

Significance
Commensal microbes affect autoimmunity, but it is not clear how. Type 1 diabetes is an organ-specific autoimmune disorder, and it too can be influenced by commensal microbiota. However, the complexity of the microbiota makes it difficult to connect specific microbes with disease progression or prevention. Studies of signaling pathways that microbes stimulate may shed light on disease pathogenesis and provide tools to interfere with it. We found that different Toll-like receptors can induce both pro- and antidiabetogenic signals. Both signals are triggered by commensal microbes, but the former signals control the microbiota, whereas the latter induce tolerance to self-antigens. Identification of signaling pathways that link commensal microbes with autoimmunity shortens the path to intervention with autoimmunity.
Keywords: Toll-like receptors, commensal microbiota, type 1 diabetes
Abstract
Deletion of the innate immune adaptor myeloid differentiation primary response gene 88 (MyD88) in the nonobese diabetic (NOD) mouse model of type 1 diabetes (T1D) results in microbiota-dependent protection from the disease: MyD88-negative mice in germ-free (GF), but not in specific pathogen-free conditions develop the disease. These results could be explained by expansion of particular protective bacteria (“specific lineage hypothesis”) or by dominance of negative (tolerizing) signaling over proinflammatory signaling (“balanced signal hypothesis”) in mutant mice. Here we found that colonization of GF mice with a variety of intestinal bacteria was capable of reducing T1D in MyD88-negative (but not wild-type NOD mice), favoring the balanced signal hypothesis. However, the receptors and signaling pathways involved in prevention or facilitation of the disease remained unknown. The protective signals triggered by the microbiota were revealed by testing NOD mice lacking MyD88 in combination with knockouts of several critical components of innate immune sensing for development of T1D. Only MyD88- and TIR-domain containing adapter inducing IFN β (TRIF) double deficient NOD mice developed the disease. Thus, TRIF signaling (likely downstream of Toll-like receptor 4, TLR4) serves as one of the microbiota-induced tolerizing pathways. At the same time another TLR (TLR2) provided prodiabetic signaling by controlling the microbiota, as reduction in T1D incidence caused by TLR2 deletion was reversed in GF TLR2-negative mice. Our results support the balanced signal hypothesis, in which microbes provide signals that both promote and inhibit autoimmunity by signaling through different receptors, including receptors of the TLR family.
It has been established that the commensal microbiota is an important environmental factor that regulates autoimmunity. The range of microbial influences varies with the type of autoimmunity from protection (1, 2), to no influence (3), to stimulation (4–6). Additionally, it has been demonstrated that host genetics plays an important role in shaping the microbiota (7). Our previous report (8) has shown that type 1 diabetes (T1D)-prone nonobese diabetic (NOD) mice that have lost signaling by the majority of Toll-like receptors (TLRs), due to genetic ablation of the myeloid differentiation primary response gene 88 (MyD88) adaptor protein, were protected from the disease. The protection required microbes, because germ-free (GF) MyD88-negative mice did develop T1D. The original explanation for these observations was that in the absence of MyD88 some particular microbial lineages were amplified and induced tolerogenic signaling. The hypothesis was later termed the “specific lineage hypothesis” (9). However, we also recognized that microbes stimulate both the microbial control mechanisms of the host and mechanisms that reduce the host’s control over the microbiota. It is possible that the balance of these signals (“balanced signal hypothesis”) is important for the initiation of autoimmunity (9). This hypothesis assumes that the negative regulatory mechanisms that pacify the antimicrobial responses also lead to tolerance to self-antigens. It also suggests that given the richness of the normal microbiota, the role of specific lineages may not be crucial in eliciting tolerizing signals. The type of receptors and signaling pathways involved in microbiota-dependent protection from T1D remained unknown.
Here, we sought to identify such pathway(s). We used genetic approaches, in which we tested NOD mice deficient in different innate signaling pathways for their ability to develop T1D as well as when these deficiencies were combined with the lack of MyD88 signaling. The results suggest that both pro- and antiinflammatory signaling in T1D is primarily regulated by TLRs.
Results
Distinct Bacterial Lineages Can Reduce Diabetes Incidence in Gnotobiotic MyD88-Negative NOD Mice.
The alternative to the balanced signal hypothesis, which explains the microbiota dependence of autoimmunity development by the competition of host-driven and microbe-driven mechanisms, and which is heavily influenced by the genetics of the host, is the specific microbial lineage hypothesis (9). The latter suggests that because genetic variations of the host can affect microbial communities (7), the expanded lineages can potentially affect the course of autoimmunity development. In fact, MyD88-negative NOD mice did show statistically significant expansion of several bacterial lineages (8), including Lactobacillacae. Because gnotobiotic NOD mice are the most useful tools for testing a putative role of bacterial lineages in T1D development, we colonized GF NOD and GF NOD MyD88-deficient mice with a probiotic mix, VSL3, containing several strains of Lactobacillacae. Histopathology of the islets was assessed at the age of 13 wk (Fig. 1). We have previously established (10) that percentage of islets with bona fide insulitis at this age quite accurately predicts overt diabetes development in the respective animal cohort. Colonization with VSL3 did not affect insulitis in the wild-type NOD mice, but suppressed it in MyD88-negative gnotobiotic NOD mice. Moreover, we have previously found (8) that a bacterial community termed “altered Schaedler’s flora” (ASF) has reduced T1D incidence and histopathology (Fig. 1) in colonized NOD MyD88-deficient mice, suggesting that various bacterial lineages can reduce T1D in MyD88-negative mice. Whereas VSL3 is a probiotic based on human isolates, ASF is a community of microbes normally present in the mouse gut. In addition to these bacterial communities, we introduced a single microbe, segmented filamentous bacteria (SFB) into GF NOD mice. SFB also reduced T1D incidence (7) and histopathology at 13 wk (Fig. 1) in MyD88-negative mice. Thus, multiple and very different bacterial lineages can contribute to protection of MyD88-negative, but not wild-type NOD mice. None of these lineages offered a complete protection compared with specific pathogen-free (SPF) MyD88-negative animals, indicating that more than one pathway, likely triggered by different microbial agonists, could contribute to protection when the adaptor MyD88 is disabled. These results argued in favor of the balanced signal hypothesis.
Fig. 1.

Distinct bacterial lineages reduce diabetes incidence in gnotobiotic MyD88-negative NOD mice. Bona fide insulitis (percentage of total islets) in 13-wk-old female MyD88+/+ or MyD88−/− mice in SPF and GF conditions and colonized with ASF, VSL3, or SFB. n = number of mice per group. P values for histopathology were determined by Student’s t test. *P < 0.05, **P < 0.01, ***P < 0.001.
Testing the Balanced Signal Hypothesis: Several Non-TLR Bacteria-Sensing Mechanisms Do Not Protect from Diabetes.
To test the balanced signal hypothesis, it was important to uncover MyD88-independent bacteria-sensing mechanisms that may contribute to protection from T1D. We used a genetic approach in which we first tested a specific pathway for its requirement for T1D development per se, and then determined whether its inactivation rescues T1D development in MyD88-deficient NOD mice.
Signaling through a complex of signaling molecules termed “the inflammasome” is required to produce effector cytokines. IL1/IL18-induced signaling had previously been shown to be dispensable for T1D development (11) in caspase 1-negative NODs. Because receptors for IL1 and IL18 also signal through MyD88, the classical processing of these cytokines by inflammasome-activated caspase 1 cannot lead to protection from T1D in MyD88-negative NOD mice. However, the caspase 1 knockout has been shown to be a double knockout: the caspase 11 gene has also been inactivated (12). Caspase 4/11 oligomers can bind LPS and activate yet another MyD88-independent signaling cascade (13). Thus, we have crossed Casp1/11−/− mice to MyD88−/− NOD mice to produce double knockout animals (Fig. 2A). The deletion of caspase 11 did not change the outcome of disease development: double knockout mice were still protected from T1D, indicating that the LPS–caspase 11 pathway is not involved in the microbiota-induced protection.
Fig. 2.

Non-TLR bacteria-sensing mechanisms do not protect from diabetes. (A) Diabetes incidence in NOD female caspase 1/11+/−, caspase 1/11−/− littermates, and MyD88−/− caspase 1/11+/−, MyD88−/− caspase 1/11−/− littermates. (B) Diabetes incidence in NOD female Ripk2+/−, Ripk2−/− littermates and MyD88−/− Ripk2+/−, MyD88−/− Ripk2−/− littermates. n = number of mice per group. P values for incidence were determined using Kaplan–Meier statistics. *P < 0.05, **P < 0.01, ***P < 0.001. ns, nonsignificant, P > 0.05.
The NOD-like receptor (NLR) family is another group of intracellular receptors that represent a key component of the innate immune system. NOD1 and NOD2 recognize the bacterial cell wall component peptidoglycan. Ligand-bound NOD1 and NOD2 oligomerize and signal through the serine-threonine RIP2 kinase via CARD–CARD homophilic interactions (14). Upon activation, RIP2 mediates ubiquitination of NEMO/IKKγ, which results in the activation of NF-κB and the production of inflammatory cytokines. To test the role of NLRs in generating the protective signal from the microbiota, we have backcrossed Ripk2-negative C57BL/6 mice (14) to NOD mice for 10 generations. Importantly, NOD Ripk2-negative mice were sensitive to T1D (Fig. 2B) similarly to wild-type NODs or heterozygous controls. We then bred Ripk2-negative mice to MyD88-deficient NOD mice. The double knockout of Ripk2 and MyD88 was indistinguishable from the single MyD88 knockout. Thus, we concluded that the protective signal in the absence of MyD88 was not a result of signaling through the RIP2-dependent NLR family of innate immune receptors.
Testing the Balanced Signal Hypothesis: The Signaling Adaptor TRIF Participates in Microbiota-Dependent Protection from T1D.
TLRs signal through two alternatively used adaptors, MyD88 and TRIF (encoded by the Ticam1 gene). Some reports have suggested that TRIF signaling could be a negative regulator of immunity (15–17). Thus, we have crossed TRIF-negative mice to the NOD strain for 10 generations and then to MyD88-negative NOD mice. TRIF deficiency did not affect T1D in MyD88-sufficient NOD mice, but reversed the protection significantly (although partially) in double-negative mice (Fig. 3A). Given the level of protection offered by MyD88 deficiency in SPF mice (100% at the University of Chicago facility), even 25% reversal (at 30 wk of age) was significant and important. This conclusion was strengthened by examination of islet histopathology at 30 wk of age (Fig. 3 B–D). A significantly higher number of islets with insulitis were observed in double knockout mice compared with MyD88 single knockout animals (Fig. 3B). TRIF+/− littermates of double-negative mice were treated as a separate group to ensure that the same input microbiota was acquired by experimental and control animals. Additionally, histological examination was scored to include the numbers of intact islets and islets with periinsulitis (Fig. 3C and Fig. S1). Fig. 3D shows examples of islet pathology in four groups of mice (NOD, NOD.MyD88−/−, NOD.TRIF−/−, and NOD.MyD88−/−TRIF−/−. Thus, signaling through TRIF, which is linked to TLR4 and TLR3 upstream receptors, contributes to protection from T1D in MyD88-deficient mice.
Fig. 3.

Signaling adaptor TRIF participates in microbiota-dependent protection from T1D. (A) Diabetes in NOD female TRIF+/−, TRIF−/− littermates and MyD88−/− TRIF+/−, MyD88−/− TRIF−/− littermates. (B) Bona fide insulitis (percentage of total islets) in diabetic or nondiabetic 30-wk-old female mice: NOD.MyD88+/+ TRIF+/− and NOD.MyD88+/+ TRIF−/− littermates, NOD.MyD88−/−TRIF+/+, as well as NOD.MyD88−/− TRIF+/− and NOD.MyD88−/− TRIF−/− littermates. (C) Full histopathology scores from mice shown in B. No insulitis (score 0), periinsulitis (score I), and insulitis (score II). (D) Representative images of islet histology in the nondiabetic 30-wk-old mice. n = number of mice per group. P values for incidence were determined using Kaplan–Meier statistics, for histopathology by Student’s t test. *P < 0.05, **P < 0.01, ***P < 0.001.
Fig. S1.
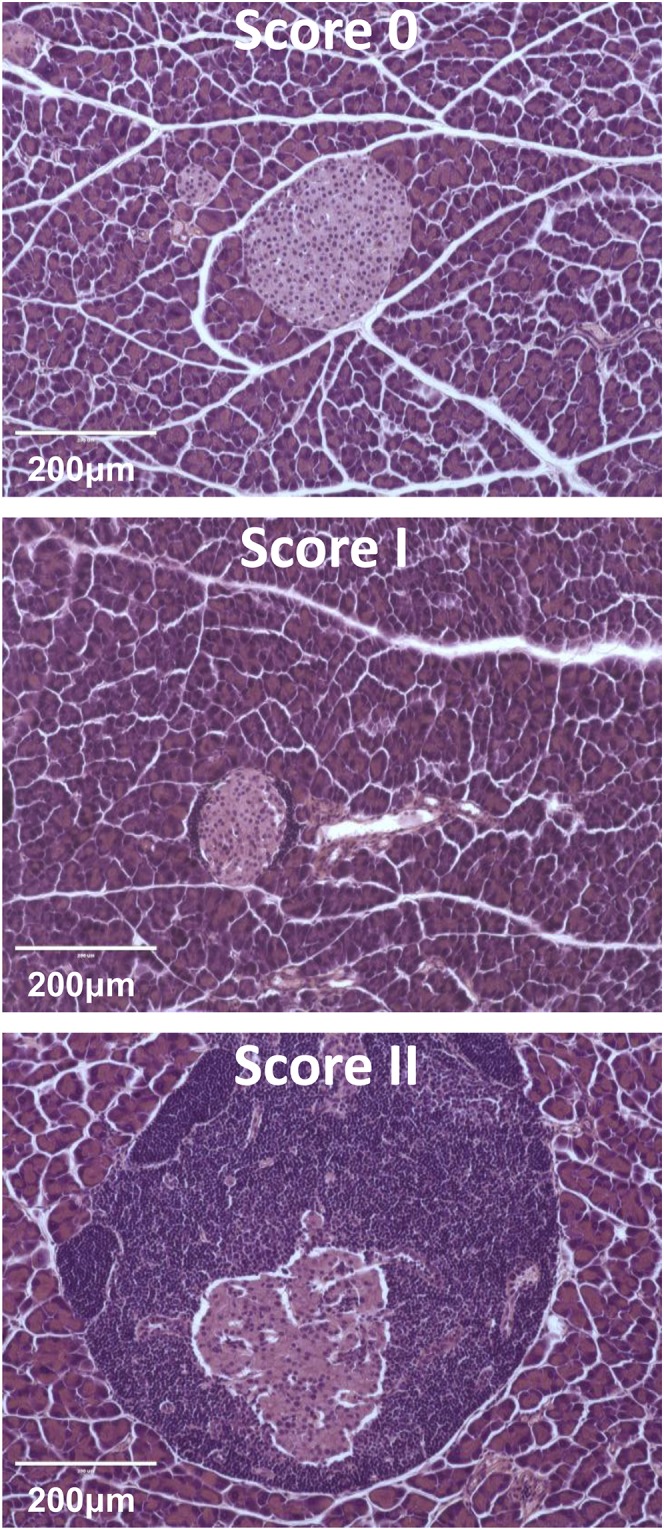
Histopathology scoring examples. Uninfiltrated islets (score 0), periinsulitis (score I), and insulitis (score II), which includes islets with variable degrees of destruction, are shown.
Testing the Balanced Signal Hypothesis: Antimicrobial TLR-Dependent Mechanisms Favoring T1D Development in NOD Mice.
To explain the protective role of the loss of MyD88 in SPF mice, we suggest that MyD88-dependent antimicrobial mechanisms are normally balancing out the pacifying signals from the commensal microbiota. Efficient T1D development in caspase 1-negative NOD mice (11) has indicated that the MyD88-dependent antimicrobial pathways in NOD mice were not based on signaling through IL1 or IL18 receptors, which involve MyD88, but rather on TLRs. Moreover, previous studies (8, 18) found that genetic ablation of different TLRs had different effects on T1D development in SPF mice. In particular, genetic ablation of TLR9 (18) and TLR2 (19) partially reduced T1D incidence. The loss of T1D in TLR2- and TLR9-deficient mice could be explained by either their direct contribution to T1D development or by their input in the overall control over microbes that reduce the microbial negative regulation of T1D. To discriminate between these possibilities, we have rederived TLR2-negative NOD mice into GF conditions and followed T1D development in these mice. TLR2-negative SPF mice had lost about one-half of their capacity to cause T1D in SPF mice as expected. However, GF TLR2-negative mice developed T1D with high incidence (Fig. 4A) and had increased histopathology (Fig. 4 B and C) compared with SPF TLR2-negative NOD. Thus, the prodiabetic effect of TLR2 was indirect and its knockout allowed microbes to reduce T1D incidence.
Fig. 4.

Antimicrobial TLR-dependent mechanisms favor T1D development in NOD mice. (A) Diabetes incidence in NOD SPF and GF female TLR2 +/− and TLR2−/− littermates. (B) Bona fide insulitis in diabetic or 30-wk-old nondiabetic female mice housed in SPF or GF conditions. NOD.TLR2+/− and NOD.TLR2−/− littermates were used in these experiments. (C) Full histopathology scores in mice shown in B. (D) Diabetes incidence in female NOD SPF and GF TLR4+/− and TLR4−/− littermates. The difference between SPF TLR4+/− (n = 21) and SPF TLR4−/− (n = 17) was significant (P = 0.04). The difference between GF TLR4+/− (n = 11) and GF TLR4−/− (n = 10) was not significant (P = 0.84). n = number of mice per group. P values for incidence were determined using Kaplan–Meier statistics, for histopathology by Student’s t test. *P < 0.05, **P < 0.01, ***P < 0.001.
Interestingly, previous reports found that ablation of TLR3 had no effect on T1D (18), whereas the knockout of TLR4 shifted T1D development curves toward earlier time points (20, 21) promoting the disease. These results suggested that TLR4 was activating a MyD88-independent (MyD88 knockout had the opposite result) signaling pathway that favored tolerance induction rather than disease promotion. To test whether TLR4 promoted tolerogenic signaling in a microbiota-dependent manner, we rederived NOD TLR4-negative mice into GF conditions and followed T1D incidence comparing it to the incidence in SPF conditions (Fig. 4D). As expected, TLR4-negative SPF mice demonstrated some acceleration of T1D, but in GF conditions they did not differ from TLR4-sufficient mice. These results indicate that TLR4 signaling actually represents a protective signal from the microbiota to reduce T1D development. On the other hand, TLR2 (and possibly TLR9, which has not been tested in GF conditions) contributes to antimicrobial control and thus indirectly to T1D development.
Discussion
Microbial regulation of autoimmunity may be explained by several potential mechanisms. These include molecular mimicry, which is based on cross-reactivity of effector lymphocytes to microbial and host peptides or proteins (in the case of antibodies); bystander activation of “adaptive” autoimmunity by antigen-presenting cells (APCs) activated by microbial stimuli and expressing self-antigens; and interference with the process of activation of innate immune mechanisms (4, 22–25). The dissection of mechanisms by which microbes can influence the development of autoimmunity is not a simple task. There are several levels of complexity. First, host-commensal symbiosis is generally mutualistic, but the loss of innate control mechanisms leads to damage by symbiotic microbes (26, 27), indicating that this type of relationship is tightly controlled. Second, microbial communities are very complex, making it difficult to identify specific lineages that could be beneficial or detrimental. Moreover, it could be specific fraternities of microbes and not specific individual lineages that exert regulatory functions in autoimmunity. Finally, microbial communities are variable between individuals, as well as between animal colonies (and over time within the same colony), or even individual cages within the same facility. These factors make the task of assigning an important role in autoimmunity to particular lineages in humans and experimental animals extremely difficult.
Because the principal goal of studies of the microbiota in autoimmunity is the ability to influence disease development by either manipulating the microbiota or by manipulating the signaling pathways that the microbiota uses to regulate autoimmunity, we reasoned that identification of the host pathways and their agonists would be the most feasible approach to address the problem. For that reason, the current study was focused on testing the role of known innate signaling receptors in microbial-mediated manipulation of T1D.
Our results indicate that TLRs play a critical role in the microbial contribution to T1D development. The presence of commensal microbes is not required for T1D development in rodents, as demonstrated for both mice and rats (8, 28, 29). Thus, TLRs play regulatory rather than causative roles. As shown here, a prodiabetic MyD88-dependent signal is provided in part through activation of TLR2, and this prodiabetic activity is lost in GF conditions. Previous work also identified TLR9 as sustaining disease incidence in SPF conditions. Thus, observation under GF conditions of TLR9 and double TLR9/2 knockouts under SPF conditions would be important to determine whether TLR2 and TLR9 together are sufficient to promote T1D by curbing tolerizing signals induced by the microbiota.
Importantly, here we report that TRIF signaling provides a protective signal in T1D. This finding suggests that protective signaling occurs by activation of either TLR3 or TLR4. The facts that TLR4-negative SPF mice (20), but not TLR3-negative SPF mice (18), have accelerated T1D and that GF TLR4-negative mice have normal T1D incidence, argue in favor of TLR4/TRIF signaling being a negative regulatory pathway and protective against T1D. TLR4 has been shown to mediate negative regulation of immunity against a retrovirus, which triggers TLR4 with bacterial LPS acquired in the gut (30). Further validation of the role of these receptors in a straightforward experiment (production of double knockouts with MyD88) is complicated by low survival rates of such double knockout mice after birth. An alternative experiment with tissue-specific conditional removal of these receptors may be more fruitful. It has to be acknowledged that the removal of TRIF in MyD88-negative mice provides significant restoration of insulitis and only partial restoration of overt T1D. This could be explained by involvement of other negative signaling pathways in addition to TRIF. A similar conclusion can be drawn from experiments with gnotobiotic mice lacking the MyD88 adaptor, which were protected from T1D to various degrees by various bacterial lineages, but never completely compared with the SPF MyD88-negative NOD mice (Fig. 1 and refs. 8, 10).
In sum, evidence collected so far implicates TLRs in both prodiabetic and antidiabetic regulatory mechanisms. Additional experiments are required to determine other important regulatory pathways and the cell types critical for protection from T1D by commensal microbes. These advances will bring us closer to the potential development of novel therapeutic approaches for autoimmunity.
Materials and Methods
Mice.
NOD/ShiLtJ (The Jackson Laboratory) mice were kept under SPF and GF conditions at the University of Chicago Animal Resource Center. TLR4-negative C57BL/10ScN mice were originally purchased from the NIH and backcrossed to NOD/ShiLtJ mice for more than 10 generations. TLR2 B6 mice were a gift from S. Akira, Osaka University, Osaka, and were backcrossed to NOD/ShiLtJ for more than 10 generations. Caspase 1/11-negative NOD mice were purchased from The Jackson Laboratory. C57BL/6J-Ticam1Lps2/J mice purchased from The Jackson Laboratory and B6.129S1-Ripk2tm1Flv were backcrossed to NOD/ShiLtJ for over 10 generations and intercrossed to produce knockout and heterozygous animals for observation of diabetes incidence and histologic examination of islet infiltration.
The chromosomal locations of the targeted genes were as follows: MyD88, Chr9(119335934–119341411); TRIF(Ticam1), Chr17 (56269319–56276786); Casp1, Chr9 (5298517–5307265); Tlr2, Chr3 (83836272–83841767); Tlr4, Chr4 (66827584–66930284); and Ripk2, Chr4 (16122733–16163647) according to the Mouse Genome Informatics database (www.informatics.jax.org/genes.shtml).
GF animals were rederived from NOD/ShiLtJ females impregnated by TLR2−/− and TLR4−/− NOD males and kept GF at the University of Chicago. GF status of mice was monitored by bacterial culture, aerobic and anaerobic fecal cultures. Additionally, PCR amplification of bacterial 16S rRNA genes from fecal DNA was performed as previously described (30). VSL3 was introduced to GF NOD mice by gavaging GF NOD mice with VSL3 probiotic mix in 1× PBS.
SPF mice were fed 7913 NIH-31 modified open formula mouse autoclavable diet (Harlan Laboratories). GF mice received autoclaved LabDiet 5K67. All experiments were performed in accordance with both The University of Chicago Animal Care and Use Committee and national guidelines.
Diabetes Testing.
Diabetes development was monitored from 10 wk of age by weekly testing of urine glucose with Diastix strips (Bayer). Mice were considered diabetic following two consecutive tests with urinary glucose concentrations over 500 mg/dL.
Histopathology of Diabetes.
Pancreas tissue was removed from mice, and hematoxylin and eosin–stained sections were made for histological analysis. The severity of islet inflammation was determined by scoring the degree of the inflammatory cellular infiltration. Briefly, at least 100 islets per group of animals were scored with pancreatic sections cut at 40-μm intervals and graded as follows: 0, no visible infiltration; I, periinsulitis; and II, bona fide insulitis. At 13 wk of age, only the percentage of islets with bona fide insulitis was found to predict overt diabetes (10).
Statistical Analysis.
Statistical analysis of diabetes incidence was performed by Kaplan–Meier with Prism 5 (GraphPad). Statistical analysis of histology scoring was performed with Prism 5. Results are expressed as means ± SEM. The statistical difference between two groups was determined by Student’s t test. For multiple groups, the statistical difference was determined with one-way ANOVA. A P value of <0.05 was considered statistically significant.
Acknowledgments
We thank the University of Chicago Gnotobiotic research facility staff: Betty Theriault, Kristin Kolar, and Alan Vest and Dr. Claudio De Simone for his help with VSL3. A.V.C. is supported by NIH Grant AI082418, Juvenile Diabetes Research Foundation Grant 17-2011-519, and NIH/National Institute of Diabetes and Digestive and Kidney Diseases Digestive Disease Research Core Center Grant DK42086. M.P.B. was supported by the Clinical Translational Science Award Training Grant TL1TR000432.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1508740112/-/DCSupplemental.
References
- 1.Markle JGM, et al. Sex differences in the gut microbiome drive hormone-dependent regulation of autoimmunity. Science. 2013;339(6123):1084–1088. doi: 10.1126/science.1233521. [DOI] [PubMed] [Google Scholar]
- 2.Kriegel MA, et al. Naturally transmitted segmented filamentous bacteria segregate with diabetes protection in nonobese diabetic mice. Proc Natl Acad Sci USA. 2011;108(28):11548–11553. doi: 10.1073/pnas.1108924108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gray DHD, Gavanescu I, Benoist C, Mathis D. Danger-free autoimmune disease in Aire-deficient mice. Proc Natl Acad Sci USA. 2007;104(46):18193–18198. doi: 10.1073/pnas.0709160104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ivanov II, et al. Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell. 2009;139(3):485–498. doi: 10.1016/j.cell.2009.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mazmanian SK, Round JL, Kasper DL. A microbial symbiosis factor prevents intestinal inflammatory disease. Nature. 2008;453(7195):620–625. doi: 10.1038/nature07008. [DOI] [PubMed] [Google Scholar]
- 6.Wu H-J, et al. Gut-residing segmented filamentous bacteria drive autoimmune arthritis via T helper 17 cells. Immunity. 2010;32(6):815–827. doi: 10.1016/j.immuni.2010.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Goodrich JK, et al. Human genetics shape the gut microbiome. Cell. 2014;159(4):789–799. doi: 10.1016/j.cell.2014.09.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wen L, et al. Innate immunity and intestinal microbiota in the development of Type 1 diabetes. Nature. 2008;455(7216):1109–1113. doi: 10.1038/nature07336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chervonsky AV. Microbiota and autoimmunity. Cold Spring Harb Perspect Biol. 2013;5(3):a007294. doi: 10.1101/cshperspect.a007294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yurkovetskiy L, et al. Gender bias in autoimmunity is influenced by microbiota. Immunity. 2013;39(2):400–412. doi: 10.1016/j.immuni.2013.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schott WH, et al. Caspase-1 is not required for type 1 diabetes in the NOD mouse. Diabetes. 2004;53(1):99–104. doi: 10.2337/diabetes.53.1.99. [DOI] [PubMed] [Google Scholar]
- 12.Kayagaki N, et al. Non-canonical inflammasome activation targets caspase-11. Nature. 2011;479(7371):117–121. doi: 10.1038/nature10558. [DOI] [PubMed] [Google Scholar]
- 13.Kayagaki N, et al. Noncanonical inflammasome activation by intracellular LPS independent of TLR4. Science. 2013;341(6151):1246–1249. doi: 10.1126/science.1240248. [DOI] [PubMed] [Google Scholar]
- 14.Kobayashi K, et al. RICK/Rip2/CARDIAK mediates signalling for receptors of the innate and adaptive immune systems. Nature. 2002;416(6877):194–199. doi: 10.1038/416194a. [DOI] [PubMed] [Google Scholar]
- 15.Seregin SS, et al. TRIF is a critical negative regulator of TLR agonist mediated activation of dendritic cells in vivo. PLoS One. 2011;6(7):e22064. doi: 10.1371/journal.pone.0022064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Baetz A, Frey M, Heeg K, Dalpke AH. Suppressor of cytokine signaling (SOCS) proteins indirectly regulate toll-like receptor signaling in innate immune cells. J Biol Chem. 2004;279(52):54708–54715. doi: 10.1074/jbc.M410992200. [DOI] [PubMed] [Google Scholar]
- 17.Rathinam VAK, et al. TRIF licenses caspase-11-dependent NLRP3 inflammasome activation by gram-negative bacteria. Cell. 2012;150(3):606–619. doi: 10.1016/j.cell.2012.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wong FS, et al. The role of Toll-like receptors 3 and 9 in the development of autoimmune diabetes in NOD mice. Ann N Y Acad Sci. 2008;1150:146–148. doi: 10.1196/annals.1447.039. [DOI] [PubMed] [Google Scholar]
- 19.Kim D-H, et al. Inhibition of autoimmune diabetes by TLR2 tolerance. J Immunol. 2011;187(10):5211–5220. doi: 10.4049/jimmunol.1001388. [DOI] [PubMed] [Google Scholar]
- 20.Dong B, et al. TLR4 regulates cardiac lipid accumulation and diabetic heart disease in the nonobese diabetic mouse model of type 1 diabetes. Am J Physiol Heart Circ Physiol. 2012;303(6):H732–H742. doi: 10.1152/ajpheart.00948.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gülden E, et al. Toll-like receptor 4 deficiency accelerates the development of insulin-deficient diabetes in non-obese diabetic mice. PLoS One. 2013;8(9):e75385. doi: 10.1371/journal.pone.0075385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Malkiel S, Liao L, Cunningham MW, Diamond B. T-cell-dependent antibody response to the dominant epitope of streptococcal polysaccharide, N-acetyl-glucosamine, is cross-reactive with cardiac myosin. Infect Immun. 2000;68(10):5803–5808. doi: 10.1128/iai.68.10.5803-5808.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lang KS, et al. Toll-like receptor engagement converts T-cell autoreactivity into overt autoimmune disease. Nat Med. 2005;11(2):138–145. doi: 10.1038/nm1176. [DOI] [PubMed] [Google Scholar]
- 24.Rashid T, Ebringer A. Autoimmunity in rheumatic diseases is induced by microbial infections via crossreactivity or molecular mimicry. Autoimmune Dis. 2012;2012:539282. doi: 10.1155/2012/539282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gaboriau-Routhiau V, et al. The key role of segmented filamentous bacteria in the coordinated maturation of gut helper T cell responses. Immunity. 2009;31(4):677–689. doi: 10.1016/j.immuni.2009.08.020. [DOI] [PubMed] [Google Scholar]
- 26.Shiloh MU, et al. Phenotype of mice and macrophages deficient in both phagocyte oxidase and inducible nitric oxide synthase. Immunity. 1999;10(1):29–38. doi: 10.1016/s1074-7613(00)80004-7. [DOI] [PubMed] [Google Scholar]
- 27.Agarwal S, Mayer L. Gastrointestinal manifestations in primary immune disorders. Inflamm Bowel Dis. 2010;16(4):703–711. doi: 10.1002/ibd.21040. [DOI] [PubMed] [Google Scholar]
- 28.Suzuki T, et al. In: Immune-Deficient Animals in Biomedical Research. Rygaard N, Graem N, Sprang-Thomsen M, editors. Karger; Basel: 1985. [Google Scholar]
- 29.Rossini AA, Williams RM, Mordes JP, Appel MC, Like AA. Spontaneous diabetes in the gnotobiotic BB/W rat. Diabetes. 1979;28(11):1031–1032. doi: 10.2337/diab.28.11.1031. [DOI] [PubMed] [Google Scholar]
- 30.Kane M, et al. Successful transmission of a retrovirus depends on the commensal microbiota. Science. 2011;334(6053):245–249. doi: 10.1126/science.1210718. [DOI] [PMC free article] [PubMed] [Google Scholar]