

Abstract
A population drug-disease model was developed to describe the time course of influenza virus with and without oseltamivir treatment and to investigate opportunities for antiviral combination therapy. Data included viral titers from 208 subjects, across 4 studies, receiving placebo and oseltamivir at 20 to 200 mg twice daily for 5 days. A 3-compartment mathematical model, comprising target cells infected at rate β, free virus produced at rate p and cleared at rate c, and infected cells cleared at rate δ, was implemented in NONMEM with an inhibitory Hill function on virus production (p), accounting for the oseltamivir effect. In congruence with clinical data, the model predicts that the standard 75-mg regimen initiated 2 days after infection decreased viral shedding duration by 1.5 days versus placebo; the 150-mg regimen decreased shedding by an additional average 0.25 day. The model also predicts that initiation of oseltamivir sooner postinfection, specifically at day 0.5 or 1, results in proportionally greater decreases in viral shedding duration of 5 and 3.5 days, respectively. Furthermore, the model suggests that combining oseltamivir (acting to subdue virus production rate) with an antiviral whose activity decreases viral infectivity (β) results in a moderate additive effect dependent on therapy initiation time. In contrast, the combination of oseltamivir with an antiviral whose activity increases viral clearance (c) shows significant additive effects independent of therapy initiation time. The utility of the model for investigating the pharmacodynamic effects of novel antivirals alone or in combination on emergent influenza virus strains warrants further investigation.
INTRODUCTION
Oseltamivir is an orally active antiviral which inhibits the neuraminidase enzyme necessary for the release of newly replicated influenza virus from infected cells. Oseltamivir has a wide spectrum of activity, acting against a range of influenza A and B subtypes. The Centers for Disease Control and Prevention reported that 99.6% of the 2009 H1N1 viral strains tested were susceptible to oseltamivir (see http://www.cdc.gov/H1N1flu/recommendations.htm). The standard regimen of 75 mg twice a day (b.i.d.) for 5 days is used for treatment of seasonal influenza as previous studies in adult subjects showed that this regimen results in plasma levels sufficient to inhibit neuraminidase enzyme activity from all the tested seasonal influenza virus strains (1, 2). The same treatment regimen was applied in the most recent H1N1 pandemic in 2009-2010 without a prospective dose optimization trial, due to the impracticality of performing such a study on an emergent viral strain during a pandemic. The World Health Organization has recommended use of higher doses (150-mg regimen) for treatment of highly virulent strains such as H5N1 (3). In addition, there were reported concerns regarding the ability of oseltamivir manufacturing capacity to fully meet global stockpiling demands if a rapid pandemic ensues (4, 5). This factor, in addition to opportunities for enhanced therapeutic efficacy, is prompting interest in the combined pharmacodynamic (PD) effects of neuraminidase inhibitors with other antivirals currently under development.
A mathematical model-based approach offers the advantage of leveraging prior knowledge of seasonal influenza viral dynamics by characterizing the time course of influenza progression in placebo-treated patients to then isolate the effect of the antiviral therapy on influenza progression in treated patients. Additionally, if relationships between in vitro viral characteristics and model-based viral dynamic parameters are identified, they could be used to scale the viral titer curve of an emerging virus whose properties are characterized in vitro and then used to predict the PD antiviral effect in vivo. A drug-influenza model can also be used in simulation mode to generate a hypothesis on the single and combined PD effects of oseltamivir with other experimental antivirals acting on other targets in the influenza virus life cycle. The influenza model by Baccam et al. (6) is the most parsimonious in vivo viral dynamics model reported and is based on the fundamental predator-prey concept: a pool of free virus infecting a susceptible pool of target respiratory epithelial cells. It is similar to the earlier models used to describe human immunodeficiency virus dynamics (7), but differs in that no turnover is assumed for the target cells because the duration of acute influenza virus infection (approximately 7 days) is much shorter than the life span of the target respiratory epithelial cells, an assumption which does not hold true for chronic infections such as AIDS or hepatitis C (7–9).
In experimental inoculation studies (10), in which patients are intranasally injected with influenza virus, the time of infection relative to treatment initiation is known, unlike in seasonal studies. Moreover, the data generated from such studies are optimal for drug-influenza modeling because such studies typically incorporate a wide range of doses and involve more frequent viral titer sampling, especially at early times postinfection, which allows fuller characterization of the viral titer curve. The current study aims to develop a mechanistic drug-disease model using the viral dynamics framework of Baccam et al. (6) with the purposes of (i) describing the time course of influenza progression under placebo and oseltamivir therapy, (ii) investigating the PD effects of dose and time on treatment postinfection, (iii) identifying covariates relating in vitro viral characteristics to influenza model parameters, and (iv) investigating additive PD effects on top of oseltamivir's by modulating other drug targets in the influenza viral life cycle.
MATERIALS AND METHODS
Data.
Data from a total of 208 subjects who participated in 4 clinical studies were used to develop the influenza and oseltamivir PD models. All viral shedding data were collected from nasal washings obtained by nasal swab with the concentration of virus measured in units of 50% tissue culture infective dose per milliliter (TCID50/ml) of nasal wash on MDCK cells. This concentration was assumed to be proportional to the concentration of free virus at the site of infection (biophase) at the time of nasal wash. A general method for the collection of nasal washings was common for all studies where a subject extended his or her neck approximately 30° from the horizontal while in a sitting position. Then 5 ml of lactated Ringer's solution or normal saline (0.9%) at room temperature was instilled into each nostril using a 10-ml syringe while the volunteer made a hard K sound to close off the back of the throat. After approximately 10 s, the volunteer bent their head forward and gently expelled the mucus and saline into a cup. A 4-ml sample of nasal wash was put into a collecting broth and kept on ice until transportation to the laboratory for viral culture. The generalizability of the assay allowed pooling of data for model building, and a description of viral titer data collected across all studies is shown in Table 1. Three influenza A virus experimental inoculation studies (PV15616, PV15615, and Baccam) included densely sampled viral titer data with at least one positive viral titer sample measured every day over a 1-week duration (at least 7 viral titers per subject). Study PV15616 was the only study that contributed oseltamivir treatment data considered appropriate for modeling. This is because a wide range of doses (20 to 200 mg) was used, and viral titers were densely sampled, allowing for better PD parameter estimation. In study PV15616, a phase II experimental influenza study, oseltamivir therapy was initiated 28 h after intranasal inoculation with human influenza A virus. In the phase III study (WV15670), subjects with naturally acquired influenza were enrolled within 48 h of first experiencing influenza symptoms. Viral titer data obtained from study WV15670 were sparsely measured (2 to 4 viral titers measured per subject). Inspection of individual plots of viral titer versus time showed no significant differences in shape between the inoculation studies and the phase III study WV15670 (see Fig. S1 in the supplemental material). All studies were performed in accordance with the Declaration of Helsinki. All subjects provided written, informed consent to participate in each clinical study, and the relevant study protocols were approved by the institutional review board at each study site.
TABLE 1.
Description of the influenza viral titer data used for model building
| Study reference | Study type | Intervention(s) used for modeling (no. of subjects) | Sampling scheme |
|---|---|---|---|
| Baccam | Inoculation of influenza A/Hong Kong/123/77 (H1N1) | Placebo (n = 6) | Nasal washings collected once daily for the first week of infection on days 1–8 |
| PV15616 | Inoculation of influenza A/Texas/36/91 (H1N1); phase II study | Placebo (n = 13); treatment (n = 56) with oseltamivir administered orally as multiple doses of 20, 100, or 200 mg b.i.d. or 200 mg q.d.a for 5 days | Nasal washings collected once prior to inoculation, then b.i.d. on days 1 and 2, and then once daily on days 3, 4, 5, 6, 7, and 8 |
| PV15615 | Inoculation of influenza A/Texas/36/91 (H1N1) | Placebo (n = 6) | Nasal washings collected once prior to inoculation and once daily for the first week of infection on days 1–8 |
| WV15670 | Naturally acquired influenza virus infection; phase III study | Placebo (n = 127) | Nasal washings collected once daily on days 2, 4, and 8 (some sites) and day 6 (all sites) |
q.d., once a day.
Influenza model and oseltamivir pharmacodynamics.
Drug-disease modeling was performed in NONMEM (version 7.2; ICON Development Solutions, Dublin, Ireland) using the ADVAN 9 integration subroutine for stiff ordinary differential equation systems (11). The NONMEM methodology was described elsewhere (12). The influenza model as described by Baccam et al. (6) was used to characterize the natural time course of influenza viral titers in the absence of drug (placebo model) by first fitting the model to data from placebo-treated patients (Table 1). The influenza model was described by the following differential equations:
| (1) |
| (2) |
| (3) |
where T is the number of uninfected target cells, I is the number of infected cells producing virus, and V is the pool of free virus (viral titer) expressed in TCID50/ml of nasal wash (6). The compartment V is initialized by the viral titer lower limit of quantification, i.e., it is assumed that infection of the respiratory tract is initiated by a viral titer equivalent to the lower limit of quantification of the influenza virus (V0), as determined by Baccam et al. (6). The lower limit of quantification of influenza viral titers was 100.25 TCID50/ml. The initial number of target cells (T0) is approximately 4 × 108 cells, which was estimated by Baccam et al. (6) from the area of epithelial cells lining the upper respiratory tract (160 cm2) (13) and the surface area per epithelial cell, 2 × 10−11 to 4 × 10−11 m2/cell (14). The model assumes that target cells become infected by the virus with a second-order rate constant β. Infected cells shed virus, increasing the viral titer at rate p per cell per day and die at rate δ per day. Free virus is cleared at rate c per day. Since the values of β and p are numerically small (on the order of 10−4), the exponent of base 10 for these two parameters was estimated in NONMEM to allow easier numerical estimation and to stabilize the model runs (for the NONMEM code, see the supplemental material). For example, p was modeled as 10θ, where θ is a fixed-effects parameter. As an extension of the model, a delay in the production of free virus was also tested by defining an eclipse compartment, which is a population of infected target cells not yet producing virus.
The final modeling step involved inclusion of drug treatment data from study PV15616 only (as this study was a dose-ranging study with well-controlled experimental conditions) and estimation of drug effect and viral kinetics parameters in the model. Since oseltamivir binds to the influenza virus surface neuraminidase, which is important for viral release from the host cell, the antiviral effect of oseltamivir was described using an inhibitory Hill function acting on the model parameter p, production of virus per infected cell, as described in equation 4:
| (4) |
where θ is a fixed-effects parameter describing the exponent of base 10 for the parameter p without treatment, the maximum effect (Emax) is related to the maximal inhibition of viral production per infected cell, ED50* is a fixed-effects parameter related to the ED50, and ED50 (the effective dose producing 50% of maximal inhibition of viral production per infected cell) is given by the equation ED50 = ED50*/([Emax/log10(2)] − 1). Dose here refers to the dose administration records in the NONMEM data set as per study PV15616 in Table 1.
Unexplained interindividual variability in a model parameter P was estimated using an exponential model with the random effect ηj, as follows: Pj = TVP exp(ηj). In this model, TVP is the population mean parameter P (typical value in the population), Pj is the individual post hoc estimate for the parameter P in the jth individual, and ηj is a normally distributed random variable with a mean of zero and variance ω2p. For calculation of the NONMEM extended least-squares objective function (12), viral titer data were log10 transformed; therefore, an additive error model (which corresponds to a proportional model in the untransformed domain) was used to describe the residual variability.
Model refinement was data driven and based on goodness-of-fit indicators, including the visual inspection of diagnostic scatter plots, the plausibility and precision of parameter estimates, the minimization of the objective function (MOF) value, and the number of estimated parameters. For hierarchical models, a decrease in the MOF of at least 10.8 upon addition of a parameter was considered statistically significant (P < 0.001, 1 degree of freedom) according to a chi-square distribution (NONMEM Users Guide, version 7.2). A visual predictive check (VPC) was used to diagnose the final model and qualify the model for simulation. The basic idea of a VPC is to simulate predictions from the final model while including all random effects and to compare a confidence range of predictions with observed data (15).
Relating model parameters to viral growth curve characteristics.
To prepare the cells for investigation of viral growth characteristics, five 96-well plates with MDCK-SIAT cells (MDCK cells engineered to have more 2,6-linked sialic acid for better growth of human viruses) (16) were incubated in a humidified incubator (37°C, 5% CO2) for 1 day until cells reached a confluent monolayer. To prepare the viruses, H3N2 A/Sydney-like influenza virus strains were isolated at baseline from nasal swabs (one swab per patient) taken from 26 placebo-treated patients in study WV15670. A 2.5-ml virus challenge stock of each virus was prepared by diluting samples in infection medium, so that all stocks were the same titer. An aliquot of each stock was plated onto five 96-well plates, which also included a positive (A/Panama/20/99 with known TCID50/ml) and negative (infection medium) control.
MDCK-SIAT cells in each 96-well plate were challenged by removing the culture medium and replacing it with 150 μl of either virus challenge stock or infection medium, before returning them to the incubator (the remaining 100 μl of virus challenge stock was stored at −80°C until titration). At each of the 5 time points postchallenge (8, 24, 32, 48, and 96 h), one 96-well plate was frozen at −80°C and stored until titration. Standard virus titration assays were performed using MDCK-SIAT cells and a starting dilution of 1:10 for postchallenge samples. Viral growth curves were generated by plotting virus titer (expressed as TCID50/ml) against time.
The following in vitro viral growth characteristics were measured: initial growth rate, calculated as (VT24 h − VT8 h)/16, where VT8 h and VT24 h are the viral titers measured at 8 h and 24 h, respectively; area under the viral growth curve (AUCVT) from 0 to 48 h and 0 to 96 h; and peak viral titers at 48 h and 96 h. The covariate relationships between model parameters describing the viral growth curve and the in vitro viral growth characteristics were explored; for each of the 26 patients, the unique set of post hoc model parameters (e.g., p) was plotted separately against their isolated in vitro viral growth characteristics.
Pharmacodynamic model simulations.
Modeling was performed using NONMEM; however, all model simulations were performed deterministically (i.e., without interindividual variability) using Berkeley Madonna software (version 8.3.18). Influenza and drug parameters of the final pharmacodynamic model (generated by NONMEM) were used to simulate the natural progression of the influenza viral load over time without treatment and following administration of oseltamivir twice daily for 5 days at doses of 20, 75, 100, 150, and 200 mg. These doses were investigated in phase II and III clinical trials (1, 2).To explore the maximal drug effect, a hypothetical full-inhibition scenario in which the oseltamivir dose was assumed to be infinite was also simulated. In a separate simulation exercise, the influenza viral load was simulated after administration of standard oseltamivir treatment therapy alone by varying the time of treatment start relative to infection (i.e., at 0.5, 1, 2, and 3 days after infection). The joint PD effects of targeting different parts of the viral life cycle, as a proxy for combining the standard oseltamivir treatment with an additional hypothetical antiviral agent over the course of infection, were simulated empirically by considering the effects of (i) decreasing the infection rate, β, from baseline, (ii) further decreasing the production rate, p, from baseline, (iii) increasing the viral clearance, c, from baseline, and (iv) increasing the death rate of infected cells, δ, from baseline. All parameters were changed by a factor of 10 from baseline.
RESULTS
The placebo data consisted of 573 positive viral titer time points in total, while the oseltamivir treatment data consisted of 298 positive viral titer time points. The final influenza model is shown in Fig. 1; the data set, including the NONMEM output file and simulation code, is provided as supplemental material. Addition of an eclipse compartment to account for delayed viral production did not improve the fit of the model or result in a significant decrease in the MOF and, as such, was excluded (results not shown). Final model goodness-of-fit plots showed no bias in the concordance of the observed viral titers versus the population-predicted and individual-predicted viral titers as shown by random scatter around the line of identity; the plots of weighted residuals versus time and population predictions showed random scatter of residuals and no systematic bias or obvious outliers (data not shown). Inspection of the correlation matrix of estimates showed that the model was devoid of high correlations (|correlation coefficient| of >0.8) with the exception of the negative correlation between the parameters β and p. The VPC of the final model (Fig. 2) shows the ability of the model to capture the central tendency of the influenza viral titer time course for the placebo and oseltamivir dose groups (20, 100, and 200 mg) as shown by the random scatter of observed data around the simulated median (solid line). The interindividual variability is also well described as shown by the ability of the simulated 5th and 95th percentiles (dashed lines) to capture the spread of the observed viral titer data.
FIG 1.

A simple mechanistic model describing influenza virus progression. A pool of target respiratory epithelial cells (T) are infected by a pool of free virus (V) described by a second-order rate constant β. Infected cells (I) shed virus at a production rate p. Free virus is cleared by a rate c, and infected cells are cleared by rate δ. Oseltamivir acts by inhibiting viral production from infected cells. INH, inhibition.
FIG 2.
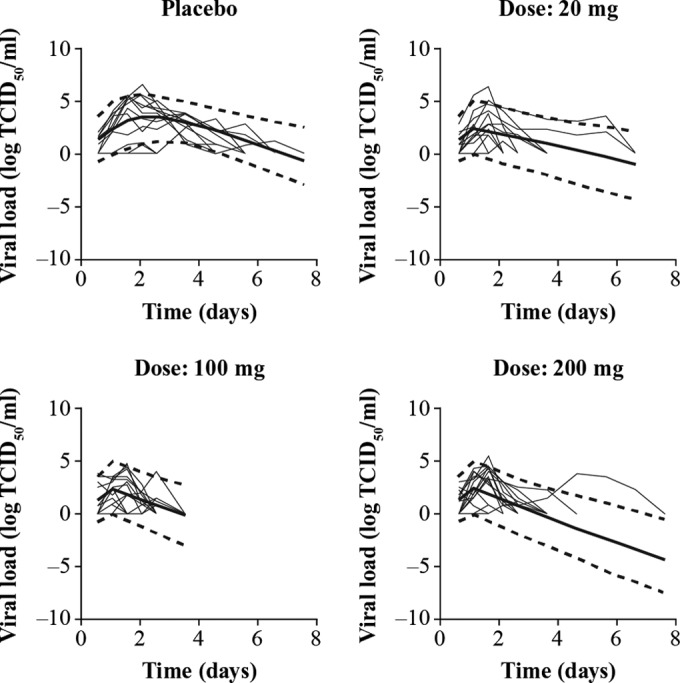
VPCs showing the central tendency and variability in influenza viral progression of placebo and oseltamivir treatment cohorts (study PV15616). Viral load is measured from nasopharyngeal swabs as log10 TCID50/ml where TCID is the tissue culture infective dose. The thin solid lines indicate observed individual viral load data, while the thick solid line indicates the model simulated median and dotted lines the simulated 5th and 95th percentiles.
Table 2 shows the influenza, drug, and random-effect parameter estimates of the final PD model. All influenza parameters were estimated with adequate precision as shown by the relatively low percent standard error of the mean (SEM), while the percent SEM of the ED50 (oseltamivir dose producing 50% of maximal p inhibition) was relatively high (69%). The interindividual variability (IIV) of viral production was high (percent IIV of 65%) as well as the oseltamivir pharmacodynamics (percent IIV on Emax of 82%). The proportional residual variability, a composite measure of intrasubject variability, model misspecification, and assay error in measuring viral titers, was relatively low (coefficient of variance [CV] percent of σerror of 14%).
TABLE 2.
Final parameter estimates of the influenza-oseltamivir PD model
| Parameter | Definition | Unit | Estimate | % SEMa |
|---|---|---|---|---|
| Influenza parameters | ||||
| β | Target cell infection rate | (TCID50/ml)−1 · day−1 | 7.41 × 10−4 | 10 |
| p | Viral production rate | (TCID50/ml) · day−1 | 2.0 × 10−4 | 9 |
| c | Viral clearance rate | day−1 | 3.33 | 22 |
| δ | Infected cell clearance rate | day−1 | 2.49 | 28 |
| Drug parameters | ||||
| Emax | Maximum drug effect on p inhibition | 2.35 | 25 | |
| ED50b | Drug sensitivity on p inhibition | mg | 3.2 | 69 |
| Random-effect parameters | ||||
| IIVp | Interindividual variability of p | CVc % | 65 | 29 |
| IIVEmax | Interindividual variability of Emax | CV % | 82 | 72 |
| σerror | Proportional error term | CV % | 14 | 9 |
% SEM is percent standard error of the mean calculated as SE/mean × 100.
ED50 on p inhibition is derived using the equation ED50*/([Emax/log10(2)] − 1), where ED50* is a fixed-effects parameter related to ED50 as shown in equation 4.
CV, coefficient of variation.
From the influenza virus growth curves from SIAT cells, the initial growth rate (IGR) was the only viral characteristic which showed associations with the influenza model parameters β (negative correlation, P = 0.087, n = 26) and p (positive correlation, P = 0.084, n = 26); however, neither relationship achieved statistical significance. Figure 3 shows the linear relationship between the log10 of the p parameter and IGR.
FIG 3.

Exploring the relationship between in vivo viral production (p) and the in vitro initial (8 to 24 h) viral growth rate (IGR) in MDCK-SIAT cells from 26 placebo-treated patients from study WV15670.
Figure 4 shows p (Fig. 4A) and log10 of p (Fig. 4B) as a function of the oseltamivir dose. Inspection of the dose-response curves shows an ED50 of approximately 5 mg b.i.d. (calculated from the model parameters in Table 2 as 3.2 mg) with the standard clinical 75-mg b.i.d. dose near the plateau of the curve. Figure 4 also shows how the dose response translates to effect on the viral titer curve with simulations of the time course of influenza viral titer without treatment (placebo) and with oseltamivir phase II study doses of 20, 100, and 200 mg (Fig. 4C) and phase III doses of 75 mg and 150 mg (Fig. 4D) administered b.i.d. for 5 days. Treatment is assumed to start 2 days after infection. As shown in Fig. 4, with no treatment, influenza viral titers decreased to below the limit of quantification at approximately 6.5 days after infection. At a 20-mg dose, the duration of viral shedding was approximately 5.6 days, whereas with the oseltamivir standard treatment at the 75-mg dose the duration of viral shedding was approximately 5 days, and with the 150-mg dose the duration was approximately 4.75 days.
FIG 4.

Oseltamivir dose-response relationships. Dose-response curves of oseltamivir action on the production of virus (p) (A) and the log10 of p (B). (C) Influenza viral titer curves simulated under no treatment, at phase II study doses and full inhibition (oseltamivir dose set to infinity). (D) Influenza viral titer curves under no treatment, at phase III study doses and full inhibition. Oseltamivir therapy was initiated 2 days after infection and administered b.i.d. for 5 days. OP, oseltamivir phosphate.
Figure 5 shows that the earlier oseltamivir therapy is initiated postinfection, the shorter the duration of viral shedding (solid line simulations). With the reference time point of 6.5 days as the mean duration of influenza virus infection without treatment (Fig. 4), initiation of oseltamivir therapy 0.5 day postinfection results in an approximate 5-day decrease in the duration of viral shedding. Initiation of treatment 1, 2, or 3 days after infection results in mean decreases in the infection duration of 3.5, 1.5, or 1 days, respectively.
FIG 5.

Simulations predicting the combined in vivo pharmacologic effects of 75-mg b.i.d. oseltamivir standard therapy on influenza viral dynamics, decreasing viral production (p), decreasing infection rate (β), increasing viral clearance rate (c), and increasing clearance rate (δ) of infected cells. Treatment is started at 0.5 (red line), 1 (green line), 2 (blue line), and 3 (black line) days after infection. Solid lines depict the single pharmacologic effect of the standard oseltamivir 75-mg b.i.d. regimen administered over 5 days. Dashed lines show the combined pharmacologic effects. All secondary effects were changed 10-fold from their baseline values in Table 2.
The effect of targeting other phases of the influenza virus life cycle (Fig. 1) in combination with targeting virus production through oseltamivir therapy was also investigated in Fig. 5. When the infectivity or virus infection rate, β, is decreased to 1/10 its value in Table 2, a moderate additive effect is observed that is dependent on the time elapsed between infection and the initiation of therapy. The model suggests that combining oseltamivir with an antiviral agent targeting β can reduce the duration of viral shedding by 0.5 days, but only if this dual-combination therapy is initiated within 1 day of infection, whereas this therapy has little or no additive effect if it is initiated 2 days postinfection or later. The model predicts that the same additive effect, which is dependent on the time elapsed since infection and initiation of therapy, would also be observed for combination therapy with an antiviral that further decreases production of virus or an antiviral that potentiates clearance δ of infected cells. On the other hand, when the virus clearance rate, c, is increased to 10 times its value in Table 2, a more pronounced additive effect (approximate 2-day decrease in the duration of viral shedding) is observed, irrespective of the time postinfection at which the therapy is initiated.
DISCUSSION
This study is the first to use a population approach to implement a mathematical influenza model in NONMEM, which incorporates antiviral therapy using both placebo and drug treatment data. Oseltamivir inhibits the action of neuraminidase, an enzyme which cleaves sialic acid residues, enabling the release of newly produced virus progeny from the surface of infected epithelial cells of the human respiratory tract. In the mathematical model employed herein, the pharmacologic action of oseltamivir is captured as a reduction in the production rate of virus (p) by influenza virus-infected cells. Our model, as well as our data, shows that oseltamivir inhibits viral shedding from productive cells manifested as a decrease in the duration of viral shedding in oseltamivir- versus placebo-treated patients (Fig. 2). This is also shown in the inhibition of virus production versus the oseltamivir dose (Fig. 4). The dose-response relationship shows that the clinical dose of 75 mg lies near the plateau of the curve. This clinical dose produced a decrease in the duration of viral shedding of 1.5 days when treatment was started 2 days after infection, consistent with phase III results, which showed a similar decrease in viral shedding on average and employed an inclusion criterion of reporting influenza symptoms within 48 h (1, 2). The 150-mg b.i.d. dose showed an additional decrease of only 0.25 days relative to the standard therapy, whereas the phase III study results (which employed very sparse viral titer measurements) showed no significant difference on the duration of viral shedding or the symptom scores between the 75-mg and 150-mg b.i.d. doses.
Most of the viral titer data from the dose-ranging phase II study PV15616 were measured experimentally near the plateau and not in the lower range of the log-linear part of the dose response (Fig. 4A). As such, the precision of the ED50 estimate, as measured by the percent SEM (Table 2), was lower than optimal. Regardless, the analysis suggests that the exact ED50 for oseltamivir is low (<20 mg) and confirms that the selection of doses during the phase II clinical trials was intended to well characterize the ED90 (and higher) of the dose response, which is the clinically more relevant endpoint for an antiviral drug. As oseltamivir concentrations were measured sparsely in the dose-ranging study PV15616 that was used to build the drug effect part of the model, the current analysis used dose and did not incorporate oseltamivir pharmacokinetics (PK), which is a limitation of the study; however, some of the PK variability is inherently captured in the PD parameters estimated (Table 2). A semiparametric analysis recently published (17) showed a PK-PD relationship between the oseltamivir area under the curve and both the duration of viral shedding and the time to resolution of composite symptom scores, suggesting increases in the effects on both endpoints (0.5-day faster time to event) with higher exposures than that produced by the standard 75-mg dose. This analysis also showed that both the virologic (viral shedding) and clinical (symptoms) endpoints were synchronized by exposure. Further efforts are needed to better understand the relationship between virologic and symptom endpoints as the latter is a subjective measure and not always easily determined, e.g., in pediatric studies. While the analysis by Rayner et al. (17) and the current parametric analysis suggest a modest increase in the PD effect by increasing the dose/exposure beyond standard exposures (75-mg b.i.d. dose), these observations are made under ideal conditions where treatment initiation time relative to infection is controlled by virtue of the experimental inoculation study design; however, this factor cannot be controlled in phase III studies. Further exposure-response analysis using phase III data is warranted (18).
One of the advantages of the experimental inoculation design in controlling treatment initiation time relative to infection is that it allows for investigation of the impact of treatment initiation time on viral dynamics in the model simulation mode. It is recommended that oseltamivir therapy be started prior to the time of the peak viral titer for the maximal benefit. The peak of the viral titer occurs at day 2 to 3 (Fig. 4). The model served as a platform to investigate the impact of varying the treatment initiation time relative to infection (19). Our model clearly shows that as oseltamivir therapy is started earlier, the impact on viral dynamics is increased, with cessation of viral shedding occurring earlier. The effect of the treatment initiation time relative to the infection (time to treatment) has never been quantified in a controlled manner in a seasonal influenza study since it is difficult to stratify prospectively on this endpoint. The model predicts that starting therapy 0.5 or 1 day after infection would decrease viral shedding durations by factors of 3 and 2, respectively, compared with starting therapy 2 days after infection. Several studies have reported decreases in the viral shedding durations by oseltamivir ranging from 1 to 4 days (1, 2, 10, 19, 20). This wide range of reported drug effects is likely due to different study inclusion criteria and enrollment of various proportions of subjects with different times since infection and first reporting symptoms. For high-risk patients such as infants, clinicians are advised to start oseltamivir therapy immediately upon suspicion of influenza, even before laboratory confirmation (see http://www.cdc.gov/H1N1flu/recommendations.htm). The pharmacologic findings in our study suggest that starting therapy earlier has a greater impact on oseltamivir PD than increasing the dose beyond the standard 75-mg dose. The findings reinforce the need for rapid point-of-care diagnostics and mobilization of drug stockpiles quickly during pandemics. It is important to note that these results are based on otherwise healthy adults with influenza virus infection and may not extrapolate to all treatment populations, including, for example, hospitalized patients with severe influenza or immunocompromised patients, who may exhibit prolonged viral shedding and hence the potential for a larger therapeutic window (21, 22).
Various parts of the influenza virus replication life cycle were modulated in simulation mode as a proxy to empirically investigate the joint PD effects of oseltamivir in combination with hypothetical antivirals and identify attractive targets for adjunctive therapy. The model assumes that the system parameters (Table 2) represent independent processes, and, as such, no interaction is assumed in the combined simulations in Fig. 5. The parameter β, which describes the rate of infection, was decreased by a factor of 10 from its baseline estimate as a proxy to investigate the effect of combining an M2 inhibitor (such as amantadine or rimantadine) or of a hypothetical protease inhibitor of viral RNA replication with oseltamivir therapy at the clinical dose of 75 mg b.i.d. The simulation suggests that an additional decrease of approximately 0.5 days in viral shedding is expected if this dual therapy is initiated within a day of infection, while no significant additional effect is apparent if treatment is started thereafter (days 2 and 3). Similar conclusions can be drawn for oseltamivir combinations with antivirals that act on viral production (e.g., other neuraminidase inhibitors) or antivirals that potentiate clearance of infected cells (parameter δ) through immunomodulatory effects. If, however, the parameter c, which describes clearance of free virus, is increased, a significant additive impact on influenza viral load is observed regardless of when therapy is initiated. In theory, a drug such as the hemagglutinin A monoclonal antibody (23) may indirectly increase the clearance of free virus by inhibiting viral entry into the host cell and hence increase the extracellular exposure of free virus to antibodies, which would bind and remove free virus. However, evidence suggests that the humoral immune response to influenza starts at day 3 postinfection and peaks at day 10 (24).
An exploratory sensitivity analysis in Berkeley Madonna showed that modulation of β had an effect on the ascending slope of the viral titer curve, while that of p affected the peak of the curve. The system parameters c and δ affected the terminal slope (8). This information, coupled with the need to utilize the model for investigating oseltamivir pharmacodynamics on emerging influenza virus strains, prompted the need to study relationships between influenza model parameters and viral growth characteristics in vitro. Identifying a relationship particularly on the production of virus, p, may allow an extrapolation of the viral titer curve of an emerging influenza virus strain and pharmacodynamics by exploring the effect of oseltamivir on the shape of the curve. It is acknowledged that in vitro viral curves of strains of different pathophysiology may look rather similar yet confer different morbidity/mortality profiles in vivo. Nonetheless, an exploratory covariate analysis was conducted and showed a moderate positive relationship between IGR and p, the viral production rate constant. The results (Fig. 3) suggest that a virus with a higher initial growth rate may require increased inhibition of virus production by antivirals. While these results are interesting, the sample size (n = 26) was small, and these relationships did not reach statistical significance. These results are therefore exploratory and require further investigation as to their merit.
While the model adequately describes the time course of influenza viral dynamics, it assumes that target cell limitation (i.e., a finite pool of target cells that can be infected) is the key in stopping influenza virus infection rather than the effects of the host immune response. Although the model includes the parameters c, which represents clearance of free virus by antibodies, and δ, removal of infected cells, which occurs by T lymphocytes, the model's main limitation is that it does not explicitly capture the features of the host immune response. The work by Canini and Carrat (25) incorporates certain aspects of the human innate and acquired immune response, including cytokines and natural killer cells within the viral dynamic framework presented, while also linking clinical symptom score data. The model does not include drug effect and describes the host immune effects using hypothetical compartments and no measured data (25), leading to inflated estimates of the interindividual variability of immune system parameters. This results in a wide prediction interval and confers an artificial flexibility to the model. In subsequent work, Canini et al. did attempt to incorporate the oseltamivir drug effect but did so only in simulation mode without fitting their model to treatment data as performed in the current analysis (18). It is clear that further studies should be planned to measure the immune response components longitudinally during the course of a clinical study. However, it is presently unclear which immune effectors correlate most with influenza severity and hence would be ideal to measure (26). Moreover, the optimal frequency and duration of sampling are also unclear since cell-mediated and humoral immune responses such as T lymphocytes and antibodies peak after viral infection ends, i.e., beyond 7 days postinfection.
In conclusion, a parsimonious PD model describing the effect of oseltamivir on influenza viral progression has been presented. The time of initiation of oseltamivir treatment after infection had a larger impact, relative to dose, on the magnitude of oseltamivir PD effect with respect to decreasing the duration of viral shedding. Drugs that directly or indirectly increase viral clearance may show a significant combined antiviral effect with oseltamivir, regardless of the time of treatment initiation, while drugs that affect the rate of infection, viral production, and clearance of infected cells would have modest to moderate additive effects, the magnitude of which is dependent on the time of treatment initiation. Future directions for model enhancement include incorporating drug pharmacokinetics, clinical symptom score and host immune response data, and strengthening in vitro (virus)-in vivo (viral load) relationships.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by funding from F. Hoffmann-La Roche Ltd. Support for third-party writing assistance for the manuscript, furnished by Lucy Carrier at Gardiner-Caldwell Communications, United Kingdom, was provided by F. Hoffmann-La Roche Ltd.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.00069-15.
REFERENCES
- 1.Nicholson KG, Aoki FY, Osterhaus AD, Trottier S, Carewicz O, Mercier CH, Rode A, Kinnersley N, Ward P. 2000. Efficacy and safety of oseltamivir in treatment of acute influenza: a randomised controlled trial. Neuraminidase Inhibitor Flu Treatment Investigator Group. Lancet 355:1845–1850. doi: 10.1016/S0140-6736(00)02288-1. [DOI] [PubMed] [Google Scholar]
- 2.Treanor JJ, Hayden FG, Vrooman PS, Barbarash R, Bettis R, Riff D, Singh S, Kinnersley N, Ward P, Mills RG. 2000. Efficacy and safety of the oral neuraminidase inhibitor oseltamivir in treating acute influenza: a randomized controlled trial. US Oral Neuraminidase Study Group. JAMA 283:1016–1024. doi: 10.1001/jama.283.8.1016. [DOI] [PubMed] [Google Scholar]
- 3.Writing Committee of the Second World Health Organization Consultation on Clinical Aspects of Human Infection with Avian Influenza A (H5N1) Virus, Abdel-Ghafar AN, Chotpitayasunondh T, Gao Z, Hayden FG, Nguyen DH, de Jong MD, Naghdaliyev A, Peiris JS, Shindo N, Soerso S, Uyeki TM. 2008. Update on avian influenza A (H5N1) virus infection in humans. N Engl J Med 358:261–273. doi: 10.1056/NEJMra0707279. [DOI] [PubMed] [Google Scholar]
- 4.Butler D. 2005. Wartime tactic doubles power of scare bird-flu drug. Nature 438:6. doi: 10.1038/438006a. [DOI] [PubMed] [Google Scholar]
- 5.Howton JC. 2006. Probenecid with oseltamivir for human influenza A (H5N1) virus infection? N Engl J Med 354:879–880. doi: 10.1056/NEJMc052422. [DOI] [PubMed] [Google Scholar]
- 6.Baccam P, Beauchemin CA, Macken CA, Hayden FG, Perelson AS. 2006. Kinetics of influenza A virus infection in humans. J Virol 80:7590–7599. doi: 10.1128/JVI.01623-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Beauchemin CA, Handel A. 2011. A review of mathematical models of influenza A infections within a host or cell culture: lessons learned and challenges ahead. BMC Public Health 11(Suppl 1):S7. doi: 10.1186/1471-2458-11-S1-S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Perelson AS. 2002. Modelling viral and immune system dynamics. Nat Rev Immunol 2:28–36. doi: 10.1038/nri700. [DOI] [PubMed] [Google Scholar]
- 9.Snoeck E, Chanu P, Lavielle M, Jacqmin P, Jonsson EN, Jorga K, Goggin T, Grippo J, Jumbe NL, Frey N. 2010. A comprehensive hepatitis C viral kinetic model explaining cure. Clin Pharmacol Ther 87:706–713. doi: 10.1038/clpt.2010.35. [DOI] [PubMed] [Google Scholar]
- 10.Hayden FG, Treanor JJ, Fritz RS, Lobo M, Betts RF, Miller M, Kinnersley N, Mills RG, Ward P, Straus SE. 1999. Use of the oral neuraminidase inhibitor oseltamivir in experimental human influenza: randomized controlled trials for prevention and treatment. JAMA 282:1240–1246. doi: 10.1001/jama.282.13.1240. [DOI] [PubMed] [Google Scholar]
- 11.Gibiansky L, Gibiansky E, Bauer R. 2012. Comparison of Nonmem 7.2 estimation methods and parallel processing efficiency on a target-mediated drug disposition model. J Pharmacokinet Pharmacodyn 39:17–35. doi: 10.1007/s10928-011-9228-y. [DOI] [PubMed] [Google Scholar]
- 12.Sheiner LB, Beal SL. 1980. Evaluation of methods for estimating population pharmacokinetics parameters. I. Michaelis-Menten model: routine clinical pharmacokinetic data. J Pharmacokinet Biopharm 8:553–571. [DOI] [PubMed] [Google Scholar]
- 13.Ménache MG, Hanna LM, Gross EA, Lou SR, Zinreich SJ, Leopold DA, Jarabek AM, Miller FJ. 1997. Upper respiratory tract surface areas and volumes of laboratory animals and humans: considerations for dosimetry models. J Toxicol Environ Health 50:475–506. doi: 10.1080/009841097160366. [DOI] [PubMed] [Google Scholar]
- 14.Fedoseev GB, Geharev SS. 1989. Basic defence mechanisms of bronchio-lung system, p 112–144. In Outov NV. (ed), General pulmonology, vol 1 Medicina, Moscow, Russia. [Google Scholar]
- 15.Karlsson MO, Holford N. 2008. A tutorial on visual predictive checks, abstr 1434 Abstr 17th Annu Meet Population Approach Group, Marseille, France. [Google Scholar]
- 16.Matrosovich M, Matrosovich T, Carr J, Roberts NA, Klenk HD. 2003. Overexpression of the alpha-2,6-sialyltransferase in MDCK cells increases influenza virus sensitivity to neuraminidase inhibitors. J Virol 77:8418–8425. doi: 10.1128/JVI.77.15.8418-8425.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rayner CR, Bulik CC, Kamal MA, Reynolds DK, Toovey S, Hammel JP, Smith PF, Bhavnani SM, Van Wart SA, Ambrose PG, Forrest A. 2013. Pharmacokinetic-pharmacodynamic determinants of oseltamivir efficacy using data from phase 2 inoculation studies. Antimicrob Agents Chemother 57:3478–3487. doi: 10.1128/AAC.02440-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Canini L, Conway JM, Perelson AS, Carrat F. 2014. Impact of different oseltamivir regimens on treating influenza A virus infection and resistance emergence: insights from a modelling study. PLoS Comput Biol 10:e1003568. doi: 10.1371/journal.pcbi.1003568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Aoki FY, Macleod MD, Paggiaro P, Carewicz O, El Sawy A, Wat C, Griffiths M, Waalberg E, Ward P, IMPACT Study Group. 2003. Early administration of oral oseltamivir increases the benefits of influenza treatment. J Antimicrob Chemother 51:123–129. doi: 10.1093/jac/dkg007. [DOI] [PubMed] [Google Scholar]
- 20.Heinonen S, Silvennoinen H, Lehtinen P, Vainionpää R, Vahlberg T, Ziegler T, Ikonen N, Puhakka T, Heikkinen T. 2010. Early oseltamivir treatment of influenza in children 1-3 years of age: a randomized controlled trial. Clin Infect Dis 51:887–894. doi: 10.1086/656408. [DOI] [PubMed] [Google Scholar]
- 21.Lee N, Chan PK, Hui DS, Rainer TH, Wong E, Choi KW, Lui GC, Wong BC, Wong RY, Lam WY, Chu IM, Lai RW, Cockram CS, Sung JJ. 2009. Viral loads and duration of viral shedding in adult patients hospitalized with influenza. J Infect Dis 200:492–500. doi: 10.1086/600383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Memoli MJ, Athota R, Reed S, Czajkowski L, Bristol T, Proudfoot K, Hagey R, Voell J, Fiorentino C, Ademposi A, Shoham S, Taubenberger JK. 2014. The natural history of influenza infection in the severely immunocompromised versus non-immunocompromised hosts. Clin Infect Dis 58:214–224. doi: 10.1093/cid/cit725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Du L, Jin L, Zhao G, Sun S, Li J, Yu H, Li Y, Zheng BJ, Liddington RC, Zhou Y, Jiang S. 2013. Identification and structural characterization of a broadly neutralizing antibody targeting a novel conserved epitope on the influenza virus H5N1 hemagglutinin. J Virol 87:2215–2225. doi: 10.1128/JVI.02344-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dobrovolny HM, Gieschke R, Davies BE, Jumbe NL, Beauchemin CA. 2011. Neuraminidase inhibitors for treatment of human and avian strain influenza: a comparative modeling study. J Theor Biol 269:234–244. doi: 10.1016/j.jtbi.2010.10.017. [DOI] [PubMed] [Google Scholar]
- 25.Canini L, Carrat F. 2011. Population modeling of influenza A/H1N1 virus kinetics and symptom dynamics. J Virol 85:2764–2770. doi: 10.1128/JVI.01318-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dobrovolny HM, Reddy MB, Kamal MA, Rayner CR, Beauchemin CAA. 2013. Assessing mathematical models of influenza infections using features of the immune response. PLoS One 28:e57088. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.