

Significance
Consumption of red meat is associated with increased colorectal cancer risk. We show that the gut microbiota is pivotal in this increased risk. Mice receiving a diet with heme, a proxy for red meat, show a damaged gut epithelium and a compensatory hyperproliferation that can lead to colon cancer. Mice receiving heme together with antibiotics do not show this damage and hyperproliferation. Our data indicate that microbial hydrogen sulfide opens the protective mucus barrier and exposes the epithelium to cytotoxic heme. Antibiotics block microbial sulfide production and thereby maintain the mucus barrier that prevents heme-induced hyperproliferation. Our study indicates that fecal trisulfide is a novel biomarker of mucus barrier integrity, which could be of relevance in human colon disease diagnostics.
Keywords: colorectal cancer, red meat, mucus barrier, mucolysis, (tri)sulfides
Abstract
Colorectal cancer risk is associated with diets high in red meat. Heme, the pigment of red meat, induces cytotoxicity of colonic contents and elicits epithelial damage and compensatory hyperproliferation, leading to hyperplasia. Here we explore the possible causal role of the gut microbiota in heme-induced hyperproliferation. To this end, mice were fed a purified control or heme diet (0.5 μmol/g heme) with or without broad-spectrum antibiotics for 14 d. Heme-induced hyperproliferation was shown to depend on the presence of the gut microbiota, because hyperproliferation was completely eliminated by antibiotics, although heme-induced luminal cytotoxicity was sustained in these mice. Colon mucosa transcriptomics revealed that antibiotics block heme-induced differential expression of oncogenes, tumor suppressors, and cell turnover genes, implying that antibiotic treatment prevented the heme-dependent cytotoxic micelles to reach the epithelium. Our results indicate that this occurs because antibiotics reinforce the mucus barrier by eliminating sulfide-producing bacteria and mucin-degrading bacteria (e.g., Akkermansia). Sulfide potently reduces disulfide bonds and can drive mucin denaturation and microbial access to the mucus layer. This reduction results in formation of trisulfides that can be detected in vitro and in vivo. Therefore, trisulfides can serve as a novel marker of colonic mucolysis and thus as a proxy for mucus barrier reduction. In feces, antibiotics drastically decreased trisulfides but increased mucin polymers that can be lysed by sulfide. We conclude that the gut microbiota is required for heme-induced epithelial hyperproliferation and hyperplasia because of the capacity to reduce mucus barrier function.
Colorectal cancer, the second leading cause of cancer death in Western countries, is associated with diets high in red meat (1), whereas consumption of white meat does not have this association (2). Heme, the iron-porphyrin pigment, is present at much higher levels in red compared with white meat. Epidemiological studies show that heme intake is related to colon cancer risk (3, 4). Our previous studies show that when rodents consume heme, their colonic contents become more cytotoxic (5, 6). This increased cytotoxicity injures the colonic epithelial surface cells. To replace the injured surface cells, hyperproliferation from the stem cells in the crypts is initiated. Together with inhibition of apoptosis, this compensatory hyperproliferation leads to hyperplasia (6), which eventually can develop into colorectal cancer.
Dietary heme is poorly absorbed in the small intestine; ∼90% of dietary heme enters the colon (7). Besides the toxic effect of heme on the colonic mucosa, dietary heme affects the microbiota. The relationship between intestinal microbiota and colon cancer has long been suspected (8). In humans, a red meat diet increases Bacteroides spp. in feces (9). We recently showed that in mice, a heme diet changed the microbiota drastically, majorly increasing the Gram-negative bacteria (mainly Bacteroidetes, Proteobacteria, and Verrucomicrobia) (10). The gut microbiota can induce hyperproliferation via mechanisms occurring in the colon lumen, such as modulation of oxidative and cytotoxic stress or by influencing the mucus barrier. Oxidative stress induces the formation of peroxidized lipids, which react with heme to form the cytotoxic heme factor (CHF), thereby increasing cytotoxic stress (5, 11). In a time course study, we showed that there is a lag time in the formation of CHF and in the induction of hyperproliferation when mice are transferred from a control to heme diet (11). This lag time could be due to a time-dependent adaptation of the microbiota to the heme diet. Notably, heme does not increase cytotoxicity and epithelial hyperproliferation in the small intestine (12), indicating that formation of CHF only occurs in the colon where bacterial density is high. Moreover, these experiments suggested that CHF-induced hyperproliferation coincided with a reduced mucus barrier function (11), leading to enhanced contact of colonocytes with microbiota and toxic substances. In the present study, we investigate whether bacteria play a causal role in heme-induced cytotoxicity and hyperproliferation by using broad-spectrum antibiotics (Abx). Our results illustrate the crucial role of the gut microbiota in heme-induced hyperproliferation and indicate the involvement of microbial hydrogen sulfide formation in reduction of the polymeric mucin network to degrade the protective mucus barrier and expose the colon mucosa to CHF.
Results
Heme- and Abx-Induced Changes in the Colonic Lumen.
Mice were divided into four groups receiving either a control diet (C-group), a heme diet (H-group), a control diet with Abx treatment (CA-group), or a heme diet with Abx treatment (HA-group). After 2 wk of intervention the H- and HA-group had a lower body weight compared with their controls (Table 1). Fecal dry and wet weight was significantly increased in the HA-group.
Table 1.
Effects of heme and Abx on body weight and fecal parameters
| Variable | Control | Heme | Control + Abx | Heme + Abx |
| Body weight (g) | 27.7 ± 0.5a | 24.9 ± 0.5b | 27.2 ± 0.3a | 24.8 ± 0.4b |
| Fecal wet weight (g/d) | 0.49 ± 0.08a | 0.60 ± 0.05a | 0.62 ± 0.09a | 1.20 ± 0.19b |
| Fecal dry weight (g/d) | 0.12 ± 0.01a | 0.11 ± 0.01a | 0.12 ± 0.01a | 0.19 ± 0.02b |
| TBARS (MDA equivalents, µmol/L) | 12.70 ± 1.43a | 59.84 ± 2.46b | 11.06 ± 1.63a | 46.06 ± 3.92c |
| Cytotoxicity (% lysis) | 1.09 ± 0.45a | 66.90 ± 10.45b | 0.05 ± 0.04a | 31.30 ± 8.98c |
Data are represented as mean ± SEM (n = 9/group). Differences between the groups were tested by ANOVA with a Bonferroni post hoc test and different superscripts indicate significant differences (P < 0.05).
To confirm that Abx decreased the abundance of bacteria, quantitative PCR (qPCR) analyses with specific primers targeting total bacteria, Bacteroidetes, or Firmicutes were performed (Fig. 1A). Abx treatment significantly reduced the abundance of total bacteria and Firmicutes ∼100-fold and Bacteroidetes ∼1,000-fold. Moreover, the H-group had a significantly increased abundance of Bacteroidetes compared with the C-group, which corroborates previous observations (10). Abx thus drastically decreased the bacterial density, affecting both Gram-positive Firmicutes and Gram-negative Bacteroidetes. To study how this impacts the normal microbial modification of host compounds, we determined the fecal bile acid composition. No conjugated bile acids were detected in fecal water in the C- and H-group, where unconjugated and secondary bile acids were predominant (Fig. 1B). However, with Abx almost all bile acids were primary and conjugated with glycine or taurine (ratio about 3), showing that Abx blocked microbial bile deconjugation and dehydroxylation almost completely.
Fig. 1.
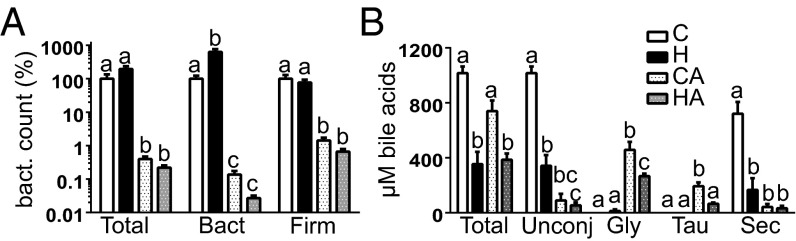
(A) Counts of total bacteria, Bacteroidetes, and Firmicutes measured by qPCR. Control bars are set at 100%, and other bars are relative to controls; mean ± SEM (n = 9/group). (B) Bile acid profiles determined by HPLC; mean ± SEM (n = 3/group). Letters indicate significant different groups (P < 0.05), ANOVA with Bonferroni post hoc test. Gly, glycine conjugated; Sec, secondary; Tau, taurine conjugated.
Heme increases oxidative and cytotoxic stress in the colon (10, 11). Reactive oxygen species (ROSs) induce the formation of lipid peroxides that react with heme to form CHF, thereby increasing the cytotoxicity of luminal contents (5, 11). We determined lipid peroxidation product levels by measuring thiobarbituric acid reactive substances (TBARS) in fecal water. TBARS were low in the C- and CA-group (Table 1) and increased significantly and to a similar extent in the H- and HA-group, implying that heme, both in presence and absence of Abx, induced ROS stress. Analogously, fecal water cytotoxicity (Table 1) was significantly increased in the H- and HA-group compared with their controls. Because Abx drastically reduced microbiota density (100- to 1,000-fold), but only slightly reduced cytotoxicity (2-fold) and TBARS (1.3-fold), it is unlikely that bacteria play a major role in the formation of TBARS and cytotoxicity.
Heme- and Abx-Induced Changes in the Colonic Mucosa.
Morphology analyses of H&E-stained colon tissue (Fig. 2A) confirmed the previously reported heme-induced increased crypt depth (H- vs. C-group). Abx treatment did not affect the colon morphology in the CA- vs. C-group but completely restored tissue morphology in the HA- vs. H-group. The crypt depth increase in the H-group did not result from inflammation because neutrophil and macrophage infiltration in the lamina propria was comparable to the C-group. Analogous to earlier reports (6), cell proliferation quantification using Ki67 staining (Fig. 2 B and C) shows that the heme diet strongly induced cell proliferation (H- vs. C-group), leading to expansion of the proliferative compartment and increased crypt depth. Abx treatment led to slightly reduced numbers of cells per crypt in the CA- vs. C-group, but did not significantly affect their labeling index or amount of proliferative cells. However, Abx treatment in the heme diet (HA- vs. H-group) completely suppressed heme-induced hyperproliferation and hyperplasia to levels observed in the C- and CA-groups (Fig. 2C). In conclusion, heme-induced hyperproliferation and hyperplasia in mouse colon only occurs in the presence of the gut microbiota.
Fig. 2.
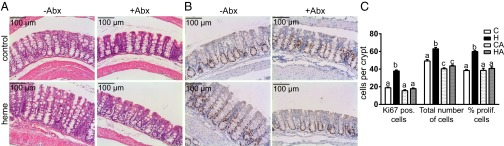
(A) Histochemical H&E staining and (B) immunohistochemical Ki67 staining of colon of control and heme-fed mice. (C) Quantification of Ki67-positive cells per crypt, total number of cells per crypt, and labeling index (percentage of proliferative cells per crypt); mean ± SEM (n = 9/group). Letters indicate significant different groups (P < 0.05), ANOVA with Bonferroni post hoc test.
Abx Block the Heme-Induced Expression of Cell Cycle Genes.
Using whole genome transcriptomics we investigated whether the physiological changes were reflected in gene expression profiles. The heme diet (H- vs. C-group) led to 5,507 differentially expressed genes (q < 0.01), of which almost 90% (4,859) were not significantly affected in the HA vs. CA comparison (Fig. S1A). The 4,859 genes specific for the H-group were analyzed by GSEA, indicating that mainly cell cycle related processes were affected by heme (Fig. S1B). Moreover, mining of these genes for the involved transcription factors (Fig. S1C), revealed that Cdkn2a, Smarcb1, and the tumor suppressors Tp53 and Rb1 were inhibited, whereas oncogenes such as Myc and Foxm1, as well as cell cycle regulators E2f1 and Tbx2, were activated by heme. Importantly, these processes and transcription factors were not modulated in the HA-group compared with the CA-group. There were only 369 differentially expressed genes unique for the HA-group (Fig. S1A). Notably, none of the modulated processes identified in the HA-group related to the end points of our study. Because of the specific heme-Abx interaction, the Abx-mediated differential gene expression profiles and processes were substantially different in the heme diet background (Tables S1 and S2) compared with the control diet background (Tables S3 and S4). These observations indicate that heme-induced mucosal gene expression changes of cell cycle-related processes require the presence of the microbiota, which is in agreement with the microbiota requirement for the increased labeling index (Fig. 2C).
Fig. S1.

(A) Venn diagram showing the numbers of heme and heme plus Abx-specific regulated genes and of overlapping genes (q < 0.01). (B) GSEA showing positive enriched processes. Sources of gene sets are indicated in superscript and represent (1) gene ontology, (2) reactome, and (3) KEGG. Size represents the number of genes in the gene-set and NES is normalized enrichment score, which is the enrichment score for the gene set after it has been normalized to account for variations in gene set size. (C) Ingenuity analysis showing activated or inhibited transcription factors (z-score > 2 and P < 0.05) in both treatments specifically and in overlapping genes.
Table S1.
Fifty highest up-regulated genes (q < 0.01) in mouse colon scrapings for the comparison of heme Abx vs. heme
| Gene symbol | Gene description | SI ± SEM | Fold change (heme + Abx vs. heme) | SI ± SEM | Fold change (control + Abx vs. control) | ||||||
| Heme | Heme + Abx | Control | Control + Abx | ||||||||
| SI | SEM | SI | SEM | SI | SEM | SI | SEM | ||||
| Gbp2 | Guanylate binding protein 2 | 96 | 24 | 412 | 102 | 4.0 | 303 | 77 | 962 | 234 | NS |
| Slc6a19 | Solute carrier family 6 (neurotransmitter transporter), member 19 | 81 | 14 | 320 | 53 | 3.9 | 21 | 2 | 60 | 6 | 2.8 |
| Enpep | Glutamyl aminopeptidase | 145 | 10 | 557 | 41 | 3.8 | 209 | 21 | 265 | 18 | NS |
| Slc5a4b | Solute carrier family 5 (neutral amino acid transporters, system A), member 4b | 52 | 5 | 197 | 23 | 3.7 | 14 | 0 | 16 | 1 | NS |
| Abcc2 | ATP-binding cassette, subfamily C (CFTR/MRP), member 2 | 115 | 15 | 425 | 49 | 3.7 | 40 | 5 | 42 | 3 | NS |
| Ace2 | Angiotensin I converting enzyme (peptidyl-dipeptidase A) 2 | 67 | 7 | 239 | 32 | 3.5 | 93 | 6 | 165 | 4 | 1.8 |
| Cyp3a25 | Cytochrome P450, family 3, subfamily a, polypeptide 25 | 21 | 1 | 79 | 14 | 3.5 | 21 | 3 | 15 | 1 | NS |
| Sis | Sucrase isomaltase (alpha-glucosidase) | 29 | 3 | 105 | 23 | 3.3 | 117 | 19 | 147 | 16 | NS |
| 2010106E10Rik | RIKEN cdna 2010106E10 gene | 60 | 7 | 191 | 13 | 3.2 | 70 | 5 | 82 | 3 | NS |
| Trac | T-cell receptor alpha constant | 97 | 14 | 274 | 27 | 2.8 | 127 | 10 | 320 | 24 | 2.5 |
| 1810030J14Rik | RIKEN cDNA 1810030J14 gene | 666 | 76 | 1,664 | 82 | 2.5 | 2,451 | 249 | 2,909 | 158 | NS |
| Cyp4f39 | Cytochrome P450, family 4, subfamily f, polypeptide 39 | 38 | 4 | 92 | 6 | 2.5 | 23 | 2 | 23 | 1 | NS |
| Dbp | D site albumin promoter binding protein | 111 | 22 | 274 | 40 | 2.4 | 608 | 33 | 399 | 60 | NS |
| Naaladl1 | N-acetylated alpha-linked acidic dipeptidase-like 1 | 522 | 7 | 1,275 | 71 | 2.4 | 880 | 88 | 925 | 50 | NS |
| Slc13a1 | Solute carrier family 13 (sodium/sulfate symporters), member 1 | 141 | 22 | 336 | 25 | 2.4 | 786 | 120 | 974 | 71 | NS |
| Rsad2 | Radical S-adenosyl methionine domain containing 2 | 57 | 9 | 139 | 21 | 2.4 | 56 | 1 | 152 | 15 | 2.7 |
| Npc1l1 | NPC1-like 1 | 37 | 3 | 100 | 23 | 2.4 | 33 | 2 | 49 | 4 | NS |
| Gcg | Glucagon | 586 | 25 | 1,354 | 60 | 2.3 | 491 | 22 | 580 | 7 | NS |
| Sstr1 | Somatostatin receptor 1 | 112 | 8 | 255 | 8 | 2.3 | 234 | 19 | 226 | 6 | NS |
| Gm10768 | Predicted gene 10768 | 10 | 1 | 24 | 3 | 2.2 | 7 | 1 | 8 | 0 | NS |
| Per3 | Period homolog 3 (Drosophila) | 81 | 10 | 174 | 19 | 2.1 | 286 | 14 | 198 | 27 | NS |
| Cyp2f2 | Cytochrome P450, family 2, subfamily f, polypeptide 2 | 107 | 5 | 223 | 13 | 2.1 | 426 | 32 | 218 | 13 | −2.0 |
| Ddx60 | DEAD (Asp-Glu-Ala-Asp) box polypeptide 60 | 83 | 14 | 173 | 26 | 2.1 | 115 | 10 | 486 | 78 | 4.1 |
| Gbp6 | Guanylate binding protein 6 | 90 | 9 | 191 | 29 | 2.0 | 188 | 27 | 323 | 49 | NS |
| Ccl5 | Chemokine (C-C motif) ligand 5 | 99 | 5 | 203 | 20 | 2.0 | 163 | 7 | 267 | 18 | 1.6 |
| Tgm3 | Transglutaminase 3, E polypeptide | 1,397 | 179 | 2,766 | 166 | 2.0 | 4,496 | 132 | 3,340 | 110 | NS |
| Ang4 | Angiogenin, ribonuclease A family, member 4 | 158 | 6 | 320 | 27 | 2.0 | 570 | 126 | 581 | 81 | NS |
| Oasl2 | 2'-5′ Oligoadenylate synthetase-like 2 | 217 | 39 | 432 | 66 | 2.0 | 247 | 12 | 905 | 123 | 3.6 |
| Cml5 | Camello-like 5 | 16 | 0 | 31 | 2 | 2.0 | 20 | 1 | 19 | 1 | NS |
| Rtp4 | Receptor transporter protein 4 | 93 | 14 | 187 | 29 | 2.0 | 111 | 10 | 331 | 42 | 3.0 |
| Slc37a2 | Solute carrier family 37 (glycerol-3-phosphate transporter), member 2 | 329 | 41 | 641 | 58 | 2.0 | 933 | 67 | 830 | 76 | NS |
| Apob | Apolipoprotein B | 501 | 34 | 982 | 67 | 1.9 | 752 | 26 | 797 | 41 | NS |
| Gm129 | Predicted gene 129 | 50 | 5 | 98 | 11 | 1.9 | 160 | 15 | 104 | 13 | NS |
| Slc6a20a | Solute carrier family 6 (neurotransmitter transporter), member 20A | 98 | 5 | 190 | 6 | 1.9 | 234 | 12 | 228 | 10 | NS |
| Rnf212 | Ring finger protein 212 | 124 | 5 | 245 | 23 | 1.9 | 132 | 16 | 117 | 6 | NS |
| Ces1d | Carboxylesterase 1D | 343 | 21 | 654 | 25 | 1.9 | 335 | 32 | 284 | 8 | NS |
| Slc10a2 | Solute carrier family 10, member 2 | 169 | 20 | 321 | 25 | 1.9 | 376 | 63 | 536 | 46 | NS |
| Gsdmc3 | Gasdermin C3 | 1,006 | 92 | 1,906 | 101 | 1.9 | 2744 | 138 | 2,631 | 181 | NS |
| Ces1g | Carboxylesterase 1G | 1,947 | 44 | 3,710 | 177 | 1.9 | 757 | 138 | 766 | 47 | NS |
| Edn1 | Endothelin 1 | 212 | 43 | 380 | 20 | 1.9 | 495 | 22 | 267 | 32 | −1.9 |
| Cck | Cholecystokinin | 47 | 2 | 88 | 5 | 1.9 | 44 | 1 | 42 | 2 | NS |
| Gdpd2 | Glycerophosphodiester phosphodiesterase domain containing 2 | 78 | 4 | 145 | 7 | 1.9 | 97 | 14 | 114 | 8 | NS |
| Ifit3 | IFN-induced protein with tetratricopeptide repeats 3 | 55 | 3 | 106 | 13 | 1.9 | 79 | 3 | 210 | 45 | 2.5 |
| G6pc | Glucose-6-phosphatase, catalytic | 19 | 2 | 37 | 5 | 1.9 | 14 | 1 | 16 | 1 | NS |
| Slc7a8 | Solute carrier family 7 (cationic amino acid transporter, y+ system), member 8 | 135 | 6 | 250 | 14 | 1.8 | 296 | 25 | 235 | 21 | NS |
| Bmp3 | Bone morphogenetic protein 3 | 270 | 8 | 498 | 21 | 1.8 | 564 | 49 | 395 | 21 | −1.4 |
| Trim30d | Tripartite motif-containing 30D | 96 | 10 | 176 | 13 | 1.8 | 248 | 16 | 369 | 47 | NS |
| Irf7 | IFN regulatory factor 7 | 355 | 21 | 649 | 26 | 1.8 | 495 | 19 | 1,052 | 97 | 2.1 |
| Slc35e3 | Solute carrier family 35, member E3 | 156 | 8 | 285 | 14 | 1.8 | 167 | 12 | 106 | 5 | −1.6 |
| Ccl8 | Chemokine (C-C motif) ligand 8 | 30 | 3 | 55 | 6 | 1.8 | 22 | 3 | 81 | 8 | 3.7 |
Table includes only genes with a signal intensity > 20 in at least one condition. n = 4 per group for Control, Control Abx, and Heme; n = 6 per group for Heme Abx. SI, signal intensity.
Table S2.
Fifty highest down-regulated genes (q < 0.01) in mouse colon scrapings for the comparison of heme Abx vs. heme
| Gene symbol | Gene description | SI ± SEM | Fold change (heme + Abx vs. heme) | SI ± SEM | Fold change (control + Abx vs. control) | ||||||
| Heme | Heme + Abx | Control | Control + Abx | ||||||||
| SI | SEM | SI | SEM | SI | SEM | SI | SEM | ||||
| Reg3b | Regenerating islet-derived 3 beta | 1,385 | 309 | 66 | 7 | −19.5 | 566 | 297 | 84 | 19 | NS |
| Reg3g | Regenerating islet-derived 3 gamma | 1,115 | 170 | 118 | 10 | −9.2 | 337 | 167 | 125 | 20 | NS |
| Slpi | Secretory leukocyte peptidase inhibitor | 511 | 58 | 167 | 14 | −3.1 | 60 | 4 | 118 | 28 | 1.9 |
| AI747448 | Expressed sequence AI747448 | 570 | 94 | 225 | 49 | −2.7 | 71 | 12 | 187 | 21 | 2.7 |
| Capg | Capping protein (actin filament), gelsolin-like | 724 | 32 | 272 | 13 | −2.7 | 164 | 11 | 196 | 18 | NS |
| Dhrs9 | Dehydrogenase/reductase (SDR family) member 9 | 639 | 56 | 243 | 17 | −2.6 | 227 | 15 | 190 | 29 | NS |
| Trim29 | Tripartite motif-containing 29 | 197 | 8 | 76 | 6 | −2.6 | 29 | 1 | 38 | 4 | NS |
| Spp1 | Secreted phosphoprotein 1 | 51 | 5 | 20 | 1 | −2.6 | 17 | 0 | 22 | 2 | NS |
| Fabp1 | Fatty acid binding protein 1, liver | 606 | 99 | 249 | 22 | −2.4 | 19 | 1 | 17 | 1 | NS |
| Mfsd2a | Major facilitator superfamily domain containing 2A | 138 | 20 | 60 | 6 | −2.3 | 51 | 5 | 71 | 11 | NS |
| Mcpt2 | Mast cell protease 2 | 130 | 17 | 57 | 5 | −2.3 | 26 | 2 | 33 | 3 | NS |
| Nov | Nephroblastoma overexpressed gene | 654 | 64 | 305 | 35 | −2.2 | 695 | 79 | 598 | 16 | NS |
| Lpcat4 | Lysophosphatidylcholine acyltransferase 4 | 460 | 28 | 215 | 9 | −2.1 | 244 | 6 | 281 | 13 | NS |
| Nt5dc2 | 5′-nucleotidase domain containing 2 | 232 | 12 | 110 | 5 | −2.1 | 100 | 3 | 112 | 9 | NS |
| Mcpt1 | Mast cell protease 1 | 65 | 9 | 31 | 3 | −2.1 | 24 | 2 | 29 | 2 | NS |
| Fer1l4 | Fer-1-like 4 (C. Elegans) | 83 | 9 | 41 | 2 | −2.0 | 27 | 2 | 46 | 4 | 1.7 |
| Il1rl1 | Interleukin 1 receptor-like 1 | 63 | 5 | 31 | 1 | −2.0 | 20 | 1 | 39 | 3 | 2.0 |
| Tnfrsf8 | Tumor necrosis factor receptor superfamily, member 8 | 66 | 6 | 34 | 1 | −1.9 | 23 | 1 | 42 | 6 | 1.8 |
| Duoxa2 | Dual oxidase maturation factor 2 | 137 | 4 | 73 | 3 | −1.9 | 177 | 27 | 151 | 22 | NS |
| Fads3 | Fatty acid desaturase 3 | 316 | 17 | 173 | 6 | −1.8 | 162 | 6 | 150 | 12 | NS |
| Cpn1 | Carboxypeptidase N, polypeptide 1 | 402 | 22 | 226 | 18 | −1.8 | 264 | 28 | 256 | 19 | NS |
| Hist1h2ab | Histone cluster 1, h2ab | 205 | 19 | 115 | 10 | −1.8 | 68 | 5 | 117 | 12 | 1.7 |
| E2f2 | E2F transcription factor 2 | 191 | 6 | 109 | 5 | −1.8 | 60 | 3 | 110 | 12 | 1.8 |
| Rdh9 | Retinol dehydrogenase 9 | 127 | 4 | 73 | 5 | −1.8 | 103 | 6 | 78 | 3 | NS |
| Pnpla1 | Patatin-like phospholipase domain containing 1 | 74 | 9 | 42 | 3 | −1.7 | 57 | 5 | 49 | 5 | NS |
| Fkbp11 | FK506 binding protein 11 | 69 | 2 | 40 | 1 | −1.7 | 43 | 3 | 49 | 1 | NS |
| 4632434I11Rik | RIKEN cdna 4632434I11 gene | 89 | 5 | 52 | 3 | −1.7 | 38 | 1 | 57 | 5 | 1.5 |
| Plau | Plasminogen activator, urokinase | 68 | 7 | 40 | 4 | −1.7 | 48 | 7 | 60 | 5 | NS |
| Pla2g2a | Phospholipase A2, group IIA (platelets, synovial fluid) | 103 | 7 | 60 | 3 | −1.7 | 43 | 3 | 68 | 3 | 1.6 |
| Spon2 | Spondin 2, extracellular matrix protein | 114 | 13 | 66 | 5 | −1.7 | 71 | 3 | 78 | 7 | NS |
| Tat | Tyrosine aminotransferase | 191 | 21 | 111 | 6 | −1.7 | 439 | 31 | 268 | 6 | −1.6 |
| Plvap | Plasmalemma vesicle associated protein | 171 | 5 | 101 | 5 | −1.7 | 114 | 17 | 111 | 10 | NS |
| 2310016C08Rik | RIKEN cdna 2310016C08 gene | 108 | 16 | 62 | 4 | −1.7 | 63 | 2 | 58 | 3 | NS |
| Nrg1 | Neuregulin 1 | 255 | 9 | 155 | 14 | −1.7 | 83 | 10 | 199 | 14 | 2.4 |
| Myl7 | Myosin, light polypeptide 7, regulatory | 55 | 3 | 33 | 2 | −1.7 | 33 | 2 | 45 | 4 | NS |
| Expi | Extracellular proteinase inhibitor | 53 | 3 | 32 | 1 | −1.7 | 43 | 3 | 33 | 2 | NS |
| Nek2 | NIMA (never in mitosis gene a)-related expressed kinase 2 | 288 | 13 | 177 | 11 | −1.6 | 98 | 6 | 174 | 10 | 1.8 |
| Duox2 | Dual oxidase 2 | 391 | 25 | 237 | 6 | −1.6 | 611 | 66 | 490 | 56 | NS |
| Bmp8b | Bone morphogenetic protein 8b | 152 | 7 | 93 | 5 | −1.6 | 66 | 4 | 68 | 3 | NS |
| Cdca3 | Cell division cycle associated 3 | 465 | 18 | 285 | 13 | −1.6 | 168 | 7 | 282 | 14 | 1.7 |
| Ripk3 | Receptor-interacting serine-threonine kinase 3 | 315 | 16 | 194 | 7 | −1.6 | 166 | 6 | 237 | 4 | 1.4 |
| Rbp4 | Retinol binding protein 4, plasma | 209 | 1 | 129 | 4 | −1.6 | 178 | 18 | 189 | 13 | NS |
| 4930547N16Rik | RIKEN cdna 4930547N16 gene | 144 | 6 | 90 | 6 | −1.6 | 54 | 3 | 86 | 8 | 1.6 |
| Aurka | Aurora kinase A | 290 | 10 | 180 | 10 | −1.6 | 111 | 5 | 178 | 19 | 1.6 |
| Igsf10 | Ig superfamily, member 10 | 142 | 7 | 88 | 3 | −1.6 | 110 | 14 | 134 | 16 | NS |
| Eef1g | Eukaryotic translation elongation factor 1 gamma | 143 | 13 | 88 | 3 | −1.6 | 76 | 7 | 108 | 10 | 1.4 |
| Dlgap5 | Discs, large (Drosophila) homolog-associated protein 5 | 171 | 5 | 107 | 6 | −1.6 | 77 | 4 | 122 | 10 | 1.6 |
| Cdc25c | Cell division cycle 25 homolog C (S. Pombe) | 85 | 4 | 53 | 3 | −1.6 | 34 | 1 | 54 | 4 | 1.6 |
| Dkkl1 | Dickkopf-like 1 | 34 | 1 | 21 | 1 | −1.6 | 23 | 0 | 25 | 1 | NS |
| Cenpa | Centromere protein A | 393 | 16 | 247 | 11 | −1.6 | 170 | 9 | 256 | 13 | 1.5 |
Table includes only genes with a signal intensity > 20 in at least one condition. n = 4 per group for Control, Control Abx, and Heme; n = 6 per group for Heme Abx.
Table S3.
Fifty highest up-regulated genes (q < 0.01) in mouse colon scrapings for the comparison of control Abx vs. control
| Gene symbol | Gene description | SI ± SEM | Fold change (heme + Abx vs. heme) | SI ± SEM | Fold change (control + Abx vs. control) | ||||||
| Heme | Heme + Abx | Control | Control + Abx | ||||||||
| SI | SEM | SI | SEM | SI | SEM | SI | SEM | ||||
| Ighv1-85 | Ig heavy variable 1–85 | 99 | 40 | 64 | 16 | NS | 20 | 8 | 135 | 62 | 5.7 |
| Ighv8-12 | Ig heavy variable V8-12 | 561 | 61 | 439 | 35 | NS | 196 | 41 | 1,111 | 246 | 5.6 |
| Cxcl9 | Chemokine (C-X-C motif) ligand 9 | 75 | 12 | 236 | 59 | NS | 62 | 18 | 326 | 47 | 5.6 |
| Gm12250 | Predicted gene 12250 | 32 | 6 | 81 | 22 | NS | 37 | 9 | 190 | 39 | 5.1 |
| Ifi44 | IFN-induced protein 44 | 70 | 7 | 124 | 21 | NS | 66 | 6 | 340 | 82 | 4.8 |
| Oas3 | 2'-5′ oligoadenylate synthetase 3 | 50 | 5 | 97 | 14 | NS | 51 | 5 | 241 | 41 | 4.6 |
| Ddx60 | DEAD (Asp-Glu-Ala-Asp) box polypeptide 60 | 83 | 14 | 173 | 26 | 2.1 | 115 | 10 | 486 | 78 | 4.1 |
| Usp18 | Ubiquitin specific peptidase 18 | 67 | 9 | 89 | 16 | NS | 48 | 4 | 204 | 39 | 4.1 |
| Ccl8 | Chemokine (C-C motif) ligand 8 | 30 | 3 | 55 | 6 | 1.8 | 22 | 3 | 81 | 8 | 3.7 |
| Gm5431 | Predicted gene 5431 | 17 | 3 | 61 | 15 | NS | 49 | 11 | 185 | 48 | 3.7 |
| Oas2 | 2'-5′ oligoadenylate synthetase 2 | 63 | 8 | 94 | 13 | NS | 56 | 3 | 215 | 42 | 3.6 |
| Oasl2 | 2'-5′ oligoadenylate synthetase-like 2 | 217 | 39 | 432 | 66 | 2.0 | 247 | 12 | 905 | 123 | 3.6 |
| Ifi47 | IFN gamma inducible protein 47 | 69 | 14 | 128 | 28 | NS | 79 | 16 | 265 | 44 | 3.4 |
| H2-DMa | Histocompatibility 2, class II, locus dma | 112 | 8 | 175 | 17 | NS | 99 | 9 | 340 | 50 | 3.3 |
| Gm9740 | Predicted gene 9740 | 116 | 50 | 191 | 43 | NS | 76 | 8 | 251 | 37 | 3.3 |
| Igkv1-117 | Ig kappa chain variable 1–117 | 522 | 13 | 518 | 54 | NS | 355 | 57 | 1,201 | 259 | 3.2 |
| Apol9b | Apolipoprotein L 9b | 22 | 2 | 37 | 3 | NS | 21 | 4 | 66 | 11 | 3.2 |
| Ifit1 | IFN-induced protein with tetratricopeptide repeats 1 | 129 | 21 | 255 | 39 | NS | 188 | 13 | 610 | 94 | 3.1 |
| Apol7c | Apolipoprotein L 7c | 43 | 7 | 41 | 6 | NS | 29 | 3 | 89 | 7 | 3.1 |
| Apol9a | Apolipoprotein L 9a | 73 | 4 | 110 | 13 | NS | 68 | 7 | 213 | 34 | 3.1 |
| H2-Q6 | Histocompatibility 2, Q region locus 6 | 185 | 8 | 303 | 25 | 1.6 | 178 | 13 | 561 | 91 | 3.1 |
| Ubd | Ubiquitin D | 18 | 1 | 28 | 4 | NS | 17 | 1 | 58 | 15 | 3.0 |
| H2-Aa | Histocompatibility 2, class II antigen A, alpha | 525 | 29 | 791 | 55 | NS | 482 | 47 | 1,484 | 213 | 3.0 |
| H2-Q9 | Histocompatibility 2, Q region locus 9 | 104 | 11 | 160 | 16 | NS | 88 | 3 | 279 | 48 | 3.0 |
| Igtp | IFN gamma induced gtpase | 47 | 7 | 107 | 26 | NS | 71 | 13 | 222 | 44 | 3.0 |
| Rtp4 | Receptor transporter protein 4 | 93 | 14 | 187 | 29 | 2.0 | 111 | 10 | 331 | 42 | 3.0 |
| Gm4951 | Predicted gene 4951 | 28 | 2 | 54 | 10 | NS | 35 | 6 | 118 | 37 | 2.9 |
| Slc6a19 | Solute carrier family 6 (neurotransmitter transporter), member 19 | 81 | 14 | 320 | 53 | 3.9 | 21 | 2 | 60 | 6 | 2.8 |
| H2-Ab1 | Histocompatibility 2, class II antigen A, beta 1 | 698 | 30 | 965 | 62 | NS | 607 | 68 | 1,677 | 189 | 2.8 |
| Herc6 | Hect domain and RLD 6 | 69 | 8 | 135 | 19 | NS | 118 | 9 | 340 | 65 | 2.7 |
| Cd74 | CD74 antigen (invariant polypeptide of major histocompatibility complex, class II antigen-associated) | 877 | 38 | 1160 | 94 | NS | 775 | 70 | 2,112 | 221 | 2.7 |
| Gbp3 | Guanylate binding protein 3 | 43 | 5 | 71 | 12 | NS | 56 | 9 | 157 | 31 | 2.7 |
| AI747448 | Expressed sequence AI747448 | 570 | 94 | 225 | 49 | −2.7 | 71 | 12 | 187 | 21 | 2.7 |
| Zbp1 | Z-DNA binding protein 1 | 98 | 13 | 148 | 23 | NS | 142 | 21 | 395 | 76 | 2.7 |
| Psmb8 | Proteasome (prosome, macropain) subunit, beta type 8 (large multifunctional peptidase 7) | 70 | 7 | 120 | 14 | NS | 88 | 11 | 249 | 47 | 2.7 |
| Rsad2 | Radical S-adenosyl methionine domain containing 2 | 57 | 9 | 139 | 21 | 2.4 | 56 | 1 | 152 | 15 | 2.7 |
| Tap1 | Transporter 1, ATP-binding cassette, subfamily B (MDR/TAP) | 134 | 11 | 243 | 36 | NS | 161 | 18 | 439 | 72 | 2.6 |
| Gm5571 | Predicted gene 5571 | 429 | 102 | 439 | 71 | NS | 250 | 29 | 691 | 159 | 2.6 |
| Dnase1l3 | Dnase 1-like 3 | 59 | 3 | 104 | 9 | 1.7 | 53 | 2 | 138 | 5 | 2.6 |
| Nlrc5 | NLR family, CARD domain containing 5 | 45 | 3 | 68 | 7 | NS | 42 | 3 | 110 | 16 | 2.5 |
| Trac | T-cell receptor alpha constant | 97 | 14 | 274 | 27 | 2.8 | 127 | 10 | 320 | 24 | 2.5 |
| Gbp8 | Guanylate-binding protein 8 | 43 | 3 | 73 | 8 | NS | 47 | 6 | 120 | 17 | 2.5 |
| Ifit3 | IFN-induced protein with tetratricopeptide repeats 3 | 55 | 3 | 106 | 13 | 1.9 | 79 | 3 | 210 | 45 | 2.5 |
| Ifit2 | IFN-induced protein with tetratricopeptide repeats 2 | 28 | 1 | 39 | 4 | NS | 32 | 1 | 87 | 21 | 2.5 |
| Upp1 | Uridine phosphorylase 1 | 322 | 46 | 542 | 66 | NS | 518 | 53 | 1,280 | 148 | 2.5 |
| Tfrc | Transferrin receptor | 493 | 105 | 567 | 38 | NS | 288 | 2 | 708 | 30 | 2.4 |
| Dhx58 | DEXH (Asp-Glu-X-His) box polypeptide 58 | 81 | 7 | 111 | 10 | NS | 74 | 4 | 184 | 23 | 2.4 |
| Nrg1 | Neuregulin 1 | 255 | 9 | 155 | 14 | −1.7 | 83 | 10 | 199 | 14 | 2.4 |
| H2-DMb1 | Histocompatibility 2, class II, locus Mb1 | 28 | 1 | 44 | 2 | NS | 30 | 5 | 73 | 12 | 2.4 |
| Irgm2 | Immunity-related gtpase family M member 2 | 146 | 22 | 343 | 68 | NS | 250 | 34 | 597 | 87 | 2.3 |
Table includes only genes with a signal intensity > 20 in at least one condition. n = 4 per group for Control, Control Abx, and Heme; n = 6 per group for Heme Abx.
Table S4.
Fifty highest down-regulated genes (q < 0.01) in mouse colon scrapings for the comparison of control Abx vs. control
| Gene symbol | Gene description | SI ± SEM | Fold change (heme + Abx vs. heme) | Signal intensity (SI) ± SEM. | Fold change (control + Abx vs. control) | ||||||
| Heme | Heme + Abx | Control | Control + Abx | ||||||||
| SI | SEM | SI | SEM | SI | SEM | SI | SEM | ||||
| Des | Desmin | 622 | 84 | 933 | 220 | NS | 2,474 | 559 | 674 | 195 | −3.8 |
| Cnn1 | Calponin 1 | 629 | 95 | 955 | 216 | NS | 2,419 | 531 | 680 | 205 | −3.7 |
| Tacr2 | Tachykinin receptor 2 | 87 | 9 | 128 | 23 | NS | 330 | 77 | 93 | 21 | −3.4 |
| Calb2 | Calbindin 2 | 24 | 3 | 41 | 6 | NS | 102 | 23 | 29 | 5 | −3.4 |
| Synpo2 | Synaptopodin 2 | 132 | 23 | 188 | 38 | NS | 512 | 123 | 143 | 27 | −3.3 |
| Actg2 | Actin, gamma 2, smooth muscle, enteric | 653 | 81 | 1,020 | 213 | NS | 2,343 | 483 | 748 | 219 | −3.3 |
| Pgm5 | Phosphoglucomutase 5 | 286 | 42 | 445 | 89 | NS | 978 | 211 | 295 | 68 | −3.2 |
| Hspb8 | Heat shock protein 8 | 78 | 11 | 111 | 18 | NS | 236 | 47 | 73 | 12 | −3.1 |
| Hspb7 | Heat shock protein family, member 7 (cardiovascular) | 51 | 4 | 65 | 8 | NS | 183 | 39 | 56 | 8 | −3.1 |
| Chrm2 | Cholinergic receptor, muscarinic 2, cardiac | 95 | 13 | 124 | 26 | NS | 280 | 56 | 93 | 27 | −3.1 |
| Kcnmb1 | Potassium large conductance calcium-activated channel, subfamily M, beta member 1 | 88 | 8 | 108 | 15 | NS | 255 | 52 | 80 | 15 | −3.1 |
| Kcnip4 | Kv channel interacting protein 4 | 21 | 3 | 32 | 6 | NS | 86 | 23 | 26 | 6 | −3.1 |
| Stmn2 | Stathmin-like 2 | 50 | 5 | 89 | 16 | NS | 210 | 40 | 67 | 10 | −3.1 |
| Flnc | Filamin C, gamma | 92 | 6 | 114 | 18 | NS | 285 | 63 | 88 | 14 | −3.0 |
| Hspb6 | Heat shock protein, alpha-crystallin-related, B6 | 263 | 21 | 325 | 48 | NS | 784 | 166 | 255 | 57 | −3.0 |
| Myl9 | Myosin, light polypeptide 9, regulatory | 780 | 90 | 1,206 | 215 | NS | 2,647 | 502 | 885 | 181 | −3.0 |
| Tagln | Transgelin | 319 | 40 | 457 | 89 | NS | 1,035 | 222 | 338 | 65 | −2.9 |
| Atp2b4 | Atpase, Ca++ transporting, plasma membrane 4 | 125 | 15 | 198 | 39 | NS | 465 | 103 | 152 | 25 | −2.9 |
| Myh11 | Myosin, heavy polypeptide 11, smooth muscle | 573 | 83 | 886 | 183 | NS | 2,068 | 413 | 698 | 135 | −2.9 |
| Nmu | Neuromedin U | 17 | 1 | 27 | 4 | NS | 73 | 13 | 26 | 6 | −2.9 |
| Chrna3 | Cholinergic receptor, nicotinic, alpha polypeptide 3 | 41 | 4 | 70 | 11 | NS | 165 | 37 | 54 | 9 | −2.9 |
| Bves | Blood vessel epicardial substance | 41 | 4 | 53 | 7 | NS | 125 | 27 | 42 | 6 | −2.8 |
| Grp | Gastrin releasing peptide | 28 | 2 | 49 | 8 | NS | 125 | 28 | 43 | 8 | −2.8 |
| Cd59a | CD59a antigen | 69 | 9 | 105 | 20 | NS | 231 | 55 | 76 | 10 | −2.8 |
| Popdc2 | Popeye domain containing 2 | 40 | 4 | 56 | 9 | NS | 126 | 26 | 45 | 11 | −2.8 |
| Htr3a | 5-hydroxytryptamine (serotonin) receptor 3A | 20 | 1 | 26 | 2 | NS | 53 | 10 | 18 | 3 | −2.8 |
| Scube2 | Signal peptide, CUB domain, EGF-like 2 | 37 | 4 | 48 | 8 | NS | 112 | 28 | 36 | 4 | −2.7 |
| Rit2 | Ras-like without CAAX 2 | 21 | 1 | 28 | 3 | NS | 67 | 16 | 23 | 3 | −2.7 |
| Klk15 | Kallikrein related-peptidase 15 | 23 | 2 | 31 | 4 | NS | 95 | 12 | 35 | 4 | −2.7 |
| Pcp4l1 | Purkinje cell protein 4-like 1 | 103 | 9 | 152 | 27 | NS | 319 | 54 | 119 | 24 | −2.7 |
| Pdlim3 | PDZ and LIM domain 3 | 394 | 42 | 522 | 88 | NS | 1,175 | 237 | 417 | 63 | −2.7 |
| Cpe | Carboxypeptidase E | 100 | 10 | 139 | 16 | NS | 344 | 60 | 126 | 19 | −2.7 |
| Syt1 | Synaptotagmin I | 35 | 3 | 56 | 7 | NS | 132 | 27 | 47 | 8 | −2.7 |
| Cacnb2 | Calcium channel, voltage-dependent, beta 2 subunit | 44 | 3 | 65 | 10 | NS | 148 | 29 | 54 | 9 | −2.7 |
| Ppyr1 | Pancreatic polypeptide receptor 1 | 24 | 2 | 32 | 6 | NS | 75 | 16 | 26 | 4 | −2.7 |
| Myom1 | Myomesin 1 | 43 | 4 | 55 | 7 | NS | 133 | 31 | 45 | 3 | −2.6 |
| Rbpms2 | RNA binding protein with multiple splicing 2 | 52 | 1 | 78 | 10 | NS | 144 | 30 | 52 | 8 | −2.6 |
| Snap25 | Synaptosomal-associated protein 25 | 73 | 6 | 114 | 13 | NS | 259 | 45 | 98 | 15 | −2.6 |
| Cryab | Crystallin, alpha B | 70 | 4 | 87 | 13 | NS | 218 | 44 | 83 | 15 | −2.5 |
| Penk | Preproenkephalin | 70 | 9 | 110 | 17 | NS | 264 | 53 | 99 | 12 | −2.5 |
| Art3 | ADP ribosyltransferase 3 | 49 | 6 | 67 | 9 | NS | 132 | 26 | 50 | 6 | −2.5 |
| Mir145 | Microrna 145 | 100 | 18 | 131 | 22 | NS | 325 | 69 | 124 | 12 | −2.5 |
| Slc30a10 | Solute carrier family 30, member 10 | 47 | 3 | 49 | 4 | NS | 256 | 34 | 105 | 17 | −2.5 |
| Cckar | Cholecystokinin A receptor | 21 | 1 | 31 | 5 | NS | 68 | 13 | 27 | 4 | −2.5 |
| Slc2a4 | Solute carrier family 2 (facilitated glucose transporter), member 4 | 42 | 5 | 69 | 9 | NS | 122 | 23 | 48 | 5 | −2.5 |
| Tmem90b | Transmembrane protein 90B | 36 | 3 | 48 | 6 | NS | 112 | 25 | 42 | 4 | −2.4 |
| Myocd | Myocardin | 65 | 7 | 79 | 9 | NS | 176 | 30 | 70 | 9 | −2.4 |
| Slc7a14 | Solute carrier family 7 (cationic amino acid transporter, y+ system), member 14 | 30 | 2 | 46 | 7 | NS | 90 | 19 | 35 | 4 | −2.4 |
| Htr2b | 5-hydroxytryptamine (serotonin) receptor 2B | 14 | 1 | 22 | 3 | NS | 49 | 12 | 19 | 2 | −2.4 |
| Ckb | Creatine kinase, brain | 525 | 31 | 486 | 70 | NS | 931 | 183 | 372 | 51 | −2.4 |
Table includes only genes with a signal intensity > 20 in at least one condition. n = 4 per group for Control, Control Abx, and Heme; n = 6 per group for Heme Abx.
Abx Do Not Affect the Heme-Induced Antioxidant Response.
A set of 648 genes was significantly regulated in both the H- and HA-group compared with their controls (Fig. S1A). Of those shared genes, 599 were similarly regulated in both groups. Notably, this group of genes included the activation of several transcription factors, including the PPARs, involved in fatty acid metabolism, and Nrf2, involved in antioxidant response (Fig. S1 B and C). These data imply that oxidative stress and lipid peroxidation products induced the antioxidant response and the induction of PPAR target genes in the mucosa, irrespective of the addition of Abx to the heme diet.
Abx Block the Mucosal Sensing of Heme-Induced Luminal Cytotoxicity.
In the colon, the mucus preserves the epithelial barrier by protecting the surface epithelial cells through creating a diffusion barrier for toxic micelles (13). In the HA-group, substantial luminal cytotoxicity was present but did not induce hyperproliferation, indicating that CHF did not reach the surface epithelial cells. We investigated epithelial sensing of cytotoxicity using the marker genes survivin (Birc5), immediately early response 3 (Ier3), the necrosis facilitator Ripk3, and secretory leukocyte protease inhibitor (Slpi), which are up-regulated by cell injury as described earlier (11, 14). In the present study, heme up-regulated the expression of these cytotoxicity sensors only in the H-group but not in the HA-group (Fig. 3A). In addition, Fig. 3B shows that heme only injured surface cells, which corroborates our laser capture analysis (6). Taken together, these results indicate that the microbiota is required for the cytotoxic micelles to reach the surface epithelium, as the diffusion barrier of the mucus layer was maintained by Abx.
Fig. 3.

(A) Gene expression of injury markers Birc5, Ier3, Ripk3, and Slpi. (B) immunohistochemical colonic Slpi staining of control and heme-fed mice. (C) Gene expression of mucin genes 1–4 and Galnt 3 and 12. Expression levels of control is set at 1. Expression of other bars is relative to controls; mean ± SEM (n = 4 for C, H, and CA; n = 6 for HA). Letters indicate significant differences (P < 0.05), ANOVA with Bonferroni post hoc test.
An increased mucus barrier could be caused by increased mucin synthesis or by decreased mucin degradation. Expression levels of secreted Muc2 and cell-associated Muc4 were decreased in the HA- compared with the H-group (Fig. 3C). Moreover, the Kyoto Encyclopedia of Genes and Genomes pathway mucin type O-glycan biosynthesis was significantly repressed in the HA- compared with the H-group, according to GSEA analysis (q = 0.049). For instance, Galnt 3 and 12, involved in the first step of o-glycosylation, were down-regulated by Abx (Fig. 3C), indicating that mucus layer production is most probably decreased by Abx. Regarding mucus degradation, it is well established that the microbiota degrades mucins and uses their carbohydrates and amino acids as substrates for growth. In addition, sulfate-reducing bacteria (SRBs) use mucin-derived sulfate as electron acceptor in anaerobic respiration forming sulfides. As the Abx treatment drastically reduced the microbiota density, we hypothesized that Abx increased the mucus barrier by preventing microbial degradation of mucin. Notably, the abundance of the mucin-degrading Akkermansia muciniphila was eightfold increased by heme, whereas Abx reduced the abundance of this mucin degrader more than 1,000-fold (Fig. 4A). This result implies that the relative reduction of the Akkermansia population by Abx exceeded the overall effect of Abx on the total microbial load, supporting that Abx treatment could increase mucus barrier function by strongly reducing the levels of mucin degraders such as Akkermansia.
Fig. 4.

(A) Bacterial counts of A. muciniphila and SRB determined by qPCR. Controls are set at 100%, and other bars are relative to controls; mean ± SEM (n = 8–9/group). Letters indicate significant different groups (P < 0.05). (B) Rate and extent of S-S bond splitting and synthesis of trisulfide bonds; mean ± SD (n = 3–6/group). *Significant difference with thiol groups (P < 0.05). (C) Reaction scheme by which sulfide splits S-S bonds. (D) Concentrations of sulfides in fecal water; mean ± SEM (n = 8–9/group). (E) Excretion of fecal mucins, expressed as µmoles O-glycan per day (n = 6–9/group). (F) Western blot analysis of fecal mucin with or without DTT as reducing agent and with sulfide. Samples were stained with anti-Muc2 antibody. Each lane represents a pool of n = 9/group. Letters indicate significant different groups (P < 0.05), ANOVA with Bonferroni post hoc test.
Sulfide Reduces S-S Bonds, Leading to Mucolysis.
Mucus is high in intra- and intermolecular S-S bonds, stabilizing its polymeric, network-like structure. When S-S bonds are broken, mucin monomers dissociate and/or denaturate, leading to decreased viscosity and higher mucus accessibility for bacterial degradation (15). SRBs can use mucus-derived sulfate as oxidant in anaerobic respiration, generating sulfide. We hypothesized that sulfide could have a mucolytic effect by reducing the intermolecular S-S bonds, which contributes to an enhanced mucus barrier in the Abx groups due to the decreased abundance of SRBs (Fig. 4A) and the corresponding decrease of luminal sulfide production. We tested the reducing potency of several sulfur containing compounds on the model compound DTNB [5,5′-dithiobis-(2-nitrobenzoic acid)] containing a central S-S bond. Splitting of this S-S bond in DTNB leads to increased absorbance at 412 nm, and the assay allows the determination of the overall S-S splitting extent as well as the initial S-S splitting rate (Fig. 4B). Cysteine, glutathione, and N-acetylcysteine (NAC) were able to split the S-S bond with a similar rate between 24 and 31 µM/min, whereas the negative control Na2SO4 did not affect DTNB integrity. Importantly, sulfide gave a significantly higher rate of S-S bond splitting, 62.4 ± 5.9 µM/min, indicating that sulfide has a twofold more potent mucolytic effect compared with the amino acid thiols that have been shown to split S-S bonds and make mucins less viscous (16). Moreover, the overall extent of S-S bond splitting by sulfide was also twofold higher compared with the other amino acid thiols. Based on Ellman's mechanism (17), this indicates that a reactive persulfide anion originates from the splitting of the first S-S bond, which can subsequently target a second S-S bond, creating a trisulfide bond. We developed a method to quantitatively determine trisulfide bonds based on the difference between total bound sulfides and acid labile sulfides (18). Indeed, trisulfide bonds were generated when sulfide was used to reduce S-S bonds in DTNB (Fig. 4B), but not on amino acid thiol (NAC, GSH, or cysteine) treatment of DTNB. The theoretical (Fig. 4C) and measured ratio (Fig. 4B) of sulfide-dependent formation of thiol to trisulfide is 2, indicating that all sulfide reacts with DNTB to form trisulfide bonds.
To test whether similar redox reactions also occurred in vivo, total sulfides and trisulfides were determined in mice fecal water (Fig. 4D). Heme increased the levels of total bound sulfides, which is in agreement with literature showing that heme addition to the growth medium stimulates bacterial reduction of sulfate to sulfide (19). Concentrations of trisulfide in fecal water of mice not receiving Abx were much higher (with H > C) than those of mice receiving Abx, indicating that Abx suppressed sulfide-dependent splitting of S-S bonds and thus colonic mucolysis. In line with this, Abx drastically increased fecal excretion of mucin (Fig. 4E), because the slightly lower steady-state mucin synthesis (Fig. 3C) is not anymore balanced by its bacterial degradation. Fecal mucin was present as a high-molecular-weight form of Muc2 that could not penetrate the gel in SDS/PAGE analysis (Fig. 4F), whereas with DTT as a reducing agent most of the Muc2 appeared as a band of low MW in the gel. To corroborate the reducing effect of sulfide, Muc2 Western blotting was repeated for the CA- and HA-groups and showed that also sulfide lysed the polymeric Muc2 almost completely (Fig. 4F). Overall, these results indicate that in normal colon physiology, microbial sulfide opens the polymeric Muc2 network to bacterial degradation resulting in a lumen-to-surface permeability gradient through the mucus layer. Abx block these microbial processes and thereby increase the mucus barrier.
Discussion
This study shows that dietary heme changes the microbiota and increases ROS and cytotoxicity in the colonic lumen. In addition, heme injures the surface epithelium leading to compensatory hyperproliferation, hyperplasia, and differential expression of tumor suppressor and oncogenes, which increases colorectal cancer risk (20, 21). These luminal and epithelial effects of heme are similar to those detailed in our recent studies (6, 10, 11). Also the Abx effects on the microbiota replicate those shown by us in an earlier study (22). The crucial finding of the present study is that when the microbial abundance is drastically reduced by Abx, heme does not injure the surface epithelium and does not induce the carcinogenic changes in the crypts, mentioned above. The absence of carcinogenic changes is not due to the slightly lower levels of cytotoxicity and ROS in the HA-group, which can be explained by the higher luminal dilution factor because of the increased fecal wet weight. Recently, we reported that heme diets induce, in the colon lumen, covalent heme modification resulting in the very lipophilic and toxic CHF, which is solubilized in mixed micelles (11). Abx block the mucosal sensing of these cytotoxic micelles, as they prevent the heme-induced changes in epithelial histology and in up-regulation of injury markers such as Slpi. In contrast, Abx do not block mucosal sensing of luminal ROS, as the Nrf2-mediated antioxidant response was initiated and PPARs were activated by oxidized lipids in both heme diet groups. This differential mucosal sensing shows that, with Abx, the mucus layer is still permeable to small molecules, such as oxidized lipids, but no longer to larger micellar aggregates containing CHF. The absence of hyperproliferation in the HA-group also shows that mucosal exposure to ROS does not cause hyperproliferation. This result is in line with our previous observation that ROS is instantly formed after consumption of the heme diet, whereas there is a delay in the appearance of luminal cytotoxicity and the induction of hyperproliferation (11).
Our study implies that the colon microbiota facilitates heme-induced epithelial injury and hyperproliferation by opening the mucus barrier by the concerted action of hydrogen sulfide-producing and mucin-degrading bacteria. The principal steps of this hypothesis (Fig. 5) are (i) mucolysis by hydrogen sulfide to open the compact, protective mucus layer for (ii) further bacterial degradation, thereby (iii) allowing diffusion of luminal, cytotoxic micelles to the mucosal surface. Consequently, surface epithelial cells are less protected against luminal cytotoxicity, leading to induction of compensatory hyperproliferation. The diffusion barrier function of the mucus layer is illustrated by Muc2 KO mice, which display colitis and epithelial hyperproliferation, as well as spontaneous development of colorectal cancer (23). In addition, an in vitro study shows that apically applied mucin creates a diffusion barrier, preventing the contact between cytotoxic micelles and colonocytes (13). That microbiota increase the permeability of mucus barrier is illustrated by a study of recolonized vs. Abx-treated rats showing that bacteria colonizing the isolated colonic segment increase epithelial injury by luminally added toxic compounds (24). Unfortunately, there are no established methods to determine the permeability of the mucus layer in vivo. Although the mucus layer can be stained in appropriately fixed intestinal samples, this does not provide information about its permeability, as its thickness and permeability are not inversely related (15).
Fig. 5.

Proposed mechanism of how microbiota facilitates heme-induced compensatory hyperproliferation. (Upper) Processes when normal microbiota is present (i.e., without Abx) leading to compensatory hyperproliferation. (Lower) How Abx cause the mucus layer to be protective against cytotoxic micelles. R-S-S-R indicates native intra- and intermolecular disulfide bonds in the mucus that can be reduced by H2S to thiols (R-S-H) and trisulfides (R-S-S-S-R).
Central to our hypothesis is that hydrogen sulfide can reduce and thus split S-S bonds, which opens the mucus layer. The compactness of this layer is determined by disulfide bond-stabilized polymer Muc2 network of C-terminal dimers and N-terminal trimers (15). Partial proteolysis by host proteases, which is visible in our denaturating gels of fecal mucin, does not change the structure of this network (15). However, splitting of S-S bonds dissolves it (i.e., mucolysis) resulting in reduced viscosity and increased permeability of the mucus layer (15). Typical S-S breaking agents are N-acetyl-cysteine (NAC), used in the treatment of cystic fibrosis, l-cysteine, and 2-mercaptoethanol, all known to decrease mucin viscosity in vivo and in vitro (16). Our DTNB results show that sulfide, compared with these thiols, is twofold more potent in breaking S-S bonds. The pKa of hydrogen sulfide is about 1 unit lower than that of the thiols, implying that the concentration of the nucleophilic agent (i.e., the anion) in the splitting of S-S is higher with hydrogen sulfide. Moreover, as sulfide donates two electrons, it splits two S-S bonds, whereas thiols only split one. Elaborating on Ellman’s mechanism of S-S splitting (17), we reasoned that the highly nucleophlic persulfide, formed in the first reaction, generates a trisulfide bond in the second one. Our in vitro results show that trisulfide formation is indeed specific for S-S reduction by sulfide. Our fecal analysis shows that trisulfide is also formed in vivo and stimulated by dietary heme, probably because bacterial sulfate reduction is heme dependent (19). Abx strongly reduce overall bacterial abundance and suppress this trisulfide formation almost completely, supporting our mechanism that trisulfide is formed by bacterial sulfide. In line with this, Abx greatly increased fecal excretion of Muc2 in a high-molecular-weight polymeric form, as shown by nonreducing SDS/PAGE. Moreover, this polymeric Muc2 dissociates almost completely after reduction by DTT or sulfide, supporting the hypothesis that S-S bond splitting by sulfide opens the mucus barrier. This hypothesis is supported further by the recent finding that increasing the number of S-S bonds in the Muc2 network increases the mucus barrier in mouse colon (25).
Our mechanism of S-S bond splitting by sulfide is corroborated by a recent nutritional study by Devkota et al. (26), showing that monoassociation of germ-free mice with the sulfide-producing proteobacterium Bilophila wardsworthia, in the presence of taurocholate, results in breaking of the mucus barrier. The authors suggest that this is either due to sulfide (produced from taurine) or to unconjugated deoxycholate. However, the authors did not find barrier breaking in the presence of glycocholate, which is also metabolized to deoxycholate but does not generate sulfide. Therefore, we feel that their results can only be explained by the action of taurine-derived sulfide opening the mucus barrier via a mechanism analogous to what we propose here. Thus, it would be worthwhile to measure fecal trisulfides in that study.
Also in humans the colonic mucus layer functions as a barrier. As in mice, it prevents bacterial colonization of the epithelial surface and protects the surface cells from exposure to luminal toxic compounds (27). Three prevalent microbial profiles, so-called “enterotypes,” have been proposed to exist in human microbiota (28). Interestingly, for two of those enterotypes, mucin-degrading bacteria are identified as microbial drivers. One enterotype is rich in Prevotella and the co-occurring Desulfovibrio. Prevotella degrades mucin and Desulfovibrio may enhance the rate-limiting sulfatase step by hydrolyzing glycosyl-sulfate esters. The second mucin-degrading enterotype is rich in Ruminococcus and Akkermansia, both able to degrade mucins. We showed previously that dietary heme drastically increases the abundance of Prevotella and Akkermansia (10), which may be of relevance for these two enterotypes. The third enterotype is rich in Bacteroides using carbohydrates and proteins as substrates for fermentation (28). It would be of interest to see whether mucus barrier differences between different enterotypes exist or that diseases of the gut, such as colorectal cancer and IBD, are associated with mucin-degrading enterotypes.
Overall, we conclude that the microbiota facilitates the heme-induced hyperproliferation by opening the mucus barrier. Bacterial hydrogen sulfide can reduce the S-S bonds in polymeric mucin, thereby increasing the mucus layer permeability for mucin-degrading bacteria and for cytotoxic micelles. Consequently, epithelial surface cells are injured by the cytotoxic heme and compensatory hyperproliferation is initiated. This hyperproliferation might eventually lead to colorectal cancer (20). Our model and our results imply that fecal trisulfides can serve as a suitable marker of colonic mucolysis. Therefore, it would be of interest to measure levels of trisulfide in the human enterotypes, mentioned above, and in gut diseases in which the mucus barrier is compromised, such as irritable bowel syndrome (29).
Materials and Methods
Animal Handling and Design of the Study.
Experiments were approved by the Ethical Committee on Animal Testing of Wageningen University and were in accordance with national law. Eight-week-old male C57BL6/J mice (Harlan) were housed individually in a room with controlled temperature (20–24 °C) and relative humidity (55 ± 15%) and a 12-h light dark cycle. Mice were fed diets and demineralized water ad libitum. We designed our study as a 2 × 2 factorial experiment with heme and antibiotics as independent factors/treatments. Mice (n = 9/group) received either a “Westernized” control diet [40 energy% fat (mainly palm-oil), low calcium (30 µmol/g)] or this diet supplemented with 0.5 µmol/g heme for 14 d, as previously described (30). Broad-spectrum Abx, containing ampicillin (1 g/L), neomycin (1 g/L), and metronidazole (0.5 g/L), were administered in drinking water during the time of intervention. There were four experimental groups: control, heme, control plus Abx, and heme plus Abx. Feces were quantitatively collected during days 11–14, frozen at −20 °C, and subsequently freeze dried. After 14 d, the colon was excised, mesenteric fat was removed, and the colon was opened longitudinally, washed in PBS, and cut into three parts. The middle 1.5 cm of colon tissue was formalin-fixed and paraffin embedded for histology. The remaining proximal and distal parts were scraped, pooled per mouse, snap-frozen in liquid nitrogen, and stored at −80 °C until further analysis. Colonic contents were sampled for microbiota analysis. Chemicals were from Sigma-Aldrich, unless indicated otherwise.
Fecal Analyses.
Fecal water was prepared by reconstituting freeze-dried feces with double distilled water to obtain a physiological osmolarity of 300 mOsm/L, as described previously (5). Cytotoxicity of fecal water was quantified by potassium release from human erythrocytes after incubation (5) and validated with human colon carcinoma-derived Caco-2 cells (31). See SI Materials and Methods for TBARS, bile acids, and fecal mucin measurements.
Immunohistochemistry.
Histological H&E and immunohistochemical Ki67 (6) and Slpi (32) stainings were performed on paraffin-embedded colon sections as described previously. To quantify Ki67-positive colonocytes, 15 crypts per animal were counted. The number of Ki67-positive cells per crypt, total number of cells per crypt, and labeling index were determined.
RNA Isolation and Microarray Analysis.
RNA was isolated from colon scrapings and hybridized on Affymetrix GeneChip Mouse Gene 1.1 ST arrays (SI Materials and Methods). Genes satisfying the criterion of false discovery rate <1% (q < 0.01) were considered significantly expressed. Array data were submitted to the Gene Expression Omnibus with accession no. GSE40670.
Bacterial DNA Extraction and qPCR.
DNA was extracted from ∼0.1 g fresh fecal pellet from the colon using the method described by Salonen et al. (33). By qPCR, total bacteria were quantified using generic 16S rRNA primers and Bacteroidetes and Firmicutes using phylum-specific 16S rRNA primers (SI Materials and Methods).
Reduction of Disulfide (S-S) Bonds.
S-S splitting potency of sodium sulfide (Na2S) and thiols was determined using DTNB as the model disulfide compound. Western blot analysis of fecal mucin was performed to determine whether DTT and sulfide reduces disulfide bonds in Muc2 (SI Materials and Methods).
Measurement of Trisulfide Bonds.
Trisulfides were calculated as the difference between total bound sulfides and acid-labile sulfides measured in individual fecal waters by GC-MS (SI Materials and Methods).
Statistics.
In vivo data are presented as mean ± SEM. Differences between groups were tested for main effects by two-way ANOVA. In vitro data are given as mean ± SD, and differences between groups were tested by one-way ANOVA with the Bonferroni multiple comparison test. P < 0.05 was considered significant.
SI Materials and Methods
TBARS.
To determine lipid peroxidation products in the gut lumen, TBARS in fecal water were quantified. The assay determines lipid peroxidation by quantifying the concentration of malondialdehyde (MDA) in fecal water. Briefly, fecal water was diluted fourfold with double-distilled water. To 100 µL of this dilution, 100 µL of 8.1% (wt/vol) SDS and 1 mL of 0.11 mol/L 2,6-di-tert-butyl-p-cresol, 0.5% TBA in 10% (vol/vol) acetic acid (pH 3.5) was added. To correct for background, TBA was omitted from the assay. TBARS were extracted, after heating for 75 min at 82 °C, with 1.2 mL l-butanol. The absorbance of the extracts was measured at 540 nm. The amount of TBARS was calculated as MDA equivalents using 1,1,3,3,-tetramethoxypropane as standard.
Bile acids were determined in fecal water of n = 3/group. Fecal waters were evaporated under nitrogen and resolubilized in 20% (vol/vol) acetonitrile in water. Samples were analyzed using reversed-phase HPLC combined with simultaneous amperometric detection of free and conjugated bile acids (34). Fecal mucins were quantified by means of their oligosaccharide side chains, as described in detail earlier (35), and expressed as µmoles O-glycans.
RNA Isolation.
Total RNA was isolated by using TRIzol reagent (Invitrogen) according to the manufacturer’s protocol. For microarray hybridization the isolated RNA was further column purified (SV total RNA isolation system Promega, Leiden, The Netherlands). RNA concentration was measured on a nanodrop ND-1000 UV-Vis spectrophotometer (Isogen) and analyzed on an Agilent 2100 bioanalyzer (Agilent Technologies) with 6000 Nano Chips, according to the manufacturer’s protocol. RNA was judged suitable for array hybridization only if samples exhibited intact bands corresponding to the 18S and 28S ribosomal RNA subunits and displayed no chromosomal peaks or RNA degradation products (RNA integrity number > 8.0).
Microarray Analysis.
The Ambion WT Expression kit (P/N 4411974; Life Technologies) in conjunction with the Affymetrix GeneChip WT Terminal Labeling kit (P/N 900671; Affymetrix) was used for the preparation of labeled cDNA from 100 ng of total RNA without rRNA reduction. Labeled samples were hybridized on Affymetrix GeneChip Mouse Gene 1.1 ST arrays, provided in plate format. Hybridization, washing, and scanning of the array plates was performed on an Affymetrix GeneTitan Instrument, according to the manufacturer’s recommendations. RNA labeling was performed in two rounds using a complete block design. Normalized expression estimates were obtained from the raw intensity values applying the robust multiarray analysis (RMA) preprocessing algorithm available in the Bioconductor library AffyPLM with default settings. Subsequently, an empirical Bayes method, called ComBat (36), was used to correct for the systematic error (batch effect) introduced during labeling. Probe sets were redefined according to Dai et al. (37). In this study, probes were reorganized based on the Entrez Gene database, build 37, version 1 (remapped CDF v14). Probe sets that satisfied the criterion of a false discovery rate <1% (q < 0.01) were considered significantly regulated and used for bioinformatics analysis by Ingenuity (Ingenuity Systems; www.ingenuity.com) and gene set enrichment analysis (GSEA; www.broad.mit.edu/gsea/). The number included in the array analysis were n = 4 per group for control, control plus Abx, and heme, and n = 6 for heme plus Abx. Genes with signal intensities below 20 in all mice from all treatments were considered not expressed and excluded from further analysis.
Bacterial DNA Extraction and qPCR.
Approximately 0.1 g fresh fecal pellet was subjected to mechanical and chemical lysis using 0.5 mL buffer [500 mM NaCl, 50 mM Tris⋅HCl (pH 8), 50 mM EDTA, and 4% (wt/vol) SDS] and 0.25 g of 0.1-mm zirconia beads and 3-mm glass beads. Nucleic acids were precipitated by addition of 130 μL, 10 M ammonium acetate, using one volume of isopropanol. Subsequently, DNA pellets were washed with 70% (vol/vol) ethanol. Further purification of DNA was performed using the QiaAmp DNA Mini Stool Kit (Qiagen). Finally, DNA was dissolved in 200 μL Tris/EDTA buffer, and its purity and quantity were checked spectrophotometrically (ND-1000; NanoDrop Technologies).
Total DNA was used for the quantitative estimation of total bacterial numbers per sample by real-time PCR amplification using generic 16S rRNA primers: forward Eub338F, ACTCCTACGGGAGGCAGCAG; and reverse Eub518R, ATTACCGCGGCTGCTGG. Bacteroidetes and Firmicutes populations were quantified by real-time amplification using phylum-specific 16S rRNA primers: Bacteroidetes forward Bact934F, GGARCATGTGGTTTAATTCGATGAT; reverse Bact1060R, AGCTGACGACAACCATGCAG and Firmicutes forward Firm934F, GGAGYATGTGGTTTAATTCGAAGCA; reverse Firm1060R, AGCTGACGACAACCATGCAC, respectively. Akkermansia muciniphila was quantified using genus-specific 16S rRNA primers S-St-Muc-1129-a-a-20 (AM1): CAGCACGTGAAGGTGGGGAC and S-St-Muc-1437-a-A-20 (AM2) CCTTGCGGTTGGCTTCAGAT. The sulfate-reducing capacity of the microbiota was estimated on basis of the gene-specific PCR that targets the dsrA gene that is associated with microbial sulfate respiration, using the following primers: forward RH1dsr, GCCGTTACTGTGACCAGCC and reverse RH3-dsr GGTGGAGCCGTGCATGTT. Bacterial counts are calculated and expressed in percentages relative to the control without Abx.
In Vitro Splitting of Disulfide (S-S) Bonds.
The potency of sodium sulfide (Na2S), N-acetylcysteine (NAC), glutathione (GSH), and cysteine to split S-S bonds were determined using 5,5′-dithiobis-(2-nitrobenzoic acid) (DTNB) as model disulfide compound. Splitting of the S-S bond of DTNB can be quantified by measuring absorption at 412 nm (17). Analyses were performed at 20 °C in a Perkin-Elmer spectrophotometer using a 1.5-mL quartz cuvette containing 100 mM potassium phosphate buffer (pH 7.0) and 100 µM DTNB. The reaction was started by adding sulfide or the thiols (final concentration, 25 µM). Reaction rates were determined as the slope of progress curve immediately after mixing. The extent of the overall reaction was determined from the final, stable, absorption, and corrected for blank absorption, using the milimolar extinction coefficient of 14.15. Sodium sulfate (Na2SO4) was used as negative control.
Western Blot Analysis.
Homogenates of freeze-dried feces were made by adding 1 mL PBS containing protease inhibitors (CompleteMini; Roche Diagnostics) to 10 mg freeze-dried pooled feces (n = 9/group). Samples were incubated for 5 min at 100 °C with nonreducing sample buffer (without DTT), with reducing sample buffer (with DTT), or with nonreducing sample buffer with Na2S. Samples were applied to SDS/PAGE [10% (wt/vol) gel] and transferred to polyvinylidene fluoride membranes under 350 mA for 4 h on ice. Twenty micrograms total protein was loaded. The blot was probed overnight at 4 °C with rabbit anti-mouse Muc2 antibody (38) (1:2,500) and then incubated with peroxidase-conjugated goat anti-rabbit antibody (1:5,000; Thermo Scientific). The signals were detected with the enhanced chemiluminescence detection system (Amersham ECL, GE Healthcare).
Measurement of Trisulfide Bonds.
From each individual fecal water 20 µL was added to 2-mL headspace vials containing 450 µL of Tris buffer (90 mM; pH 8.0) with or without 0.9 mM cysteine. Capped vials were incubated 30 min at 35 °C to facilitate cysteine-driven reduction of the central sulfur of the trisulfide bond to inorganic sulfide, as described earlier (18). Subsequently, 20 µL of 6 M HCL was added to the vials to force the sulfide as H2S into the headspace. With a headspace sampler this H2S was then measured on a GC-MS (TraceGC ulta; ThermoFinnigan) using a VF-1ms 30 × 0.25 column (Varian) with helium as carrier gas, and quantified with standard solutions of Na2S × 9 H2O treated identical to the samples. Trisulfide was calculated as the difference in H2S between the vial containing cysteine (i.e., total bound sulfide) and the vial without it (i.e., acid-labile sulfide). To validate this method control samples containing dimethyltrisulfide were analyzed identically and showed a recovery >90%. It should be noted that our assay is not confounded by free inorganic sulfides (such as HS− and S2−) in feces as these are removed during freeze drying. Samples from the in vitro incubations were analyzed identically. H2S was only detected in the cysteine vials containing the samples from the Na2S incubations, illustrating the specificity the trisulfide measurement.
Acknowledgments
We thank Jan van Riel and Kees Olieman for trisulfide analysis; Nicole de Wit for discussions; Jenny Jansen, Shohreh Keshtkar, Arjan Schonewille, Rob Dekker, Sandra ten Bruggencate, and Bert Weijers for technical assistance; and Dicky Lindenbergh-Kortleve and Janneke Samsom for Slpi immunohistochemistry. This work was financially supported by Top Institute Food and Nutrition Grant A-1001 (Wageningen, The Netherlands).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
Data deposition: Array data have been deposited in the Gene Expression Omnibus (GEO) database, www.ncbi.nlm.nih.gov/geo (accession no. GSE40670).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1507645112/-/DCSupplemental.
References
- 1.World Cancer Research Fund . Food, Nutrition, Physical Activity and the Prevention of Cancer: A Global Perspective. American Institute for Cancer Research; Washington, DC: 2007. [Google Scholar]
- 2.Giovannucci E, et al. Intake of fat, meat, and fiber in relation to risk of colon cancer in men. Cancer Res. 1994;54(9):2390–2397. [PubMed] [Google Scholar]
- 3.Lee DH, Anderson KE, Harnack LJ, Folsom AR, Jacobs DR., Jr Heme iron, zinc, alcohol consumption, and colon cancer: Iowa Women’s Health Study. J Natl Cancer Inst. 2004;96(5):403–407. doi: 10.1093/jnci/djh047. [DOI] [PubMed] [Google Scholar]
- 4.Balder HF, et al. Heme and chlorophyll intake and risk of colorectal cancer in the Netherlands cohort study. Cancer Epidemiol Biomarkers Prevent. 2006;15(4):717–725. doi: 10.1158/1055-9965.EPI-05-0772. [DOI] [PubMed] [Google Scholar]
- 5.Sesink AL, Termont DS, Kleibeuker JH, Van der Meer R. Red meat and colon cancer: The cytotoxic and hyperproliferative effects of dietary heme. Cancer Res. 1999;59(22):5704–5709. [PubMed] [Google Scholar]
- 6.IJssennagger N, et al. Dietary haem stimulates epithelial cell turnover by downregulating feedback inhibitors of proliferation in murine colon. Gut. 2012;61(7):1041–1049. doi: 10.1136/gutjnl-2011-300239. [DOI] [PubMed] [Google Scholar]
- 7.Young GP, Rose IS, St John DJ. Haem in the gut. I. Fate of haemoproteins and the absorption of haem. J Gastroenterol Hepatol. 1989;4(6):537–545. doi: 10.1111/j.1440-1746.1989.tb00858.x. [DOI] [PubMed] [Google Scholar]
- 8.McGarr SE, Ridlon JM, Hylemon PB. Diet, anaerobic bacterial metabolism, and colon cancer: A review of the literature. J Clin Gastroenterol. 2005;39(2):98–109. [PubMed] [Google Scholar]
- 9.Zimmer J, et al. A vegan or vegetarian diet substantially alters the human colonic faecal microbiota. Eur J Clin Nutr. 2012;66(1):53–60. doi: 10.1038/ejcn.2011.141. [DOI] [PubMed] [Google Scholar]
- 10.IJssennagger N, et al. Dietary heme alters microbiota and mucosa of mouse colon without functional changes in host-microbe cross-talk. PLoS One. 2012;7(12):e49868. doi: 10.1371/journal.pone.0049868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ijssennagger N, et al. Dietary heme induces acute oxidative stress, but delayed cytotoxicity and compensatory hyperproliferation in mouse colon. Carcinogenesis. 2013;34(7):1628–1635. doi: 10.1093/carcin/bgt084. [DOI] [PubMed] [Google Scholar]
- 12.Sesink AL, Termont DS, Kleibeuker JH, Van der Meer R. Red meat and colon cancer: Dietary haem-induced colonic cytotoxicity and epithelial hyperproliferation are inhibited by calcium. Carcinogenesis. 2001;22(10):1653–1659. doi: 10.1093/carcin/22.10.1653. [DOI] [PubMed] [Google Scholar]
- 13.Kindon H, Pothoulakis C, Thim L, Lynch-Devaney K, Podolsky DK. Trefoil peptide protection of intestinal epithelial barrier function: Cooperative interaction with mucin glycoprotein. Gastroenterology. 1995;109(2):516–523. doi: 10.1016/0016-5085(95)90340-2. [DOI] [PubMed] [Google Scholar]
- 14.Nakamura A, et al. Increased susceptibility to LPS-induced endotoxin shock in secretory leukoprotease inhibitor (SLPI)-deficient mice. J Exp Med. 2003;197(5):669–674. doi: 10.1084/jem.20021824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Johansson ME, Larsson JM, Hansson GC. The two mucus layers of colon are organized by the MUC2 mucin, whereas the outer layer is a legislator of host-microbial interactions. Proc Natl Acad Sci USA. 2011;108(Suppl 1):4659–4665. doi: 10.1073/pnas.1006451107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sheffner AL. The reduction in vitro in viscosity of mucoprotein solutions by a new mucolytic agent, N-acetyl-L-cysteine. Ann N Y Acad Sci. 1963;106:298–310. doi: 10.1111/j.1749-6632.1963.tb16647.x. [DOI] [PubMed] [Google Scholar]
- 17.Ellman GL. A colorimetric method for determining low concentrations of mercaptans. Arch Biochem Biophys. 1958;74(2):443–450. doi: 10.1016/0003-9861(58)90014-6. [DOI] [PubMed] [Google Scholar]
- 18.Jespersen AM, et al. Characterisation of a trisulphide derivative of biosynthetic human growth hormone produced in Escherichia coli. Eur J Biochem FEBS. 1994;219(1-2):365–373. doi: 10.1111/j.1432-1033.1994.tb19948.x. [DOI] [PubMed] [Google Scholar]
- 19.Tracy O. Pigment production in bacteroides. J Med Microbiol. 1969;2(3):309–315. doi: 10.1099/00222615-2-3-309. [DOI] [PubMed] [Google Scholar]
- 20.Kinzler KW, Vogelstein B. Lessons from hereditary colorectal cancer. Cell. 1996;87(2):159–170. doi: 10.1016/s0092-8674(00)81333-1. [DOI] [PubMed] [Google Scholar]
- 21.Pierre F, Taché S, Petit CR, Van der Meer R, Corpet DE. Meat and cancer: Haemoglobin and haemin in a low-calcium diet promote colorectal carcinogenesis at the aberrant crypt stage in rats. Carcinogenesis. 2003;24(10):1683–1690. doi: 10.1093/carcin/bgg130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lichtenstein L, et al. Angptl4 protects against severe proinflammatory effects of saturated fat by inhibiting fatty acid uptake into mesenteric lymph node macrophages. Cell Metab. 2010;12(6):580–592. doi: 10.1016/j.cmet.2010.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Velcich A, et al. Colorectal cancer in mice genetically deficient in the mucin Muc2. Science. 2002;295(5560):1726–1729. doi: 10.1126/science.1069094. [DOI] [PubMed] [Google Scholar]
- 24.García-Lafuente A, et al. Derangement of mucosal barrier function by bacteria colonizing the rat colonic mucosa. Eur J Clin Invest. 1998;28(12):1019–1026. doi: 10.1046/j.1365-2362.1998.00405.x. [DOI] [PubMed] [Google Scholar]
- 25.Bel S, et al. Reprogrammed and transmissible intestinal microbiota confer diminished susceptibility to induced colitis in TMF-/- mice. Proc Natl Acad Sci USA. 2014;111(13):4964–4969. doi: 10.1073/pnas.1319114111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Devkota S, et al. Dietary-fat-induced taurocholic acid promotes pathobiont expansion and colitis in Il10-/- mice. Nature. 2012;487(7405):104–108. doi: 10.1038/nature11225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Johansson ME, et al. Bacteria penetrate the normally impenetrable inner colon mucus layer in both murine colitis models and patients with ulcerative colitis. Gut. 2014;63(2):281–291. doi: 10.1136/gutjnl-2012-303207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Arumugam M, et al. MetaHIT Consortium Enterotypes of the human gut microbiome. Nature. 2011;473(7346):174–180. doi: 10.1038/nature09944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Swidsinski A, et al. Comparative study of the intestinal mucus barrier in normal and inflamed colon. Gut. 2007;56(3):343–350. doi: 10.1136/gut.2006.098160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.de Vogel J, Jonker-Termont DS, van Lieshout EM, Katan MB, van der Meer R. Green vegetables, red meat and colon cancer: Chlorophyll prevents the cytotoxic and hyperproliferative effects of haem in rat colon. Carcinogenesis. 2005;26(2):387–393. doi: 10.1093/carcin/bgh331. [DOI] [PubMed] [Google Scholar]
- 31.Lapré JA, Termont DS, Groen AK, Van der Meer R. Lytic effects of mixed micelles of fatty acids and bile acids. Am J Physiol. 1992;263(3 Pt 1):G333–G337. doi: 10.1152/ajpgi.1992.263.3.G333. [DOI] [PubMed] [Google Scholar]
- 32.Menckeberg CL, et al. Human buccal epithelium acquires microbial hyporesponsiveness at birth, a role for secretory leukocyte protease inhibitor. Gut. 2015;64(6):884–893. doi: 10.1136/gutjnl-2013-306149. [DOI] [PubMed] [Google Scholar]
- 33.Salonen A, et al. Comparative analysis of fecal DNA extraction methods with phylogenetic microarray: Effective recovery of bacterial and archaeal DNA using mechanical cell lysis. J Microbiol Methods. 2010;81(2):127–134. doi: 10.1016/j.mimet.2010.02.007. [DOI] [PubMed] [Google Scholar]
- 34.Dekker R, Van der Meer R, Olieman C. Sensitive pulsed amperometric detection of free and conjugated bile-acids in combination with gradient reversed-phase Hplc. Chromatographia. 1991;31(11-12):549–553. [Google Scholar]
- 35.Bovee-Oudenhoven IM, Termont DS, Heidt PJ, Van der Meer R. Increasing the intestinal resistance of rats to the invasive pathogen Salmonella enteritidis: additive effects of dietary lactulose and calcium. Gut. 1997;40(4):497–504. doi: 10.1136/gut.40.4.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Johnson WE, Li C, Rabinovic A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics. 2007;8(1):118–127. doi: 10.1093/biostatistics/kxj037. [DOI] [PubMed] [Google Scholar]
- 37.Dai M, et al. Evolving gene/transcript definitions significantly alter the interpretation of GeneChip data. Nucleic Acids Res. 2005;33(20):e175. doi: 10.1093/nar/gni179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.van Klinken BJ, et al. Gastrointestinal expression and partial cDNA cloning of murine Muc2. Am J Physiol. 1999;276(1 Pt 1):G115–G124. doi: 10.1152/ajpgi.1999.276.1.G115. [DOI] [PubMed] [Google Scholar]