

Abstract
New organometallic gold(III) and platinum(II) complexes containing iminophosphorane ligands are described. Most of them are more cytotoxic to a number of human cancer cell lines than cisplatin. Cationic Pt(II) derivatives 4 and 5, which differ only in the anion, Hg2Cl62– or PF6– respectively, display almost identical IC50 values in the sub-micromolar range (25–335-fold more active than cisplatin on these cell lines). The gold compounds induced mainly caspase-independent cell death, as previously reported for related cycloaurated compounds containing IM ligands. Cycloplatinated compounds 3, 4, and 5 can also activate alternative caspase-independent mechanisms of death. However, at short incubation times cell death seems to be mainly caspase dependent, suggesting that the main mechanism of cell death for these compounds is apoptosis. Mercury-free compound 5 does not interact with plasmid (pBR322) DNA or with calf thymus DNA. Permeability studies of 5 by two different assays, in vitro Caco-2 monolayers and a rat perfusion model, have revealed a high permeability profile for this compound (comparable to that of metoprolol or caffeine) and an estimated oral fraction absorbed of 100%, which potentially makes it a good candidate for oral administration.
Introduction
Cisplatin and the follow-on drugs carboplatin (paraplatin) and oxaliplatin (eloxatin) have been used to treat different cancers for the past 40 years.1 However, their effectiveness is still hindered by clinical problems, including acquired or intrinsic resistance, a limited spectrum of activity, and high toxicity, leading to side effects.1,2 In the search for more effective and selective potential anticancer metallodrugs,3 different approaches have been pursued, including the study of organometallic compounds. Evidence showing that organometallic compounds of platinum perform better than their non-organometallic derivatives was reported.4 In general, organometallic compounds are kinetically more inert and lipophilic than coordination metal complexes, which may offer opportunities in the design of anticancer metallodrugs with improved properties. Several reviews on the anticancer activity of organometallic compounds from a number of different transition metals have appeared in the past 5 years.5−17
More specifically, gold(III)3,6 and platinum(II)3,4,18−20 organometallic compounds have been studied as potential anticancer agents. A number of complexes containing the [Pt(COD)] fragment and different ligands, such as alkyls, alkynyls, and nucleosides, have been described.21−23 Platinum COD alkynyl compounds showed high toxicity against HT-29 colon carcinoma and MCF-7 breast adenocarcinoma cell lines,24,25 while [PtMe(R-COD)L] compounds26 with different ligands (halides, alkyl, aryl, alkynyl) revealed higher toxicity to HeLa cells in comparison to that of cisplatin. In the case of gold(III), it is well known that pincer ligands containing carbon and nitrogen stabilize the metal center against reduction to gold(I) and gold(0) species in physiological media.27 The anticancer activities of cyclometalated gold(III) and platinum(II) compounds with bidentate C,N- or terdentate C,N,N-pincer ligands have been recently reviewed.27−29 Some cyclometalated gold(III) complexes28,30,31 based on C,N,N- and C,N,C-pincer complexes have displayed impressive anticancer activity in vitro and in vivo by a mode of action different from that shown by cisplatin. It has been proposed that, for these complexes, the presence of the σ(M–C) bond increases the stability of the compounds allowing the organometallic fragment to reach the cell unaltered. In addition, it has been postulated that in platinum compounds the presence of aromatic groups in the cyclometalated ligand might favor intercalative binding to DNA (π–π stacking), while the labile positions in the coordination sphere may favor covalent coordination for DNA as in cisplatin. Very recently, a luminescent DNA intercalator cyclometalated platinum(II)complex, [Pt(C^N^N)(C-NtBu)]ClO4 (HC^N^N = 6-phenyl-2,2′-bipyridyl) with a potent inhibitory effect in human cancer cells in vitro and in a xenograft model in mice has been described.32 The stabilization of the topoisomerase I–DNA complex with resulting DNA damage by the cyclometalated compound is suggested to contribute to its anticancer activity. Multinuclear (SSCs) fluorescent rhomboidal Pt(II) metallacycles have also been reported recently18 showing a potent tumor growth inhibitory effect on MDA-MB-231 xenograft models in mice as well as high stability in media and in cancer cells in vitro.
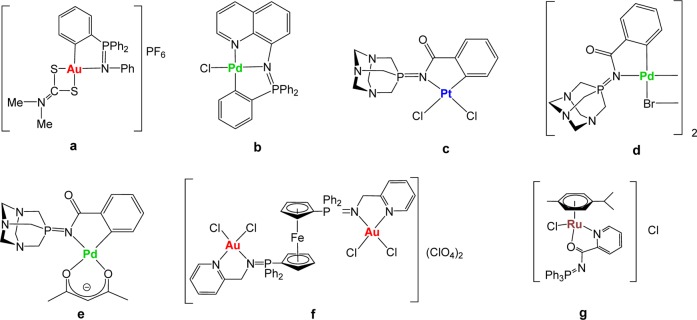
We have reported that nontoxic iminophosphorane or iminophosphane (IM) compounds (R3P=N-R′, IM) are useful precursors for the preparation of coordination (N,N-) or cyclometalated (C,N-) complexes of d8 (Au(III), Pd(II), and Pt(II)) and d6 (Ru(II)) metals (selected compounds a–g in Chart 1). These IM metal complexes display high cytotoxicity in vitro (low micromolar to nanomolar) against a variety of human cancer cell lines with different degrees of selectivity.33−39 Organogold(III) complexes containing iminophosphorane ligands (e.g., a) exert cell death with pathways involving mitochondrial production of reactive oxygen species.33,34 We have studied the interaction of the IM metal compounds with (pBR322) DNA, calf thymus (CT) DNA, and human serum albumin (HSA).33,35−39 We have confirmed that some compounds (such as f) inhibit PARP-1 proteins.37 More recently,39 we have described a water-soluble ruthenium(II) IM compound (g in Chart 1) which has displayed high activity against a number of cancer cell lines in vitro. This compound was also highly active on MDA-MB-231 xenografts in mice, with an impressive tumor reduction (shrinkage) of 56% after 28 days of treatment (14 doses of 5 mg/kg every other day), with low systemic toxicity, quick absorption in plasma, and preferential accumulation in breast tumor tissues.39 In most cases (including some Pd(II) and Pt(II) derivatives), we have demonstrated that DNA is not the target for these compounds and that most complexes are highly active against cisplatin-resistant cancer cell lines, pointing to a mode of action different from that of cisplatin.34−39 We also evaluated the stability of the compounds in solution and proved that, for gold(III) and palladium(II) metal centers, cyclometalated C,N-IM compounds were more stable than those in which the IM ligand was N,N-coordinated.35−38
Chart 1. Selected Iminophosphorane (IM) d8 and d6 Transition Metal Complexes with Significant Anticancer Properties Prepared in Our Group33−39.
In this context, we aimed to prepare cyclometalated IM compounds of gold(III) and platinum(II) in which the aryl group of the imino fragment is coordinated to the metal center (exo derivatives such as palladium compounds d and e in Chart 1) as opposed to an aryl group of the phosphine fragment (endo derivatives like a and b) in order to expand the range of phosphines incorporated into the final molecule to tune electronic/steric properties of the resulting complexes. The synthesis of the exo cyclometalated palladium starting material containing a water-soluble phosphine (d) was achieved by oxidative addition of Pd(0) to the C–Br bond in the IM bromide-containing ligand,36 a method that cannot be used to generate gold(III) and platinum(II) analogues.
We report here on the synthesis of novel exo cyclometalated C,N-IM compounds of gold(III) and platinum(II) containing the water-soluble phosphine 1,3,5-triaza-7-phosphaadamantane (PTA) and the synthesis of endo-C,N-IM compounds of platinum(II) derivatives never described before. All these complexes, along with the previously described exo derivative [Au(2-C6H4C(O)N=PPh3)Cl2] (1)40 and cisplatin, have been evaluated against a number of human cancer cell lines in vitro, and initial cell death mechanistic insights are discussed. We have studied the interaction of these compounds with relevant biomolecules such as plasmid (pBR322) DNA as a model for nucleic acids and HSA (the most abundant carrier protein in plasma). The platinum(II) compounds for which effects on DNA may be expected (3–5) have been further evaluated for their interaction with CT DNA by circular dichroism (CD). All these studies point out that cationic cycloplatinated compounds 4 and 5 (differing only in the anion) are the most active and have a mode of action different from that of cisplatin. Additionally, we report on the permeability of 4 and 5 evaluated by two different assays, in vitro Caco-2 monolayers and rat perfusion assay, in order to make comparisons with cisplatin and drugs or compounds that can be orally administered.
Results and Discussion
Synthesis and Characterization of the Cyclometalated Compounds
The synthesis of the exo cyclometalated gold(III) and platinum(II) compounds was based on the preparation of [Hg(Ph3P=N-CO-2-C6H4)Cl] by Nicholson et al.40 The C–H activation at the N-CO-Ph fragments takes place at a manganese center; thus, by transmetalation of the resulting cyclometalated iminophosphorane manganese compounds to HgCl2, the organomercury derivatives with PPh3, [Hg(Ph3P=N-CO-2-C6H4)Cl],40 or water-soluble phosphine PTA, [Hg(PTA=N-CO-2-C6H4)Cl],37 described by us, are obtained in high yields (Scheme 1).
Scheme 1. Previously Described Synthesis of Organomercury Compounds Containing the Semi-stabilized IM Ligand PR3=N-CO-2-C6H439,40.

Transmetalation reactions of [Hg(PR3=N-CO-2-C6H4)Cl] (PR3 = PPh3;40 PTA39) with NMe4[AuCl4] or [PtCl2(COD)] afforded previously described compound [Au(2-C6H4C(O)N=PPh3)Cl2] (1)40 and new cyclometalated exo-iminophosphorane complexes of gold(III) and platinum(II) of the type [Au(2-C6H4C(O)N=PTA)Cl2] (2) and [Pt(2-C6H4C(O)N=PTA)(COD)]2[Hg4Cl10] (3) (Scheme 2) in moderate to high yields.
Scheme 2. Synthesis of Gold(III) and Platinum(II) Cyclometalated exo-Iminophosphorane Complexes.

Compound [Au(2-C6H4C(O)N=PPh3)Cl2] (1) was previously reported.40
The reaction of [Hg(PPh3=N-CO-2-C6H4)Cl] with [PtCl2(COD)] did not afford a pure cycloplatinated compound. Different synthetic conditions were tried, and in most cases abundant Pt(0) decomposition took place, while unreacted [Hg(PPh3=N-CO-2-C6H4)Cl] and PPh3=O were the observed products along with free COD. Longer refluxing times in polar solvents afforded small amounts (4–10%) of a possible cyclometalated product along with [Hg(PPh3=N-CO-2-C6H4)Cl] and PPh3=O.
New compounds 2 and 3 are obtained as air-stable yellow and white solids, respectively. Compound 2 is neutral, whereas the Pt(II) derivative 3 is cationic (2:1 ions), as confirmed by conductivity measurements (see Experimental Section). Compound 3 is only soluble in solvents such as DMSO or DMF. We found that the COD ligand in 3 is immediately exchanged by DMSO molecules in DMSO-d6 solution at RT and that the new IM-cycloplatinated species did not change in DMSO-d6 over time (see Supporting Information (SI)). This was surprising since a COD ligand is not easily replaceable (usually requires thermal activation). The structures of these compounds have been proposed on the basis of elemental analysis, NMR and IR spectroscopy, and mass spectrometry (MS). Both compounds are soluble in mixtures of 1:99 DMSO:H2O at micromolar concentrations (relevant for biological studies).
The structure of 2 has been determined by an X-ray analysis, and it is very similar to that previously reported40 for compound [Au(2-C6H4C(O)N=PPh3)Cl2] (1),40 with very similar distances and angles. The molecular structure of 2 is depicted in Figure 1, while selected structural parameters are collected in Table 1.
Figure 1.
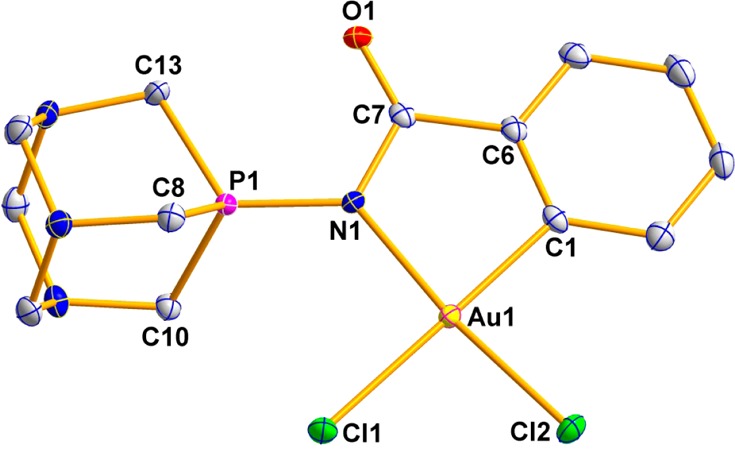
Molecular structure of compound 2.
Table 1. Selected Structural Parameters of Complex 2 Obtained from Single-Crystal X-ray Diffraction Studies (Bond Lengths in Angstroms and Angles in Degrees).
| Au(1)–Cl(1) | 2.3834(5) | N(1)–Au(1)–Cl(2) | 173.33(5) |
| Au(1)–Cl(2) | 2.2798(5) | N(1)–Au(1)–Cl(1) | 97.32(5) |
| Au(1)–C(1) | 2.020(2) | Cl(2)–Au(1)–Cl(1) | 88.62(2) |
| Au(1)–N(1) | 2.0497(18) | P(1)–N(1)–Au(1) | 126.07(10) |
| P(1)–N(1) | 1.6658(18) | P(1)–N(1)–C(7) | 119.25(15) |
| N(1)–C(7) | 1.404(3) | N(1)–C(7)–C(6) | 112.16(19) |
| C(7)–O(1) | 1.213(3) | N(1)–C(7)–O(1) | 123.72(19) |
| C(7)–C(6) | 1.478(3) | C(7)–N(1)–Au(1) | 114.68(14) |
| C(6)–C(1) | 1.385(3) | C(7)–C(6)–C(1) | 118.0(2) |
| C(6)–C(1)–Au(1) | 113.24(16) | ||
| C(1)–Au(1)–N(1) | 81.68(8) | N(1)–P(1)–C(10) | 113.99(10) |
| C(1)–Au(1)–Cl(2) | 92.44(6) | N(1)–P(1)–C(8) | 116.45(10) |
| C(1)–Au(1)–Cl(1) | 178.28(6) | N(1)–P(1)–C(13) | 117.81(10) |
The analysis confirms the square-planar arrangement around the gold(III) center with a bite angle of 81.68(8)°. Like in other C,N-IM cycloaurated complexes,33,40−42 the Au–Cl(1) bond trans to the carbon is longer (2.3834(5) Å) than the Au–Cl(2) bond trans to the nitrogen (2.2798(5) Å) due to the higher trans influence of the C donor atom. As observed in compound 1, upon coordination to the gold there is an increase in both the P–N and N–C bond lengths when compared to the uncoordinated ligand43 (P–N: 1.626(3) Å in ligand, 1.6658 (18) Å in 2; N–C: 1.353(5) Å in ligand, 1.401(3) Å in 2). This effect is also observed in the IR spectra of compound 2, for which the band corresponding to the P–N bond appears at a lower frequency than that for the free ligand (1289 cm–1 versus 1374 cm–1). As described in the structure of compound 1, a decrease of the C=O bond length was observed (from 1.245(5) Å in the ligand to 1.213(3) Å in the cycloaurated complex).
We had described the biological activity of the endo-iminophosphorane compound [Au{κ2-C,N-C6H4(PPh2=N(C6H5))-2}Cl2]42 and some of its cationic derivatives, like [Au{κ2-C,N-C6H4(PPh2=N(C6H5)-2}(S2CN(CH3)2)]PF6 (a in Chart 1),33 but we had never synthesized Pt(II) endo compounds with the IM Ph-N=PPh3 ligand. We carried out the reaction of [Hg{C6H4(PPh2=N(C6H5))-2}Cl]41,42 with [PtCl2(COD)] and obtained (as in the case of the exo compound 3) a cationic species (4) with a mercury chloride-containing anion (in this case [Hg2Cl6]2–). In order to avoid the use of organomercury compounds and the presence of mercury in the resulting compound, a “greener” synthetic approach44 based on transmetalation with an organogold(I)phosphine compound [Au{C6H4(PPh2=N(C6H5))-2}(PPh3)], described previously,42 was employed (Scheme 3).
Scheme 3. Synthesis of the New Platinum(II) Cyclometalated endo-Iminophosphorane Complexes 4 and 5.

We had used this mercury-free approach to obtain the endo gold(III) cyclometalated compound [Au{κ2-C,N-C6H4(PPh2=N(C6H5))-2}Cl2].42 The reaction proceeds much faster and in much milder conditions than that for the synthesis of 4 (25 min at RT in CH2Cl2 instead of 5 days in refluxing acetone), and compound 5 is obtained in moderate yield (58%). In order to avoid the formation of a neutral platinum(II) dimer with chloride bridges, [Pt{C6H4(PPh2=N(C6H5))-2}Cl]2, observed while performing this reaction, NH4PF6 was added. In this way, we obtained compound 5, an analogue of cationic compound 4 with a mercury-free anion (PF6–). The structures of these compounds have been confirmed by elemental analysis, NMR (including 195Pt NMR) and IR spectroscopy, and MS studies. In this case the compounds do not exchange the COD ligand by DMSO molecules at RT in DMSO-d6 solution as it happened for compound 3, which may have some connotations for the biological activity of the compounds. Compounds 4 and 5 are soluble in mixtures of 1:99 DMSO:H2O solutions at micromolar concentrations (relevant for biological studies). A mercury-free analogue of compound 3 could not be obtained, since the preparation of the appropriate Au(I) transmetalation agent from the organomanganese compound (Scheme 1) was not successful.
The number of cycloplatinated iminophosphorane compounds described previously is limited to two examples of endo neutral derivatives, [Pt(C6H4-2-PPh2=N-C(O)-2-NC5H4-κ-C,N,N)Cl]45 and [Pt{κ3-C,N,N-C6H4(PPh2=N-8-C9H6N}Cl],38 in which the iminophosphorane fragment acts as a C,N,N-pincer ligand. In compounds 3–5, the IM ligand is cyclometalated in either an exo (3) or endo (4, 5) position, acting as a C,N-pincer ligand. The other two coordination positions for the Pt(II) center are occupied by the COD ligand. The molecular structure for compound 4 was determined by X-ray crystallography, confirming the proposed structure. The molecular structure of the cation in 4 is depicted in Figure 2, while selected structural parameters are collected in Table 2. A complete drawing of the crystal structure of 4, including the [Hg2Cl6]2– anion and crystallization molecules, along with a more complete table of distances and angles are provided in the SI. The coordination geometry around the platinum atom is slightly distorted from square-planarity, with the C(1)–Pt(1)–N(1) angle of 85.31(9)° suggesting a rigid “bite” angle. The X(1)–Pt(1)–N(1) angle also deviates (94.14(9)°). The distance Pt–N(1) 2.039(2) Å is shorter than those for other C,N-cyclometalated Pt(II) derivatives, such as dimethylbenzylamine (dmba) compounds (ca. 2.1230–2.1340 Å) (see, e.g., refs (19, 46, and 47)). The distances Pt–N(1) and Pt–C(1), both 2.039(2) Å, are almost identical to those found for Au–N(1) and Au–C(1), both 2.035(4) Å, in [Au{C6H4(PPh2=N(C6H5))-2}(PPh3)].42 The distances Pt–X(1) and Pt–X(2) to the centroids of the COD ligand are 2.169(3) and 2.039(3) Å, respectively, which reflects the higher trans influence of C versus N (longer Pt–X(1) distance).
Figure 2.
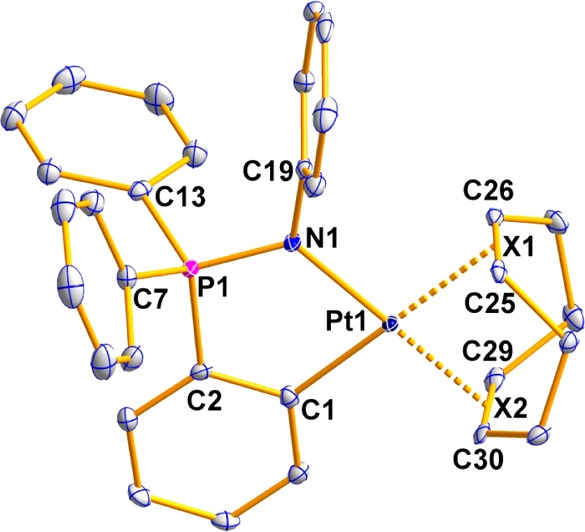
Molecular structure of the cation in compound 4. The anion [Hg2Cl6]2– is omitted for clarity.
Table 2. Selected Structural Parameters of the Cation in Complex 4 Obtained from Single-Crystal X-ray Diffraction Studies (Bond Lengths in Angstroms and Angles in Degrees).
| Pt(1)–C(1) | 2.039(2) | N(1)–Pt(1)–C(1) | 85.31(9) |
| Pt(1)–N(1) | 2.039(2) | C(1)–Pt(1)–X(1) | 178.95(9) |
| Pt(1)–X(1) | 2.169(3) | C(1)–Pt(1)–X(2) | 94.79(10) |
| Pt(1)–X(2) | 2.039(2) | N(1)–Pt(1)–X(1) | 94.14(9) |
| P(1)–N(1) | 1.622(2) | N(1)–Pt(1)–X(2) | 179.01(9) |
| P(1)–C(2) | 1.773(2) | X(1)–Pt(1)–X(2) | 85.78(10) |
| P(1)–C(7) | 1.797(3) | C(19)–N(1)–Pt(1) | 125.82(16) |
| P(1)–C(13) | 1.797(3) | C(19)–N(1)–P(1) | 116.21(16) |
| N(1)–C(19) | 1.444(3) | C(19)–N(1)–P(1) | 116.21(16) |
| C(1)–C(2) | 1.409(3) |
The stability of 1 and of the new compounds 2–5 was evaluated in DMSO-d6 solution by 31P{1H} and 1H NMR spectroscopy. All the complexes are stable for months in DMSO-d6 solution (see spectra and stability table in the SI). As mentioned before, compound 3 exchanges the COD ligand by DMSO immediately when dissolved in DMSO-d6 (free COD is clearly visible along with coordinated DMSO). In the case of compounds 4 and 5, this exchange is extremely slow, and after 1 week the percentage of free un-coordinated COD observed is around 6% (see SI). Compounds 4 and 5 are stable in mixtures of 1:99 DMSO:PBS for 24 h, as established by vis–UV spectroscopy (see SI). The stability of mercury-free compound 5 in acidic media over time was studied by 1H and 31P{1H} NMR spectroscopy. Due to the lack of solubility of 5 in mixtures of 1:99 DMSO:PBS in concentrations high enough to obtain a meaningful 31P{1H} NMR spectrum, these experiments were performed in a 2:1 DMSO-d6/PBS-1X(D2O) solution at pH 6 (see Experimental Section for details). In these conditions compound 5 is stable for at least 5 days, as can be observed by comparison with its 1H and 31P{1H} NMR spectra in the same deuterated mixture at pH 7.4 (Figures S9–S12 in the SI).
Biological Activity in Vitro
Anti-proliferative Studies In Vitro
The anti-proliferative properties of the gold(III) and platinum(II) complexes 1–5 and ligand COD were assessed by monitoring their ability to inhibit cell growth using the MTT assay (see Experimental Section). The cytotoxicity activity of the compounds was determined in several human cancer cell lines, i.e., leukemia Jurkat-T, lung A549, prostate DU-145, pancreas MiaPaca2, and triple-negative breast MDA-MB-231, in comparison to cisplatin. The results are summarized in Table 3. The COD ligand is poorly cytotoxic in all tested cell lines (IC50 > 125 μM). The IM ligands are known to be poorly cytotoxic (IC50 >100–500 μM in different cell lines).35−38
Table 3. IC50 (μM) of Metal Complexes 1–5, Ligand COD, and Cisplatin in Human Cell Linesa.
| Jurkat | A549 | DU-145 | MiaPaca2 | MDA-MB-231 | HEK-293T | |
|---|---|---|---|---|---|---|
| 1 | 3.4 ± 0.5 | 85.3 ± 5.9 | 40 ± 8.1 | 81.8 ± 2.6 | 101.8 ± 16 | 14.6 ± 1.4 |
| 2 | 9.5 ± 0.07 | >125 | >125 | >125 | >125 | >125 |
| 3 | 2.13 ± 0.24 | 20.8 ± 1.7 | 22.5 ± 4.2 | 7.53 ± 5.0 | 14.6 ± 3.7 | 4.0 ± 0.42 |
| 4 | 0.43 ± 0.06 | 0.85 ± 0.29 | 0.93 ± 0.43 | 0.79 ± 0.09 | 0.39 ± 0.05 | 1.25 ± 0.25 |
| 5 | 0.53 ± 0.13 | 2.01 ± 0.89 | 0.81 ± 0.07 | 1.03 ± 0.06 | 0.84 ± 0.29 | 0.94 ± 0.07 |
| COD | >125 | >125 | >125 | >125 | >125 | >125 |
| cisplatin | 10.8 ± 1.2 | 114.2 ± 9.1 | 112.5 ± 33 | 76.5 ± 7.4 | 131.2 ± 18 | 69.0 ± 6.7 |
All compounds were dissolved in 1% DMSO and diluted with water before addition to cell culture medium for a 24 h incubation period. Cisplatin was dissolved in H2O. Data are expressed as mean ± SD (n = 4).
Cyclometalated neutral gold(III) showed cytotoxicity similar to that of cisplatin, while compound 2 was less cytotoxic for all the studied cell lines, with the exception of the leukemia Jurkat cell line. We have found previously that replacement of PPh3 by PTA in IM-cyclometalated complexes decreases the cytotoxicity.39 The IC50 value for Jurkat for compound 1 is very similar to that obtained for the neutral iminophosphorane endo derivative [Au{κ2-C,N-C6H4(PPh2=N(C6H5))-2}Cl2].42 Cationic gold(III) complexes containing IM ligands are more cytotoxic than neutral derivatives.33,34 The cationic cyclometalated platinum compounds described here, 3 and especially 4 and 5, were considerably more cytotoxic than cisplatin in all the cell lines studied. 4 and 5 (same cation) display almost identical IC50 values, with the exception of A549 and MDA-MB-231, for which compound 4 containing the Hg2Cl62– anion is twice as active than 5. The data indicate that cytotoxicity for these compounds comes mainly from the cationic platinum fragment.
In order to assess the compounds’ selectivity for cancerous cells with respect to normal cell lines, they were also screened for their anti-proliferative effects on the non-tumorigenic human embryonic kidney cells HEK293T. In most cases the cytotoxicity is comparable for the cancerous and HEK293T cells. All compounds are more toxic to leukemia than to HEK293T cell lines (2–12 times), and compound 4 is more toxic to all the cell lines than to the HEK293T cell lines. The toxicity of mercury-free compound 5 to HEK293T is comparable to that in the human cancer cell lines. As HEK293T cell lines (immortalized cells) can display a higher sensitivity to chemicals, we measured the effect of compound 5 on human renal proximal tubular cells (RPTC). RPTCs in primary culture have been described as an in vitro model to study nephrotoxicity.39 The IC50 value (XTT assay 24 h; see SI for details) for 5 in this cell line was 2.77 ± 0.83 μM, making 5 more sensitive to cancerous cell lines than to RPTCs. In addition, we have described recently an IM ruthenium compound, [(η6-p-cymene)Ru{(Ph3P=N-CO-2-N-C5H4)-κ-N,O}Cl]Cl, which displayed similar IC50 values in vitro for all the human cancer cell lines described above and HEK293T but which was very effective in vivo on MDA-MB-231 xenografts in NOD.CB17-Prkdc SCID/J mice while having low toxicity.
Mechanism of Cell Death for the New Compounds
The mechanism of cell death induced by mercury-free cytotoxic cycloplatinated compound 5 was analyzed in two cell lines of different origin: A549 lung carcinoma and Jurkat T-cell leukemia. Phosphatidyl serine exposure, plasma membrane damage, and nuclear morphology were assessed in both cell lines after treatment with 5. Caspase implication in the toxicity of 5 was studied using the general caspase inhibitor z-VAD-fmk. In A549 cells we found that z-VAD-fmk protected cells from 5 at doses up to 0.5 μM (Figure 3), inhibiting both phosphatidylserine exposure (annexin V binding) and plasma membrane permeabilization (7-ADD uptake). As expected, phosphatidylserine exposure was more dependent on caspase activity. At higher concentrations 7-AAD staining, but not annexin V binding, increased, suggesting that cell death was necrotic.
Figure 3.

Role of caspases on cell death induced by compound 5 in A595 cells. Cells were cultured for 24 h in the presence of 5 at the indicated concentrations, alone (solid lines) or combined with the general caspase inhibitor z-VAD-fmk (dashed lines). Subsequently, phosphatidylserine exposure (triangles) and cell membrane permeabilization (squares) were analyzed by flow cytometry after staining with annexin V-DY634 and 7-AAD, respectively. Results are mean ± SD of two independent experiments with duplicates.
The apparent decrease in the percentage of annexin V-positive cells could reflect cell disintegration caused by necrosis. Consistently, z-VAD-fmk did not inhibit cell death at 1 μM (Figure 3). When an early event of apoptosis, loss of mitochondrial transmembrane potential, was analyzed, we also observed that caspase inhibition by z-VAD-fmk only partially reduced ΔΨm loss caused by 5 (Figure 4), further suggesting that compound 5 can induce caspase-dependent and caspase-independent cell death in A549 cells.
Figure 4.

Caspase implication in mitochondrial effects of compound 5 in A549 cells. Cells were cultured for 24 h in the presence of compound 5 at the indicated concentrations, alone (solid line) or combined with the general caspase inhibitor z-VAD-fmk (dashed line). Then, transmembrane mitochondrial potential was analyzed by flow cytometry after cells were stained with the probe DiOC6(3). Results are mean ± SD of two independent experiments with duplicates.
Jurkat cells were more sensitive to 5 than A549 cells, with an IC50 of 0.6 μM, even though this cell line does not express functional p53, discarding an essential role of this protein in the activity of compound 5. In these cells, the percentages of 7-AAD (Figure 5) and annexin V-positive (data not shown) cells were the same in every assay. High sensitivity of Jurkat cells was confirmed in short-term experiments, as we observed that 5 at 0.5 μM induced cell death in almost 100% of the cells even at 6 h. Caspase inhibition by z-VAD-fmk completely avoided cell death at 6 h. However, longer treatment with 5 induced both caspase-dependent and caspase-independent cell death (Figure 5).
Figure 5.

Implication of caspases in cell death induced by compound 5 in Jurkat cells. Cells were treated with compound 5 for 6 or 24 h in the presence or in the absence of the general caspase inhibitor z-VAD-fmk. Membrane integrity was analyzed by flow cytometry after the cells were stained with 7-AAD, as indicated in the Experimental Section. Results are mean ± SD of two independent experiments.
Mitochondrial damage was also analyzed in Jurkat cells (Figure 6). At 24 h, treatment with 5 caused a decrease in ΔΨm in 80% of cells, compared to 32% in cells treated with 5 in the presence of the general caspase inhibitor z-VAD-fmk. In order to determine whether mitochondrial damage caused by 5 was irreversible and committed cells to death, cells were washed and resuspended in fresh medium. After a further 24 h incubation in fresh medium, ΔΨm collapse was observed in nearly 100% of cells (Figure 6). These results indicate that caspase inhibition only delays cell death in Jurkat cells, and 5 induces cell damage, leading to cell death independently of caspase activation. Thus, these experiments confirm that alternative caspase-independent cell death mechanisms are activated by this compound, as observed in A549 cells.
Figure 6.

Jurkat cells were treated with 5 or 5+z-VAD for 24 h and then harvested, washed, and seeded in fresh medium. After further 24 h in fresh medium, mitochondrial transmembrane potential (ΔΨm) was analyzed as indicated in the Experimental Section. Results are mean ± SD of three independent experiments.
On the other hand, analysis of nuclear morphology indicated that 5 induced typical apoptotic features (chromatin condensation and fragmentation) that were prevented by z-VAD-fmk in both cell lines (Figure 7). However, some nuclei of cells treated with 5+zVAD displayed an altered morphology when compared to controls. This morphology could be caused by necroptosis48 or AIF-mediated cell death.49
Figure 7.

Compound 5 induces apoptosis in Jurkat (upper panels) and A549 cells (bottom panels). Cells were cultured for 24 h in the presence of compound 5 (0.5 μM), alone or combined with the general caspase inhibitor z-VAD-fmk or left untreated (Control). Nuclei were stained with Hoechst 33342 (10 μg/mL), and cells were photographed under UV light. Magnification ×400.
Finally, we analyzed the implication of mitochondria in the toxicity of compound 5. We used Jurkat-shBak cells, obtained by RNAi of Bak.49 Since Jurkat cells do not express Bax, the Jurkat-shBak cell line constitutes a model of human leukemia cells deficient in the intrinsic (mitochondrial) pathway of apoptosis. A cell line transfected with a nonspecific shRNA was used as a control (Jurkat pLVTHM). As shown in Figure 8, Jurkat-shBak cells were less sensitive to 5 than control cells. However, high concentrations of 5 induced Bax/Bak-independent cell death in Jurkat-shBak cells, suggesting that this compound could be useful in the treatment of tumors with alterations in the intrinsic pathway of apoptosis.
Figure 8.

Jurkat-pLVTHM (control) and Jurkat-shBak cells were treated with compound 5 for 24 h. Mitochondrial transmembrane potential was analyzed as indicated in the Experimental Section. Results are mean ± SD of three independent experiments.
We also analyzed the type of cell death for the cycloaurated exo compounds 1 and 2, the cycloplatinated exo compound 3, and the cycloplatinated endo compound 4 (analogue of 5 containing the mercury anion Hg2Cl62–) in Jurkat cells in the presence or absence of z-VAD-fmk (caspase inhibitor). This analysis showed that the gold compounds 1 and 2 were less toxic than platinum complexes 3 and 4, as already indicated by the IC50 values (Table 3). Moreover, these results show that gold compounds 1 and 2 induced mainly caspase-independent cell death. We had found that iminophosphorane–organogold(III) endo compounds also activated caspase-independent pathways that lead to cell death, as the addition of z-VAD-fmk did not significantly reduce the percentage of annexin V-PE+ or PI+ in Jurkat cells treated with these derivatives.34 Thus, the behavior of iminophosphorane–organogold(III) (both endo and exo) compounds is basically the same and also similar to that of other cyclometalated gold(III) anticancer agents.6
The toxicity for the cycloplatinated exo compound 3 (Figure 9) and for the endo compound 4, differing from 5 only in the anion (data not shown), was only partially dependent on caspase activity. At 24 h, 3 caused cell death in around 50% of total Jurkat cells, and caspase inhibition reduced this percentage to 25%. These data indicate that cycloplatinated compounds 3, 4, and 5 can activate alternative caspase-independent mechanisms of death. However, at short incubation times, cell death seems to be mainly caspase dependent (Figure 5), suggesting that the main mechanism of cell death for these compounds is apoptosis.
Figure 9.

Jurkat cells were treated with DMSO (Control) or compound 1 (10 μM), 2 (20 μM), or 3 (10 μM) for 24 h, in the absence or in the presence of 50 μM z-VAD-fmk. Cell death was analyzed by annexin V-FITC binding and flow cytometry. Results are mean ± SD of two independent experiments.
To summarize, from these initial mechanistic studies it seems clear that the cell death type for the most active mercury-free cycloplatinated compound 5 is mainly through caspase-dependent apoptosis but that 5 triggers caspase-independent cell death when apoptosis is blocked, pointing to a mode of action different from that of cisplatin.
Lipophilicity and Permeability Assays
The lipophilicity of the most active cycloplatinated compounds 4 and 5 was determined by calculating the partition coefficients (see Table 4 and Experimental Section) between n-octanol and phosphate buffer (pH 7.00). Partition coefficients have been used to predict the permeability of drugs since there is a good correlation between intestinal permeability and physicochemical parameters such as lipophilicity. We wanted to study the influence of the two different anions (Hg2Cl62–, 4, and PF6– in 5) on the lipophilicity and permeability of these cationic cycloplatinated compounds.
Table 4. Partition Coefficients (Ratio n-Octanol:Phosphate Buffer) of Compounds 4 and 5 and Reference Metoprolol.
| compound | P | log P |
|---|---|---|
| metoprolol | 0.20 ± 0.02 | –0.68 |
| 4 | 0.54 ± 0.03 | –0.26 |
| 5 | 1.05 ± 0.05 | 0.02 |
Metoprolol was chosen as the reference compound for permeability since it is known that 95% of the drug is absorbed from the gastrointestinal tract. Thus, drugs that exhibit partition coefficients and human intestinal permeability values greater than or equal to the corresponding values for metoprolol are considered high-permeability drugs. Drugs with estimated partition coefficients and human intestinal permeability values less than the corresponding values for metoprolol are classified as low-permeability drugs. This type of correlation is a suitable source of information on the passive and also possible carrier-mediated absorption mechanism. From these data we can state that compound 5 is more lipophilic than 4 and metropolol.
Subsequently, the permeabilities of cisplatin as commercialized parent compound, cycloplatinated 4 and 5 as test compounds, and metoprolol, cimetidine, and atenolol/Lucifer Yellow as reference compounds of high, intermediate, and low permeability, respectively, were determined using an in vitro cell model based on the measurement of the permeabilities of the compounds through Caco-2 monolayers50,51 and an in situ method by performing a rat perfusion assay.52,53 Results from the in vitro cell assay are shown in Figure 10 and data collected in Table 5, while the results in the rat model are depicted in Figure 11 and data collected in Table 6.
Figure 10.
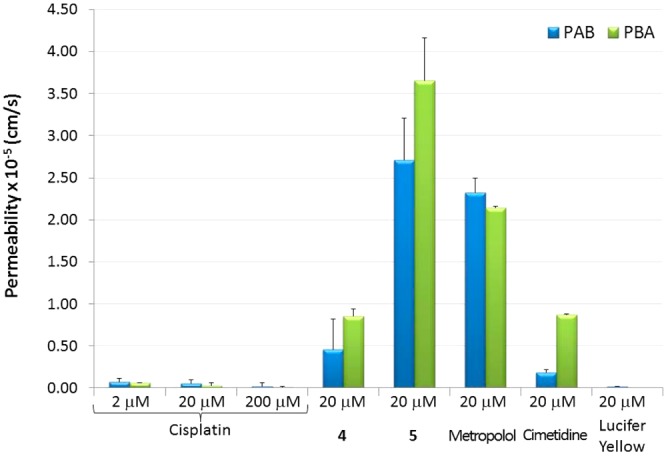
Permeability values obtained from apical to basal (PAB) and from basal to apical (PBA) of cisplatin (at different concentrations), cycloplatinated 4 and 5, and permeability reference compounds metoprolol, cimetidine, and Lucifer Yellow at 20 μM in Caco-2 cells. Data correspond to the averaged values for three independent experiments.
Table 5. Permeability Values Obtained by the Caco-2 Cell Monolayers Assaya.
| compound (20 μM) | Peff (cm/s) | SD |
|---|---|---|
| cisplatin | 5.44 × 10–7 | 4.66 × 10–7 |
| 4 | 4.62 × 10–6 | 3.54 × 10–6 |
| 5 | 2.71 × 10–5 | 5.00 × 10–6 |
| metoprolol | 2.32 × 10–5 | 1.75 × 10–6 |
| cimetidine | 1.86 × 10–6 | 3.71 × 10–7 |
| Lucifer Yellow | 1.90 × 10–7 | 4.98 × 10–8 |
Metoprolol, cimetidine, and Lucifer Yellow were used as model compounds of high, medium, and low oral permeability, respectively. Data correspond to the averaged values for three independent experiments.
Figure 11.
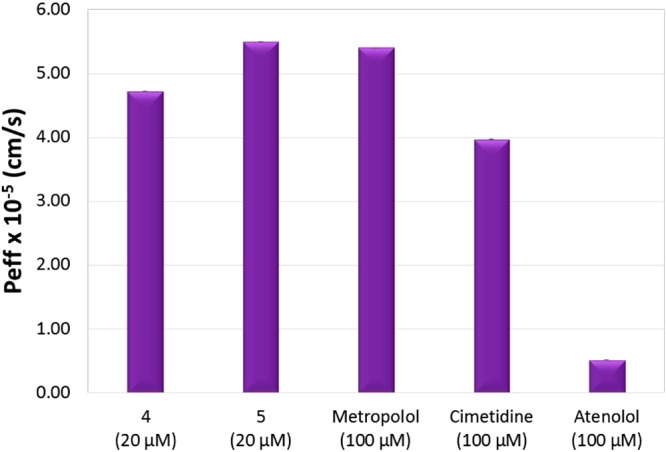
Absorption rate coefficients in rats.
Table 6. Absorption Rate Coefficients, Ka, and Permeability Values Obtained from in Situ Rat Assaysa.
| compound | Ka1 (h–1) | SD | Peff (cm/s) | SD |
|---|---|---|---|---|
| cisplatinb | ND | – | ND | – |
| 4b | 2.00 | ±0.11 | 4.72 × 10–5 | ±2.60 × 10–6 |
| 5b | 2.12 | ±0.22 | 5.50 × 10–5 | ±5.40 × 10–6 |
| metoprololc | 2.30 | ±0.15 | 5.40 × 10–5 | ±3.54 × 10–6 |
| cimetidinec | 1.68 | ±0.12 | 3.97 × 10–5 | ±3.04 × 10–6 |
| atenololc | 0.22 | ±0.02 | 5.19 × 10–6 | ±4.72 × 10–7 |
Metoprolol, cimetidine, and atenolol were used as model compounds of high, medium, and low oral permeability, respectively. Data correspond to values of six independent experiments. ND = not detectable.
20 μM.
100 μM.
PAB is the value corresponding to the permeability from the apical to the basolateral chamber that simulates the permeability in the physiological sense from intestine to plasma. The PBA value corresponds to the permeability from the basolateral to apical chamber. This PBA value would be the hypothetical value for the permeability “from plasma to intestine”. Although the PBA value has no physiological sense, this parameter and the ratio PAB/PBA can help to elucidate the mechanism of drug transport across the intestinal barrier.54
Cell transport assays reveal that cisplatin is a compound with very low permeability. This permeability value (5.44 × 10–7cm/s) indicates that cisplatin is not a suitable drug for oral administration if the objective is to obtain therapeutic plasma values. However, organoplatinum compounds 4 and 5 show higher permeability values than cisplatin. In fact, the permeability value of compound 4 is 10-fold higher than that of cisplatin at the same concentration, and the permeability of compound 5 is 50-fold higher. The permeability of compound 4 is higher than those of Lucifer Yellow and cimetidine but lower than that of metoprolol. 4 can be considered a compound of medium oral permeability. However, the permeability of compound 5 is higher than that of compound 4 and even higher than that of metoprolol (in accordance with the lipophilicity data), indicating that it can be considered a highly permeable compound. The high permeability of active principles is a crucial condition for oral administration.
Results from the in situ rat model assays confirm those obtained by the in vitro cell experiments. The permeability of compound 4 is higher than that of atenolol, slightly higher that that of cimetidine, but lower than that of metoprolol. Compound 4 can be considered a compound of intermediate permeability. Compound 5 exhibits permeability higher than that of compound 4 and slightly higher than that of metoprolol, indicating that 5 is a highly permeable compound. Both compounds 4 and 5 display a much better absorption profile than cisplatin.
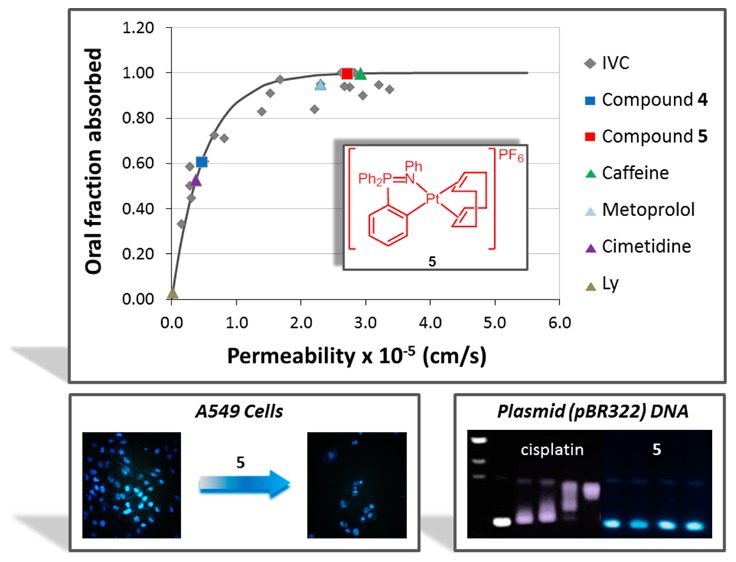
In addition, we have validated the relationship between Caco-2 cells’ permeability and oral fraction absorbed in our experimental system (represented in Figure 12) and previously used for fraction absorbed predictions.54 The permeabilities of cisplatin and derivatives 4 and 5 have been included in this correlation. The predicted oral fraction absorbed is more than 60% for compound 4 and almost 100% for compound 5, demonstrating its improved absorbability properties with respect to cisplatin. In the absence of solubility or dissolution limitations the absorption of these compounds would be almost complete; thus, with the adequate formulation strategy, they represent promising candidates for oral administration.
Figure 12.
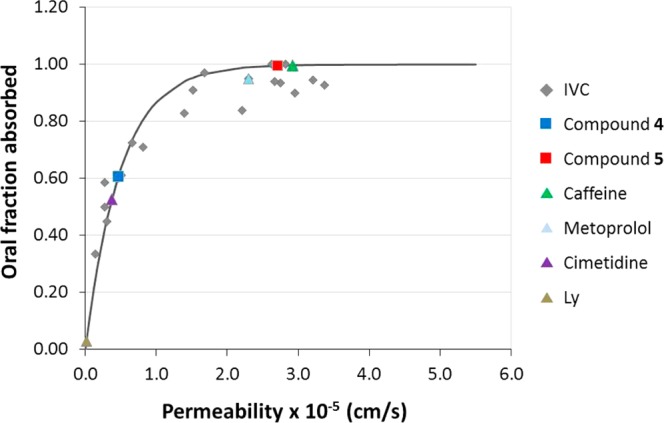
Correlation between oral fractions absorbed vs permeability values obtained from Caco-2 cell monolayers transport assay in apical to basal direction (PAB). Gray diamonds correspond to the internally validated correlation (IVC).54 Triangles correspond to permeability reference compounds (metoprolol/caffeine for high permeability, cimetidine for intermediate permeability, and Lucifer Yellow for low permeability). Light gray squares correspond to tested compounds 4 and 5.
3. Reactivity with Biomolecules
Interactions with DNA
Since DNA replication is the key event for cell division, it is among the critically important targets in cancer chemotherapy. Most cytotoxic platinum drugs form strong covalent bonds with DNA bases.55 However, a variety of platinum compounds act as DNA intercalators upon coordination to the appropriate ancillary ligands.56 It has been reported that most gold(III) compounds display reduced affinity for DNA,33 although there are a number of Au(III) porphyrin complexes6,27,57 and cyclometalated species with C,N,C-pincer ligands6,27,58 that act as DNA intercalators and, in some cases, as DNA topoisomerase inhibitors. We investigated the interactions of the gold(III) and platinum(II) complexes with plasmid pB322 DNA and with CT DNA and directly compared them to the same interactions of cisplatin.
Interaction of Complexes 1–5 with Plasmid pBR322 DNA
To gain insight into the nature of the compound–DNA interactions, gel electrophoresis studies were performed with gold(III) (1 and 2) and platinum(II) (3–5) complexes on plasmid (pBR322) DNA (Figure 13). Plasmid pBR322 presents two main forms, OC (open circular or relaxed) and CCC (covalently closed or supercoiled), which display different electrophoretic mobility. Changes in the electrophoretic mobility of any of the forms upon incubation of the plasmid with a compound are usually interpreted as evidence of interaction. Generally, a drug that induces unwinding of the CCC form will produce a retardation of the electrophoretic mobility, while coiling of the OC form will result in increased mobility. Figure 13 shows the effect of cisplatin and compounds 1–5 on plasmid (pBR322) DNA after incubation at 37 °C for 20 h in Tris-HCl buffer at different drug/DNA ratios. As previously reported, cisplatin is able to both increase and decrease the mobility of the OC and the CCC forms, respectively.59 Treatments with increasing amounts of compounds 1, 2, 4, and 5 do not cause any shift for either form, consistent with no unwinding or other changes in topology under the chosen conditions. Treatment with increasing amounts of 3 retards the mobility of the faster-running supercoiled form (Form I), especially at higher molar ratios. In order to understand the interaction of 3 with DNA, platinum compounds 3–5 were incubated with CT DNA and analyzed by CD.
Figure 13.
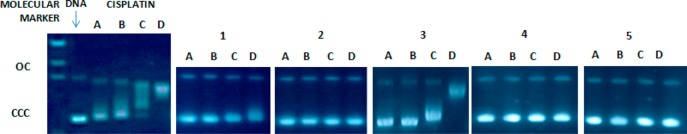
Electrophoresis mobility shift assays for cisplatin and compounds 1–5 (see Experimental Section for details). DNA refers to untreated plasmid pBR322. A, B, C, and D correspond to metal/DNAbp ratios of 0.25, 0.5, 1.0, and 2.0, respectively.
Interaction with Calf Thymus DNA
More detailed DNA conformational changes can be detected by means of CD spectroscopy. CD spectral technique is very sensitive to diagnose alteration on the secondary structure of DNA that results from DNA–drug interactions. A typical CD spectrum of CT DNA shows two conservative bands, a positive band with a maximum at 273 nm due to base stacking and a negative band with a minimum at 242 nm due to helipticity, characteristic of the B conformation of DNA.60 Therefore, changes in the CD signals can be assigned to corresponding changes in DNA secondary structures. In addition, it is known that simple grove binding or electrostatic interaction of small molecules causes little or no alteration to any of the CD bands when compared to major perturbation induced by covalent binding or intercalation. The most dramatic changes in CT DNA can be observed with compound 3 (Figure 14A). Upon addition of increasing amounts of the complex, the intensity of the positive band diminishes, and a new negative band at 287 nm and a positive band at 251 nm appear. This type of modification in the CD spectrum of CT DNA is characteristic of conformational changes in DNA from B, the usual right-handed form of DNA, to Z, the left-handed form of DNA.61 The formation of left-handed helix of Z-form DNA structure is similar to the transition seen in purely electrostatic environments such as those provided by HgCl262 and Hg(ClO4)2.63 Thus, the presence of [Hg4Cl10]2– anion in compound 3 seems to lead to the conformational change from B form to Z form.
Figure 14.

CD spectra of CT DNA (195 μM) and CT DNA incubated with 0.1, 0.25, 0.5, and 1.0 equiv of compounds 3 (A), 4 (B), 5 (C), and cisplatin (D) for 20 h at 37 °C.
Organoplatinum endo compound 4 leads to minor changes of the B-type CD spectrum (Figure 14B) with slight decrease of the intensities of the positive bands and with no modification in the negative region. This points out that the DNA binding of complex 4 induces conformational changes including conversion from a more B-like to a more C-like structure within the DNA molecule.64 This conformational change is indicative of a non-intercalative mode of binding of the complex and offers support that the complex is either grove binding or electrostatic in nature,65,66 and the change might be due to the lower concentration of Hg2+ released by compound 4, [Hg2Cl6]2– (compared to that released by compound 3 containing a [Hg4Cl10]2– anion), although the influence of the more lipophilic Pt(II) cation in compound 4, could not be completely ruled out.
Finally, as shown in Figure 14C, compound 5 does not lead to any modification of the DNA bands with respect to untreated CT DNA, suggesting that the interaction of compound 5 with CT DNA is almost nonexistent. This is in good agreement with our findings described above about the influence of the mercury anion in compounds 3 and 4 in their interaction with CT DNA since the anion in compound 5 is PF6–.
In conclusion, the experiments of DNA–drug interactions have shown that compound 3 induces the formation of left-handed helix of Z-form DNA through strong electrostatic interactions and compound 4 appears to be either grove binding or electrostatic in nature. This is supported by two main facts: (1) retardation of the plasmid (pBR322) DNA electrophoretic mobility observed only for compound 3 and (2) results obtained by CD spectroscopy. Importantly, the mercury-free cationic organoplatinum compound 5 does not seem to interact with DNA, indicating that, as for other transition-metal IM complexes,33−39 its antitumor properties are due to non-DNA-related mechanisms/factors.
Interactions with Human Serum Albumin
HSA is the most abundant carrier protein in plasma and is able to bind a variety of substrates, including metal cations, hormones, and most therapeutic drugs. It has been demonstrated that the distribution, the free concentration, and the metabolism of various drugs can be significantly altered as a result of their binding to this protein.67 HSA possesses three fluorophores, namely tryptophan (Trp), tyrosine (Tyr), and phenylalanine (Phe) residues, with Trp214 being the major contributor to the intrinsic fluorescence of HSA. This Trp fluorescence is sensitive to the environment and binding of substrates, as well as changes in conformation that can result in quenching (either dynamic or static).
Thus, the fluorescence spectra of HSA in the presence of increasing amounts of the compounds 1–5 and cisplatin were recorded in the 300–450 nm range upon excitation of the tryptophan residue at 295 nm. The compounds caused a concentration-dependent quenching of fluorescence without changing the emission maximum or the shape of the peak, as seen in Figure 15A for compound 3. All these data indicate an interaction of the compounds with HSA. The fluorescence data were analyzed by the Stern–Volmer equation (Figure 15B). While a linear Stern–Volmer plot is indicative of a single quenching mechanism, either dynamic or static, the positive deviation observed in the plots of F0/F versus [Q] of compounds 2–4 (Figure 15C) suggests the presence of different binding sites in the protein with different binding affinities.69 Of note, a similar behavior was observed in the case of coordination iminophosphorane complexes of d8 metals, for which we also reported a concentration-dependent fluorescence quenching.35−39 On the other hand, the Stern–Volmer plot for complexes 1 and 5 shows a linear relationship (Figure 15D), suggesting the existence of a single quenching mechanism, most likely dynamic, and a single binding affinity. The Stern–Volmer constants for complexes 1 and 5 are 4.58 × 106 and 3.67 × 106 M–1, respectively.
Figure 15.

(A) Fluorescence titration curve of HSA for compound 3. Arrow indicates the increase of quencher concentration (10–100 mM). Stern–Volmer plot for HSA fluorescence quenching observed with compounds 1–5 and cisplatin (B), 2–4 and cisplatin (C), and 1, 5, and cisplatin (D).
In general, higher quenching by the iminophosphorane complexes was observed compared to that of cisplatin under the chosen conditions, most likely due to the faster reactivity of our compounds with HSA, as compared to cisplatin.
Conclusions
We have reported on the synthesis and anticancer properties of cyclometalated neutral gold(III) and cationic platinum(II) derivatives containing iminophosphorane ligands. Most compounds are more cytotoxic to a number of human cancer cell lines than cisplatin. The gold compounds induced mainly caspase-independent cell death, as previously described for related cycloaurated iminophosphorane compounds. Cycloplatinated compounds 3, 4, and 5 can also activate alternative caspase-independent mechanisms of death. However, at short incubation times cell death seems to be mainly caspase dependent, suggesting that the main mechanism of cell death for these compounds is apoptosis. The most promising candidate is the mercury-free lipophilic cationic cycloplatinated compound 5. This derivative is much more active (25–300-fold) than cisplatin against a number of cancer cell lines while being less toxic on human renal proximal tubular cell lines. These facts, along with the lack of interaction observed for 5 with plasmid (pBR322) and CT DNA, point to a mode of action different from that of cisplatin. Permeability studies of 5 by two different assays, in vitro Caco-2 monolayers and a rat perfusion model, have revealed its high permeability profile (comparable to that of metoprolol or caffeine) and an estimated oral fraction absorbed of 100%, which potentially makes it a good candidate for oral administration. The results described for 5 and those recently reported for a ruthenium–iminophosphorane compound highly active in vivo against breast cancer39 warrant further advanced preclinical studies with selected organometallic iminophosphorane compounds. The work described in this paper supports the idea that nontoxic iminophosphorane molecules are excellent ligands for the synthesis of organometallic compounds of d6 and d8 metals (especially cationic species) with relevant anticancer properties, high permeability, and, in some cases, water-solubility.
Experimental Section
All manipulations involving air-free syntheses were performed using standard Schlenk-line techniques under a nitrogen atmosphere or in a glovebox (MBraun MOD system). Solvents were purified by use of a PureSolv purification unit from Innovative Technology, Inc. The phosphine substrates TPA and PPh3 were purchased from Sigma-Aldrich, [Mn2(CO)10] and [PtCl2(COD)] were purchased from Strem Chemicals, and Na/Hg were purchased from Fisher Scientific and used without further purification. Compounds [PhCH2Mn(CO)5],68 [Hg(2-C6H4C(O)N=PPh3)Cl],40 and [Hg(2-C6H4C(O)N=PTA)Cl]39 and IM ligands Ph3P=N-CO-2-N-C5H469 were prepared by reported methods. The purity of the compounds, based on elemental analysis, is ≥99.5%. NMR spectra were recorded on a Bruker AV400 instrument (1H NMR at 400 MHz, 13C NMR at 100.6 MHz, 31P NMR at 161.9 MHz, 195Pt NMR at 85.7 Hz). Chemical shifts (δ) are given in ppm using CDCl3 or DMSO-d6 as solvent, unless otherwise stated. Elemental analyses were performed on a PerkinElmer 2400 CHNS/O analyzer, Series II. High-resolution electrospray ionization (HR-ESI) and matrix-assisted laser desorption/ionization (MALDI) mass spectra were obtained on an Agilent analyzer or a Bruker analyzer. Conductivity was measured in an Oakton pH/conductivity meter in acetone solution (10–3 M). X-ray collection was performed at room temperature (RT) using graphite-monochromated and 0.5 mm MonoCap-collimated Mo Kα radiation (λ = 0.71073 Å) with the ω scan method. CD spectra were recorded using a Chirascan CD spectrometer equipped with a thermostated cuvette holder. Electrophoresis experiments were carried out in a Bio-Rad Mini subcell GT horizontal electrophoresis system connected to a Bio-Rad Power Pac 300 power supply. Photographs of the gels were taken with an Alpha Innotech FluorChem 8900 camera. Fluorescence intensity measurements were carried out on a PTI QM-4/206 SE spectrofluorometer (PTI, Birmingham, NJ) with right angle detection of fluorescence using a 1 cm path length quartz cuvette.
Synthesis
[Au(2-C6H4C(O)N=PTA)Cl2] (2)
[Hg(2-C6H4C(O)N=PTA)Cl] (0.15 g, 0.3 mmol), [NMe4][AuCl4] (0.12 g, 0.2 mmol), and [NMe4]Cl (0.035 g, 0.32 mmol) were stirred at RT in CH2Cl2 (15 mL) for 1 day in a foil-covered flask. The solvent was removed under reduced pressure. The fraction containing compound 2 was then extracted from the solid residue with CHCl3 (3 × 10 mL), and the resulting yellow solution was filtered through Celite. The volume was reduced (<3 mL), and upon addition of Et2O (20 mL) a pale yellow solid was precipitated. This solid was finally isolated by filtration and dried in vacuo. Yield: 0.15 g (93%). Anal. Calcd for C13H16N4OPCl2Au (543.14): C, 28.75; H, 2.97; N, 10.32. Found: C, 28.32; H, 3.07; N, 9.93. ESI-MS: m/z 507.04 (100%, [M–Cl]+, calcd 507.04). 31P{1H} NMR (CDCl3): δ −7.66 (s); (DMSO-d6): δ −2.68 (s). 1H NMR (CDCl3): δ 4.54 (6H, AB system, NCH2N), 5.10 (6H, d, 2JPH = 9.1 Hz, PCH2N), 7.34 (1H, d, 3JHH = 7.0 Hz, 6-C6H4), 7.38 (dd, 3JHH = 7.8, 3JHH = 7.8 Hz, 4-C6H4), 7.42 (dd, 3JHH = 7.2, 3JHH = 7.1 Hz, 5-C6H4), 8.04 (d, 3JHH = 8.1 Hz, 3-C6H4). 13C{1H} NMR: δ 55.78 (d, 1JPC = 36.8 Hz, PCH2N), 72.37 (d, 3JPC = 10.8 Hz, NCH2N), 128.64 (s, 2-C6H4), 129.73 (s, 3-C6H4), 130.74 (s, 5-C6H4), 134.42 (s, 4-C6H4), 143.16 (s, Au–C) ppm. Signals corresponding to NC=O and C1 not observable. IR (cm–1): ν 352 (Au–Cl), 1299 (N=P) 1654 (C=O). Conductivity: 37.66 μS/cm (acetone) (neutral).
[Pt(2-C6H4C(O)N=PTA)(COD)]2Hg4Cl10 (3)
[Hg(2-C6H4C(O)N=PTA)Cl] (0.225 g, 0.44 mmol) and [PtCl2(COD)] (0.165 g, 0.44 mmol) were refluxed in CH3CN (20 mL) for 2 h, affording a white solid that was filtrated off and washed with Et2O (3 × 10 mL), benzene (2 × 5 mL), and hexane (2 × 10 mL). After drying in vacuo, complex 3 was isolated as a white powder. Yield: 0.097 g (40%). Anal. Calcd for C42H56Cl10Hg4N8O2P2Pt2 (2313.94): C, 21.80; H, 2.44; N, 4.84. Found: C, 21.72; H, 2.58; N, 4.72. ESI-MS: m/z 470.1 ([M–COD–Hg4Cl10]+, calcd 470.4), 578.2 (100%, [M–Hg4Cl10]+, calcd 577.9), 1275.0 (100%, [2M–Hg4Cl6]2+ + CCl3–, calcd 1275.4). 31P{1H} NMR (CDCl3): δ −7.66 (s); (DMSO-d6): −10.25 (s). 195Pt{1H} NMR (DMSO-d6): δ −3652.87 (s). 1H NMR (DMSO-d6): δ 2.30 (8H, s, COD), 4.39 (6H, s, NCH2N), 4.91 (6H, d, 2JPH = 10.3 Hz, PCH2N), 5.51 (4H, s, COD), 7.07 (1H, m, 4-C6H4), 7.16 (1H, m, 5-C6H4), 7.29 (1H, d, 3JHH = 7.3 Hz, 6-C6H4), 8.15 (1H, d, 3JHH = 8.0 Hz, 3-C6H4). 13C{1H} NMR (DMSO-d6): δ 27.98 (s, COD), 54.27 (d, 1JPC = 39.2 Hz, PCH2N), 71.30 (d, 3JPC = 10.2 Hz, NCH2N), 124.6 (s, 4-C6H4), 128.1 (s, 6-C6H4), 128.9 (s, COD), 131.9 (s, 5-C6H4), 132.7 (s, 3-C6H4), 135.0 (s, 1-C6H4), 138.0 (d, 2JCPtc = 10.2 Hz, 2-PtC), 182.1 (d, 2JPC = 5.18 Hz, C=O) ppm. IR (cm–1): ν 1300 (N=P), 1643 (C=O). Conductivity: 129 μS/cm (DMF) (1:2 electrolyte).
[Pt{κ2-C,N-C6H4(PPh2=N(C6H5)(COD)]2(Hg2Cl6) (4)
[Hg{C6H4(PPh2=N(C6H5)}Cl] (0.18 g, 0.3 mmol) and [PtCl2(COD)] (0.11 g, 0.3 mmol) were refluxed in acetone (30 mL) for 5 d. The solvent was removed under reduced pressure. The final product was extracted with CH2Cl2 and the resulting solution filtered through Celite, giving a light yellow solution. The solution was concentrated (<3 mL), and upon addition of Et2O (20 mL) the final product was precipitated as a white solid, isolated by filtration, and dried in vacuo. Yield: 0.20 g (72%). Anal. Calcd for C64H62N2P2Cl6Pt2Hg2·CH2Cl2 (1961.24): C, 38.84; H, 3.21; N, 1.39. Found: C, 38.84; H, 3.42; N, 1.30. ESI-MS: m/z 655.18 (100%, [M]+, calcd 655.18). 31P{1H} NMR (CDCl3): δ 64.51 (s); (DMSO-d6): δ 63.19 (s). 195Pt{1H} NMR (CDCl3): δ −3622.47 (d, 2JPPt = 404 Hz). 1H NMR (CDCl3): δ 2.66 (8H, m, COD), 5.12–5.30 (4H, m, COD), 6.88 (2H, m, 2-,6-NAr), 7.12 (1H, m, 4-NAr), 7.21 (2H, m, 3-,5-NAr), 7.35 (1H, m, 4-C6H4), 7.56–7.67 (10H, m, o-,m-,p-C6H5), 7.75 (2H, m, 5-C6H4). 13C{1H} NMR (CDCl3): δ 28.24 (s, COD), 31.23 (s. COD), 89.30 (s, COD), 116.12 (s, COD), 124.3 (d, 1JPC = 91.6 Hz, Ci), 127.1 (d, 5JPC = 2.7 Hz, 4-NAr), 127.8 (d, 3JPC = 13.9 Hz, 4-C6H4), 129.5 (d, 3JPC = 2.9, Hz, m-C6H5), 129.7 (s, 3-,5-NAr), 130.1 (d, J = 4.8 Hz, 2-,6-NAr), 132.7 (d, 2JPC = 14.3 Hz, 3-C6H4), 133.3 (m, o-,p-C6H5), 134.3 (d, 4JPC = 2.3 Hz, 5-C6H4), 147.8 (s, Pt–C). IR (cm–1): ν 529 (Pt–N), 1310 (N=P). Conductivity: 102.3 μS/cm (acetone) (1:1 electrolyte).
[Pt{κ2-C,N-C6H4(PPh2=N(C6H5)(COD)](PF6) (5)
[Au{C6H4(PPh2=N(C6H5))-2}(PPh3)] (0.28 g, 0.3 mmol) and PtCl2(COD) (0.11 g, 0.3 mmol) were stirred in CH2Cl2 (20 mL) for 25 min at RT, follow by addition of NH4PF6 (0.049 g, 0.3 mmol). The resulting solution was stirred for an additional 1 h. The solution was concentrated (<3 mL), dry Et2O (20 mL) was added, affording a gray precipitate, and the solution was stirred for 10 min. The precipitated gray solid was isolated by filtration and washed with water (4 × 2 mL) and a cold mixture (1:8) of CH2Cl2/Et2O (4 × 5 mL), yielding a white solid that was dried in vacuo. Yield: 0.14 g (58%). Anal. Calcd for C32H31F6NP2Pt (800.63): C, 48.01; H, 3.90; N, 1.75. Found: C, 47.74; H, 4.11; N, 1.74. ESI-MS: m/z 655.18 (100%, [M]+, calcd 655.18). 31P{1H} NMR (CDCl3): δ 64.65 (s); (DMSO-d6): δ 63.19 (s). 195Pt{1H} NMR (CDCl3): δ −3614.48 (d, 2JPPt = 406.1 Hz). 1H NMR (CDCl3): δ 2.64 (8H, m, COD), 5.20 (4H, m, COD), 6.88 (2H, m, 2-,6-NAr), 7.09 (1H, m, 4-NAr), 7.20 (2H, m, 3-,5-NAr), 7.23 (1H, m, 3-C6H4), 7.33 (1H, m, 4-C6H4), 7.49–7.66 (10H, m, o-,m-,p-C6H5), 7.72 (2H, m, 5-C6H4). 13C{1H} NMR (CDCl3): δ 28.05 (s, COD), 31.02 (s, COD), 89.22 (s, COD), 116.49 (s, COD), 124.3 (s, Cipso), 125.2 (s, Cipso), 127.0 (d, 5JPC = 2.9 Hz, 4-NAr), 127.6 (d, 5JPC = 13.9 Hz, 4-C6H4), 129.5 (d, 3JPC = 2.5, Hz, m-C6H5), 129.6 (s, 3-,5-NAr), 130.1 (d, 3JPC = 4.9 Hz, 2-,6-NAr), 132.6 (d, 2JPC = 13.8 Hz, 3-C6H4), 133.3 (m, o-,p-C6H5), 134.2 (d, 4JPC = 2.9 Hz, 5-C6H4), 147.8 (s, Pt–C). IR (cm–1): ν 566 (Pt–N), 838 (br, PF6–), 1298 (N=P). Conductivity: 106.5 μS/cm (acetone) (1:1 electrolyte). The stability of 5 in acidic media over time was studied by 1H and 31P{1H} NMR in a 2:1 DMSO-d6/PBS-1X(D2O) solution at pH 6. The PBS-1X solution was prepared using D2O as solvent and adjusting the pH to 7.4 by addition of 0.1 N HCl. The deuterated PBS-1X solution was then used to prepare a 2:1 DMSO-d6/PBS-1X(D2O) solution for which the pH was adjusted to 6 by addition of 0.1 N HCl. Complex 5 was then dissolved in the 2:1 DMSO-d6/PBS-1X(D2O) solution (pH 6). Compound 5 dissolved completely in this medium, affording a colorless solution. A 2:1 DMSO-d6/PBS-1X(D2O) solution at pH 7.4 was prepared to run control NMR experiments.
X-ray Crystallography
A gold block-like crystal with the size of 0.10 × 0.18 × 0.18 mm3 was selected for geometry and intensity data collection with a Bruker SMART APEXII CCD area detector on a D8 goniometer at 100 K. The temperature during the data collection was controlled with an Oxford Cryosystems Series 700+ instrument. Preliminary lattice parameters and orientation matrices were obtained from three sets of frames. Data were collected using graphite-monochromated and 0.5 mm MonoCap-collimated Mo Kα radiation (λ = 0.71073 Å) with the ω and φ scan method. Data were processed with the INTEGRATE program of the APEX2 software for reduction and cell refinement. Multiscan absorption corrections were applied by using the SCALE program for the area detector. The structure was solved by the direct method and refined on F2 (SHELXTL).2 Non-hydrogen atoms were refined with anisotropic displacement parameters, and hydrogen atoms were placed in idealized positions (C–H = 0.95–0.99 Å) and included as riding with Uiso(H) = 1.2 or 1.5 Ueq(non-H).
Cell Culture, Inhibition of Cell Growth, and Cell Death Analysis
MTT Toxicity Assays
For toxicity assays, cells (5 × 104 for Jurkat cells and 104 for adherent cell lines) were seeded in flat-bottom 96-well plates (100 μL/well) in complete medium. Adherent cells were allowed to attach for 24 h prior to addition of cisplatin or tested compounds. Compounds were added at different concentrations in triplicate. Cells were incubated with cisplatin or compounds for 24 h, and then cell proliferation was determined by a modification of the MTT-reduction method. Briefly, 10 μL/well of MTT (5 mg/mL in PBS) was added, and plates were incubated for 1–3 h at 37 °C. Finally, formazan crystal was dissolved by adding 100 μL/well iPrOH (0.05 M HCl) and gently shaking. The optical density was measured at 570 nm using a 96-well multiscanner autoreader (ELISA).
Cell Death Analysis
Apoptosis/necrosis hallmarks of cells treated with compound 5 were analyzed by measuring mitochondrial membrane potential, plasma membrane integrity, and exposure of phosphatidylserine. Cells were treated with different concentrations and at different incubation times as indicated in figure legends. In some experiments the general caspase inhibitor z-VAD-fmk was added at 50 μM, 1 h before compounds. For mitochondrial membrane potential determination, cells (2.5 × 105 in 200 μL) after treatment with 5 were incubated at 37 °C for 15 min in medium containing 5 nM DiOC6(3) (Molecular Probes). Phosphatidylserine exposure was quantified by labeling cells with annexin V-DY634 (Invitrogen) after treatment with 5. Annexin V was added at a concentration of 0.5 μg/mL in Annexin Binding Buffer (ABB), and cells were incubated at room temperature for 15 min. Plasma membrane integrity was evaluated by staining with 7-amino-actinomycin D (7-AAD, Inmunostep). At the end of the treatment with 5, cells were incubated for 15 min in 200 μL of PBS containing 50 ng/μL 7-AAD. In all cases, cells were diluted to 1 mL with ABB or phosphate buffered saline (PBS) to be analyzed by flow cytometry (FACScan, BD Bioscience, Spain).
Permeability Determinations
Cell Culture and Transport Assays
Caco-2 cells were grown in Dubelcco’s Modified Eagle’s Medium containing l-glutamine, fetal bovine serum, and penicillin–streptomycin. To obtain cells, monolayers of 250 000 cells/cm2 were seeded on each well with polycarbonate membrane with 4.2 cm2 area. Plates were incubated at standard conditions of 37 °C temperature, 90% humidity, and 5% CO2 until confluence. After 19–21 days, the integrity of the each cell monolayer was evaluated by measuring the trans-epithelial electrical resistance (TEER). Values ranging 500–750 Ω·cm2 were considered appropriate.
Transport studies were performed using an orbital environmental shaker at constant temperature (37 °C) and agitation rate (50 rpm). Hank’s balanced salt solution (HBSS) supplemented with HEPES was used to fill the receiver chamber and to prepare the drug solution placed in the donor chamber. Four samples of 200 μL each were taken from the receiver chamber side at predefined times (15, 30, 60, and 90 min) and replaced with the same volume of fresh buffer. Moreover, two samples of the donor side were taken at the beginning and the end of the experiment. The amount of compound in cell membranes and inside the cells was determined at the end of the experiments in order to check the mass balance, and the percentage of compound retained in the cell compartment was always less than 5%.
Transport studies were performed in both directions, from apical-to-basal (A-to-B) and from basal-to-apical (B-to-A) sides. The volume of donor compartment was 2 mL in A-to-B direction and 3 mL in B-to-A direction.
Analysis of the Samples
Samples were analyzed by HPLC using a 5 μm, 4 × 200 mm Novapack C18 column. Samples of cisplatin and compound 4 were analyzed with UV detection (λ = 240 nm). The mobile phase was 95:5 acetonitrile:water, with a flow rate of 1 mL/min, and the injected sample volume was 50 μL. Samples of compound 5 were analyzed similarly but using a UV detector at λ = 215 nm and a mobile phase of 80:20 acetonitrile:water.
Data Analysis
The apparent permeability coefficient was calculated following the equation
| 1 |
where Creceiver,t is concentration of compound in the receiver chamber at time t, Qtotal is the total amount of drug in both chambers, Vreceiver and Vdonor are the volumes corresponding to receiver and donor compartment, respectively, in each chamber, Creceiver,t–1 is the concentration of compound in the receiver chamber at the previous time, f is the sample dilution factor due to replaced volume, S is the surface area of the monolayer, Δt is the time interval, and P is the permeability coefficient. This equation takes into account the continuous change of the donor and receiver concentrations, i.e., non-sink conditions. However, when the transport rate is low, there are not significant changes between the donor and the receiver concentrations with time. Sink conditions are assumed, and a simpler expression can be used to estimate the permeability coefficient:
| 2 |
where dQ/dt is the apparent appearance rate of drug in the receiver side calculated using linear regression of amounts in the receiver chamber versus time, S is the surface area of the monolayer, and C is the drug concentration in the donor chamber.
The permeability coefficient estimations in sink and non-sink conditions were carried out in an Excel worksheet. Studies were performed in triplicate, and the data are presented as mean ± SD. Student’s t test was performed with SPSS 16.0 (SPSS Inc.) in order to determine statistically significant differences between A-to-B and B-to-A permeabilities.
In Situ Absorption Experiments
The absorption experiments were performed using a Doluisio in situ loop technique.52 The study was approved by the Scientific Committee of the Faculty of Pharmacy and followed the guidelines described in the EC Directive 86/609, the Council of the Europe Convention ETS 123, and Spanish national laws governing the use of animals in research (Real Decreto 223/1988, BOE 67, 18-3-98: 8509-8511). Male Wistar rats weighing 280–320 g were used after 8 h of fasting. Previously to surgical procedure, animals were anesthetized with diazepam (Valium, Roche) (1.67 mg/kg), ketamine (Ketolar, Parke-Davis) (50 mg/kg), and atropine (atropine sulfate, Braun) (1 mg/kg). The body temperature was maintained during the procedure by heating with a lamp. Therefore, a midline abdominal incision were performed, and a loop was isolated from the duodenal and ileal region of each rat. The proximal ligatures of the duodenal and ileal regions were placed approximately 1 cm from the pylorus and 2 cm above the ileocecal junction. The bile duct was tight up in all experiments. First, 50 mL of cleaning solution (Solution A (pH 7.4): 9.2 g of NaCl, 0.34 g of KCl, 0.19 g of CaCl2·H2O, and 0.76 g of NaH2PO4·2 H2O per liter) was used to flushed out the content of the loop, and then 20 mL of solution B (NaCl g, NaH2PO4·2H2O 1/15 M 3.9 mL, Na2HPO4 1/15 M 6.1 mL, and water up to 1 L) was perfused to condition the intestinal mucosa prior to the experiments. A catheter was tight up at both intestinal ends and connected to a glass syringe by the use of a stopcock type valve. Under this setup, the intestinal segment is an isolated compartment, and the drug solution can be perfused. The drug solutions were prepared freshly each day at 20 μM using solution B as solvent and perfused into the loop, and then the entire intestine was restored into the abdominal cavity. Samples of the perfusate were taken every 5 min for 30 min.
Permeability Calculations
The apparent first-order absorption rate coefficients (kapp) were obtained by nonlinear fitting of a monoexponential equation to the luminal concentrations versus time:
| 3 |
where C is the drug concentration remaining in the lumen, kapp is the apparent absorption rate constant, and C0 corresponds to a calculated fraction of the initial perfusion concentration. Test solutions suffer a slight dilution in the intestinal lumen due to the remaining cleaning solution, the adsorption to the membrane, and the loading process in the enterocyte. So, the intercept, C0, is lower than the perfusion concentration. The quasi-steady-state is achieved in the membrane when this process is finished. Under these conditions, the disappearance of the compound from the lumen can be considered as a first-order process during the sampling time interval. For these reasons, only the concentrations obtained after 5 min were used for regression analysis. In order to obtain good prediction data, water re-absorption correction was introduced for the concentration calculations.
The intestinal permeability values were calculated taking into account the relationship between ka and Peff:
| 4 |
where R is the radius of the intestinal segment, calculated as area/volume ratio. The effective intestinal permeabilities (Peff) of the tested compounds (means of at least of three animals) were used as indexes of the absorption effectiveness.
Interaction of Compounds 1–5 and Cisplatin with Plasmid (pBR322) DNA by Electrophoresis (Mobility Shift Assay)
First, 10 μL aliquots of pBR322 plasmid DNA (20 μg/mL) in buffer (5 mM Tris-HCl, 50 mM NaClO4, pH 7.39) were incubated with different concentrations of the compounds 1–5 (in the range 0.25–2.0 metal complex:DNAbp) at 37 °C for 20 h in the dark. Samples of free DNA and cisplatin–DNA were prepared as controls. After the incubation period, the samples were loaded onto 1% agarose gel. The samples were separated by electrophoresis for 1.5 h at 80 V in Tris-acetate/EDTA buffer (TAE). Afterward, the gel was stained for 30 min with a solution of GelRed Nucleic Acid stain.
Interaction of Compounds 3–5 and Cisplatin with Calf Thymus DNA by Circular Dichroism
Stock solutions (5 mM) of each complex were freshly prepared in water prior to use. The right volume of those solutions was added to 3 mL samples of an also freshly prepared solution of CT DNA (195 μM) in Tris-HCl buffer (5 mM Tris-HCl, 50 mM NaClO4, pH 7.39) to achieve molar ratios of 0.1, 0.25, 0.5, and 1.0 drug/DNA. The samples were incubated at 37 °C for a period of 20 h. All CD spectra of DNA and of the DNA–drug adducts were recorded at 25 °C over a range 220–330 nm and finally corrected with a blank and noise reduction. The final data are expressed in molar ellipticity (millidegrees).
Interaction of Compounds 1–5 and Cisplatin with Human Serum Albumin by Fluorescence Spectroscopy
A solution of each compound (8 mM) in DMSO was prepared, and 10 aliquots of 2.5 μL were added successively to a solution of HSA (10 μM) in phosphate buffer (pH 7.4) to achieve final metal complex concentrations in the range 10–100 μM. The excitation wavelength was set to 295 nm, and the emission spectra of HSA samples were recorded at room temperature in the range of 300–450 nm. The fluorescence intensities of all the metal compounds, the buffer, and the DMSO are negligible under these conditions. The fluorescence was measured 240 s after each addition of compound solution. The data were analyzed using the classical Stern–Volmer equation, F0/F = 1 + KSV[Q].
Acknowledgments
Research at Brooklyn College was supported by a grant from the National Cancer Institute (NCI), 1SC1CA182844 (M.C.), and a grant from the National Institute of General Medical Sciences (NIGMS), SC2GM082307 (M.C.). The Spanish authors thank the Ministerio de Economía y Competitividad project, SAF2010-1490 (I.M.), for support, and the Ministerio of Educacion y Ciencia/Universidad Miguel Hernandez for a Ph.D. fellowship, FPU AP2010-2372 (V.M.-S.). A.S. thanks the AECC (Asociación Española Contra el Cáncer) for a fellowship. We thank Mr. Tomer Madar (Brooklyn College undergraduate student at the time) for the initial preparation of compound 4 and Benelita T. Elie (Brooklyn College-Graduate Center CUNY Ph.D. student) for testing the IC50 of 5 in RPTC cell lines.
Glossary
Abbreviations Used
- ABB
Annexin Binding Buffer
- CD
circular dichroism
- COD
cyclooctadiene
- CT
calf thymus
- DMSO
dimethyl sulfoxide
- HSA
human serum albumin
- HBSS
Hank’s balanced salt solution
- HEPES
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- IM
iminophosphorane
- ITC
isothermal titration calorimetry
- MTT
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- PARP-1
Poly(ADP-ribose) Polymerase-1
- PBS
phosphate buffered saline
- PTA
1,3,5-triaza-7-phosphaadamantane
- RPTC
renal proximal tubular cells
- TAE
Tris-acetate/EDTA buffer
- TEER
trans-epithelial electrical resistance
- T-Jurkat
human acute lymphoblastic leukemia cells
- T-Jurkat sh Bak
human acute lymphoblastic leukemia cells which do not express the Bak gene
- XTT
2,3-bis(2-methoxy-4-nitro-5-sulfophenyl)-2H-tetrazolium-5-carboxanilide
Supporting Information Available
Crystallographic data for compounds 2 and 4, including complete drawing of the structure and table of selected distances and angles for compound 4; stability of compounds 1–5 by 31P{1H} spectroscopy in DMSO-d6 solution; selected 1H and 31P{1H} NMR spectra for compounds 1–5 in DMSO-d6; stability of compound 5 in DMSO-d6/PBS-1X (2:1) at pH 7.4 and pH 6 over time; stability of compounds 4 and 5 in DMSO:PBS (1:99) determined by vis–UV. The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jmedchem.5b00427.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Thayer A. M. Platinum drugs take their roll. Chem. Eng. News 2010, 882624–28 10.1021/cen-v088n026.p024. [DOI] [Google Scholar]
- Kelland L. The resurgence of platinum-based cancer chemotherapy. Nat. Rev. Cancer 2007, 7, 573–584 10.1038/nrc2167. [DOI] [PubMed] [Google Scholar]
- Medici S.; Peana M.; Nurchi V. M.; Lachowicz J. I.; Crisponi G.; Zoroddu M. A. Noble metals in medicine: latests advances. Coord. Chem. Rev. 2015, 284, 329–350 10.1016/j.ccr.2014.08.002. [DOI] [Google Scholar]
- Cullinane C.; Deacon G. B.; Drago P. R.; Hambley T. W.; Nelson K. T.; Webster L. K. Preparation and cell growth inhibitory activity of [PtR2L2] (R=polyfluorophenyl, L2=diene, cyclohexane-1,2-diamine (chxn) or cis-(dimethyl sulfoxide)2) and the X-ray crystal structure of [Pt(C6F5)2(cis-chxn)]. J. Inorg. Biochem. 2002, 89, 293–301 10.1016/S0162-0134(02)00363-X. [DOI] [PubMed] [Google Scholar]
- Leonidova A.; Gasser G. Underestimated potential of organometallic rhenium complexes as anticancer agents. ACS Chem. Biol. 2014, 9, 2180–2193 10.1021/cb500528c. [DOI] [PubMed] [Google Scholar]
- Bertrand B.; Casini A. A golden future in medicinal inorganic chemistry: the promise of anticancer gold organometallic compounds. Dalton Trans. 2014, 43, 4209–4219 10.1039/C3DT52524D. [DOI] [PubMed] [Google Scholar]
- Liu W.; Gust R. Metal N-heterocyclic carbene complexes as potential antitumor metallodrugs. Chem. Soc. Rev. 2013, 42, 755–773 10.1039/C2CS35314H. [DOI] [PubMed] [Google Scholar]
- Oehninger L.; Rubbiani R.; Ott I. N-heterocyclic carbene metal complexes in medicinal chemistry. Dalton Trans. 2013, 42, 3269–3284 10.1039/C2DT32617E. [DOI] [PubMed] [Google Scholar]
- Leung C.-H.; Zhong H.-J.; Chan D.S.-H.; Ma D. L. Bioactive iridium and rhodium complexes as therapeutic agents. Coord. Chem. Rev. 2013, 257, 1764–1776 10.1016/j.ccr.2013.01.034. [DOI] [Google Scholar]
- Noffke A. L.; Habtemariam A.; Pizarro A. M.; Sadler P. J. Designing organometallic compounds for catalysis and therapy. Chem. Commun. 2012, 48, 5219–5246 10.1039/c2cc30678f. [DOI] [PubMed] [Google Scholar]
- Therrien B. Drug delivery by water-soluble organometallic cages. Top. Curr. Chem. 2012, 319, 35–55 10.1007/128_2011_272. [DOI] [PubMed] [Google Scholar]
- Gasser G.; Ott I.; Metzler-Nolte N. Organometallic anticancer complexes. J. Med. Chem. 2011, 54, 3–25 10.1021/jm100020w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ang W. H.; Casini A.; Sava G.; Dyson P. J. Organometallic ruthenium-based antitumor compounds. J. Organomet. Chem. 2011, 696, 989–998 10.1016/j.jorganchem.2010.11.009. [DOI] [Google Scholar]
- Metzler-Nolte N. Biomedical applications of organometal-peptide conjugates. Top. Organomet. Chem. 2010, 32, 195–217 10.1007/978-3-642-13185-1_8. [DOI] [Google Scholar]
- Hillard E. A.; Vessieres A.; Jaouen G. Ferrocene functionalized endocrine modulators as anticancer agents. Top. Organomet. Chem. 2010, 32, 81–117 10.1007/978-3-642-13185-1_4. [DOI] [Google Scholar]
- Pizarro A. M.; Habtemarian A.; Sadler P. Activation mechanisms for organometallic anticancer complexes. Top. Organomet. Chem. 2010, 32, 21–56 10.1007/978-3-642-13185-1_2. [DOI] [Google Scholar]
- Olszewski U.; Hamilton G. Mechanisms of cytotoxicity of anticancer titanocenes. Anti-cancer Agents Med. Chem. 2010, 10, 302–311 10.2174/187152010791162261. [DOI] [PubMed] [Google Scholar]
- Grishagin I. V.; Pollock J. B.; Kushal S.; Cook T. R.; Stang P. T.; Olenyuk B. Z. In vivo anticacer activity of rhomboidal Pt(II) metallacycles. Proc. Natl. Acad. Sci. U. S. A. 2014, 111, 18448–18453 10.1073/pnas.1418712111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamora A.; Perez S. A.; Rodriguez V.; Janiak C.; Yellol G. S.; Ruiz J. On the Dual Antitumor and Anti-angiogenic Activity of Organoplatinum(II) Complexes. J. Med. Chem. 2015, 58, 1320–1336 10.1021/jm501662b. [DOI] [PubMed] [Google Scholar]
- Chen Z.-F.; Quin Q.-P.; Quin J.-L.; Liu Y.-C.; Huang K.-B.; Li Y.-L.; Meng T.; Zhang G.-H.; Peng Y.; Luo X.-J.; Liang H. Stabilization of G-quadruplex DNA, inhibition of telomerase activity and tumor cell apoptosis of organoplatinum(II) complexes with oxoisoaporphine. J. Med. Chem. 2015, 10.1021/jm40111t. [DOI] [PubMed] [Google Scholar]
- Butsch K.; Elmas S.; Gupta N. S.; Gust R.; Heinrich F.; Klein A.; von Mering Y.; Neugebauer M.; Ott I.; Schafer M.; Scherer H.; Schurr T. Organoplatinum(II) and -palladium(II) complexes of nucleobases and their derivatives. Organometallics 2009, 28, 3906. 10.1021/om900208r. [DOI] [Google Scholar]
- Klein A.; Schurr T.; Scherer H.; Gupta N. S. Cytosine Binding in the Novel Organoplatinum(II) Complex [(COD)PtMe(cytosine)](SbF6). Organometallics 2007, 26, 230. 10.1021/om060856y. [DOI] [Google Scholar]
- Klein A.; Luning A.; Ott L.; Hamel L.; Neugebauer M.; Butsch K.; Lingen V.; Heinrich F.; Elmas S. Organometallic palladium and platinum complexes with strongly donating alkyl coligands – Synthesis, structures, chemical and cytotoxic properties. J. Organomet. Chem. 2010, 695, 1898. 10.1016/j.jorganchem.2010.04.027. [DOI] [Google Scholar]
- Luning A.; Schur J.; Hamel L.; Ott I.; Klein A. Strong Cytotoxicity of Organometallic Platinum Complexes with Alkynyl Ligands. Organometallics 2013, 32, 3662. 10.1021/om400293u. [DOI] [Google Scholar]
- Luning A.; Neugebauer M.; Lingen V.; Krest A.; Stirnat K.; Deacon G. B.; Drago P. R.; Ott I.; Schur J.; Pantenburg I.; Meyer G.; Klein A. Platinum Diolefin Complexes-Synthesis, Structures, and cytotoxicity. Eur. J. Inorg. Chem. 2015, 2015, 226–239 10.1002/ejic.201402954. [DOI] [Google Scholar]
- Enders M.; Gorling B.; Braun A. B.; Seltenreich J. E.; Reichenbach L. F.; Rissanen K.; Nieger M.; Luy B.; Schepers U.; Brase S. Cytotoxicity and NMR studies of platinum complexes with cyclooctadiene ligands. Organometallics 2014, 33, 4027–4034 10.1021/om500540x. [DOI] [Google Scholar]
- Cutillas N.; Yellol G. S.; de Haro C.; Vicente C.; Rodriguez V.; Ruiz J. Anticancer cyclometalated complexes of platinum group metals and gold. Coord. Chem. Rev. 2013, 257, 2784–2797and references therein 10.1016/j.ccr.2013.03.024. [DOI] [Google Scholar]
- Omae I. Applications of five-membered ring products of cyclometalation reactions as anticancer agents. Coord. Chem. Rev. 2014, 280, 84–95and references therein 10.1016/j.ccr.2014.07.019. [DOI] [Google Scholar]
- To W.-P.; Zou T.; Sun R.W.-Y.; Che C.-M. Light-induced catalytic and cytotoxic properties of phosphorescent transition metal compounds with a d8 electronic configuration. Philos. Trans. R. Soc., A 2013, 371199520120126/1–20120126/19and references therein 10.1098/rsta.2012.0126. [DOI] [PubMed] [Google Scholar]
- Selected example:Casini A.; Kelter G.; Gabbiani C.; Cinellu M. A.; Minghetti G.; Fregona D.; Fiebig H.-H.; Messori L. Chemistry, antiproliferative properties, tumor selectivity, and molecular mechanisms of novel gold(III) compounds for cancer treatment: a systematic study. JBIC, J. Biol. Inorg. Chem. 2009, 14, 1139–1149 10.1007/s00775-009-0558-9. [DOI] [PubMed] [Google Scholar]
- Selected example:Sun R.W.-Y.; Lok C.-N.; Fong T.T.-H.; Li C.K.-L.; Yang Z. F.; Zou T.; Siu F.-M.; Che C.-M. A dinuclear cyclometalated gold(III)–phosphine complex targetingthioredoxin reductase inhibits hepatocellular carcinoma in vivo. Chem. Sci. 2013, 4, 1979–1988 10.1039/c3sc21972k. [DOI] [Google Scholar]
- Zou T.; Liu J.; Lum C. T.; Ma C.; Chan R.C.-T.; Lok C.-N.; Kwok W.-M.; Che C.-M. Luminescent cyclometalated platinum(II) complex forms emissive intercalating adducts with double-stranded DNA and RNA: differential emissions and anticancer activities. Angew. Chem., Int. Ed. 2014, 53, 10119–10123 10.1002/anie.201405384. [DOI] [PubMed] [Google Scholar]
- Shaik N.; Martínez A.; Augustin I.; Giovinazzo H.; Varela-Ramirez A.; Aguilera R.; Sanaú M.; Contel M. Synthesis of Apoptosis-Inducing Iminophosphorane Organogold(III) Complexes and Study of Their Interactions with Biomolecular Targets. Inorg. Chem. 2009, 48, 1577–1587 10.1021/ic801925k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vela L.; Contel M.; Palomera L.; Azaceta G.; Marzo I. Iminophosphorane-organogold(III) complexes induce cell death through mitochondrial ROS production. J. Inorg. Biochem. 2011, 105, 1306–1313 10.1016/j.jinorgbio.2011.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carreira M.; Calvo-Sanjuán R.; Sanaú M.; Zhao X.; Magliozzo R. S.; Marzo I.; Contel M. Cytotoxic Hydrophilic iminophosphorane coordination compounds of d8 metals. Studies of their Interactions with DNA and HSA. J. Inorg. Biochem. 2012, 116, 204–214 10.1016/j.jinorgbio.2012.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carreira M.; Calvo-Sanjuán R.; Sanaú M.; Marzo I.; Contel M. Organometallic Palladium Complexes with a Water-Soluble Iminophosphorane Ligand as Potential Anticancer Agents. Organometallics 2012, 31, 5772–5781 10.1021/om3006239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lease N.; Vasilevski V.; Carreira M.; de Almeida A.; Sanau M.; Hirva P.; Casini A.; Contel M. Potential Anticancer Heterometallic Fe-Au and Fe-Pd Agents: Initial Mechanistic Insights. J. Med. Chem. 2013, 56, 5806–5818 10.1021/jm4007615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frik M.; Jimenez J.; Vasilevski V.; Carreira M.; de Almeida A.; Gascon E.; Sanaú M.; Casini A.; Contel M. Luminescent iminophosphorane gold, palladium and platinum complexes as potential anticancer agents. Inorg. Chem. Front. 2014, 1, 231–241 10.1039/C4QI00003J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frik M.; Martinez A.; Elie B. T.; Gonzalo O.; Ramirez de Mingo D.; Sanaú M.; Sanchez-Delgado R.; Sadhukha T.; Prabha S.; Ramos J.; Marzo I.; Contel M. In vitro and in vivo evaluation of water-soluble iminophosphorane ruthenium(II) compounds. A potential chemotherapeutic agent for triple negative breast cancer. J. Med. Chem. 2014, 57, 9995–10012 10.1021/jm5012337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilpin K. J.; Linklater R.; Henderson W.; Nicholson B. K. Synthesis and characterization of isomeric cycloaurated complexes derived from the iminophosphorane PP3=NC(O)Ph. Inorg. Chim. Acta 2010, 363, 1021–1030 10.1016/j.ica.2009.12.019. [DOI] [Google Scholar]
- Brown S. D. J.; Henderson W.; Kilpin K. J.; Nicholson B. K. Orthomercurated and cycloaurated derivatives of the iminophosphorane Ph3P=NPh. Inorg. Chim. Acta 2007, 360, 1310. 10.1016/j.ica.2006.02.009. [DOI] [Google Scholar]
- Aguilar D.; Contel M.; Navarro R.; Urriolabeitia E. P. Organogold(III) Iminophosphorane Complexes as Efficient Catalysts in the Addition of 2-Methylfuran and Electron-Rich Arenes to Methyl Vinyl Ketone. Organometallics 2007, 26, 4604. 10.1021/om700453s. [DOI] [Google Scholar]
- Bar I.; Bernstein J. N(triphenylphosphoranylidene)benzamide. Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem. 1980, 36, 1962. 10.1107/S0567740880007650. [DOI] [Google Scholar]
- Contel M.; Stol M.; Casado M. A.; van Klink G. P. M.; Ellis D. D.; Spek A. L.; van Koten G. A Bis(ortho-amine)aryl–Gold(I) Compound as an Efficient, Nontoxic, Arylating Reagent. Organometallics 2002, 21, 4556. 10.1021/om020286b. [DOI] [Google Scholar]
- Bielsa R.; Larrea R.; Navarro R.; Soler T.; Urriolabeitia E. P. Synthesis, Structure, Reactivity, and Catalytic Activity of C,N- and C,N,N-Orthopalladated Iminophosphoranes. Eur. J. Inorg. Chem. 2005, 2005, 1724. 10.1002/ejic.200400907. [DOI] [Google Scholar]
- Ruiz J.; Lorenzo J.; Vicente C.; Lopez G.; Lopez-De-Luzuriaga J. M.; Monge M.; Aviles F. X.; Bautista D.; Moreno V.; Laguna A. New Palladium(II) and Platinum(II) Complexes with 9-Aminoacridine: Structures, Luminiscence, Theoretical Calculations, and Antitumor Activity. Inorg. Chem. 2008, 47, 6990–7001 10.1021/ic800589m. [DOI] [PubMed] [Google Scholar]
- Cutillas N.; Martinez A.; Yellol G. S.; Rodríguez V.; Zamora A.; Pedreno M.; Donaire A.; Janiak C.; Ruiz J. Anticancer C,N-cycloplatinated(II) complexes containing fluorinated phosphine ligands: synthesis, structural characterization and biological activity. Inorg. Chem. 2013, 52, 13529–13535 10.1021/ic401973k. [DOI] [PubMed] [Google Scholar]
- Galluzzi L.; Vitale I.; Abrams J. M.; Alnemri E. S.; Baehrecke E. M.; Blagosklonny M. V.; Dawson T. M.; El-Deiry W. S.; Fulda S.; Gottlieb E.; et al. Molecular definitions of cell death subroutines: recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Differ. 2012, 19, 107–120 10.1038/cdd.2011.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- López-Royuela N.; Pérez-Galán P.; Galán-Malo P.; Yuste V. J.; Anel A.; Susín S. A.; Naval J.; Marzo I. Different contribution of BH3-only proteins and caspases to doxorubicin-induced apoptosis in p53-deficient leukemia cells. Biochem. Pharmacol. 2010, 79, 1746–58 10.1016/j.bcp.2010.02.010. [DOI] [PubMed] [Google Scholar]
- Oltra-Noguera D.; Mangas-Sanjuan V.; Centelles-Sangüesa A.; Gonzalez-Garcia I.; Sanchez-Castaño G.; Gonzalez-Alvarez M.; Casabo V.; Merino V.; Gonzalez-Alvarez I.; Bermejo M. Variability of permeability estimation from different protocols of subculture and transport experiments in cell monolayers. J. Pharmacol. Toxicol. Methods 2015, 2671C21–32 10.1016/j.vascn.2014.11.004. [DOI] [PubMed] [Google Scholar]
- Mangas-Sanjuan V.; González-Álvarez I.; González-Álvarez M.; Casabó V. G.; Bermejo M. Modified nonsink equation for permeability estimation in cell monolayers: comparison with standard methods. Mol. Pharmaceutics 2014, 1151403–1414 10.1021/mp400555e. [DOI] [PubMed] [Google Scholar]
- Bermejo M.; Merino V.; Garrigues T. M.; Pla Delfina J. M.; Mulet A.; Vizet P.; Trouiller G.; Mercier C. Validation of a biophysical drug absorption model by the PATQSAR system. J. Pharm. Sci. 1999, 88, 398–405 10.1021/js980370+. [DOI] [PubMed] [Google Scholar]
- Mangas-Sanjuan V.; Oláh J.; Gonzalez-Alvarez I.; Lehotzky A.; Tőkési N.; Bermejo M.; Ovádi J. Tubulin acetylation promoting potency and absorption efficacy of deacetylase inhibitors. Br. J. Pharmacol. 2015, 1723829–40 10.1111/bph.12946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodríguez-Berna G.; Mangas-Sanjuán V.; Gonzalez-Alvarez M.; Gonzalez-Alvarez I.; García-Giménez J. L.; Díaz Cabañas M. J.; Bermejo M.; Corma A. A promising camptothecin derivative: Semisynthesis, antitumor activity and intestinal permeability. Eur. J. Med. Chem. 2014, 83, 366–373 10.1016/j.ejmech.2014.06.050. [DOI] [PubMed] [Google Scholar]
- Dabrowiak J. C.Metals in medicine; John Wiley and Sons, Ltd.: Chichester, UK, 2009; Chap. 4, pp 109–114. [Google Scholar]
- Liu H.-K.; Sadler P. Metal complexes as DNA intercalators. Acc. Chem. Res. 2011, 44, 349–359 10.1021/ar100140e. [DOI] [PubMed] [Google Scholar]
- Che C.-M.; Sun R. W.-Y. Therapeutic applications of gold complexes: lipophilic gold(III) cations and gold(I) complexes for anti-cancer treatment. Chem. Commun. 2011, 47, 9554–9560and references therein 10.1039/c1cc10860c. [DOI] [PubMed] [Google Scholar]
- Li C.K.-L.; Sun R.W.-Y.; Kui S.C.-F.; Zhu N.; Che C.-M. Anticancer Cyclometalated [AuIIIm(C^N^C)mL]n+ Compounds: Synthesis and Cytotoxic Properties. Chem. - Eur. J. 2006, 12, 5253. 10.1002/chem.200600117. [DOI] [PubMed] [Google Scholar]
- Timerbaev A. R.; Hartinger C. G.; Aleksenko S. S.; Keppler B. K. Interactions of antitumor metallodrugs with serum proteins: advances in characterization using modern analytical methodology. Chem. Rev. 2006, 106, 2224–2248 10.1021/cr040704h. [DOI] [PubMed] [Google Scholar]
- Sherman S. E.; Lippard S. J. Structural aspects of platinum anticancer drug interactions with DNA. Chem. Rev. 1987, 87, 1153–1181 10.1021/cr00081a013. [DOI] [Google Scholar]
- McGregor T. D.; Bousfield W.; Qu Y.; Farrell N. Circular dichroism study of the irreversibility of conformational changes induced by polyamine-linked dinuclear platinum compounds. J. Inorg. Biochem. 2002, 91, 212–219 10.1016/S0162-0134(02)00398-7. [DOI] [PubMed] [Google Scholar]
- Ueda K.; Makino R.; Tobe T.; Okamoto Y.; Kojima N. Effects of organic and inorganic mercury(II) on gene expression via DNA conformational changes. Fundam. Toxicol. Sci. 2014, 1, 73–79 10.2131/fts.1.73. [DOI] [Google Scholar]
- Walter A.; Luck G. Interactions of Hg(ll) ions with DNA as revealed by CD measurements. Nucleic Acids Res. 1977, 4, 539–550 10.1093/nar/4.3.539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shahabadi N.; Heidari L. Synthesis, characterization and multi-spectroscopic DNA interaction studies of a new platinum complex containing the drug metformin. Spectrochim. Acta, Part A 2014, 128, 377–385 10.1016/j.saa.2014.02.167. [DOI] [PubMed] [Google Scholar]
- Vaidyanathan G.-V.; Nair U.-B. Synthesis, characterization, and binding studies of chromium(III) complex containing an intercalating ligand with DNA. J. Inorg. Biochem. 2003, 95, 334–342 10.1016/S0162-0134(03)00132-6. [DOI] [PubMed] [Google Scholar]
- Marcon G.; O’Connell T.; Orioli P.; Messori L. Comparative analysis of [Au(en)2]3+ and [Pt(en)2]2+ non covalent binding to calf thymus DNA. Metal Based Drugs 2000, 7, 253–256 10.1155/MBD.2000.253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacowicz J. R.Principles of Fluorescence Spectroscopy; Kluwer Academic/Plenum Publishers: New York, 1999; Chap. 8, pp 238–264. [Google Scholar]
- Kaesz H. D. Benzylpentacarbonylmanganese. Inorg. Synth. 1989, 26, 172. 10.1002/9780470132579. [DOI] [Google Scholar]
- Bittner S.; Assaf Y.; Krief P.; Pomerantz M.; Ziemnicka B. T.; Smith C. G. Synthesis of N-acyl, N-sulfonyl, and N-phosphinylphospha-λ5-azenes by a redox-condensation reaction using amides, triphenylphosphine, and diethyl azocarboxylate. J. Org. Chem. 1985, 50, 1712. 10.1021/jo00210a027. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.