

Significance
Aire is an enigmatic transcription factor that controls immunologic tolerance by inducing, specifically in the thymus, a battery of transcripts encoding proteins not usually encountered until the periphery, thereby promoting negative selection of self-reactive thymocytes and positive selection of regulatory T cells. We document a striking correspondence between those genes induced by Aire and those inhibited by a small-molecule inhibitor of the bromodomain protein Brd4. Aire and Brd4 directly interact, dependent on an orchestrated series of phosphorylation and acetylation events. Aire:Brd4 engagement draws in P-TEFb, mobilizing the transcription and splicing machineries and inducing transcription. Blocking the Aire:Brd4 interaction inhibits negative selection of self-reactive T cells in mice, and point-mutations of Aire that abrogate this association give rise to autoimmune disease.
Keywords: immunological tolerance, thymus, Aire, bromodomain protein, transcriptional elongation
Abstract
Aire controls immunologic tolerance by inducing a battery of thymic transcripts encoding proteins characteristic of peripheral tissues. Its unusually broad effect is achieved by releasing RNA polymerase II paused just downstream of transcriptional start sites. We explored Aire’s collaboration with the bromodomain-containing protein, Brd4, uncovering an astonishing correspondence between those genes induced by Aire and those inhibited by a small-molecule bromodomain blocker. Aire:Brd4 binding depended on an orchestrated series of posttranslational modifications within Aire’s caspase activation and recruitment domain. This interaction attracted P-TEFb, thereby mobilizing downstream transcriptional elongation and splicing machineries. Aire:Brd4 association was critical for tolerance induction, and its disruption could account for certain point mutations that provoke human autoimmune disease. Our findings evoke the possibility of unanticipated immunologic mechanisms subtending the potent antitumor effects of bromodomain blockers.
One of the more recently discovered mechanisms of immunologic tolerance is centered on the transcriptional regulator, Aire (reviewed in refs. 1 and 2). This intriguing protein is expressed primarily in medullary epithelial cells (MECs) of the thymus, where it controls the expression of a battery of genes, the products of which are typically associated with fully differentiated parenchymal cells residing in the periphery, so-called peripheral-tissue antigens (PTAs) (3). Aire-dependent PTA transcripts number in the thousands, including representatives from a score of organs (4, 5). These transcripts are translated into proteins, and derivative peptides are presented by major histocompatibility complex (MHC) molecules displayed on the MEC surface. MHC:PTA-peptide complexes mold the T-cell repertoire by inducing negative selection of self-reactive specificities (6–8) or by promoting positive selection of regulatory T cells (9, 10). By a still-obscure mechanism, Aire-induced MEC-generated peptides are also cross-presented by local dendritic cells (8, 10). As a consequence, humans and mice harboring a defective AIRE/Aire locus develop multiorgan autoimmunity (1, 2).
Aire’s molecular mechanism of action has been enigmatic. Several of its structural domains are found in other transcriptional regulators; for example, a CARD (caspase activation and recruitment domain), a nuclear localization signal, a SAND (Sp100, Aire-1, NucP41/75, Deaf-1) domain, and two PHD (plant homeodomain) zinc fingers (1, 2). However, the large number of genes whose expression Aire affects and the vast geographic, temporal, and quantitative variety of these genes’ expression in nonthymic cells suggest that it may not operate as a classical transcription factor, binding to a promoter or enhancer element and simply turning transcription up or down. Indeed, recent data derived from a variety of experimental approaches argue that Aire’s major modus operandi is to release promoter-proximal RNA polymerase II (Pol-II) pausing (11–13).
It has become increasingly clear that the regulation of Pol-II pausing is a major nexus of transcriptional control (14), and the focus of transcription factors as diverse as NF-κB (15), c-Myc (16), and HIF1A (17). Upon Pol-II recruitment to a gene promoter, serine-5 residues within a repeated motif in its carboxyl-terminal domain are phosphorylated by the general transcription factor TFIIH, launching transcription (reviewed in ref. 18). Pol-II continues for 20–60 base pairs, but is then blocked from proceeding further by the action of dominant pause factors. For transcription to continue, the block must be released by the elongation factor P-TEFb, composed of a kinase (CDK9) and a cyclin (CycT1/T2/K). P-TEFb phosphorylates the pause factors, surmounting their blockade, and also phosphorylates serine-2 residues in the repeated motif of Pol-II’s carboxyl-terminal domain, further facilitating transcription by creating a platform for the binding of chromatin modifying factors, RNA processing enzymes, nuclear export factors, and so on.
Where does Aire fit into this scenario? Here we focus on its interaction with a critical regulator of Pol-II pausing, the bromodomain-containing protein, Brd4 (19). As a member of the bromodomain and extraterminal domain (BET) family of proteins, Brd4 contains tandem bromodomains, BD1 and BD2, which bind to acetylated lysine residues on diverse proteins, and also harbors an extraterminal domain at the carboxy end that recruits P-TEFb. Brd4 exploits diverse tethers to recruit P-TEFb to transcribable chromatin: acetylated H3 and H4 histones, the Mediator complex, or particular nonhistone proteins; notably, NF-κB (20, 21), p53 (22), and Twist (23). Our findings argue that Brd4, recruited in response to an orchestrated series of posttranslational modifications of Aire’s CARD, bridges Aire and P-TEFb to release promoter-proximal Pol-II pausing and induce transcription of a broad swath of genes. As a consequence, pharmacologic inhibition of the Aire:Brd4 interaction compromises immunologic tolerance, and abrogation of this association can explain certain mutations in patients with autoimmune polyendocrinopathy/candidiasis/ectodermal dystrophy (APECED).
Results
The BET Protein Inhibitor, I-BET151, Selectively Suppresses the Expression of Aire-Induced Genes in MECs
To determine whether Aire’s effect on MEC gene expression depends on Brd4, we examined the effect of a small-molecule bromodomain blocker on the MEC transcriptome. I-BET151 is a selective and potent inhibitor of BET proteins, displacing them from chromatin by competing with histone and nonhistone acetyl-lysine residues for bromodomain binding (24).
Mice were injected intraperitoneally (ip) with 10 mg/kg I-BET151 every day for 3 d, MECs (CD45−Ly51loMHC-IIhi cells) were sorted by flow cytometry, and RNA was isolated and profiled using microarrays. Under these conditions, the overall effect of I-BET151 on the MEC transcriptome was surprisingly mild, with the expression of few genes changing more than twofold (Fig. 1A). However, a volcano plot comparing MEC transcripts from mice treated with the inhibitor versus vehicle (DMSO) alone revealed the known set of Aire-induced transcripts to be selectively down-regulated (P = 5.1 × 10−51) and, conversely, Aire-repressed transcripts to be preferentially up-regulated (P = 4.9 × 10−5) (Fig. 1B). That expression of Aire-induced genes was specifically targeted by the inhibitor was highlighted by a fold-change/fold-change (FC/FC) plot comparing the transcriptional effect of I-BET151 with that of the Aire-knockout (KO) mutation: the more a locus was up-regulated by Aire, the more it was down-regulated by BET protein blockade (Fig. 1C). The correlation was less clear for Aire-repressed genes, raising the question of whether many of the suppressive effects might not be secondary. Performing the same series of analyses on Aire-KO mice demonstrated that most of I-BET151’s influence on the MEC transcriptome was mediated through Aire: the mutant mice showed a reduced overall effect (Fig. 1D) and little evidence of a preferential influence on loci responsive to Aire (Fig. 1 E and F). These results also indicate that I-BET151’s effects were directly on Aire+ MECs, and not through a thymocyte intermediary.
Fig. 1.
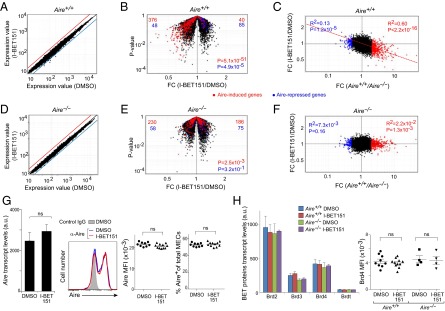
I-BET151-provoked transcriptome changes in MECs. Mice were ip-injected with 10 mg/kg I-BET151 in 50% DMSO in PBS or vehicle alone every day for 3 d. MECs were sorted as CD45−Ly51loMHC-IIhi cells, and their RNA was isolated and profiled. (A–F) Global transcript changes in MECs of Aire-WT (A–C) or Aire-KO (D–F) mice. (A and D) Comparison plots of normalized expression values for mice treated with vehicle alone (n = 4) (x axis) or with I-BET151 (n = 3) (y axis). Red and blue lines indicate twofold differences. (B and E) Volcano plots of the same data as in A and D. Aire-induced genes (more than twofold more in vehicle-treated Aire-WT vs. Aire-KO) highlighted in red; Aire-repressed genes (more than twofold less) in blue. The number of Aire-induced and Aire-suppressed genes up- (Right) or down-regulated (Left) by I-BET151 are at the top in red and blue, respectively. (Lower right) P values from a χ2 test. (C and F) FC values for vehicle-treated Aire-WT or Aire-KO mice (n = 4) (x axis) versus FC values for I-BET151-treated or vehicle-treated mice (n = 3) (y axis). Dashed red line indicates linear regression for Aire-induced genes. Correlation coefficient (R2) and P value by the F-test for Aire-induced and Aire-repressed genes are indicated in red and blue, respectively. (G) Aire expression. Same mouse treatment protocol as for (A–F). (Left) Microarray-derived expression values (n = 3 or 4). P value from the two-tailed unpaired Student’s t test. (Left-center) representative cytofluorimetric data on intracellular Aire protein expression by purified MECs from the designated mice. (Right-center) MFI of Aire in Aire+ MECs. Each dot corresponds to an individual mouse. P values calculated using the two-tailed unpaired Student’s t test. (Right) Summary data on intracellular Aire expression by MECs. Percentage of Aire+ cells from individual mice (mean ± SD). (H, Left) Quantification of transcripts encoding BET proteins. Normalized microarray-determined expression values for MECs using data from A–F (mean ± SD). (Right) Summary of intracellular expression of Brd4 in MECs determined from flow cytometry data. MFI in individual mice (mean ± SD). In G and H, ns = not significant.
The strikingly selective effect of I-BET151 on Aire-dependent genes did not simply reflect an effect on Aire expression itself. Levels of Aire transcripts and protein, and the fraction of Aire-expressing MECs, were all indistinguishable for mice treated with inhibitor versus those treated with DMSO only (Fig. 1G). Neither did I-BET151 appear to influence transcription of the BET family proteins expressed in MECs. Transcripts encoding BRDs 2, 3, and 4 could be detected above background, and their levels did not vary in the presence or absence of Aire with or without inhibitor treatment (Fig. 1H, Left). It was possible to visualize Brd4 protein expression by flow cytometry and, again, neither the Aire nor the inhibitor status affected its expression levels (Figs. S1 and 1H, Right). Thus, blocking the function of BET family proteins, including Brd4, in MECs selectively inhibited the expression of Aire-dependent genes.
Fig. S1.

No change in BET protein levels with or without Aire, with or without I-BET151. Typical cytofluorimetric dot-plots used to generate the data in Fig. 1H.
Aire Interacts with BD1 of Brd4.
Next, we examined Aire interactions with individual BET family members. Because one can cleanly isolate only about 50,000 Aire-expressing MECs per mouse, it was not feasible to perform standard coimmunoprecipitation (co-IP) experiments on ex vivo cells. Therefore, for most of the experiments described here, we used 4D6 cells transfected with a plasmid driving expression of Flag-tagged Aire. 4D6 is a human MEC line that proved useful in many past studies on Aire function (e.g., refs. 12, 13, 25, and 26). The amount of plasmid transfected was carefully titered to ensure a physiologic level of expression and the usual punctate distribution of wild-type (WT) Aire within the nucleus (see following).
IP analysis of micrococcal-nuclease-treated nuclear extracts from Aire/Flag-transfected 4D6 cells using antibodies (Abs) recognizing Brd2–Brd4 confirmed that all of these BET family members were expressed in MECs, but co-IP analysis revealed that only Brd4 interacted substantially with Aire (Fig. 2A). This association was inhibited when the cells were pretreated with 3 μM I-BET151 for 6 h (Fig. 2B). To identify which of Brd4’s two bromodomains was involved in its interaction with Aire, we cotransfected 4D6 cells with plasmids driving expression of Flag-tagged Aire and WT or mutant V5-tagged Brd4 (see map in Fig. 2C, Upper). Point-mutations of both BD1 and BD2 known to block their binding to acetyl-lysine residues, but not to compromise Brd4’s overall structure (27), or the BD1 mutation alone, largely abolished the Aire:Brd4 interaction, whereas the single BD2 mutation did not (Fig. 2C, Middle and Lower).
Fig. 2.

Interaction between Aire and Brd4 via the latter’s amino-terminal bromodomain. (A) Co-IP of Aire with Brd4. 4D6 cells expressing Flag-tagged Aire were lysed and their nuclear extracts incubated with Abs recognizing Brds 2, 3, or 4 or with control IgG. Control quantification of BET proteins was via sequential blotting. Association between Aire and the BET proteins revealed using an anti-Flag Ab. (Upper) Typical Western blot. (Lower) Summary quantification of co-IPed Aire, comparing with the relevant input control. Mean ± SD from four experiments. (B) Inhibition of Aire:Brd4 association by I-BET151. 4D6 cells expressing Aire/Flag were treated with I-BET151 or vehicle for 6 h before lysis. Their nuclear extracts were incubated with anti-Brd4, followed by Western blotting for the indicated proteins. (Upper) Sample Western blot. (Lower) Quantification of co-IPed Aire. Percentage input quantified for each condition and compared with percentage input from untreated cells. Mean ± SD from six experiments. (C) Interaction between Aire and mutant Brd4s. (Top) Domain structure of Brd4. BD, bromodomain; ET, extraterminal domain; CTD, carboxyl-terminal domain. (Middle) 4D6 cells expressing Aire/Flag and V5-tagged WT Brd4 or Brd4 bearing the indicated mutation or mutations were lysed and their nuclear extracts incubated with an anti-V5 Ab, followed by sequential immunoblotting for Aire and Brd4. dm contains both the point-mutations. (Bottom) Summary quantification of co-IPed Aire from three experiments. Percentage input quantified for each mutant and compared with percentage input for WT Brd4, normalized by the expression level in respect to Brd4. In all relevant panels: ***P < 0.001, determined by two-tailed unpaired Student’s t test.
The Aire:Brd4 Interaction Depends on a Series of Acetylated Lysine Residues in Aire’s CARD.
Because Aire is known to undergo acetylation (28, 29), we wondered whether it binds directly to Brd4 via one or more of its acetyl-lysine residues. Co-IP of Aire with anti-Brd4 was diminished by pretreatment of Aire/Flag-transfected 4D6 cells with C646, a specific inhibitor of the acetyl-transferase, CBP/p300, and was augmented by the addition of trichostatin-A, a broad-range deacetylase inhibitor (Fig. 3A). In addition, Aire could be co-IPed from 4D6 nuclear extracts with an anti-CBP Ab, consistent with data on other cell types (28, 30), and this interaction appeared to be “upstream” (or independent) of Brd4 recruitment, as it could not be inhibited by I-BET151 (Fig. 3B).
Fig. 3.
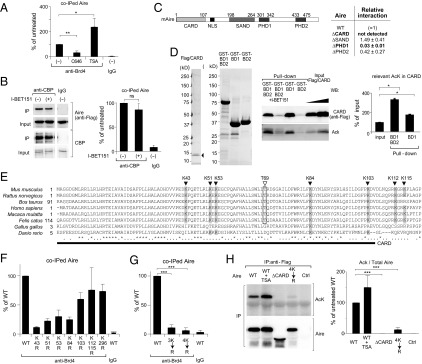
Multiple Aire CARD lysines involved in its interaction with Brd4. (A) Acetylation-dependent interaction between Aire and Brd4. 4D6 cells expressing Aire/Flag were treated with C646, trichostatin-A (TSA), or vehicle for 6 h before lysis. Co-IP and quantification carried out as in Fig. 2B. Mean ± SD from four experiments. (B) Interaction between Aire and CBP. 4D6 cells expressing Aire/Flag were treated with I-BET151 or vehicle for 6 h before lysis; nuclear extracts were incubated with an anti-CBP Ab, followed by immunoblotting for the indicated proteins. (Left) representative Western blot. (Right) Summary quantification of co-IPed Aire. Mean ± SD from three experiments. (C) Identification of Aire domains required for its interaction with Brd4. (Left) Domain structure of murine Aire. CARD, nuclear localization signal, SAND, PHD1, and PHD2. (Right) 4D6 cells expressing WT Aire/Flag or a domain-deletion mutant were lysed and their nuclear extracts incubated with anti-Brd4, followed by immunoblotting. Summary quantification. Percentage input for each co-IPed mutant Aire quantified and compared with that for WT Aire to calculate the “relative interaction.” Mean ± SD from three experiments. An example of primary data is in Fig. S2B. (D) Direct binding assay. Purified Flag/CARD [containing Aire (1–107)] (Left) was incubated with beads displaying a Brd4 bromodomain-GST fusion protein (Left-center). Specifically bound Aire fragment was eluted, followed by SDS/PAGE (Right-center). Summary quantification for acetyl lysine (AcK) in Flag/CARD (Right). Mean ± SD from three experiments. (E) Sequence alignment of Aire CARD from diverse species. Filled triangles, conserved lysines; open triangle, conserved threonine residue; bar, CARD truncation in ΔCARD Aire. (F and G) Effects of various K→R substitutions on the Aire:Brd4 interaction. 3K→R is the triple K43, 51, 53R mutant, and 4K→R the quadruple K43, 51, 53, 54R mutant. Co-IPs and quantifications carried out as in Fig. 2B from cells expressing WT Aire/Flag or Aire/Flag with the indicated mutation or mutations. Summary quantification. Mean ± SD from three experiments. Examples of primary data are in Fig. S2 C and D. (H) Acetylated lysine levels. 4D6 cells expressing WT vs. mutant Aire/Flags were lysed, their nuclear extracts IPed with anti-Flag M2 magnetic beads, and immunoblots probed with the indicated Abs. (Left) Example Western blot. (Right) Summary quantification. Mean ± SD for four experiments. In all relevant panels: P = ns, not significant; *P < 0.05; **P < 0.01; ***P < 0.001; determined by two-tailed unpaired Student’s t test. NLS, nuclear- localization signal.
As a first step in localizing the Aire acetyl-lysine residues critical for its binding to Brd4, we made use of an existing set of domain-deletion expression plasmids (26) (see map in Fig. 3C, Left). Each of the Aire mutants could be demonstrated in the nucleus of 4D6 cells by fluorescence microscopy, although only PHD2-deleted Aire assembled into punctate structures similar to those of the WT molecule (Fig. S2A). Expression of plant homeodomain-1 (PHD1)-deleted Aire was comparatively low, and CARD-deleted and SAND-domain-deleted Aire were somewhat overexpressed (Fig. S2B), suggesting that certain of the mutants had altered stabilities. Nonetheless, careful quantification of the amount of mutant protein co-IPed with Brd4 indicated that the Aire:Brd4 association did not depend critically on the SAND or PHD2 domains and that, above all, the CARD was essential (Figs. 3C, Right, and S2B).
Fig. S2.

Mapping of acetyl-lysine residues critical for Aire:Brd4 interaction. (A) Fluorescence microscopy of typical 4D6 cells 30 h after transfection with plasmids expressing WT Aire or the designated deletion mutants, localized in Fig. 3C. Blue, DAPI staining to reveal the nucleus; green, staining for Aire. (B–D) Typical Western blots used to generate the summary data corresponding to Fig. 3 C, F, and G, respectively. (E) As per A, except depicting a series of Aire-CARD point-mutations.
The importance of the CARD was confirmed using a direct-binding assay. We added a Flag-tag to the Aire (1–107) fragment (Flag/CARD), expressed it in 4D6 cells, and purified it using anti-Flag magnetic beads (Fig. 3D, Left). Each individual or the tandem Brd4 bromodomains were expressed in Escherichia coli as GST fusion proteins and were purified on glutathione Sepharose (Fig. 3D, Left-center). Then, we incubated the isolated Flag/CARD with the bead-bound bromodomain-GST fusions and quantified specific binding by GST pull-down, followed by blotting with anti-Flag. In agreement with the above-cited results on Brd4 bromodomain point-mutants (Fig. 2C) and on Aire domain-deletion mutants (Fig. 3C), BD1, but not BD2, of Brd4 directly interacted with Aire CARD (Fig. 3D, Right-center). This association was inhibited by I-BET151, thereby suggesting that BD1 binds to one or more acetyl-lysines in the CARD. Indeed, probing the same blot, we could detect an enrichment in acetylated lysine residues in the CARD:BD1 co-IPed material vis a vis input CARD (Fig. 3D, Right-center and Right).
Hence, the CARD became our focus of attention, especially given that PHD1 does not contain any conserved lysine residues (Fig. 3E). We generated a series of Aire mutants harboring a lysine→arginine (K→R) substitution, which prevents acetylation but maintains the positive charge, including mutations of K43, K51, K53, K84, and K103, the conserved lysines within the CARD; K112 and K115, also conserved but just outside the CARD; and the conserved K296 residue, close to PHD1. Acetylation of several of these lysine residues, including within the CARD, has been verified by mass spectrometry of AIRE-transfected COS-1 cells (29), which we subsequently confirmed for 4D6 cells. Co-IP analysis of 4D6 cells transfected with expression constructs corresponding to these mutants revealed a minimal effect of targeting the more carboxy-terminal lysines (K103, K112, K115, and K296) (Figs. 3F and S2C); stronger effects were observed with substitutions of the more amino-terminal CARD lysines (K43, K51, K53, and K84). Mutation of multiple of these four lysines had even greater effects, culminating in near-complete abrogation of the Aire:Brd4 interaction with the quadruple K43,51,53,84R (4K→R) mutant (Figs. 3G and S2D). The lysine-residue mutants, including that with a quadruple substitution, all localized to the nucleus and accumulated in the typical nuclear speckles (Fig. S2E). We also tested a set of lysine substitutions within and near the SAND domain (K245, K247, and K255) because these residues had previously been reported to be acetylated and to influence Aire’s transcriptional potential (28) and found no significant effect of these mutations on Aire’s ability to interact with Brd4. Probing anti-Flag IPs of mutant Aire/Flag-transfected cells with an Ab that recognizes acetyl-lysines indicated that acetylation within the CARD, in particular at the amino-terminal lysines, was an important site of this modification under our conditions (Fig. 3H).
Phosphorylation of the Aire CARD Facilitates Nearby Acetylation to Promote Recruitment of Brd4.
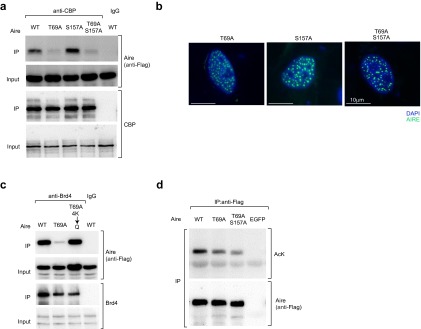
Aire is also known to be phosphorylated; specifically, T69 and S157 are targeted by DNA-dependent protein-kinase (DNA-PK) (31), which has been shown by multiple approaches to be a major Aire-interacting protein in both MECs and model cell lines (31–33). Co-IP experiments using Aire/Flag-transfected 4D6 cells pretreated or not with the specific DNA-PK inhibitor, NU7441, revealed Aire’s interaction with Brd4 to depend on a phosphorylation event mediated by this kinase (Fig. 4A). The same was true for Aire’s association with CBP (Fig. 4B), raising the question of whether, analogous to what has been seen in other contexts [e.g., with Rel-A (20, 34)], the CBP-binding site on Aire might include a required phosphorylated residue. In accord with this notion, a double-alanine substitution at Aire’s T69 and S157 positions and the single T69A mutation substantively reduced Aire’s ability to associate with CBP; the single S157A mutation did not, however (Figs. 4C and S3A). The three mutant Aire proteins were expressed at similar levels, localized to the nucleus, and accumulated in the characteristic nuclear speckles (Fig. S3B). In contrast, co-IP experiments using an Ab recognizing the catalytic subunit of DNA-PK (DNA-PKcs) showed no significant dampening of the Aire:DNA-PK interaction when either acetylation or phosphorylation was prevented by either inhibition or mutation (Fig. 4D). This result is not surprising, given that Aire’s binding to DNA-PKcs is thought to depend primarily on the PHD1 of Aire (31), and suggests that the Aire:DNA-PK interaction is “upstream” of the Aire:CBP association.
Fig. 4.

Interplay between phosphorylation and acetylation of the Aire CARD to promote interaction with Brd4. (A and B) DNA-PK-dependent association of Aire and Brd4 or CBP. 4D6 cells expressing WT Aire/Flag were treated with NU7441 or vehicle for 6 h before lysis. Nuclear extracts were incubated with anti-Brd4 (A) or anti-CBP (B), followed by immunoblotting for co-IPed Aire. Mean ± SD from three experiments. Example data are in Fig. S3A. (C) Association between CBP and Aire mutated at phosphorylation sites known to be targeted by DNA-PK. 4D6 cells expressing WT or mutant Aire/Flag were lysed and their nuclear extracts incubated with anti-CBP, followed by immunoblotting for co-IPed Aire. Mean ± SD from three experiments. (D) Association between DNA-PK and Aire, independent of Aire’s CARD’s posttranslational modifications. 4D6 cells expressing WT or mutant Aire/Flag were treated or not with the indicated inhibitors for 6 h before lysis. Nuclear extracts were incubated with an anti-DNA-PKcs Ab, followed by immunoblotting for the indicated proteins. (Left) Representative blot. (Right) Quantification of co-IPed Aire. Mean ± SD from three experiments. (E) Partial rescue of the Aire CARD phosphorylation-site mutant by substitutions that mimic acetylated-lysine residues. Co-IP and quantifications carried out as in Fig. 2B from cells expressing WT or mutant Aire/Flag. Summary quantification. Mean ± SD from four experiments. Example data are in Fig. S3C. (F) Significant effect of Aire phosphorylation on Aire acetylation levels. As in Fig. 3H, except that the effect of T69A single- and T69A/S157A double-mutations on levels of Aire acetyl-lysine was quantified. Mean ± SD from three experiments. Example data are in Fig. S3D. All relevant panels: *P < 0.05; **P < 0.001; ***P < 0.001; determined by two-tailed unpaired Student’s t test.
Fig. S3.
Aire:Brd4 interaction promoted by phosphorylation/acetylation interplay. (A) Example of a Western blot corresponding to the summary data of Fig. 4C. (B) Fluorescence microscopy of typical 4D6 cells 30 h after transfection with plasmids expressing the designated mutants. Blue, DAPI staining to reveal the nuclei; green, staining for Aire. (C and D) Examples of Western blots corresponding to the summary data depicted in Fig. 4 E and F, respectively.
We hypothesized that phosphorylation of the T69 position in Aire’s CARD by DNA-PK enables binding of CBP, which, in turn, acetylates multiple lysines in the CARD, promoting recruitment of Brd4. The T69A mutation greatly diminished Aire’s ability to recruit Brd4 (Figs. 4E and S3C), just as the quartet of CARD K→R substitutions had (Fig. 3G). In addition, substitution of the four critical CARD lysines with a glutamine residue (4K→Q), which is noncharged and has been seen to mimic acetylated lysine in other contexts (e.g., refs. 28 and 35), partially rescued the T69A mutation (Figs. 4E and S3C). The incomplete rescue might reflect only approximate mimicry of acetyl-lysine by glutamine in this context, or a role for T69 beyond binding of CBP. Similarly, mutation of T69 significantly, although partially, recapitulated the reduction in overall Aire acetylation seen with the 4K→R mutation (Figs. 4F and S3D). In summary: Aire’s recruitment of Brd4 requires both phosphorylation and acetylation of its CARD, likely in sequence.
Mutations That Disrupt the Aire:Brd4 Interaction Also Impinge on the Aire:P-TEFb Association and on Mobilization of the Elongation and Splicing Machineries.
Brd4 can be an important regulator of Pol-II pausing, releasing P-TEFb from an inactive complex with the transcriptional regulator, HEXIM1, and the 7SK small nuclear ribonuclear protein (snRNP) and bridging its interaction with chromatin by bromodomain-mediated binding to acetylated histones (19, 36). We investigated whether, in an analogous fashion, Brd4 links Aire and P-TEFb, and thereby promotes release of paused Pol-II to induce Aire-dependent transcripts. First, we showed that Aire can be co-IPed from Aire/Flag-transfected 4D6 cells by an anti-CDK9 Ab (Fig. 5A). The Aire:CDK9 association was sensitive to I-BET151, as well as to the CBP and DNA-PK small-molecule inhibitors (Fig. 5A). The triple and quadruple Aire CARD K→R substitutions and the phosphorylation-site T69A mutation also compromised Aire’s interaction with CDK9 (Figs. 5B and S4).
Fig. 5.

Bridging of Aire and P-TEFb by Brd4, thereby mobilizing the transcription and splicing machineries. (A) Disruption of Aire:P-TEFb association using inhibitors. 4D6 cells expressing Aire/Flag were treated or not with the indicated inhibitors for 6 h before lysis, and their nuclear extracts were incubated with anti-CDK9, followed by immunoblotting for the indicated proteins. (Left) Representative blot. (Right) Summary quantification of co-IPed Aire. Mean ± SD from three experiments. (B) Disruption of Aire:P-TEFb interactions through mutations of the Aire CARD. Co-IPs and quantifications were carried out as in A from 4D6 cells expressing WT or mutant Aire/Flag. Summary data from three experiments. Example data are in Fig. S4. (C) Known Aire associates. Identified in a co-IP/mass spectrometry screen and validated by reverse co-IP, shRNA knock-down, and so on. Reprinted from ref. 32, with permission from Elsevier; www.sciencedirect.com/science/journal/00928674. (D) Aire/Flag-transfected HEK293T cells were incubated with I-BET151 or vehicle 24 h before lysis, nuclear extracts were IPed with anti-Flag, and immunoblotting was performed using the designated panel of Abs. Example strips (Left) and summary quantification (Right) from three experiments. Mean ± SD. In A–D, *P < 0.05; **P < 0.01; ***P < 0.001 from the Student’s t test. (E and F) Effect of Aire on chromatin-bound Brd4. 4D6 cells were transfected with an Aire expression plasmid or control plasmid; I-BET151 or vehicle was added at 6 h after transfection and at 48 h posttransfection, ChIP-seq was performed using anti-Brd4. (E) The top 5,000 Brd4-binding regions in vector-transfected, vehicle-treated cells were ranked (x-axis), and Aire’s effect on these loci was plotted as the FC of the Brd4 signal in Aire- vs. vector-transfected (all vehicle-treated) cells. The set of Brd4 signals is grouped according to location: >10 Kb upstream of the TSS (Left), <10 Kb upstream of the TSS (Middle), and around the TSS (Right). (F) Correlated effects of Aire and I-BET151 on Brd4 binding to chromatin. For the top 5,000 Brd4 signals identified in E, the FC effect of I-BET151 treatment on Brd4 binding (y axis) was plotted against the FC effect of Aire on Brd4 binding (x-axis). For loci >10 Kb upstream from the TSS (Left) or around the TSS (Right).
Fig. S4.

Effect of Aire CARD mutations on Aire:P-TEFb interactions. Typical Western blot corresponding to the summary data of Fig. 5B.
Second, we looked at the effect of I-BET151 in a broader context. We previously reported that Aire interacts, either directly or indirectly, with more than 40 proteins, falling into four functional classes: nuclear transport, chromatin binding/structure, transcription, and pre-mRNA processing (Fig. 5C, reprinted from ref. 32). Therefore, we addressed potential radiating consequences of the Aire:Brd4 interaction by choosing various members of the latter two classes of Aire associates and determining how I-BET151 influenced their partnership with Aire (in Flag/Aire-transfected HEK293T cells, as the published biochemistry had been done on this line) (Fig. 5D). Disruption of the Aire:Brd4 interaction did not substantially inhibit Aire’s association with DNA-PKcs or Ku80, consistent with the data of Fig. 4D and with the notion that DNA-PK is one of the first Aire partners to be mobilized (32). However, association of Aire with all the other proteins examined was severely compromised by bromodomain blockade, suggesting that they were recruited to Aire downstream of the Aire:Brd4 interaction. Thus, Brd4 appears to bridge Aire and P-TEFb, mobilizing the elongation and splicing machinery in consequence.
We performed ChIP-seq analysis to obtain a genome-wide view of how Aire influences the chromatin distribution of Brd4 in 4D6 cells. An Aire-expression or a control plasmid was transfected into 4D6 cells, the cells were treated with I-BET151 or DMSO, and 48 h posttransfection, the chromatin was cross-linked, sheared, and IPed with an anti-Brd4 Ab. (Note: Aire ChIP-seq on this cell line was not interpretable because of the low signals obtained, and both Brd4 and Aire ChIP-seq on ex vivo MECs was precluded by inadequate cell numbers.) Plotting the Brd4 signal rank-order versus Aire’s effect on Brd4 binding revealed, first, that robust Brd4 signals were found predominantly at the transcriptional start-sites (TSSs), but also distally, >10 kb upstream of the TSS. Second, the effect of Aire on the Brd4 signal differed strikingly according to its genomic placement: At the TSS, Aire dampened the Brd4 signal, whereas far upstream, it enhanced it (Fig. 5E). It was also of interest to incorporate I-BET151 sensitivity into the picture because, as we demonstrated in Fig. 1C, expression of Aire-induced genes is particularly sensitive to this drug and because so-called “super-enhancers” laden with Mediator, Brd4, Pol-II, and other transcriptional regulators are also highly sensitive to BET-family blockers (37–39). Interestingly, according to the FC/FC plot of Fig. 5F, the far-upstream Brd4 signals most inhibited by I-BET151 were also those most enhanced by Aire; this relationship was less evident for the Brd4 signals around the TSS. In conclusion, then, the consequences of the Aire:Brd4 interaction radiate broadly to drive transcriptional elongation and associated events.
The Aire:Brd4 Interaction Is Required for Optimum Aire-Induced Transcription in MECs and for Effective Immunologic Tolerance.
Last, we probed the immunologic consequences of disrupting the Aire:Brd4 interaction, initially examining PTA gene transcription. Increasing amounts of the plasmids driving expression of WT Aire or the quadruple K→R mutation were transfected into 4D6 cells, the resulting Aire protein levels were quantified, and expression of a set of genes known to be up-regulated by Aire in 4D6 cells (and of GAPDH as a control, Aire-independent locus) was measured by RT-PCR (Fig. 6A). A titration analysis indicated that expression of all of the Aire-induced genes, but not that of GAPDH, was inhibited by mutating the lysine residues in Aire’s CARD (Fig. 6B). A previous study also reported a strong effect of mutating the CARD’s threonine phosphorylation site on Aire-induced transcription (31).
Fig. 6.
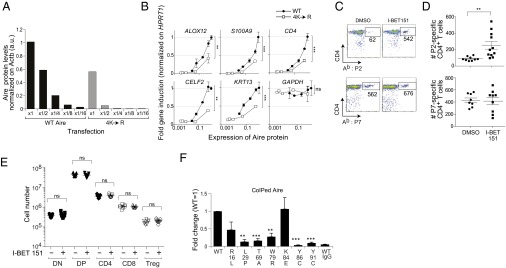
Effects of Aire CARD mutations on Aire-induced transcription. (A) Quantification of WT and mutant Aire posttransfection. WT or 4K→R mutant Aire/Flag-expressing plasmids were diluted with empty vector (fractions indicated on the x-axis) and transfected into 4D6 cells. Total cell lysates were prepared 40 h later, and Aire expression was examined by Western blotting. Approximately 5 μg protein was loaded per lane, and signal was normalized on β-actin expression. (B) Effect of the 4K→R Aire mutation on Aire-dependent gene expression. 4D6 cells were transfected with diluted WT or mutant Aire/Flag-expressing plasmids, as per A. RNA was prepared 40 h later, and the expression of Aire-induced genes was examined by qPCR and normalized on the expression of HPRT1. Relative gene induction compared with that of WT Aire ×1 (no dilution) (y axis) versus their relative protein expression examined in A (x-axis). Performed in duplicate. Mean ± SD GAPDH is a control, Aire-independent gene. Analysis of covariance was performed to exclude the influence of varying expression levels on the difference between WT and 4K→R Aire/Flag. P = ns, not significant; **P < 0.01; ***P < 0.001. (C) Effect of Brd4 blockade on thymic negative selection. Three-week-old B6 mice were ip-injected with 10 mg/kg I-BET151 or with DMSO every day for 3 d, and then every third day for another 18 d. Three days after the last injection, they were inoculated with 100 µg IRBP(P2:288–307 or P7:786–805) in Complete Freund’s Adjuvant, and 10 d later, pooled peripheral lymphoid organs were analyzed for Ab:IRBP(294–306) or Ab:IRBP(786–797) tetramer staining. Gated on CD8−CD3+ cells. Values refer to the number of cells in the gate. (D) Summary data on individual mice manipulated as per C. (E) Lack of effect of I-BET151 on thymocytes. Three-week-old B6 mice were treated with I-BET151 as per C, and the thymus was analyzed by flow cytometry. DN, double-negative; DP, double-positive. (F) Effects of APECED mutations on the Aire:Brd4 interaction. Co-IP and quantifications conducted as in Fig. 2B from cells expressing WT and point-mutated Aire/Flag. Summary quantification. Mean ± SD from ≥3 experiments. Examplar data can be found in Fig. S5B.
Next, we evaluated clonal deletion of self-reactive thymocytes, focusing on an Aire-dependent CD4+ T-cell specificity recognizing peptide 294–306 of the retina-specific protein, interphotoreceptor retinoid-binding protein (IRBP) (8). Mice were ip-injected with I-BET151 or vehicle for 3 wk; were then inoculated with IRBP(288–307) in Complete Freund’s Adjuvant; and 10 d later, peripheral lymphoid organs were dispersed, pooled, and stained with the Ab:IRBP(294–306) tetramer reagent. Clearly, treatment with I-BET151 compromised thymic negative selection of this self-reactive specificity, as significantly more Ab:IRBP(294–306)-specific T cells emerged into the periphery (Fig. 6 C and D). No effect on CD4/8 splits of thymocytes was observed (Fig. 6E), arguing against general compound-induced toxicity. This conclusion was solidified by the fact that I-BET151 had no detectable effect on selection of an Aire-independent T-cell specificity, Ab:IRBP(786–797) (8) (Fig. 6 C and D).
Finally, we wondered to what extent a defect in Aire:Brd4 binding could account for the disease-provoking effects of certain APECED point-mutations. We had already shown that the T69A substitution, which interferes with an important phosphorylation event, strongly inhibited the association of Aire and Brd4 (Fig. 4E), while permitting formation of the typical punctate nuclear structures (Fig. S3B). We now examined six additional CARD-localized point-mutations known to occur in patients with APECED. Two of the mutations, L29P and W79R, disturbed Aire's nuclear:cytoplasmic distribution, as well as its concentration within nuclear speckles (Fig. S5A). Not surprisingly, then, these alterations also inhibited Aire:Brd4 interaction (Figs. S5B and 6F). The mutations Y86C and Y91C joined T69A in strongly inhibiting the association between Aire and Brd4 while not affecting Aire’s distribution in the nucleus (Fig. 6F). Interestingly, although the K84E mutation is located at a functionally important acetylation site (Fig. 3F), it actually enhanced Aire:Brd4 interaction, perhaps because the negative charge partially mimicked acetylation of the K residue, as had been seen with the 4K→Q mutation. Thus, multiple patients with APECED carry single point-mutations that exert strong effects on Aire:Brd4 binding and thus could, in and of themselves, explain the loss of immunologic tolerance.
Fig. S5.
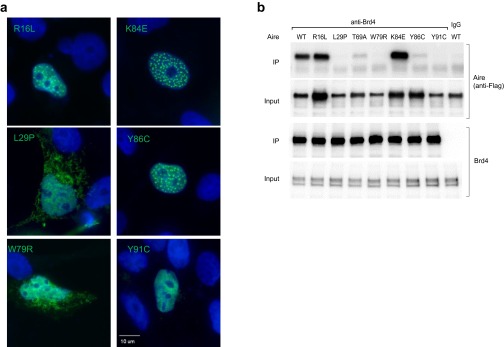
Effects of APECED mutations on Aire function. (A) Aire nuclear localization, As per Fig. S2A, except the series of APECED point-mutants examined in Fig. 6F were examined. (B) Interaction with Brd4. Typical Western blot used to generate the summary data corresponding to Fig. 6F.
Discussion
The means by which Aire induces the transcription of thousands of genes in MECS, in particular loci encoding PTAs, has intrigued investigators for more than a decade. At the outset of this study, diverse lines of evidence had argued that Aire’s primary mechanism of action is to promote elongation of Pol-II stalled just downstream of the TSS (11–13). The present study furnishes molecular insight into the mechanisms by which Aire releases polymerase pausing: Brd4 serves as a critical bridge between Aire and P-TEFb, thereby mobilizing the transcriptional elongation and splicing machineries. Three points in particular merit more profound discussion: the striking specificity of I-BET151’s effect on the MEC transcriptome, the highly orchestrated mechanistic scenario we uncovered, and the implications for immunomodulatory cancer therapy.
There was an impressive correspondence between those MEC genes whose transcription was augmented by Aire and those whose expression was inhibited by I-BET151. On first consideration, this observation is surprising, given that Brd4 is a general transcription factor that participates in the regulation of many loci in mammals. The explanation may lie in the concept of “superenhancers,” first advanced by Young et al. (40). Superenhancers are defined as remarkably long enhancers that host an exceptionally high density of transcription factors, both cell-type-specific and general, such as Mediator, p300/CBP, Brd4, and Pol-II. They are proposed to serve as depots for effective collection of regulators and their coordinate delivery to the TSS by chromosomal looping or by interchromosomal interactions. Superenhancers are preferentially associated with genes that define and control the biology of particular cell types and are characterized by an unusually high sensitivity to BET-bromodomain blockers (37–39). These two factors most likely explain how this class of drug can have unexpectedly specific effects, notably in the context of a number of cancers.
Thus, it could be that the specific targeting of Aire-induced genes by I-BET151 reflects their association with and regulation by superenhancers. This notion would be consistent with the high fraction of Aire-induced loci that encode proteins characteristic of terminally differentiated cell types. It would also be in accord with our finding that Aire’s effect on Brd4 binding to far-upstream sequences is strongest at those sites most sensitive to I-BET151 inhibition. Indeed, a scenario entailing superenhancers could explain the observation that Brd4 signals were decreased at that TSS (reflecting release of Pol-II pausing) but increased far upstream (reflecting promotion of superenhancers) in the presence of Aire. Nonetheless, other scenarios remain possible.
Traditionally, Brd4’s effect on transcription has been thought to reflect its binding to histone acetyl-lysine residues, as a so-called “histone reader” (41, 42). Analogously, the influence of bromodomain blockers such as I-BET151 on Brd4’s function has generally been attributed to perturbation of Brd4 interactions with acetylated histones (41, 42). However, our data point to a critical interaction between acetylated lysine residues in the CARD of Aire and the amino-terminal bromodomain, BD1, of Brd4. A higher-level, perhaps dynamic (22), tripartite interaction among Brd4, Aire, and acetylated histones remains possible, given that Aire’s PHD1 binds to the eight amino-terminal residues of unmodified histone H3 (26, 43). Indeed, such a structure might explain the strong dependence of the Aire:Brd4 interaction on Aire’s PHD1 (Fig. 3C). Interaction networks incorporating Brd4’s bromodomains and acetylated histones have been detected by X-ray crystallography (44).
The mechanistic scenario suggested by our studies is illustrated in Fig. S6. As we previously reported (32), and was confirmed by independent means (33), DNA-PK is an early partner in Aire’s activities. This kinase also phosphorylates Aire at T69 and S157 (31); the former residue is located within the CARD and is required for binding of CBP (Fig. 4C), an acetyl-transferase that can modify multiple nearby residues in the CARD (Fig. 3 E–G). Aire CARD multiacetylation enables the recruitment of Brd4, commandeering of P-TEFb, and mobilization of the transcriptional elongation and splicing machineries. Although a previous report (28) argued for important CBP-mediated acetylation events within and in the vicinity of the SAND domain, and we also detected these modifications in mass spectrometry analysis, we found them not to be involved in Aire’s association with Brd4.
Fig. S6.

A model for Brd4-dependent interaction between Aire and P-TEFb. After Aire is phosphorylated at T69 by DNA-PK, CBP is recruited and acetylates Aire on CARD lysine residues. These residues are recognized by Brd4’s amino-terminal bromodomain, BD1, corecruiting P-TEFb, which releases promoter-proximal Pol-II pausing, promoting transcriptional elongation and splicing.
The scenario we have constructed highlights the critical nature of Aire’s CARD beyond the oligomerization role originally attributed to it (45). This functional importance jibes with observations that the CARD is the most conserved stretch of Aire and that two-thirds of APECED mutations fall within this domain (46). Aire’s CARD has the potential for multiacetylation, with the four amino-terminal lysines seemingly most important. It is intriguing that a single bromodomain needs multiple modified lysine residues for maximum interaction. This requirement could reflect the aforementioned interaction networks (44), cooperative binding of nearby histone marks by a single bromodomain, as has been described for recognition of H4 by another BET-family member, Brdt (47), a dynamic series of acetylations, or perhaps an alteration in tertiary structure.
It is clear, then, that Aire:Brd4 interaction is at the crux of Aire control of MEC gene transcription and, thereby, immunologic tolerance. Manipulation of this association may have therapeutic applications, in particular in the context of cancer. Two recent studies highlighted Aire’s ability to regulate antitumor responses by promoting either negative selection of effector T cells (48) or positive selection of regulatory T cells (9). It will be interesting to see whether the potent antitumor effects of bromodomain blockers already documented in a number of contexts and anticipated in multiple ongoing clinical trials are at least partially the result of unleashing an immune response against the tumor previously dampened by Aire-dependent tolerance induction in the thymus.
Materials and Methods
Inhibitors.
I-BET151 (GSK1210151A) was synthesized to >99.5% purity, as previously described (24). Other inhibitors were purchased: NU7441 from Santa Cruz Biotechnology and C646 (SML0002) and Trichostatin A (T1952) from Sigma-Aldrich.
Mice.
All mice were housed under specific-pathogen-free conditions at Harvard Medical School’s Center for Animal Resources and Comparative Medicine. Relevant studies were conducted in accordance with both GlaxoSmithKline and Harvard policies (Institutional Animal Care and Use Committee protocol 2954). Aire-KO and Aire-WT littermates (3) were kept on the C57BL/6 genetic background. For Fig. 1 experiments, mice aged 4–6 wk were injected ip with 10 mg/kg I-BET151 dissolved in 50% (vol/vol) DMSO in PBS once a day for 3 d. Twelve to 16 h after the final injection, they were killed and their thymus removed. For Fig. 6 experiments, 3-wk-old B6 mice were ip-injected with 10 mg/kg I-BET151 once a day for 3 d, and then once every 3 d for another 18 d. To analyze T-cell selection, we immunized the mice with 100 μg IRBP P2 peptide (288–307) or P7 peptide (786–805) emulsified in 100 μL of 4 mg/mL Complete Freund’s Adjuvant (Chondrex, Inc) 3 d after the final I-BET151 treatment. Ten days after the immunization, mice were killed for Ab:IRBP(294–306) or Ab: IRBP(786–797) tetramer analysis.
MEC Isolation and Intracellular Aire and Brd4 Staining.
Suspensions of thymic stromal cells were prepared and stained as previously described (32) and detailed in the SI Materials and Methods.
Gene-Expression Profiling.
Detailed methods have been reported (49). Total RNA was isolated from sorted MECs using TRIzol (Life Technologies). cRNA was hybridized to Affymetrix ST1.0 microarrays. Raw data were processed with the RMA (robust multiarray average) algorithm for probe-level normalization and were analyzed using GenePattern software (Broad Institute). Genes whose expression was more than twofold up- or down-regulated in MECs from vehicle-treated Aire-WT (n = 4) versus Aire-KO (n = 4) mice constituted the Aire “up” and “down” signatures.
Cell Culture and Transfection.
4D6 cells were cultured and transfected with WT and various mutant Aire-expression constructs, as detailed in the SI Materials and Methods. In certain experiments, inhibitors were added: I-BET151 at 3 μM, C646 at 25 μM, Trichostatin A at 50 nM, or NU7441 at 1 μM. Plasmids driving expression of full-length murine Aire (Flag-tagged at the carboxyl-terminal end) or of domain-deleted Aire were described previously (26). Plasmids encoding point-mutated Aire were constructed by PCR-mediated site-directed mutagenesis (primers listed in the SI Materials and Methods) (50). PCR fragments containing the mutations were cloned into the WT Aire-expression plasmid between the NheI and EcoRI sites (except for the K296R mutant) or the BstXI and NotI sites (for K296R). All mutations and associated sequences were confirmed by DNA sequencing. For the expression of human Brd4 molecules, plasmids containing Brd4 (tagged with V5 at the carboxyl-terminal end) harboring either WT or mutant bromodomains were constructed as follows: cDNA for the human BRD4 gene was PCR amplified from human testis Marathon-ready cDNA (Clontech) and subcloned into pcDNA3.ID/TOPO V5-His vector. The gene was then mutated using splice-by-overlap extension PCR to introduce the Y97A/Y390A mutations to inactivate the bromodomains. The WT and mutated genes were finally cloned between the BamHI and NotI sites (excising EGFP) of pEGFP-N1 (Clontech). All mutations and associated sequences were confirmed by DNA sequencing.
HEK293T cells were cultured in DMEM supplemented with 10% (vol/vol) FBS, L-glutamate, and pen/strep antibiotics and maintained in a humidified atmosphere at 37 °C with 5% CO2. For transfection, around 4 × 106 cells were seeded in 10-cm tissue culture plates and transfected with pCMV-Tag1 (Clontech) carrying Flag-tagged murine Aire, using TransIT reagent (Mirus) according to the manufacturer’s instructions.
IPs and Western Blotting.
Detailed methods for 4D6 cells can be found in the SI Materials and Methods. IPs and Western blotting on HEK293T cells were performed as previously described (49). Abs are listed in the SI Materials and Methods.
Aire CARD:Brd4 bromodomain direct binding analyses are detailed in the SI Materials and Methods. In brief: Brd4 bromodomains (BD1, BD2, BD1+2) were expressed in and purified from E. coli as GST fusion proteins; Flag/CARD [Aire (1–107)] was expressed in and purified from 4D6 cells. Flag/CARD was incubated with bead-bound BD-GST fusion proteins, specifically bound material was eluted, and associated Aire fragment was visualized by Western blotting.
qPCR.
4D6 cells were harvested 40 h after transfection. Total RNA was isolated using TRIzol (Life Technologies), and was reverse-transcribed using oligo(dT) and SuperScript II (Life Technologies). qPCR was performed using Power SYBR Green PCR Master Mix (Life Technologies), using the 7900HT Fast Real-Time PCR System (Life Technologies) for PCR and signal detection. Primers are listed in the SI Materials and Methods.
SI Materials and Methods
Thymic Epithelial Cell Isolation and Intracellular Aire and Brd4 Staining.
MECs were sorted as CD45−Ly51loMHC-IIhi, using a MoFlo (Dakocytomation) and the monoclonal antibodies (mAbs): Ly51 (6C3 rat mAb; BioLegend), CD45 (30-F11 rat mAb; BioLegend), and MHC-II A and E (M5/114.15.2 rat mAb; BioLegend). Intracellular staining for Aire and Brd4 was performed using the transcription factor staining buffer set from eBioscience according to their instructions, using 5H12 (rat mAb; eBioscience) and anti-rat IgG2c (2C8F1 mouse mAb; SouthernBiotech) for Aire and rabbit polyclonal Ab (A301–985; Bethyl Laboratories) for Brd4. Data were acquired on an LSR II (BD Biosciences) and analyzed using FlowJo software (TreeStar).
Cell Culture and Transfection.
4D6 cells were cultured in RPMI1640 medium supplemented with 10% FBS/L-glutamate and were maintained in a humidified atmosphere at 37 °C with 5% CO2. For transfection, the cells were counted and seeded in six-well or 10-cm tissue-culture plates and were transfected with the specified plasmids using Lipofectamine LTX with PlusReagent (Life Technologies) according to the manufacturer’s instructions.
Primers used for mutagenesis are listed here, with the nucleotides representing mutations indicated in lowercase letters. Flanking_5′-1 and Flanking_3′-1 were used as common flanking primers for all mutants except K296R, for which Flanking_5′-2 and Flanking_3′-2 were used.
K43R_FW;GAGGACAgGTTCCAGGAGAC,
K43R_RV;GTCTCCTGGAACcTGTCCTC, K51R_FW;GCTCCGTCTGAgGGAGAAGGAAGGCTG, K51R_RV;CAGCCTTCCTTCTCCcTCAGACGGAGC, K53R_FW;GCTCCGTCTGAAGGAGAgGGAAGGCTG, K53R_RV;CAGCCTTCCcTCTCCTTCAGACGGAGC, K84R_FW;GGATTCTCTTTAgGGACTACAATCTGG, K84R_RV;CCAGATTGTAGTCCcTAAAGAGAATCC, K103R_FW;CGGCTTCCCAAgAGATGTGGACC, K103R_RV;GGTCCACATCTcTTGGGAAGCCG, K112,115R_FW;CCAGTCCCGGAgAGGGAGAAgGCCCCTTGC, K112,115R_RV;GCAAGGGGCcTTCTCCCTcTCCGGGACTGG, K51,53R_FW;GCTCCGTCTGAgGGAGAgGGAAGGCTG, K51,53R_RV;CAGCCTTCCcTCTCCcTCAGACGGAGC, K43,51,53Q_FW;GAGGACcAGTTCCAGGAGACGCTCCGTCTGcAGGAGcAGGAAGGCTG, K43,51,53Q_RV;CAGCCTTCCTgCTCCTgCAGACGGAGCGTCTCCTGGAACTgGTCCTC,
K84Q_FW;GGATTCTCTTTcAGGACTACAATCTGG,
K84Q_RV;CCAGATTGTAGTCCTgAAAGAGAATCC, T69A_FW;GCTGTCCTGGCTCCTGgCCCGGGACAGTGGGG, T69A_RV;CCCCACTGTCCCGGGcCAGGAGCCAGGACAGC, S157A_FW;GCGTCTCCAGCCCAGGCgCCCACCTGAAGACTAAGCC, S157A_RV;GGCTTAGTCTTCAGGTGGGcGCCTGGGCTGGAGACGC, K296R_FW;CCCCAGGTTAACCAGAgGAACGAGGATGAG, K296R_RV;CTCATCCTCGTTCcTCTGGTTAACCTGGGG,
Flanking_5′-1;GTGAACCGTCAGATCCGCT,
Flanking_3′-1;CAGAAGCTGCCATGGTCTGA,
Flanking_5′-2;AAGGGAGCCCAGGTCACTAT,
Flanking_3′-2;GGGAGGTGTGGGAGGTTTTT.
Immunofluorescence Analyses.
4D6 cells seeded on coverslips were transiently transfected with plasmids driving expression of Flag-tagged full-length WT Aire or of the designated domain-deletions or point-mutations of Aire. Thirty hours after transfection, cells were fixed with 4% paraformaldehyde in PBS for 10 min, followed by permeabilization with 0.2% TritonX-100 in PBS for 10 min. Cells were then blocked with 5% normal donkey serum in 0.05% Tween 20 in PBS for 2 h and were stained with anti-Flag Ab at 4 °C overnight, followed by incubation with FITC-conjugated anti-mouse IgG secondary Abs (Jackson ImmunoResearch Laboratories) in the dark for 1 h at room temperature. Cells were counterstained with DAPI (Life Technologies) for visualization of nuclei. Coverslips with cells were mounted on a slide with Fluoromount-G (SouthernBiotech), and immunofluorescent images were acquired by a fluorescence microscope (Zeiss Axio Imager M1) equipped with filters matching the spectral excitation and emission characteristics of DAPI and FITC.
Immunoprecipitations and Western Blotting.
About 30 h after the transfection, 4D6 cells were harvested by scraping. After washing them twice with cold PBS, cells were resuspended in hypotonic buffer (10 mM Hepes, 1.5 mM MgCl2, 10 mM KCl, 0.5 mM Na3VO4, 5 mM NaF, 2 mM Na4P2O7, 5 mM sodium butyrate, and cOmplete protease inhibitor mixture EDTA-free; Roche Diagnostics) and were kept on ice for 15 min. The suspension was then transferred to a glass Dounce and homogenized with 15 up-and-down strokes using the loose pestle to release the nuclei, followed by centrifugation at 3,300 × g for 15 min. Collected nuclei were extracted using hypertonic buffer containing micrococcal nuclease [20 mM Hepes, 3 mM CaCl2, 0.5 mM MgCl2, 300 mM NaCl, 20 mM KCl, 10% Glycerol, 0.5 mM Na3VO4, 5 mM NaF, 5 mM sodium butyrate, cOmplete protease inhibitor mixture EDTA-free (Roche), and 1 µg/mL nuclease S7 (Roche Diagnostics)] for 45 min. EDTA was added to a final concentration of 5 mM to stop chromatin digestion before a centrifugation at 13,000 × g for 10 min. The supernatant was isolated and used as a nuclear extract for IP or co-IP experiments.
After preclearing with Protein A and Protein G Dynabeads (Life Technologies) for 30 min, 1.5 µg of the specified Ab or IgG control Ab was added, and the extract was incubated on a rotator overnight at 4 °C. Subsequently, 15 μL of Protein A and of Protein G Dynabeads were added, and the incubation continued for another 2 h. The Dynabeads were washed three times with ice-cold PBS containing 0.1% Nonidet P-40, and once with pure ice-cold PBS. IPed proteins were eluted by boiling in sample buffer for 5 min and were separated on a 7.5% or 10% SDS polyacrylamide gel followed by transfer of proteins to an Immun-Blot PVDF membrane (Bio-Rad) by using a wet/tank blotting system. Membranes were blocked in Tris-buffered saline containing 2% BSA (Fraction V; Fisher Scientific) and were probed with primary Ab overnight at 4 °C. After several washes with Tris-buffered saline containing 0.2% Tween 20, membranes were incubated with the secondary Ab linked to horseradish peroxidase or Clean-Blot IP Detection Kit (Thermo Scientific). To detect Flag/Aire, we used anti-Flag M2-peroxidase (Sigma-Aldrich) instead of a primary and secondary Ab. Blots were developed with ECL Prime Western Blotting Detection Reagent (GE Healthcare) as per the manufacturer’s instructions. Chemiluminescent images were acquired by FluorChem Q System (ProteinSimple) and were analyzed using AlphaView Software (version 3.2.2; ProteinSimple).
For sequential detection of different targets, blots were incubated in Restore Western Blot Stripping Buffer (Thermo Scientific) for 15 min before the next blocking. For co-IP studies, Abs recognizing the following proteins were used: Brd2, Brd3, Brd4 (rabbit polyclonal Abs; Bethyl); Flag-tag (M2 mouse mAb; Sigma-Aldrich); V5-tag (SV5-Pk1 mouse mAb; Abcam); CDK9 (C-20 rabbit polyclonal Ab; Santa Cruz); CBP (A-22 rabbit polyclonal Ab; Santa Cruz); DNA-PKcs (mixture of mouse mAbs; Thermo Fisher Scientific); and acetylated-lysine (catalog #9441 rabbit polyclonal Ab; Cell Signaling). For Fig. 5D, we used Abs against SFRS3 (#H00006428-M08; Abnova), Ku80 (#ab55408; Abcam), TOP2A (#ab12318; Abcam), TOP2B (#ab72334; Abcam), Brd4 (#ab84776; Abcam), Pol-II (#SC-899; Santa Cruz), SPT5 (#SC-28678; Santa Cruz), and DDX5 (#SC-166167; Santa Cruz).
The following assay was performed to quantify direct Aire-CARD:Brd4-BD1 binding. A Flag/CARD expression plasmid was generated by inserting a PCR fragment encompassing amino acids 1–107 of murine Aire into the pCMV-Tag1 vector (Agilent Technologies). The recombinant protein was expressed via transfection in 4D6 cells and purified by anti-Flag M2 magnetic beads (Sigma-Aldrich), eluting with Flag peptide (Sigma-Aldrich). Expression plasmids for Brd4 bromodomain-GST fusion proteins were constructed by inserting the PCR fragments corresponding to BD1 (amino acids 55–168 of human Brd4), BD2 (amino acids 347–464), or both BD1 and BD2 (amino acids 55–464) into the pGEX-4T-3 vector (GE Healthcare). The purified proteins loaded onto an SDS/PAGE gel (4–20% Mini-PROTEAN TGX gel; Bio-Rad), and visualized by SimplyBlue SafeStain (Life Technologies) solution. The fusion proteins were expressed in E. coli BL21 (New England Biolabs) and were purified by Glutathione Sepharose4B (GE Healthcare). For the in vitro GST pull-down assay, ∼12 μg of each fusion protein bound to Sepharose was incubated with recombinant Flag/CARD protein in 350 μL GST pull-down buffer [20 mM Tris⋅Cl pH 8.0, 75 mM NaCl, 0.1 mM EDTA, 0.05% Tween20, 5 mM sodium butyrate, protease inhibitor mixture (cOmplete; Roche Diagnostics)] and 50 μM I-BET151 (when indicated) at 4 °C overnight. After four washes with GST pull-down buffer, the GST-BD-associated proteins were eluted into 40 μL of 1× Laemmli sample buffer and 15 μL subjected to SDS/PAGE (4–20% Mini-PROTEAN TGX Gel; Bio-Rad), immunoblotting with anti-Flag antibody.
For quantification of the expression of WT or 4K→R mutant Aire, 4D6 cells were transfected with the corresponding expression plasmids at the indicated dilutions. Forty hours after transfection, the cells were washed twice with cold PBS, collected in lysis buffer [50 mM Hepes at pH 7.2, 300 mM LiCl, 1 mM EDTA, 1% Nonidet P-40, 0.7% sodium deoxycholate, protease inhibitor mixture (cOmplete, EDTA free, Roche Diagnostics)] and lysed by brief sonication. Protein concentrations were determined using the BCA Protein Assay Reagent (Thermo Scientific). Approximately 5 μg protein was loaded per lane and separated on a 7.5% SDS polyacrylamide gel, followed by transfer of proteins to an Immun-Blot PVDF membrane (Bio-Rad). The targets were detected via sequential blotting. An anti-Actin (AC-40 mouse mAb, Sigma-Aldrich) was used to detect β-actin with an anti-mouse IgG-HRP as a secondary Ab. Chemiluminescent images were acquired and quantified as in the co-IP studies.
q-PCR.
Primers used for the reactions were as follows:
HPRT1 forward: 5-TGAAGAGCTATTGTAATGACCAGTCAAC;
HPRT1 reverse: 5-AGCAAGCTTGCGACCTTGACCA;
ALOX12 forward: 5-AGCCAGACATGGTGCCTCT;
ALOX12 reverse: 5-TTTAGCACAGCTTTGGGCTT;
S100A9 forward: CTGGTGCGAAAAGATCTGCA;
S100A9 reverse: 5-CCTTTTCATTCTTATTCTCCTTCTTGAG;
CD4 forward: 5-GGACAGGTCCTGCTGGAATC;
CD4 reverse: 5-CAATGAAAAGCAGGAGGCCG;
CELF2 forward: 5-TCCTTGACCTCTCTCGGGAC;
CELF2 reverse: 5-AAGTCCTCCATTCAGAGCCG;
KRT13 forward: 5-AGATCGCCACCTACCGCA;
KRT13 reverse: 5-AACCAATCATCTTGGCGTCC;
GAPDH forward: 5-GGGGCTGGCATTGCCCTCAACG; and
GAPDH reverse: 5-GGGGCTGGTGGTCCAGGGGT.
Brd4 ChIP-seq Analysis.
4D6 cells were transfected with pEGFP-N1 (Clontech Laboratories) or with this plasmid driving expression of murine Aire, using Lipofectamine LTX with PlusReagent (Life Technologies), according to the manufacturer’s instructions. In certain experiments, 3 μM I-BET151 was added 6 h after the transfection. For ChIP, cells were cross-linked with 1% formaldehyde 48 h after the transfection. ChIP for Brd4 (anti-Brd4 Ab A301–985; Bethyl Laboratories) and generation of Illumina sequencing libraries was performed as previously described (16, 51). Samples were sequenced on the Illumina Hiseq2000 platform for 50 cycles, and raw sequencing data were processed using CASAVA_v1.8.2 software to generate fastq files. Sequencing reads were aligned to human genome reference hg19 by using Bowtie 2 (version 2.1.0) (52). MACS (version 1.4.2) (53) was used to identify Brd4-binding regions from vehicle-treated control-plasmid-transfected 4D6 cells, and MACS2 (version 2.0.10) was used to prepare the normalized fragment pileup in bedGraph format from different conditions. Brd4 signals in the identified regions in the different conditions were counted from bedGraph, and plots were generated in R (version 3.0.2). TSS positions were taken from UCSC refGene on hg19.
Tetramer Analysis.
APC-conjugated tetramers corresponding to Ab:IRBP(294–306) and Ab:IRBP(786–797) were made as described (8). For tetramer staining, peripheral lymph nodes cells were pooled and stained for 1 h at room temperature, followed by magnetic bead purification using anti-APC MicroBeads and LS column (Miltenyi Biotech) to enrich for tetramer-positive cells. The selected cells were stained with anti-CD3 (145-2C11; BioLegend), -CD4 (GK1.5; BioLegend), -CD8 (53-6.7; BioLegend), -B220 (RA3-6B2; BioLegend), -CD19 (6D5; BioLegend), -F4/80 (BM8; BioLegend), -CD11b (M1/70; BioLegend), -CD11c (N418; BioLegend), and -CD44 (IM7; BioLegend). Stained cells were analyzed on an LSRII (BD Biosciences), and tetramer-reactive cells were gated as CD3+ and negative for CD8, CD11b, CD11c, F4/80, B220, and CD19, using FlowJo software (TreeStar). Tetramer-positive cells were quantified by counting the positively selected cells by MACSQuant (Miltenyi Biotech) and the ratio of the tetramer-reactive cells on the analysis by FlowJo.
Thymocyte Analysis.
Thymi were dissociated by passing them through a cell strainer (40 µm; BD Falcon) and were stained with anti-CD4, -CD8, -CD25 (PC61; BioLegend), -CD45 (30F-11; BioLegend), and -TCRβ (H57-597; BioLegend). Stained cells were analyzed on an LSRII, and cells in each population were gated as follows: double-negative, CD45+CD4−CD8−; double-positive, CD45+CD4+CD8+, CD4:CD45+CD4+CD8−, CD8:CD45+CD4−CD8+; and Treg, CD45+CD4+CD8−TCRβ+CD25+.
Acknowledgments
We thank Dr. M. Giraud and Dr. N. Fujikado for scientific discussion; and G. Buruzula, J. LaVecchio, J. Erickson, R. Cruse, K. Rothamel, K. Hattori, Dr. H. Paik, and Dr. A. Ortiz-Lopez for experimental assistance. This work was funded by a Sponsored Research Agreement from GlaxoSmithKline and by NIH Grants R01 AI088204 and R01 DK060027 (to D.M.). K.B. was supported by a fellowship from the American Diabetes Association.
Footnotes
Conflict of interest statement: GlaxoSmithKline has an ongoing interest in the therapeutic applications of BET-protein inhibitors.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1512081112/-/DCSupplemental.
References
- 1.Peterson P, Org T, Rebane A. Transcriptional regulation by AIRE: Molecular mechanisms of central tolerance. Nat Rev Immunol. 2008;8(12):948–957. doi: 10.1038/nri2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mathis D, Benoist C. Aire. Annu Rev Immunol. 2009;27:287–312. doi: 10.1146/annurev.immunol.25.022106.141532. [DOI] [PubMed] [Google Scholar]
- 3.Anderson MS, et al. Projection of an immunological self shadow within the thymus by the aire protein. Science. 2002;298(5597):1395–1401. doi: 10.1126/science.1075958. [DOI] [PubMed] [Google Scholar]
- 4.Johnnidis JB, et al. Chromosomal clustering of genes controlled by the aire transcription factor. Proc Natl Acad Sci USA. 2005;102(20):7233–7238. doi: 10.1073/pnas.0502670102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sansom SN, et al. Population and single-cell genomics reveal the Aire dependency, relief from Polycomb silencing, and distribution of self-antigen expression in thymic epithelia. Genome Res. 2014;24(12):1918–1931. doi: 10.1101/gr.171645.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liston A, Lesage S, Wilson J, Peltonen L, Goodnow CC. Aire regulates negative selection of organ-specific T cells. Nat Immunol. 2003;4(4):350–354. doi: 10.1038/ni906. [DOI] [PubMed] [Google Scholar]
- 7.Anderson MS, et al. The cellular mechanism of Aire control of T cell tolerance. Immunity. 2005;23(2):227–239. doi: 10.1016/j.immuni.2005.07.005. [DOI] [PubMed] [Google Scholar]
- 8.Taniguchi RT, et al. Detection of an autoreactive T-cell population within the polyclonal repertoire that undergoes distinct autoimmune regulator (Aire)-mediated selection. Proc Natl Acad Sci USA. 2012;109(20):7847–7852. doi: 10.1073/pnas.1120607109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Malchow S, et al. Aire-dependent thymic development of tumor-associated regulatory T cells. Science. 2013;339(6124):1219–1224. doi: 10.1126/science.1233913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Perry JS, et al. Distinct contributions of Aire and antigen-presenting-cell subsets to the generation of self-tolerance in the thymus. Immunity. 2014;41(3):414–426. doi: 10.1016/j.immuni.2014.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Oven I, et al. AIRE recruits P-TEFb for transcriptional elongation of target genes in medullary thymic epithelial cells. Mol Cell Biol. 2007;27(24):8815–8823. doi: 10.1128/MCB.01085-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Giraud M, et al. Aire unleashes stalled RNA polymerase to induce ectopic gene expression in thymic epithelial cells. Proc Natl Acad Sci USA. 2012;109(2):535–540. doi: 10.1073/pnas.1119351109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Giraud M, et al. An RNAi screen for Aire cofactors reveals a role for Hnrnpl in polymerase release and Aire-activated ectopic transcription. Proc Natl Acad Sci USA. 2014;111(4):1491–1496. doi: 10.1073/pnas.1323535111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Adelman K, Lis JT. Promoter-proximal pausing of RNA polymerase II: Emerging roles in metazoans. Nat Rev Genet. 2012;13(10):720–731. doi: 10.1038/nrg3293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sharma M, George AA, Singh BN, Sahoo NC, Rao KV. Regulation of transcript elongation through cooperative and ordered recruitment of cofactors. J Biol Chem. 2007;282(29):20887–20896. doi: 10.1074/jbc.M701420200. [DOI] [PubMed] [Google Scholar]
- 16.Rahl PB, et al. c-Myc regulates transcriptional pause release. Cell. 2010;141(3):432–445. doi: 10.1016/j.cell.2010.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Galbraith MD, et al. HIF1A employs CDK8-mediator to stimulate RNAPII elongation in response to hypoxia. Cell. 2013;153(6):1327–1339. doi: 10.1016/j.cell.2013.04.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fromm G, Gilchrist DA, Adelman K. SnapShot: Transcription regulation: Pausing. Cell. 2013;153(4):930–930. doi: 10.1016/j.cell.2013.04.011. [DOI] [PubMed] [Google Scholar]
- 19.Devaiah BN, Singer DS. Two faces of brd4: Mitotic bookmark and transcriptional lynchpin. Transcription. 2013;4(1):13–17. doi: 10.4161/trns.22542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huang B, Yang XD, Zhou MM, Ozato K, Chen LF. Brd4 coactivates transcriptional activation of NF-kappaB via specific binding to acetylated RelA. Mol Cell Biol. 2009;29(5):1375–1387. doi: 10.1128/MCB.01365-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang G, et al. Down-regulation of NF-κB transcriptional activity in HIV-associated kidney disease by BRD4 inhibition. J Biol Chem. 2012;287(34):28840–28851. doi: 10.1074/jbc.M112.359505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wu SY, Lee AY, Lai HT, Zhang H, Chiang CM. Phospho switch triggers Brd4 chromatin binding and activator recruitment for gene-specific targeting. Mol Cell. 2013;49(5):843–857. doi: 10.1016/j.molcel.2012.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shi J, et al. Disrupting the interaction of BRD4 with diacetylated Twist suppresses tumorigenesis in basal-like breast cancer. Cancer Cell. 2014;25(2):210–225. doi: 10.1016/j.ccr.2014.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Seal J, et al. Identification of a novel series of BET family bromodomain inhibitors: Binding mode and profile of I-BET151 (GSK1210151A) Bioorg Med Chem Lett. 2012;22(8):2968–2972. doi: 10.1016/j.bmcl.2012.02.041. [DOI] [PubMed] [Google Scholar]
- 25.Giraud M, et al. An IRF8-binding promoter variant and AIRE control CHRNA1 promiscuous expression in thymus. Nature. 2007;448(7156):934–937. doi: 10.1038/nature06066. [DOI] [PubMed] [Google Scholar]
- 26.Koh AS, et al. Aire employs a histone-binding module to mediate immunological tolerance, linking chromatin regulation with organ-specific autoimmunity. Proc Natl Acad Sci USA. 2008;105(41):15878–15883. doi: 10.1073/pnas.0808470105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Filippakopoulos P, et al. Selective inhibition of BET bromodomains. Nature. 2010;468(7327):1067–1073. doi: 10.1038/nature09504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Saare M, Rebane A, Rajashekar B, Vilo J, Peterson P. Autoimmune regulator is acetylated by transcription coactivator CBP/p300. Exp Cell Res. 2012;318(14):1767–1778. doi: 10.1016/j.yexcr.2012.04.013. [DOI] [PubMed] [Google Scholar]
- 29.Incani F, et al. AIRE acetylation and deacetylation: Effect on protein stability and transactivation activity. J Biomed Sci. 2014;21(1):85. doi: 10.1186/s12929-014-0085-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pitkänen J, et al. The autoimmune regulator protein has transcriptional transactivating properties and interacts with the common coactivator CREB-binding protein. J Biol Chem. 2000;275(22):16802–16809. doi: 10.1074/jbc.M908944199. [DOI] [PubMed] [Google Scholar]
- 31.Liiv I, et al. DNA-PK contributes to the phosphorylation of AIRE: Importance in transcriptional activity. Biochim Biophys Acta. 2008;1783(1):74–83. doi: 10.1016/j.bbamcr.2007.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Abramson J, Giraud M, Benoist C, Mathis D. Aire’s partners in the molecular control of immunological tolerance. Cell. 2010;140(1):123–135. doi: 10.1016/j.cell.2009.12.030. [DOI] [PubMed] [Google Scholar]
- 33.Žumer K, Low AK, Jiang H, Saksela K, Peterlin BM. Unmodified histone H3K4 and DNA-dependent protein kinase recruit autoimmune regulator to target genes. Mol Cell Biol. 2012;32(8):1354–1362. doi: 10.1128/MCB.06359-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yang XJ, Seto E. Lysine acetylation: Codified crosstalk with other posttranslational modifications. Mol Cell. 2008;31(4):449–461. doi: 10.1016/j.molcel.2008.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Choudhary C, et al. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science. 2009;325(5942):834–840. doi: 10.1126/science.1175371. [DOI] [PubMed] [Google Scholar]
- 36.Zhou Q, Yik JH. The Yin and Yang of P-TEFb regulation: Implications for human immunodeficiency virus gene expression and global control of cell growth and differentiation. Microbiol Mol Biol Rev. 2006;70(3):646–659. doi: 10.1128/MMBR.00011-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Parker SC, et al. NISC Comparative Sequencing Program; National Institutes of Health Intramural Sequencing Center Comparative Sequencing Program Authors; NISC Comparative Sequencing Program Authors Chromatin stretch enhancer states drive cell-specific gene regulation and harbor human disease risk variants. Proc Natl Acad Sci USA. 2013;110(44):17921–17926. doi: 10.1073/pnas.1317023110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Whyte WA, et al. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell. 2013;153(2):307–319. doi: 10.1016/j.cell.2013.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lovén J, et al. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell. 2013;153(2):320–334. doi: 10.1016/j.cell.2013.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hnisz D, et al. Super-enhancers in the control of cell identity and disease. Cell. 2013;155(4):934–947. doi: 10.1016/j.cell.2013.09.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Muller S, Filippakopoulos P, Knapp S. Bromodomains as therapeutic targets. Expert Rev Mol Med. 2011;13:e29. doi: 10.1017/S1462399411001992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Prinjha RK, Witherington J, Lee K. Place your BETs: The therapeutic potential of bromodomains. Trends Pharmacol Sci. 2012;33(3):146–153. doi: 10.1016/j.tips.2011.12.002. [DOI] [PubMed] [Google Scholar]
- 43.Org T, et al. The autoimmune regulator PHD finger binds to non-methylated histone H3K4 to activate gene expression. EMBO Rep. 2008;9(4):370–376. doi: 10.1038/embor.2008.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Vollmuth F, Blankenfeldt W, Geyer M. Structures of the dual bromodomains of the P-TEFb-activating protein Brd4 at atomic resolution. J Biol Chem. 2009;284(52):36547–36556. doi: 10.1074/jbc.M109.033712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Halonen M, et al. APECED-causing mutations in AIRE reveal the functional domains of the protein. Hum Mutat. 2004;23(3):245–257. doi: 10.1002/humu.20003. [DOI] [PubMed] [Google Scholar]
- 46.Ferguson BJ, et al. AIRE’s CARD revealed, a new structure for central tolerance provokes transcriptional plasticity. J Biol Chem. 2008;283(3):1723–1731. doi: 10.1074/jbc.M707211200. [DOI] [PubMed] [Google Scholar]
- 47.Morinière J, et al. Cooperative binding of two acetylation marks on a histone tail by a single bromodomain. Nature. 2009;461(7264):664–668. doi: 10.1038/nature08397. [DOI] [PubMed] [Google Scholar]
- 48.Khan IS, et al. Enhancement of an anti-tumor immune response by transient blockade of central T cell tolerance. J Exp Med. 2014;211(5):761–768. doi: 10.1084/jem.20131889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yang S, Bansal K, Lopes J, Benoist C, Mathis D. Aire’s plant homeodomain(PHD)-2 is critical for induction of immunological tolerance. Proc Natl Acad Sci USA. 2013;110(5):1833–1838. doi: 10.1073/pnas.1222023110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ho SN, Hunt HD, Horton RM, Pullen JK, Pease LR. Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene. 1989;77(1):51–59. doi: 10.1016/0378-1119(89)90358-2. [DOI] [PubMed] [Google Scholar]
- 51.Goldberg AD, et al. Distinct factors control histone variant H3.3 localization at specific genomic regions. Cell. 2010;140(5):678–691. doi: 10.1016/j.cell.2010.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9(4):357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhang Y, et al. Model-based analysis of ChIP-Seq (MACS) Genome Biol. 2008;9(9):R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]