype seen in these mutants.
TABLE 3.
Interaction of DNA gyrases with QnrB1 and QnrB1 mutants having loop deletionsa
| QnrB1 mutant | No. of expts | Fold change |
P valuec | |
|---|---|---|---|---|
| Median (ratiob) | Interquartile range | |||
| Interaction with GyrA | ||||
| Wild type | 14 | 404 (1) | 317–602 | |
| ΔloopA | 5 | 372 (0.92) | 161–844 | 0.893 |
| ΔloopB | 8 | 175 (0.43) | 128–294 | 0.012 |
| ΔloopA+B | 8 | 104 (0.26) | 56–230 | 0.012 |
| Interaction with GyrB | ||||
| Wild type | 14 | 3,587 (1) | 1,582–10,826 | |
| ΔloopA | 6 | 2,621 (0.73) | 1,523–6,572 | 0.028 |
| ΔloopB | 5 | 16,517 (4.60) | 6,435–18,909 | 0.080 |
| ΔloopA+B | 5 | 14,242 (3.97) | 6,797–25,222 | 0.080 |
| Interaction with GyrBA | ||||
| Wild type | 9 | 3,777 (1) | 2,368–5,436 | |
| ΔloopA | 5 | 219 (0.06) | 63–455 | 0.043 |
| ΔloopB | 9 | 3,285 (0.87) | 2,419–6,625 | 0.441 |
| ΔloopA+B | 9 | 4,926 (1.30) | 3,962–6,450 | 0.110 |
Several single-amino-acid changes in QnrB1 loops have also been associated with a reduced resistance phenotype (16). We chose five mutations in loop B (I105A, Δ105I, T106A, Δ106T, and F111A) and one in loop A (Q51A) that varied in their resistance phenotype to test for interaction with gyrase. The largest reductions in mutant interaction signal, although modest (two- to threefold), were seen with GyrA, with lesser or no effects seen with GyrB and GyrBA overall (Table 4). No significant reductions were seen with the Q51A small-loop mutant, which had a wild-type resistance phenotype. Of the loop B mutants, the I105A mutant, which had a four- to eightfold reduction in resistance, and the Δ105I mutant were particularly notable, exhibiting a reduced interaction signal with both GyrA and GyrBA, although the difference reached statistical significance only for the Δ105I mutant. The T106A mutant, which exhibited wild-type resistance, had no reduction in interaction signal with GyrA, GyrB, or GyrBA, whereas the Δ106T mutant, which exhibited an 8- to 32-fold reduction in resistance, had a 2.5-fold reduction in interaction signal with GyrA but no reduction with GyrB or GyrBA. In contrast, the F111A mutant, which like the Δ106T mutant had an 8- to 32-fold reduction in resistance, had a similar or increased interaction signal relative to the wild type. Thus, correlations of overall interaction signals of single-amino-acid loop mutants with their resistance phenotypes vary and suggest that key structural determinants of QnrB1 affecting resistance are not mediated by their determining overall physical interaction between QnrB1 and gyrase.
TABLE 4.
Interaction of DNA gyrases with QnrB1 and QnrB1 mutants having alanine substitutions or deletions in loop A or B amino acidsa
| QnrB1 mutant (ciprofloxacin MICb [μg/ml]) | No. of expts | Fold change |
P valued | |
|---|---|---|---|---|
| Median (ratioc) | Interquartile range | |||
| Interaction with GyrA | ||||
| Wild type (0.032) | 14 | 404 (1) | 317–602 | |
| Q51A (0.032) | 10 | 218 (0.54) | 63–979 | 0.799 |
| I105A (0.008) | 5 | 125 (0.32) | 51–430 | 0.345 |
| Δ105I (NA) | 5 | 197 (0.49) | 135–232 | 0.043 |
| T106A (0.064) | 5 | 465 (1.15) | 250–661 | 0.893 |
| Δ106T (0.001) | 9 | 156 (0.39) | 72–731 | 0.374 |
| F111A (0.004) | 6 | 1116 (2.76) | 240–6,599 | 0.173 |
| Interaction with GyrB | ||||
| Wild type | 14 | 3,587 (1) | 1,582–10,826 | |
| Q51A | 5 | 3,180 (0.89) | 2,247–6,223 | 0.500 |
| I105A | 8 | 2,457 (0.68) | 2,037–12,010 | 0.674 |
| Δ105I | 5 | 1,550 (0.43) | 1,364–9,722 | 0.686 |
| T106A | 5 | 7,029 (1.96) | 4,164–34,599 | 0.080 |
| Δ106T | 5 | 5,521 (1.54) | 2,550–9,529 | 0.138 |
| F111A | 6 | 3,229 (0.90) | 1,300–16,498 | 0.345 |
| Interaction with GyrBA | ||||
| Wild type | 9 | 3,777 (1) | 2,368–5,436 | |
| Q51A | 5 | 6,408 (1.70) | 4,961–8,371 | 0.043e |
| I105A | 9 | 2,377 (0.63) | 1,321–3,859 | 0.110 |
| Δ105I | 9 | 2,150 (0.57) | 1,492–3,548 | 0.038 |
| T106A | 9 | 4,494 (1.19) | 2,601–5,870 | 0.214 |
| Δ106T | 9 | 3,463 (0.92) | 2,657–5,524 | 0.953 |
| F111A | 5 | 4,180 (1.11) | 3,241–7,086 | 0.138 |
Ciprofloxacin MICs of E. coli BL21(DE3) carrying pET28a:QnrB1 derivatives in the presence of 100 μM IPTG from reference 16. NA, not available.
The ratio represents the median fold change in the mutant compared to that in the wild type.
A boldface P value indicates a statistically significant decrease in interaction.
The QnrB1:Q51A mutant showed a significantly higher fold change than the wild type.
DISCUSSION
Bacterial two-hybrid systems have been useful tools to evaluate protein-protein interactions within intact cells and to map the interaction domain of these proteins after site-directed mutagenesis (22). Using this system, we investigated the physical interactions between the quinolone resistance protein QnrB1 and its target DNA gyrase and the effects of quinolones and structural changes in QnrB1 on that interaction. This approach has the advantage of studying the protein-protein interactions in the cellular milieu in which they occur.
Purified QnrA has been shown to bind to the holoenzyme and its individual GyrA and GyrB subunits and to alter gyrase binding to DNA (20). Because of the structural similarity of QnrB and other pentapeptide repeat proteins, such as MfpA, with DNA, one model of the interaction of MfpA with GyrA has positioned MfpA in the GyrA DNA binding site (30). Direct data on the nature of interaction of the pentapeptide repeat proteins and gyrase, however, have been lacking. Notably, MfpA can inhibit gyrase function but is not able to protect gyrase from quinolone action in vitro (21), highlighting the need to explain how Qnr proteins can protect gyrase from quinolones at low concentrations without inhibiting its catalytic activity.
QnrB1 exhibited in vivo interaction signals with GyrA and GyrB, in keeping with data for the purified proteins, as well as with GyrBA, a catalytically active fusion protein capable of complementing thermosensitive gyrase mutants in vivo (Chen et al., unpublished). The GyrA interaction was notable for its complete disruption in the presence of subinhibitory concentrations of ciprofloxacin and nalidixic acid and its dependence on intact C-terminal and N-terminal domains and A and B loops of QnrB1, domains that are necessary for its resistance phenotype. GyrB interactions with QnrB1, in contrast, required substantially higher concentrations of quinolones for disruption and were not reduced by C- and N-terminal and loop deletions of QnrB1, suggesting that the interactions are less specific for the resistance phenotype. Notably, the interaction of GyrBA with QnrB1 was 7- to 11-fold higher than the interaction signal of GyrA alone but retained sensitivity to the presence of quinolones and partial dependence of binding on the C- and N-terminal domains. These findings suggest that domains of GyrB are important for stabilizing the QnrB1 interaction with the holoenzyme but that QnrB1 interaction with the domains of GyrA is most important for affecting quinolone interactions, either by blocking drug access or altering the conformation of the quinolone binding site in GyrA. Furthermore, the interaction of quinolones and QnrB1 with gyrase appears to be competitive. Such a model of competitive binding is supported by prior work (12) indicating the reciprocal relationship of quinolone concentration and Qnr protection of gyrase and specifically by our finding here that the interaction of QnrB1 with the quinolone-resistant S83L GyrA mutant, which demonstrates less quinolone binding (29), was substantially less affected by ciprofloxacin. Thus, GyrA domains determine the ability of quinolones to disrupt QnrB1 binding, a property that is inconsistent with a model in which QnrB acts simply by binding quinolones rather than by disrupting their interaction with gyrase.
The structural determinants of the ability of QnrB1 and other Qnr proteins to confer quinolone resistance have been studied previously (15, 16, 18, 31, 32). Deletion of portions of the C-terminal and N-terminal domains of QnrB1 results in four- to eightfold reductions in resistance (15). The C-terminal domains are also important for the formation of tail-to-tail Qnr dimers (17). C-terminal deletions have the greatest effect on QnrB1 binding to GyrA and GyrBA with little or no effect on binding to GyrB, which suggests that the C termini themselves, dimerization, or both are important for the overall interaction with GyrA and holoenzyme domains. A similar pattern was seen with N-terminal deletions, although the N20 deletion, in contrast to the N5 and N10 deletions, retained substantial interaction with GyrB and GyrBA, suggesting a mode of binding different from that of the shorter deletions.
The A and B external loops of QnrB and other Qnr proteins from Gram-negative bacteria are required for their ability to cause quinolone resistance (15, 31) and are absent on pentapeptide repeat proteins that do not confer quinolone resistance in vitro (17). The effects of loop deletions on QnrB1 binding to GyrA and GyrBA were, with the exception of the loop A deletion mutant interaction with GyrBA, substantially less than the effects of deletion of the C and N termini. Similarly, alanine substitutions or deletions of amino acids of a single loop had limited effects on the interaction signals. Taken together, these findings indicate that despite their importance in determining resistance, loops A and B are not strong determinants of the overall physical interaction of QnrB1 with gyrase. The findings would then further imply that the role of the loops may be instead as key determinants of the proper positioning of QnrB1 to interfere with the binding of quinolones to the gyrase-DNA complex without interfering with catalytic function. One model for such a mechanism could be envisioned to allow QnrB1 to bind specifically in the quinolone binding pocket formed in one particular enzyme conformation in such a way so as to block quinolone access but with sufficiently low affinity to be displaced upon further changes in conformation as the enzyme proceeded through its catalytic cycle.
There is a possibility that chromosome-encoded gyrase subunits could interact with plasmid-encoded gyrase subunits or QnrB1 in this study. However, the two-hybrid interaction signals are predominantly driven by the interaction of the additional domains that are fused to bait and prey plasmid-encoded proteins in order to generate transcription of the HIS3 gene in the BacterioMatch II two-hybrid system (26). These points are also borne out in Table 1 with the MIC values of ciprofloxacin and nalidixic acid for the various constructs. Cells with empty vectors have a MIC of ciprofloxacin of 0.5 μg/ml, a finding consistent with the gyrA96 resistance mutation in this background. In contrast, cells containing plasmids with both wild-type gyrA and gyrBA have MICs of 0.125 μg/ml, consistent with the known dominance of quinolone sensitivity in diploids, particularly those in which the copy number is greater for the quinolone sensitivity allele relative to the quinolone resistance alleles, and as expected, cells with a quinolone resistance gyrA allele have a MIC of 0.5 μg/ml. In addition, any such effect by chromosome-encoded gyrase subunits if present would likely be similar across QnrB mutants in this study.
Two-hybrid assays and other methods of studying protein-protein interactions are limited even with analyses of mutants, because they measure only overall protein-protein binding. Thus, more-definitive information on how Qnr proteins position themselves on gyrase to confer quinolone protection without enzymatic inhibition must await cross-linking or cocrystallization analyses.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported in part by a grant from the U.S. Public Health Service, National Institutes of Health, R01 AI057576 (to D.C.H. and G.A.J.).
We thank Ann Hochschild for kindly providing two-hybrid plasmids.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.00771-15.
REFERENCES
- 1.Hawkey PM. 2003. Mechanisms of quinolone action and microbial response. J Antimicrob Chemother 51(Suppl 1):S29–S35. [DOI] [PubMed] [Google Scholar]
- 2.Hiasa H, Shea ME. 2000. DNA gyrase-mediated wrapping of the DNA strand is required for the replication fork arrest by the DNA gyrase-quinolone-DNA ternary complex. J Biol Chem 275:34780–34786. doi: 10.1074/jbc.M001608200. [DOI] [PubMed] [Google Scholar]
- 3.Martinez-Martinez L, Pascual A, Jacoby GA. 1998. Quinolone resistance from a transferable plasmid. Lancet 351:797–799. doi: 10.1016/S0140-6736(97)07322-4. [DOI] [PubMed] [Google Scholar]
- 4.Cheung TK, Chu YW, Chu MY, Ma CH, Yung RW, Kam KM. 2005. Plasmid-mediated resistance to ciprofloxacin and cefotaxime in clinical isolates of Salmonella enterica serotype Enteritidis in Hong Kong. J Antimicrob Chemother 56:586–589. doi: 10.1093/jac/dki250. [DOI] [PubMed] [Google Scholar]
- 5.Rodriguez-Martinez JM, Pascual A, Garcia I, Martinez-Martinez L. 2003. Detection of the plasmid-mediated quinolone resistance determinant qnr among clinical isolates of Klebsiella pneumoniae producing AmpC-type β-lactamase. J Antimicrob Chemother 52:703–706. doi: 10.1093/jac/dkg388. [DOI] [PubMed] [Google Scholar]
- 6.Wang M, Tran JH, Jacoby GA, Zhang Y, Wang F, Hooper DC. 2003. Plasmid-mediated quinolone resistance in clinical isolates of Escherichia coli from Shanghai, China. Antimicrob Agents Chemother 47:2242–2248. doi: 10.1128/AAC.47.7.2242-2248.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang M, Sahm DF, Jacoby GA, Hooper DC. 2004. Emerging plasmid-mediated quinolone resistance associated with the qnr gene in Klebsiella pneumoniae clinical isolates in the United States. Antimicrob Agents Chemother 48:1295–1299. doi: 10.1128/AAC.48.4.1295-1299.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jacoby GA. 2005. Mechanisms of resistance to quinolones. Clin Infect Dis 41(Suppl 2):S120–S126. doi: 10.1086/428052. [DOI] [PubMed] [Google Scholar]
- 9.Robicsek A, Jacoby GA, Hooper DC. 2006. The worldwide emergence of plasmid-mediated quinolone resistance. Lancet Infect Dis 6:629–640. doi: 10.1016/S1473-3099(06)70599-0. [DOI] [PubMed] [Google Scholar]
- 10.Cavaco LM, Hasman H, Xia S, Aarestrup FM. 2009. qnrD, a novel gene conferring transferable quinolone resistance in Salmonella enterica serovar Kentucky and Bovismorbificans strains of human origin. Antimicrob Agents Chemother 53:603–608. doi: 10.1128/AAC.00997-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hata M, Suzuki M, Matsumoto M, Takahashi M, Sato K, Ibe S, Sakae K. 2005. Cloning of a novel gene for quinolone resistance from a transferable plasmid in Shigella flexneri 2b. Antimicrob Agents Chemother 49:801–803. doi: 10.1128/AAC.49.2.801-803.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jacoby GA, Walsh KE, Mills DM, Walker VJ, Oh H, Robicsek A, Hooper DC. 2006. qnrB, another plasmid-mediated gene for quinolone resistance. Antimicrob Agents Chemother 50:1178–1182. doi: 10.1128/AAC.50.4.1178-1182.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang M, Guo Q, Xu X, Wang X, Ye X, Wu S, Hooper DC, Wang M. 2009. New plasmid-mediated quinolone resistance gene, qnrC, found in a clinical isolate of Proteus mirabilis. Antimicrob Agents Chemother 53:1892–1897. doi: 10.1128/AAC.01400-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jacoby GA, Strahilevitz J, Hooper DC. 2014. Plasmid-mediated quinolone resistance. Microbiol Spectr 2(2):PLAS-0006-2013. doi: 10.1128/microbiolspec.PLAS-0006-2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vetting MW, Hegde SS, Wang M, Jacoby GA, Hooper DC, Blanchard JS. 2011. Structure of QnrB1, a plasmid-mediated fluoroquinolone resistance factor. J Biol Chem 286:25265–25273. doi: 10.1074/jbc.M111.226936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jacoby GA, Corcoran MA, Mills DM, Griffin CM, Hooper DC. 2013. Mutational analysis of quinolone resistance protein QnrB1. Antimicrob Agents Chemother 57:5733–5736. doi: 10.1128/AAC.01533-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vetting MW, Hegde SS, Fajardo JE, Fiser A, Roderick SL, Takiff HE, Blanchard JS. 2006. Pentapeptide repeat proteins. Biochemistry 45:1–10. doi: 10.1021/bi052130w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tavio MM, Jacoby GA, Hooper DC. 2014. QnrS1 structure-activity relationships. J Antimicrob Chemother 69:2102–2109. doi: 10.1093/jac/dku102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tran JH, Jacoby GA, Hooper DC. 2005. Interaction of the plasmid-encoded quinolone resistance protein QnrA with Escherichia coli topoisomerase IV. Antimicrob Agents Chemother 49:3050–3052. doi: 10.1128/AAC.49.7.3050-3052.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tran JH, Jacoby GA, Hooper DC. 2005. Interaction of the plasmid-encoded quinolone resistance protein Qnr with Escherichia coli DNA gyrase. Antimicrob Agents Chemother 49:118–125. doi: 10.1128/AAC.49.1.118-125.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mérens A, Matrat S, Aubry A, Lascols C, Jarlier V, Soussy CJ, Cavallo JD, Cambau E. 2009. The pentapeptide repeat proteins MfpAMt and QnrB4 exhibit opposite effects on DNA gyrase catalytic reactions and on the ternary gyrase-DNA-quinolone complex. J Bacteriol 191:1587–1594. doi: 10.1128/JB.01205-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Joung JK, Ramm EI, Pabo CO. 2000. A bacterial two-hybrid selection system for studying protein-DNA and protein-protein interactions. Proc Natl Acad Sci U S A 97:7382–7387. doi: 10.1073/pnas.110149297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Green MR, Sambrook J. 2012. Molecular cloning: a laboratory manual, 4th ed, vol 3 Cold Spring Harbor Laboratory, Cold Spring Harbor, NY. [Google Scholar]
- 24.Gubaev A, Klostermeir D. 2011. DNA-induced narrowing of the gyrase N-gate coordinates T-segment capture and strand passage. Proc Natl Acad Sci U S A 108:14085–14090. doi: 10.1073/pnas.1102100108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schoeffler AJ, May AP, Berger JM. 2010. A domain insertion in Escherichia coli GyrB adopts a novel fold that plays a critical role in gyrase function. Nucleic Acids Res 38:7830–7844. doi: 10.1093/nar/gkq665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Agilent Technology. 2008. BacterioMatch II two-hybrid system vector kit: instruction manual. Agilent Technology, Santa Clara, CA. [Google Scholar]
- 27.Tran JH, Jacoby GA. 2002. Mechanism of plasmid-mediated quinolone resistance. Proc Natl Acad Sci U S A 99:5638–5642. doi: 10.1073/pnas.082092899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Drlica K, Hooper DC. 2003. Mechanisms of quinolone action, p 19–40. In Hooper DC, Rubinstein E (ed), The quinolone antimicrobial agents, 3rd ed American Society for Microbiology, Washington, DC. [Google Scholar]
- 29.Willmott CJ, Maxwell A. 1993. A single point mutation in the DNA gyrase A protein greatly reduces binding of fluoroquinolones to the gyrase-DNA complex. Antimicrob Agents Chemother 37:126–127. doi: 10.1128/AAC.37.1.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hegde SS, Vetting MW, Roderick SL, Mitchenall LA, Maxwell A, Takiff HE, Blanchard JS. 2005. A fluoroquinolone resistance protein from Mycobacterium tuberculosis that mimics DNA. Science 308:1480–1483. doi: 10.1126/science.1110699. [DOI] [PubMed] [Google Scholar]
- 31.Xiong X, Bromley EH, Oelschlaeger P, Woolfson DN, Spencer J. 2011. Structural insights into quinolone antibiotic resistance mediated by pentapeptide repeat proteins: conserved surface loops direct the activity of a Qnr protein from a gram-negative bacterium. Nucleic Acids Res 39:3917–3927. doi: 10.1093/nar/gkq1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rodriguez-Martinez JM, Briales A, Velasco C, Conejo MC, Martinez-Martinez L, Pascual A. 2009. Mutational analysis of quinolone resistance in the plasmid-encoded pentapeptide repeat proteins QnrA, QnrB and QnrS. J Antimicrob Chemother 63:1128–1134. doi: 10.1093/jac/dkp111. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.