

Abstract
DNA helicases have essential roles in the maintenance of genomic stability. They have achieved even greater prominence with the discovery that mutations in human helicase genes are responsible for a variety of genetic disorders and are associated with tumorigenesis. A number of missense mutations in human helicase genes are linked to chromosomal instability diseases characterized by age-related disease or associated with cancer, providing incentive for the characterization of molecular defects underlying aberrant cellular phenotypes. In this chapter, we discuss some examples of clinically relevant missense mutations in various human DNA helicases, particularly those of the Iron-Sulfur cluster and RecQ families. Clinically relevant mutations in the XPD helicase can lead to Xeroderma pigmentosum, Cockayne’s syndrome, Trichothiodystrophy, or COFS syndrome. FANCJ mutations are associated with Fanconi anemia or breast cancer. Mutations of the Fe-S helicase ChlR1 (DDX11) are linked to Warsaw Breakage syndrome. Mutations in the RecQ helicases BLM and WRN are linked to the cancer-prone disorder Bloom’s syndrome and premature aging condition Werner syndrome, respectively. RECQL4 mutations can lead to Rothmund-Thomson syndrome, Baller-Gerold syndrome, or RAPADILINO. Mutations in the Twinkle mitochondrial helicase are responsible for several neuromuscular degenerative disorders. We will discuss some insights gained from biochemical and genetic studies of helicase variants, and highlight some hot areas of helicase research based on recent developments.
DNA Helicases: A Specialized Class of Molecular Motors
Helicases are molecular motors that couple nucleoside triphosphate (NTP) hydrolysis (typically ATP) to the unwinding of polynucleic acid structures [1, 2]. Helicases have multifaceted roles in virtually all aspects of nucleic acid metabolism, including replication, DNA repair, recombination, transcription, chromosome segregation, and telomere maintenance [2–6]. ATP-dependent DNA or RNA unwinding enzymes exist; however, certain helicases can act upon both DNA and RNA [6]. Although DNA helicases are conventionally known to unwind B-form DNA double helical molecules, some are specialized such that they can catalytically disrupt alternate DNA structures (e.g., D-loop, Holliday junction (HJ), triplex, G-quadruplex), strip proteins bound to either single-stranded or double-stranded DNA, perform chromatin remodeling, and/or anneal complementary single-strands [7–10] (Fig. 6.1).
Fig. 6.1.

Depending on their specialty, DNA helicases use the energy of ATP hydrolysis to perform multiple functions. (a) A DNA helicase can disrupt noncovalent hydrogen bonds between complementary strands of the DNA double helix to form transient single-stranded DNA tracts. (b) A DNA helicase can strip proteins (e.g., Rad51) off DNA to regulate HR. (c) A DNA helicase can resolve alternate DNA structures (e.g., G-quadruplex) to enable smooth progression of the replication fork
Hereditary DNA Helicase Disorders
An increasing number of genetic diseases characterized by chromosomal instability are linked to mutations in DNA helicase genes. Many of these diseases are very rare, and inherited by autosomal homozygous recessive mutations. In certain cases, mutations in human helicase genes are linked to premature aging or age-related diseases, whereas some helicase mutations have been associated with cancer. In addition to these, hereditary dominant mutations have been identified in the mitochondrial DNA helicase which lead to neuromuscular degenerative diseases. We will provide an overview on the clinical disorders associated with mutations in DNA helicase genes.
Iron-Sulfur Cluster DNA Helicase Diseases
A prominent family of DNA helicases with a linkage to human diseases that has acquired a great deal of interest is the so-called Iron-Sulfur (Fe-S) cluster helicases, named after a conserved metal binding domain in the helicase core [11, 12]. These helicases belong to the Superfamily 2 (SF2) grouping of DNA helicases which share sequence homology within the helicase core domain. Mutations in the Fe-S cluster helicase XPD, a key factor of the TFIIH complex implicated in transcription and nucleotide excision repair (NER), can give rise to multiple rare autosomal recessive clinical disorders: xeroderma pigmentosum (XP), XP combined with Cockayne’s syndrome (CS), Trichothiodystrophy (TTD), and Cerebro-oculo-facial-skeletal (COFS) syndrome (for a recent review, see ref. [13]). Photosensitivity, neurological/developmental abnormalities, and skin cancer can be used to distinguish between XP, TTD, and CS; however, related or overlapping clinical features can arise from XPD mutations. XPD unwinds duplex DNA with a 5′–3′ directionality, and together with the SF2 XPB helicase (which lacks an Fe-S cluster) is required for NER and transcription [14]. Operating as components of the TFIIH complex, two DNA helicases XPD and XPB with opposite translocation polarities are responsible for unwinding duplex DNA in the vicinity of the lesion to create a bubble containing the lesion, a prerequisite for proper removal of the damaged fragment [14]. In contrast to XPD where helicase activity is indispensable for NER, only XPB ATPase is necessary for the pathway to remove bulky lesions and UV photoproducts. XPB ATPase activity is also necessary for DNA opening for transcription to occur, but its helicase activity is important for promoter escape during transcription (for review, see ref. [15]).
Homozygous recessive mutations in the FANCJ gene encoding the FANCJ Fe-S cluster DNA helicase (also called BACH1 or BRIP1), and at least 14 other genes, are responsible for Fanconi anemia (FA), a disorder characterized by congenital defects, progressive bone marrow failure, cancer accompanied by chromosomal instability, and hypersensitivity to agents that induce DNA interstrand cross-links (ICLs) [16, 17]. Notably, FANCJ mutations have also been associated with breast cancer [18]. Indeed, FANCJ was originally discovered by its association with the tumor suppressor protein BRCA1 [19]. In addition to its role in ICL repair, FANCJ helps cells to maintain genomic stability by resolving G-quadruplex (G4) DNA structures [20, 21], suggesting a more general role of the helicase in the response to replication stress.
The latest Fe-S cluster helicase implicated in a genetic disease is ChlR1, also known as DDX11, which is linked to Warsaw Breakage syndrome (WABS), a unique disease with cellular features of both FA and the cohesinopathy Roberts syndrome [22]. The clinical features of the single WABS patient reported include severe microcephaly, pre- and postnatal growth retardation, and abnormal skin pigmentation. Cells from the WABS patient display defective sister chromatid cohesion, a finding that is consistent with studies of mutant versions of ChlR1 homologs in yeast [23] and mouse [24] which also showed cohesion defects. The Lahti lab showed that depletion of human ChlR1 by RNA interference resulted in abnormal sister chromatid cohesion and a prometaphase delay leading to mitotic failure [25]. ChlR1 has a role in heterochromatin organization [26]; however, it is still unclear how the helicase is important for the cohesion process. Since cohesion is widely thought to be coupled to cellular DNA replication, it may be that ChlR1 is necessary for smooth replication fork progression through its catalytic activity and protein interactions, and that in its absence the cohesion proteins are not assembled properly; however, this may be an over simplistic assessment. Clearly further studies are necessary to understand the precise functions of ChlR1 required for chromosomal stability.
RecQ DNA Helicase Diseases
In addition to the Fe-S cluster DNA helicases, the SF2 RecQ DNA helicases are important for genomic stability and have been implicated in hereditary disorders. Homozygous recessive mutations in the BLM gene encoding a DNA helicase are responsible for Bloom’s syndrome [27]. BS patients are highly sensitive to sunlight, immunodeficient, and display a broad spectrum of cancers early in life. The hallmark of BS is an elevated rate of sister chromatid exchange (SCE) [28]. BLM is believed to function in homologous recombination (HR) repair to maintain genomic stability. A model for the role of BLM in SCE suppression was proposed in which a BLM protein complex containing topoisomerase IIIα, RPA, RMI, and RMI2 dissolves double HJ structures which may form during recombination or the convergence of replication forks [29, 30]. BLM helicase activity working in concert with topoisomerase cleavage/ligation is required for the double HJ dissolution reaction. In addition to this function, BLM is believed to have other roles (early and late) in homologous recombination (HR) repair of double strand breaks (DSBs) [31, 32] and helping cells to deal with replication stress [33].
A second RecQ helicase disorder known as Werner syndrome (WS) is characterized by premature aging features and the early onset of age-related diseases such as cardiovascular disorders, diabetes mellitus (Type II), osteoporosis, and sarcoma and mesenchymal tumors. WS is characterized by genomic instability, sensitivity to DNA damaging agents, elevated recombination, and replication defects. The WRN gene encodes an RecQ 3′–5′ DNA helicase and 3′–5′ exonuclease that is proposed to play a role in regulation of recombination events, primarily HR [3, 34]. It may be that WRN has a specialized function when the replication fork encounters a blocking lesion or alternate DNA structure. WRN interacts with a number of nuclear proteins, and these interactions are believed to facilitate genomic stability in various capacities including telomere maintenance, replication, and DNA repair [34, 35].
Mutations in a third RecQ family member, RECQL4, can lead to three distinct genetic disorders, namely Rothmund-Thomson syndrome (RTS), Baller-Gerold syndrome (BGS), and RAPADILINO (radial hypoplasia/aplasia, patellae hypoplasia/aplasia, cleft or highly arched palate, diarrhea, dislocated joints, little size (height at least 2 S.D. smaller than the average height) and limb malformation, nose slender, and normal intelligence) syndrome [36]. RTS patients are stunted in growth, and characterized by photosensitivity with poikiloderma, early graying and hair loss, juvenile cataracts, and osteogenic sarcomas. BGS is characterized by radial aplasia/hypoplasia and craniosynostosis, but not the poikiloderma typical of RTS patients [37]. Recent work form the Bohr lab has provided evidence that RECQL4 has pleiotropic roles in cellular DNA metabolism that include DSB repair [38] and telomere maintenance [39]. RECQL4 can be found in mitochondria where it helps to preserve the mitochondrial genome [40] and recruit p53 [41].
In addition to WRN, BLM, and RECQL4, two other human RecQ helicases, RECQL1 (RECQ1) and RECQL5 (RECQ5), are not yet linked to a disease; but it seems likely that these helicases will also play a role in cancer predisposition or a hereditary disorder characterized by chromosomal instability [42]. Indeed, studies of primary fibroblasts from RECQ1-knockout mice [43] and human cells depleted of RECQ1 by RNA interference [44] show chromosomal instability. Recent evidence has implicated distinct roles of RECQ1 (and RECQL4) in DNA replication initiation [45]. RECQ5 was found to be associated with RNA polymerase and may help to maintain genomic stability during transcription [46, 47]. RECQ5 also participates in DNA decatenation through its cooperation with Topoisomerase II alpha [48].
Twinkle Mitochondrial DNA Helicase
Aside from the Fe-S and RecQ DNA helicases, mutation of the Twinkle mitochondrial DNA helicase co-segregates with a number of diseases with mitochondrial defects including adult-onset progressive external ophthalmoplegia, hepatocerebral syndrome with mtDNA depletion syndrome, and infantile-onset spinocerebellar ataxia [49]. Twinkle is required for replication of human mitochondrial DNA, and mutations in the C10orf2 gene encoding Twinkle helicase can lead to mitochondrial deletions in post-mitotic tissues. Work from several laboratories demonstrated that Twinkle is an oligomeric helicase that unwinds double-stranded DNA molecules [50–52], and can also catalyze strand annealing [51], a function that is also observed for a number of the RecQ helicases [42]. It is yet unclear what the biological significance of helicase-catalyzed strand annealing truly is, but may play a role when replication forks are stalled or during DSB repair.
Hereditary Missense Mutations in DNA Helicase Disorders
Structural and biochemical analyses of purified recombinant DNA helicase proteins has sparked tremendous interest in understanding the molecular pathology behind disease-causing mutations in this specialized class of molecular motor ATPases. Disease-causing missense mutations result in single amino acid substitutions have been identified in DNA helicase disorders, which may be insightful for understanding biochemical mechanism and cellular function. In the following section, we will discuss some basic lessons from the spectrum of clinically relevant missense mutations in human DNA helicase genes, highlighting some unique aspects and potential areas of investigation.
Disease-Causing Missense Mutations in Iron-Sulfur Cluster DNA Helicases
XPD Missense Mutations
Missense mutations in the XPD gene are linked to four hereditary diseases: XP, XP combined with CS, TTD, and COFS syndrome with some cases of partially overlapping clinical features [13] (Fig. 6.2). XPD mutations responsible for XP are located mainly in the helicase core domain, and either significantly reduce or completely inactivate helicase function. The XP-causing XPD mutations do not affect basal transcription [53], consistent with a requirement of XPD ATPase/helicase activity for NER, but not transcription. As components of the TFIIH complex, XPD helicase operating in conjunction with the opposite polarity XPB helicase unwinds double-stranded DNA in the vicinity of the helix distorting lesion to create a bubble that can be acted upon by structure-specific nucleases to remove a short (~30 nt) single-stranded fragment containing the damage. Repair synthesis and ligation complete the steps of NER.
Fig. 6.2.

Clinically relevant missense mutations in XPD helicase responsible for Xeroderma pigmentosum, Xeroderma pigmentosum combined with Cockayne syndrome, Trichothiodystrophy, and COFS Syndrome. See text for details. For discussion of XPD mutations and genetic heterogeneity, see ref. [13]. XPD-D681N mutation is associated with XP and COFS syndrome
XPD mutations responsible for XP/CS also reside within or near the conserved helicase motifs as well (Fig. 6.2). However, XPD missense mutations causing XP/CS are believed to interfere with XPD protein interactions within the TFIIH complex. Unlike the XPD mutations implicated in XP or XP/CS which are only found in or very near the helicase core domain, XPD mutations responsible for TTD can also be found in the C-terminal region of the protein (Fig. 6.2). The C-terminus of XPD is important for interaction with other proteins (e.g., p44 subunit of TFIIH), which can be critical for optimal XPD helicase activity and/or stability of the TFIIH complex (for review, see ref. [54]). Such mutations in XPD reduce DNA repair activity and basal transcription [55]. Several XPD missense mutations linked to TTD that have been examined inhibit basal transcription, suggesting a molecular defect distinct from that of XP.
The heterozygous XPD mutations identified in a COFS syndrome patient were a R616W null mutation previously observed in an XP patient and a unique D681N mutation residing on the C-terminal side of motif VI [56] (Fig. 6.2). The aspartic acid (D681) is highly conserved in all known XPD genes. UV survival assays performed with COFS syndrome patient cells showed UV sensitivity comparable to that of cells from an XP-A patient with severe XP. It is unclear how the XPD-D681N mutation can result in distinct clinical phenotypes of COFS syndrome vs. XP. The wide spectrum of XPD missense mutations resulting in heterogeneity in clinical phenotype prompt continued interest to characterize the molecular and cellular defects of XPD variants to acquire a better understanding of the relevant diseases.
FANCJ Missense Mutations
The great majority of FANCJ missense mutations genetically linked to FA or associated with breast cancer reside in the N-terminal portion of the protein where the helicase core domain is located (Fig. 6.3), suggesting that ATP-dependent DNA unwinding by FANCJ is required for its function in the FA pathway of DNA repair and its involvement as a tumor suppressor. The requirement of FANCJ helicase activity for proper operation of the FA pathway is consistent with genetic complementation studies in human cells which show that cross-link resistance is dependent on catalytically active FANCJ protein [57].
Fig. 6.3.
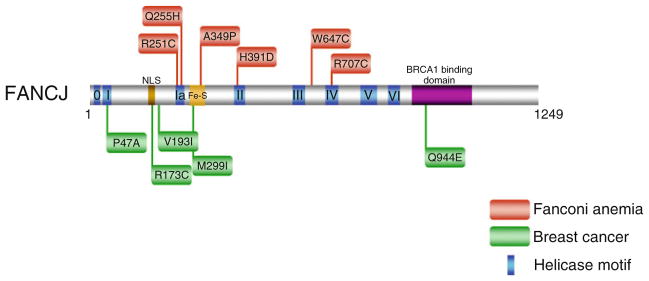
Clinically relevant missense mutations in FANCJ helicase responsible for Fanconi Anemia complementation group J and associated with breast cancer. See text for details. For a comprehensive listing of FANCJ mutations, see The Rockefeller University-Fanconi anemia mutation database www.rockefeller.edu/fanconi/mutate; also see ref. [18]
A FANCJ missense mutation linked to FA of particular interest is an alanine-to-proline substitution in the Fe-S domain at residue 349 immediately adjacent to a highly conserved cysteine that is important for binding an iron atom [58] (Fig. 6.4). Inheritance of the paternal FANCJ-A349P missense allele and a maternal truncating R798X allele resulted in phenotypic abnormalities, including intrauterine growth failure and death as a stillborn fetus with a gestational age of 22 weeks [58]. Biochemical analysis of purified recombinant FANCJ-A349P protein demonstrated that it was defective in coupling ATP-dependent DNA translocase activity to unwinding duplex DNA or displacing proteins bound to DNA [59] (Fig. 6.4). From a genetic standpoint, expression of the FANCJ-A349P mutant allele failed to rescue sensitivity of FANCJ null cells to a DNA cross-linking agent or the G-quadruplex (G4) binding drug telomestatin, indicating that it was unable to perform its role in ICL repair or resolution of G4 DNA [59]. In addition, expression of the FANCJ-A349P allele in a wild-type background exerted a dominant negative effect on resistance to DNA cross-linkers or telomestatin. Thus, the ability of FANCJ to couple ATP hydrolysis and DNA translocase activity to higher order functions such as unwinding structured nucleic acids or stripping protein from DNA is essential for its biological roles.
Fig. 6.4.

An FA complementation group J patient mutation (A349P) in the conserved Fe-S domain uncouples DNA ATPase and translocase activities from strand separation (helicase) activity. (a) FANCJ protein with the conserved helicase core domain, key protein interaction domains, and the Fe-S cluster. The conserved helicase motifs are indicated by yellow boxes, and the protein interaction domains for MLH1 and BRCA1 are shown by aqua green and blue boxes, respectively. The expanded Fe-S domain shows the locations for conserved cysteine residues in orange, and the A349P missense mutation of a FANCJ patient in bold. (b) The purified recombinant FANCJ-A349P protein fails to couple ATPase and single-stranded DNA translocase activity to unwinding duplex DNA. See text and ref. [59] for details
There is one FANCJ missense mutation, Q944E, identified in a breast cancer patient that is positioned outside the helicase core domain (Fig. 6.3). The Q944E mutation resides in the BRCA1 binding domain located in the C-terminal region of FANCJ (Fig. 6.2). Given the relatively close linear proximity of residue Q944 to S990, the site for FANCJ phosphorylation required for the physical interaction between FANCJ and BRCA1 [60], it will be of interest to determine if the BRCA1 binding to FANCJ is affected by the Q944E mutation. Such a FANCJ missense mutation may influence the molecular mechanism employed for DNA repair, as it was recently shown that disruption of the FANCJ-BRCA1 interaction blocks DNA repair by homologous recombination and promotes polη-dependent bypass [61].
DDX11 (ChlR1) Single Amino Acid Deletion Mutation
Mutations in the ChlR1 helicase (also named DDX11) are genetically linked to the chromosomal instability disorder WABS [22]. The WABS patient is characterized by a compound heterozygous K897del mutation in trans with a maternally inherited splice site mutation in an intron that leads to a truncated protein, leading to nonsense-mediated decay of the maternal allele and monoallelic expression of the K897del-containing paternal allele. The K897del mutation results in a single amino acid deletion of a lysine residue that resides ten amino acids from the C-terminus of the protein. The endogenous hChlR1-K897del protein was poorly detected by immunoblot analysis of lysates from fibroblasts or lymphoblasts of the affected individual [22]. Cells from the patient exhibit chromosomal instability characterized by sister cohesion defects, chromosomal breakage, and sensitivity to the DNA cross-linking agent mitomycin and topoisomerase inhibitor camptothecin. Based on the cellular phenotypes, it was suggested that WABS is a unique disease with cellular features of both FA and the cohesinopathy Roberts syndrome.
Biochemical analysis of the purified recombinant ChlR1 protein demonstrated that it possesses a DNA-dependent ATPase activity and a 5′–3′ helicase activity [62, 63]. The purified recombinant hChlR1-K897del protein was dramatically inhibited for DNA binding and DNA-dependent ATPase activity compared to the wild-type recombinant ChlR1 protein [64], suggesting an important role of the extreme C-terminus of the protein for its stable interaction with nucleic acid. Further cellular and biochemical studies are likely to yield insight to the proposed role of the ChlR1 helicase to allow smooth replication fork progression necessary for proper sister chromatid cohesion during mitosis [62]. The strong preference of ChlR1 to unwind G4 DNA may be important for stability at particular genomic loci [64]. Greater insight to the disease will likely be gained from more biochemical and genetic studies, including the identification and characterization of other mutant ChlR1 alleles.
Disease-Causing Missense Mutations in RecQ DNA Helicases
BLM Missense Mutations
Inspection of the clinical spectrum of Bloom’s syndrome patient missense mutations reveals that the ones identified to date reside in the helicase core domain or the adjacent RecQ C-terminal region (RQC) [65] (Fig. 6.5). This mutational pattern strongly suggests that BLM helicase activity is required for suppression of the BS disease phenotype, a result that is consistent with the observation that expression of an engineered Walker A box (motif I) catalytically inactive BLM protein failed to complement the genomic instability of BS cells; moreover, the helicase-dead BLM protein exerted a dominant negative effect on cell viability in a wild-type background [66]. In fact, all of the BS patient-derived helicase domain mutant proteins that have been biochemically characterized show defects in ATP or DNA binding, which result in low or no detectable helicase activity [66–69].
Fig. 6.5.
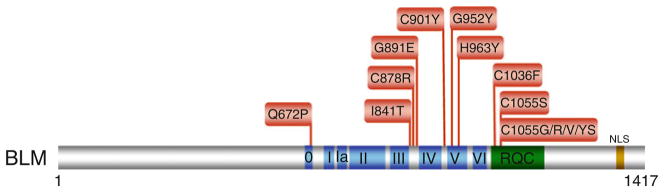
Clinically relevant missense mutations in BLM helicase responsible for Bloom’s syndrome. See text for details. For a comprehensive listing of BLM mutations, see The Bloom’s Syndrome Registry www.med.cornell.edu/bsr/; also see ref. [65]
The conserved RQC region in BLM, found in many RecQ helicases, consists of a Zn2+ binding domain and a winged helix domain. Two of the cysteine residues (C1036 and C1055) residing in the RQC region and responsible for binding Zn2+ are mutated in BS (Fig. 6.5). Five different amino acids substitutions (S, G, R, V, and Y) resulting from BLM patient mutations occur at C1055, suggesting that this particular locus in the BLM Zn2+ domain may be a hotspot for mutation and that the cysteine residue is highly important for BLM structure and function. Biochemical characterization of the purified recombinant BLM-C1055S mutant protein showed that it lacked ATPase and helicase activity [66, 67]. Expression of the BLM-C1055S mutant protein in vivo failed to rescue the p53-mediated apoptosis defect of BS fibroblasts [70]. Collectively, the location of the BLM missense mutations and their molecular analysis demonstrate that BLM helicase activity is required for its in vivo function to suppress BS phenotypes.
RECQL4 Missense Mutations
The RECQL4 helicase is distinct from other human RecQ helicases because three distinct genetic disorders can be attributed to mutations in the RECQL4 gene: RTS, RAPADILINO, and BGS [36]. Unlike BLM, missense mutations for all three RECQL4 diseases have been identified in regions outside the conserved helicase domain shared by the other RecQ helicases [36] (Fig. 6.6). RTS missense mutations are found in the C-terminal region, N-terminal region, and the helicase core domain. Only two RAPADILINO missense mutations have been reported, one in the N-terminal region and one in the helicase core domain. For BGS, a single patient missense mutation (R1021W) is known that resides in the C-terminal region. Interestingly, the C-terminal R1021W mutation was genetically linked to both BGS and RTS. The effects of the R1021W mutation, as well as all of the other mutations in the RECQL4 gene, on the biochemical functions of the RECQ4 helicase protein have not yet been determined.
Fig. 6.6.
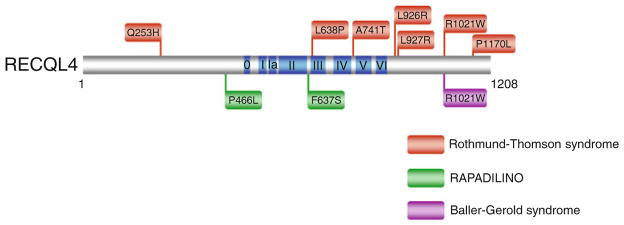
Clinically relevant missense mutations in RECQL4 helicase responsible for Rothmund-Thomson syndrome, Baller-Gerold syndrome, and RAPADILINO. See text for details. For comprehensive listing of RECQL4 mutations, see ref. [36]. RECQL4-R1021W is associated with both RTS and BGS
WRN Missense Mutations
The discovery that mutations in the WRN gene are linked to a premature aging disorder [71] sparked a tremendous amount of interest in understanding the pathological basis for the disease on a molecular level; however, the clinical spectrum of WS mutations was rather uninformative because those identified resulted in truncated proteins. Since the nuclear localization sequence (NLS) of WRN protein residing in the extreme C-terminus, it would be predicted that these WRN protein fragments would fail to localize to the nucleus where WRN is required to preserve genomic stability [72]. However, more recently a small number of WRN missense mutations have been identified (Fig. 6.6). One of these, the M1350R missense mutation would be predicted to interfere with the ability of the WRN protein to effectively localize to nuclei since it resides immediately N-terminal to the NLS of WRN [73].
Two missense mutations reside in the N-terminal exonuclease domain of WRN were shown to result in WRN protein instability [74]. From a research standpoint, this is unfortunate because if the mutant WRN proteins had been selectively inactivated for exonuclease function, they may have been useful for understanding the molecular importance of that catalytic function. Two additional WRN missense mutations were found in the helicase domain, one in the Walker A box (motif I) (G574R) [73] just three amino acids away from the invariant lysine residue implicated in nucleotide binding and the other (R637W) [75] very near helicase motif V. Although not yet biochemically characterized, these two helicase core domain mutants would be predicted to interfere with normal ATPase and helicase function. If this were true, they may constitute separation of function mutations that would be valuable for assessing the relative importance of WRN helicase vs. exonuclease activity to suppress the cellular phenotypes associated with WS. Genetic and biochemical studies suggest the dual importance of balanced and concerted WRN helicase and exonuclease activities [76–78]; however, there is still much to learn precisely how WRN helicase or exonuclease activities suppress the disease phenotypes of WS.
Disease-Causing Missense Mutations in the XPB and Twinkle DNA Helicases
XPB Missense Mutations
The XPB gene encodes a DNA helicase with opposite polarity to that of XPD that is also found in the TFIIH complex, and XPB mutations can lead to multiple clinical disorders including XP/CS, XP with neurological abnormalities, and TTD [13]. In contrast to XPD where helicase activity is indispensable for NER, only XPB ATPase is necessary for the pathway to remove bulky lesions and UV photoproducts. XPB ATPase activity is also necessary for DNA opening for transcription to occur, but its helicase activity is important for promoter escape during transcription (for review, see ref. [15]). It is generally believed that the phenotypic heterogeneity of XPB is attributed to missense mutations which only partially affect XPB biochemical and cellular function in mild XP/CS, whereas severe XP/CS is associated with nonsense mutations in both XPB alleles, resulting in altered and reduced XPB protein [79]. A causative mutation in the XPB helicase resulting in a single amino acid substitution (T119P) in the N-terminus of the protein prior to the conserved helicase motifs was identified in a patient with mild versions of both TTD and photosensitivity [80]. Cellular studies from the Sarasin lab demonstrated that the XPB-T119P allele is associated with moderately defective DNA repair [81]. This is in contrast to XPB-F99S allele (also located in the N-terminus) of an XP/CS patient which is profoundly reduced in its DNA repair function [81]. Interestingly, both the T119P and F99S mutations in XPB do not impair TFIIH helicase activity [15]; however, the F99S substitution was shown to impair the interaction of XPB with p52, one of the subunits of the TFIIH complex [55]. Collectively, the studies of TFIIH and clinical spectrum of mutations in the XPB and XPD helicases have helped to dissect the complex heterogeneity of DNA repair-transcription syndromes.
Twinkle Missense Mutations
As mentioned earlier, missense mutations in the human C10orf2 mitochondrial helicase gene encoding Twinkle DNA helicase are linked to several neuromuscular degenerative disorders. Expression of patient-derived Twinkle mutant proteins in normal cells or transgenic mice led to the accumulation of mitochondrial DNA replication intermediates and mitochondrial DNA depletion [82–84], consistent with the idea that dysfunctional Twinkle helicase can cause mitochondrial replication forks to stall. The reduction in Twinkle helicase activity for those variants that were examined correlated to the extent of mitochondrial DNA depletion and accumulation of replication intermediates [82]. Biochemical analysis of 20 disease variants of the human mitochondrial DNA helicase showed that the mutations are quite heterogeneous in terms of their molecular effects on ATPase and helicase function as well as effects in some cases on protein stability [85]. All 20 mutant Twinkle variants retained at least partial helicase activity, leading the authors to propose that these defects are consistent with the delayed presentation of mitochondrial diseases associated with the c10orf2 mutations.
Recent Advances and Research Directions for DNA Helicases Implicated in Disease
The helicase field is moving at a brisk pace, with the development of new technologies and a greater understanding of their biological roles. In the following section, we will highlight some interesting developments from our viewpoint. These emerging stories, as well as others discussed in the accompanying chapters, convey a very exciting era for the scientific community beyond the scope of DNA metabolism because there are rippling implications for the fields of aging, disease, and cancer.
Communication Between Redox Active DNA Repair Proteins
Nearly all DNA repair pathways share a few common steps: (1) initial detection of a DNA lesion; (2) processing of the damaged DNA nucleotide/strand; (3) removal of the damaged nucleotide/strand; (4) replacement of the damaged nucleotide/strand with correct nucleotide/sequence; (5) sealing the nick(s) by ligation. Of these steps, initial detection of the lesion is critical and can be rate-limiting due to low copy number of DNA damage recognition proteins and lack of preferential DNA binding affinity for the lesion compared to undamaged sequence. Recent experimental evidence supports a model that DNA charge transport is a signaling mechanism for the recruitment of redox active DNA repair proteins to the vicinity of DNA damage ([86, 87], and for review see ref. [88]). In the latest development, Sontz et al. [89] provide experimental evidence that coordinated DNA charge transport between DNA repair proteins with redox active Fe-S clusters, the XPD NER helicase and EndoIII base excision DNA glycosylase, can occur to enable efficient redistribution of the repair proteins to the location of DNA damage within a vast excess of undamaged nucleotides. Thus, DNA repair proteins (or proteins involved in other aspects of DNA metabolism such as replication or gene expression) conventionally thought to operate in distinct pathways might collaborate with each other in an unexpected way by their ability to participate in DNA charge transport chemistry. Although there is evidence for cross-talk between DNA repair pathways and also with replication [90–93], the ability of redox active proteins to collaborate by DNA charge transport broadens the scope of possibilities since the proteins do not necessarily need to physically interact with one another. This is highly provocative for the potential synergy that might exist between redox active DNA interacting proteins that influence diverse pathways previously thought to function independently of one another. Research that further explores the collaboration between proteins able to perform DNA charge transport will likely have important implications for molecular gerontology and cancer biology.
Coordination of End-Processing Proteins in Double Strand Break Repair Resection
DSBs can be highly poisonous and lethal to cells; therefore, several DNA repair pathways including nonhomologous end-joining and homologous recombination exist to join ends back together again. In replicating cells, the preferred mechanism of DSB repair is by homologous recombination because it occurs with high fidelity, using the sister chromatid duplex as a template. Based on experimental evidence, BLM is implicated in an early stage of DSB repair in which strand resection from a DNA end occurs to provide a 3′ single-stranded DNA tail for RAD51-mediated strand invasion into recipient duplex [32, 94, 95]. Strand resection is much more complex than previously thought, and requires a battalion of helicases, nucleases, DNA binding proteins, and additional accessory factors. The BLM helicase promotes access of the DNA end to either DNA2 or EXO-1, enabling processive catalytic removal of one strand to produce the 3′ single-stranded DNA tail.
Another layer of complexity for the involvement of BLM in strand resection was suggested by the discovery that BLM interacts with FANCJ [96], which is associated with BRCA1, a tumor suppressor molecule implicated in DSB repair [19]. FANCJ-deficient cells have a defect in DSB-induced HR and show delayed resolution of DSBs following ionizing radiation [97]. Moreover, FANCJ and certain other HR proteins function downstream of FANCD2/I mono-ubiquitination in ICL repair to presumably repair the processed DNA cross-link by an HR-mediated pathway. We propose a model in which strand resection is facilitated by FANCJ and BLM helicases translocating on opposite strands of the broken double-stranded DNA end to promote access to structure-specific nucleases (EXO-1, DNA2) [98]. The FANCJ-BLM partnership may facilitate resection at chemically modified blocked DNA ends or through G-rich sequences prone to form G-quadruplex (G4) structures. This model for the dual collaboration of FANCJ and BLM in DSB repair can be tested by a combination of genetic and biochemical approaches. DNA end resection assays in a reconstituted system with chromatinized templates may help to elucidate the functional importance of the FANCJ-BLM interaction in DSB repair in a biological context.
Structural Determinants of XPD Helicase Function and Translocation Polarity
Recent progress in understanding the molecular architecture of Fe-S cluster DNA helicases has come from several labs working on archaeal XPD proteins. This effort began with the discovery that XPD bears an Fe-S cluster metal binding site essential for its DNA unwinding activity, but not necessary for binding single-stranded DNA or ATP hydrolysis [99]. Crystal structures solved independently by three research groups confirmed the existence of a novel Fe-S domain [100–102]. The XPD structure contains two Rad51/RecA-like domains (HD1 and HD2) with two additional domains, the Fe-S and Arch domains, inserted between adjacent β-strands of the central β-sheet of HD1. ATP binding and hydrolysis controls the conformational state of the Fe-S and Arch domains in conjunction with the conserved helicase motifs that comprise the helicase core domain. The Fe-S domain was proposed to form a wedge with the nearby Arch domain to separate the DNA duplex as the enzyme translocates in an ATP-dependent manner. Mutations introduced to the Fe-S domain, including the conserved cysteines, abolished XPD helicase activity and/or destabilized tertiary structure [99, 100], attesting to the structural importance of the Fe-S domain. Biochemical studies demonstrated that the integrity of the XPD Fe-S domain is required for the proper folding and structural stability, and is important for coupling ATP hydrolysis to unidirectional translocation [103]. Furthermore, the Fe-S cluster serves to stabilize elements of protein secondary structure and target the helicase to the single-stranded/double-stranded DNA junction [103].
The structure of XPD in complex with a short DNA fragment was recently reported [104]. This provided a working model for the mechanism of translocation by Fe-S cluster DNA helicases. The XPD-DNA crystal structure, combined with a mutational and biochemical analysis of XPD, revealed how the 5′–3′ directionality of translocation along DNA is achieved and suggested how the XPD enzyme might act upon a DNA substrate harboring a helix-distorting lesion susceptible to NER. These conclusions on how regulation of translocation polarity by XPD helicase is achieved were further supported by another very recent study using proteolytic DNA and mutational analysis of XPD [105]. Based on these two studies, it was suggested that Fe-S domain helicases achieve a polarity of ATP-driven translocation opposite to that of 3′–5′ helicases by conformational changes within the motor domain rather than binding single-stranded DNA with an opposite orientation. It is plausible that the Fe-S redox activity may provide an additional mechanism for FANCJ and other DNA repair molecules to communicate with each other and sense DNA-mediated charge transport such that they cooperate with each other to assemble and/or translocate with a defined polarity and in a regulated manner [106]; however, this hypothesis remains to be experimentally tested.
Small Molecule Inhibitors of DNA Helicases
Understanding the precise molecular-genetic defects of helicase disorders in some cases such as the RecQ diseases can be challenging. Partial functional redundancy between DNA helicases as well as backup pathways make it difficult to establish cause and effect relationships between a helicase defect and biological outcome. Since DNA helicases provide essential functions in multiple steps of DNA damage response and repair pathways, it will be highly informative to precisely characterize their functions in vivo. This information will lead to new insight to their roles in prevention or correction of genomic DNA damage that is a causative force for cellular senescence and contributes to organismal decline associated with aging. Use of small molecules to target human helicases for inhibition (or activation) in a cell-based model system is a novel approach to the problem of assigning specific helicase functions in vivo. In addition to the new insight to the roles of helicases in DNA damage response and repair to prevent age-associated phenotypes, this work will also be informative for the development of anticancer strategies which target DNA repair proteins/processes [107].
Recently, a small molecule (NSC 19630) from the NCI Diversity Set was identified that inhibited WRN helicase activity, but not other DNA helicases tested, suggesting specificity [108]. Human cells exposed to NSC 19630 were dramatically impaired in their growth and proliferation, and displayed elevated apoptosis in a WRN-dependent manner, suggesting an effect of the small molecule that mimicked expression of a helicase-dead protein. Furthermore, cellular exposure to the WRN helicase inhibitor resulted in elevated γ-H2AX and proliferating cell nuclear antigen (PCNA) foci, and delayed S-phase progression, consistent with the accumulation of stalled replication forks. Exposure to NSC 19630 sensitized cancer cells to agents that induce replication stress or interfere with DNA repair. We anticipate that the WRN helicase inhibitor molecule may be helpful in understanding WRN-mediated pathway(s) important for the response to DNA damage and replication stress (Fig. 6.7).
Fig. 6.7.
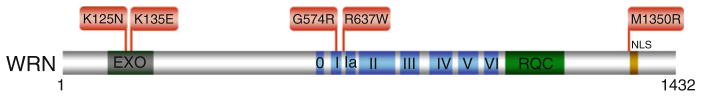
Clinically relevant missense mutations in WRN helicase-nuclease responsible for Werner syndrome. See text for details. For a comprehensive listing of WRN mutations, see The International Registry of Werner Syndrome www.wernersyndrome.org; also see ref. [73]
Summary
In this chapter, we have attempted to provide an overview of hereditary helicase disorders, with a particular emphasis on clinically relevant missense mutations as these may prove to be valuable in understanding pathway dysfunction. In addition, we have highlighted some hot topics in helicase research that we believe will be important for future studies. Clearly, progress in understanding the biochemical and genetic functions of RecQ and Fe-S cluster helicases will serve as a springboard for new investigations. In addition, the recent progress in understanding the molecular basis for Twinkle mitochondrial helicase disorders has created a lot of excitement in the field. Contributions from an increasing number of scientists will continue to unveil new insights to the roles of DNA helicases in suppressing genomic instability, cancer, and age-related diseases.
Acknowledgments
This research was supported by the Intramural Research Program of the NIH, National Institute on Aging and the Fanconi Anemia Research Fund (RMB). We apologize to helicase researchers whose work was not cited due to space limitations.
References
- 1.Lohman TM, Tomko EJ, Wu CG. Non-hexameric DNA helicases and translocases: mechanisms and regulation. Nat Rev Mol Cell Biol. 2008;9:391–401. doi: 10.1038/nrm2394. [DOI] [PubMed] [Google Scholar]
- 2.Singleton MR, Dillingham MS, Wigley DB. Structure and mechanism of helicases and nucleic acid translocases. Annu Rev Biochem. 2007;76:23–50. doi: 10.1146/annurev.biochem.76.052305.115300. [DOI] [PubMed] [Google Scholar]
- 3.Brosh RM, Jr, Bohr VA. Human premature aging, DNA repair and RecQ helicases. Nucleic Acids Res. 2007;35:7527–44. doi: 10.1093/nar/gkm1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lohman TM, Bjornson KP. Mechanisms of helicase-catalyzed DNA unwinding. Annu Rev Biochem. 1996;65:169–214. doi: 10.1146/annurev.bi.65.070196.001125. [DOI] [PubMed] [Google Scholar]
- 5.Patel SS, Donmez I. Mechanisms of helicases. J Biol Chem. 2006;281:18265–8. doi: 10.1074/jbc.R600008200. [DOI] [PubMed] [Google Scholar]
- 6.Pyle AM. Translocation and unwinding mechanisms of RNA and DNA helicases. Annu Rev Biophys. 2008;37:317–36. doi: 10.1146/annurev.biophys.37.032807.125908. [DOI] [PubMed] [Google Scholar]
- 7.Bernstein KA, Gangloff S, Rothstein R. The RecQ DNA helicases in DNA repair. Annu Rev Genet. 2010;44:393–417. doi: 10.1146/annurev-genet-102209-163602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dillingham MS, Superfamily I. helicases as modular components of DNA-processing machines. Biochem Soc Trans. 2011;39:413–23. doi: 10.1042/BST0390413. [DOI] [PubMed] [Google Scholar]
- 9.Singh DK, Ghosh AK, Croteau DL, Bohr VA. RecQ helicases in DNA double strand break repair and telomere maintenance. Mutat Res. 2012;736:15–24. doi: 10.1016/j.mrfmmm.2011.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wu Y, Suhasini AN, Brosh RM. Welcome the family of FANCJ-like helicases to the block of genome stability maintenance proteins. Cell Mol Life Sci. 2009;66:1209–22. doi: 10.1007/s00018-008-8580-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.White MF. Structure, function and evolution of the XPD family of iron-sulfur-containing 5′–>3′ DNA helicases. Biochem Soc Trans. 2009;37:547–51. doi: 10.1042/BST0370547. [DOI] [PubMed] [Google Scholar]
- 12.White MF, Dillingham MS. Iron-sulphur clusters in nucleic acid processing enzymes. Curr Opin Struct Biol. 2012;22:94–100. doi: 10.1016/j.sbi.2011.11.004. [DOI] [PubMed] [Google Scholar]
- 13.Digiovanna JJ, Kraemer KH. Shining a light on xeroderma pigmentosum. J Invest Dermatol. 2012;132:785–96. doi: 10.1038/jid.2011.426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Egly JM, Coin F. A history of TFIIH: two decades of molecular biology on a pivotal transcription/repair factor. DNA Repair (Amst) 2011;10:714–21. doi: 10.1016/j.dnarep.2011.04.021. [DOI] [PubMed] [Google Scholar]
- 15.Oksenych V, Coin F. The long unwinding road: XPB and XPD helicases in damaged DNA opening. Cell Cycle. 2010;9:90–6. doi: 10.4161/cc.9.1.10267. [DOI] [PubMed] [Google Scholar]
- 16.Crossan GP, Patel KJ. The Fanconi anaemia pathway orchestrates incisions at sites of cross-linked DNA. J Pathol. 2012;226:326–37. doi: 10.1002/path.3002. [DOI] [PubMed] [Google Scholar]
- 17.Deans AJ, West SC. DNA interstrand crosslink repair and cancer. Nat Rev Cancer. 2011;11:467–80. doi: 10.1038/nrc3088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cantor SB, Guillemette S. Hereditary breast cancer and the BRCA1-associated FANCJ/BACH1/BRIP1. Future Oncol. 2011;7:253–61. doi: 10.2217/fon.10.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cantor SB, Bell DW, Ganesan S, et al. BACH1, a novel helicase-like protein, interacts directly with BRCA1 and contributes to its DNA repair function. Cell. 2001;105:149–60. doi: 10.1016/s0092-8674(01)00304-x. [DOI] [PubMed] [Google Scholar]
- 20.London TB, Barber LJ, Mosedale G, et al. FANCJ is a structure-specific DNA helicase associated with the maintenance of genomic G/C tracts. J Biol Chem. 2008;283:36132–9. doi: 10.1074/jbc.M808152200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wu Y, Shin-Ya K, Brosh RM., Jr FANCJ helicase defective in Fanconia anemia and breast cancer unwinds G-quadruplex DNA to defend genomic stability. Mol Cell Biol. 2008;28:4116–28. doi: 10.1128/MCB.02210-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.van der LP, Chrzanowska KH, Godthelp BC, et al. Warsaw breakage syndrome, a cohesinopathy associated with mutations in the XPD helicase family member DDX11/ChlR1. Am J Hum Genet. 2010;86:262–6. doi: 10.1016/j.ajhg.2010.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Skibbens RV. Chl1p, a DNA helicase-like protein in budding yeast, functions in sister-chromatid cohesion. Genetics. 2004;166:33–42. doi: 10.1534/genetics.166.1.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Inoue A, Li T, Roby SK, et al. Loss of ChlR1 helicase in mouse causes lethality due to the accumulation of aneuploid cells generated by cohesion defects and placental malformation. Cell Cycle. 2007;6:1646–54. doi: 10.4161/cc.6.13.4411. [DOI] [PubMed] [Google Scholar]
- 25.Parish JL, Rosa J, Wang X, Lahti JM, Doxsey SJ, Androphy EJ. The DNA helicase ChlR1 is required for sister chromatid cohesion in mammalian cells. J Cell Sci. 2006;119:4857–65. doi: 10.1242/jcs.03262. [DOI] [PubMed] [Google Scholar]
- 26.Inoue A, Hyle J, Lechner MS, Lahti JM. Mammalian ChlR1 has a role in heterochromatin organization. Exp Cell Res. 2011;317:2522–35. doi: 10.1016/j.yexcr.2011.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ellis NA, Groden J, Ye TZ, et al. The Bloom’s syndrome gene product is homologous to RecQ helicases. Cell. 1995;83:655–66. doi: 10.1016/0092-8674(95)90105-1. [DOI] [PubMed] [Google Scholar]
- 28.Chaganti RS, Schonberg S, German J. A manyfold increase in sister chromatid exchanges in Bloom’s syndrome lymphocytes. Proc Natl Acad Sci USA. 1974;71:4508–12. doi: 10.1073/pnas.71.11.4508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bachrati CZ, Hickson ID. Dissolution of double Holliday junctions by the concerted action of BLM and topoisomerase IIIalpha. Methods Mol Biol. 2009;582:91–102. doi: 10.1007/978-1-60761-340-4_8. [DOI] [PubMed] [Google Scholar]
- 30.Wu L, Hickson ID. The Bloom’s syndrome helicase suppresses crossing over during homologous recombination. Nature. 2003;426:870–4. doi: 10.1038/nature02253. [DOI] [PubMed] [Google Scholar]
- 31.Chu WK, Hanada K, Kanaar R, Hickson ID. BLM has early and late functions in homologous recombination repair in mouse embryonic stem cells. Oncogene. 2010;29:4705–14. doi: 10.1038/onc.2010.214. [DOI] [PubMed] [Google Scholar]
- 32.Nimonkar AV, Genschel J, Kinoshita E, et al. BLM-DNA2-RPA-MRN and EXO1-BLM-RPA-MRN constitute two DNA end resection machineries for human DNA break repair. Genes Dev. 2011;25:350–62. doi: 10.1101/gad.2003811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chan KL, Palmai-Pallag T, Ying S, Hickson ID. Replication stress induces sister-chromatid bridging at fragile site loci in mitosis. Nat Cell Biol. 2009;11:753–60. doi: 10.1038/ncb1882. [DOI] [PubMed] [Google Scholar]
- 34.Monnat RJ., Jr Human RECQ helicases: roles in DNA metabolism, mutagenesis and cancer biology. Semin Cancer Biol. 2010;20:329–39. doi: 10.1016/j.semcancer.2010.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rossi ML, Ghosh AK, Bohr VA. Roles of Werner syndrome protein in protection of genome integrity. DNA Repair (Amst) 2010;9:331–44. doi: 10.1016/j.dnarep.2009.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Larizza L, Roversi G, Volpi L. Rothmund-Thomson syndrome. Orphanet J Rare Dis. 2010;5:2. doi: 10.1186/1750-1172-5-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Van Maldergem L, Siitonen HA, Jalkh N, et al. Revisiting the craniosynostosis-radial ray hypoplasia association: Baller-Gerold syndrome caused by mutations in the RECQL4 gene. J Med Genet. 2006;43:148–52. doi: 10.1136/jmg.2005.031781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Singh DK, Karmakar P, Aamann M, et al. The involvement of human RECQL4 in DNA double-strand break repair. Aging Cell. 2010;9:358–71. doi: 10.1111/j.1474-9726.2010.00562.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ghosh AK, Rossi ML, Singh DK, et al. RECQL4, the protein mutated in Rothmund-Thomson syndrome, functions in telomere maintenance. J Biol Chem. 2012;287:196–209. doi: 10.1074/jbc.M111.295063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Croteau DL, Rossi ML, Canugovi C, et al. RECQL4 localizes to mitochondria and preserves mitochondrial DNA integrity. Aging Cell. 2012;11(3):456–66. doi: 10.1111/j.1474-9726.2012.00803.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.De S, Kumari J, Mudgal R, et al. RECQL4 is essential for the transport of p53 to mitochondria in normal human cells in the absence of exogenous stress. J Cell Sci. 2012;125:2509–22. doi: 10.1242/jcs.101501. [DOI] [PubMed] [Google Scholar]
- 42.Sharma S, Doherty KM, Brosh RM., Jr Mechanisms of RecQ helicases in pathways of DNA metabolism and maintenance of genomic stability. Biochem J. 2006;398:319–37. doi: 10.1042/BJ20060450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sharma S, Stumpo DJ, Balajee AS, et al. RECQL, a member of the RecQ family of DNA helicases, suppresses chromosomal instability. Mol Cell Biol. 2007;27:1784–94. doi: 10.1128/MCB.01620-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sharma S, Brosh RM., Jr Human RECQ1 is a DNA damage responsive protein required for genotoxic stress resistance and suppression of sister chromatid exchanges. PLoS One. 2007;2:e1297. doi: 10.1371/journal.pone.0001297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Thangavel S, Mendoza-Maldonado R, Tissino E, et al. The human RECQ1 and RECQ4 helicases play distinct roles in DNA replication initiation. Mol Cell Biol. 2010;30:1382–96. doi: 10.1128/MCB.01290-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Aygun O, Svejstrup J, Liu Y. A RECQ5-RNA polymerase II association identified by targeted proteomic analysis of human chromatin. Proc Natl Acad Sci USA. 2008;105:8580–4. doi: 10.1073/pnas.0804424105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kanagaraj R, Huehn D, MacKellar A, et al. RECQ5 helicase associates with the C-terminal repeat domain of RNA polymerase II during productive elongation phase of transcription. Nucleic Acids Res. 2010;38:8131–40. doi: 10.1093/nar/gkq697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ramamoorthy M, Tadokoro T, Rybanska I, et al. RECQL5 cooperates with topoisomerase II alpha in DNA decatenation and cell cycle progression. Nucleic Acids Res. 2012;40:1621–35. doi: 10.1093/nar/gkr844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Copeland WC. Defects in mitochondrial DNA replication and human disease. Crit Rev Biochem Mol Biol. 2012;47:64–74. doi: 10.3109/10409238.2011.632763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Korhonen JA, Gaspari M, Falkenberg M. TWINKLE Has 5′ -> 3′ DNA helicase activity and is specifically stimulated by mitochondrial single-stranded DNA-binding protein. J Biol Chem. 2003;278:48627–32. doi: 10.1074/jbc.M306981200. [DOI] [PubMed] [Google Scholar]
- 51.Sen D, Nandakumar D, Tang GQ, Patel SS. The human mitochondrial DNA helicase TWINKLE is both an unwinding and an annealing helicase. J Biol Chem. 2012;287(18):14545–56. doi: 10.1074/jbc.M111.309468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ziebarth TD, Gonzalez-Soltero R, Makowska-Grzyska MM, Nunez-Ramirez R, Carazo JM, Kaguni LS. Dynamic effects of cofactors and DNA on the oligomeric state of human mitochondrial DNA helicase. J Biol Chem. 2010;285:14639–47. doi: 10.1074/jbc.M109.099663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dubaele S, De Proietti SL, Bienstock RJ, et al. Basal transcription defect discriminates between xeroderma pigmentosum and trichothiodystrophy in XPD patients. Mol Cell. 2003;11:1635–46. doi: 10.1016/s1097-2765(03)00182-5. [DOI] [PubMed] [Google Scholar]
- 54.Lehmann AR. XPD structure reveals its secrets. DNA Repair (Amst) 2008;7:1912–5. doi: 10.1016/j.dnarep.2008.07.008. [DOI] [PubMed] [Google Scholar]
- 55.Coin F, Oksenych V, Egly JM. Distinct roles for the XPB/p52 and XPD/p44 subcomplexes of TFIIH in damaged DNA opening during nucleotide excision repair. Mol Cell. 2007;26:245–56. doi: 10.1016/j.molcel.2007.03.009. [DOI] [PubMed] [Google Scholar]
- 56.Graham JM, Jr, Nyane-Yeboa K, Raams A, et al. Cerebro-oculo-facio-skeletal syndrome with a nucleotide excision-repair defect and a mutated XPD gene, with prenatal diagnosis in a triplet pregnancy. Am J Hum Genet. 2001;69:291–300. doi: 10.1086/321295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Peng M, Litman R, Xie J, Sharma S, Brosh RM, Jr, Cantor SB. The FANCJ/MutLalpha interaction is required for correction of the cross-link response in FA-J cells. EMBO J. 2007;26:3238–49. doi: 10.1038/sj.emboj.7601754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Levran O, Attwooll C, Henry RT, et al. The BRCA1-interacting helicase BRIP1 is deficient in Fanconi anemia. Nat Genet. 2005;37:931–3. doi: 10.1038/ng1624. [DOI] [PubMed] [Google Scholar]
- 59.Wu Y, Sommers JA, Suhasini AN, et al. Fanconi anemia Group J mutation abolishes its DNA repair function by uncoupling DNA translocation from helicase activity or disruption of protein-DNA complexes. Blood. 2010;116:3780–91. doi: 10.1182/blood-2009-11-256016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yu X, Chini CC, He M, Mer G, Chen J. The BRCT domain is a phospho-protein binding domain. Science. 2003;302:639–42. doi: 10.1126/science.1088753. [DOI] [PubMed] [Google Scholar]
- 61.Xie J, Litman R, Wang S, et al. Targeting the FANCJ-BRCA1 interaction promotes a switch from recombination to poleta-dependent bypass. Oncogene. 2010;29:2499–508. doi: 10.1038/onc.2010.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Farina A, Shin JH, Kim DH, et al. Studies with the human cohesin establishment factor, ChlR1. Association of ChlR1 with Ctf18-RFC and Fen1. J Biol Chem. 2008;283:20925–36. doi: 10.1074/jbc.M802696200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hirota Y, Lahti JM. Characterization of the enzymatic activity of hChlR1, a novel human DNA helicase. Nucleic Acids Res. 2000;28:917–24. doi: 10.1093/nar/28.4.917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wu Y, Sommers JA, Khan I, De Winter JP, Brosh RM., Jr Biochemical characterization of warsaw breakage syndrome helicase. J Biol Chem. 2012;287:1007–21. doi: 10.1074/jbc.M111.276022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.German J, Sanz MM, Ciocci S, Ye TZ, Ellis NA. Syndrome-causing mutations of the BLM gene in persons in the Bloom’s Syndrome Registry. Hum Mutat. 2007;28:743–53. doi: 10.1002/humu.20501. [DOI] [PubMed] [Google Scholar]
- 66.Neff NF, Ellis NA, Ye TZ, et al. The DNA helicase activity of BLM is necessary for the correction of the genomic instability of bloom syndrome cells. Mol Biol Cell. 1999;10:665–76. doi: 10.1091/mbc.10.3.665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bahr A, De Graeve F, Kedinger C, Chatton B. Point mutations causing Bloom’s syndrome abolish ATPase and DNA helicase activities of the BLM protein. Oncogene. 1998;17:2565–71. doi: 10.1038/sj.onc.1202389. [DOI] [PubMed] [Google Scholar]
- 68.Guo RB, Rigolet P, Zargarian L, Fermandjian S, Xi XG. Structural and functional characterizations reveal the importance of a zinc binding domain in Bloom’s syndrome helicase. Nucleic Acids Res. 2005;33(10):3109–24. doi: 10.1093/nar/gki619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Guo RB, Rigolet P, Ren H, et al. Structural and functional analyses of disease-causing missense mutations in Bloom syndrome protein. Nucleic Acids Res. 2007;35:6297–310. doi: 10.1093/nar/gkm536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wang XW, Tseng A, Ellis NA, et al. Functional interaction of p53 and BLM DNA helicase in apoptosis. J Biol Chem. 2001;276:32948–55. doi: 10.1074/jbc.M103298200. [DOI] [PubMed] [Google Scholar]
- 71.Yu CE, Oshima J, Fu YH, et al. Positional cloning of the Werner’s syndrome gene. Science. 1996;272:258–62. doi: 10.1126/science.272.5259.258. [DOI] [PubMed] [Google Scholar]
- 72.Suzuki T, Shiratori M, Furuichi Y, Matsumoto T. Diverged nuclear localization of Werner helicase in human and mouse cells. Oncogene. 2001;20:2551–8. doi: 10.1038/sj.onc.1204344. [DOI] [PubMed] [Google Scholar]
- 73.Friedrich K, Lee L, Leistritz DF, et al. WRN mutations in Werner syndrome patients: genomic rearrangements, unusual intronic mutations and ethnic-specific alterations. Hum Genet. 2010;128:103–11. doi: 10.1007/s00439-010-0832-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Huang S, Lee L, Hanson NB, et al. The spectrum of WRN mutations in Werner syndrome patients. Hum Mutat. 2006;27:558–67. doi: 10.1002/humu.20337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Uhrhammer NA, Lafarge L, Dos SL, et al. Werner syndrome and mutations of the WRN and LMNA genes in France. Hum Mutat. 2006;27:718–9. doi: 10.1002/humu.9435. [DOI] [PubMed] [Google Scholar]
- 76.Swanson C, Saintigny Y, Emond MJ, Monnat RJ., Jr The Werner syndrome protein has separable recombination and survival functions. DNA Repair (Amst) 2004;3:475–82. doi: 10.1016/j.dnarep.2004.01.002. [DOI] [PubMed] [Google Scholar]
- 77.Chen L, Huang S, Lee L, et al. WRN, the protein deficient in Werner syndrome, plays a critical structural role in optimizing DNA repair. Aging Cell. 2003;2:191–9. doi: 10.1046/j.1474-9728.2003.00052.x. [DOI] [PubMed] [Google Scholar]
- 78.Opresko PL, Otterlei M, Graakjaer J, et al. The Werner syndrome helicase and exonuclease cooperate to resolve telomeric D loops in a manner regulated by TRF1 and TRF2. Mol Cell. 2004;14:763–74. doi: 10.1016/j.molcel.2004.05.023. [DOI] [PubMed] [Google Scholar]
- 79.Oh KS, Khan SG, Jaspers NG, et al. Phenotypic heterogeneity in the XPB DNA helicase gene (ERCC3): xeroderma pigmentosum without and with Cockayne syndrome. Hum Mutat. 2006;27:1092–103. doi: 10.1002/humu.20392. [DOI] [PubMed] [Google Scholar]
- 80.Weeda G, Eveno E, Donker I, et al. A mutation in the XPB/ERCC3 DNA repair transcription gene, associated with trichothiodystrophy. Am J Hum Genet. 1997;60:320–9. [PMC free article] [PubMed] [Google Scholar]
- 81.Riou L, Zeng L, Chevallier-Lagente O, et al. The relative expression of mutated XPB genes results in xeroderma pigmentosum/Cockayne’s syndrome or trichothiodystrophy cellular phenotypes. Hum Mol Genet. 1999;8:1125–33. doi: 10.1093/hmg/8.6.1125. [DOI] [PubMed] [Google Scholar]
- 82.Goffart S, Cooper HM, Tyynismaa H, Wanrooij S, Suomalainen A, Spelbrink JN. Twinkle mutations associated with autosomal dominant progressive external ophthalmoplegia lead to impaired helicase function and in vivo mtDNA replication stalling. Hum Mol Genet. 2009;18:328–40. doi: 10.1093/hmg/ddn359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Tyynismaa H, Mjosund KP, Wanrooij S, et al. Mutant mitochondrial helicase Twinkle causes multiple mtDNA deletions and a late-onset mitochondrial disease in mice. Proc Natl Acad Sci USA. 2005;102:17687–92. doi: 10.1073/pnas.0505551102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Wanrooij S, Goffart S, Pohjoismaki JL, Yasukawa T, Spelbrink JN. Expression of catalytic mutants of the mtDNA helicase Twinkle and polymerase POLG causes distinct replication stalling phenotypes. Nucleic Acids Res. 2007;35:3238–51. doi: 10.1093/nar/gkm215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Longley MJ, Humble MM, Sharief FS, Copeland WC. Disease variants of the human mitochondrial DNA helicase encoded by C10orf2 differentially alter protein stability, nucleotide hydrolysis, and helicase activity. J Biol Chem. 2010;285:29690–702. doi: 10.1074/jbc.M110.151795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Boal AK, Genereux JC, Sontz PA, Gralnick JA, Newman DK, Barton JK. Redox signaling between DNA repair proteins for efficient lesion detection. Proc Natl Acad Sci USA. 2009;106:15237–42. doi: 10.1073/pnas.0908059106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Romano CA, Sontz PA, Barton JK. Mutants of the base excision repair glycosylase, endonuclease III: DNA charge transport as a first step in lesion detection. Biochemistry. 2011;50:6133–45. doi: 10.1021/bi2003179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Merino EJ, Boal AK, Barton JK. Biological contexts for DNA charge transport chemistry. Curr Opin Chem Biol. 2008;12:229–37. doi: 10.1016/j.cbpa.2008.01.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Sontz PA, Mui TP, Fuss JO, Tainer JA, Barton JK. DNA charge transport as a first step in coordinating the detection of lesions by repair proteins. Proc Natl Acad Sci USA. 2012;109:1856–61. doi: 10.1073/pnas.1120063109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Constantinou A. Rescue of replication failure by Fanconi anaemia proteins. Chromosoma. 2012;121:21–36. doi: 10.1007/s00412-011-0349-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Hakem R. DNA-damage repair; the good, the bad, and the ugly. EMBO J. 2008;27:589–605. doi: 10.1038/emboj.2008.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Lagerwerf S, Vrouwe MG, Overmeer RM, Fousteri MI, Mullenders LH. DNA damage response and transcription. DNA Repair (Amst) 2011;10:743–50. doi: 10.1016/j.dnarep.2011.04.024. [DOI] [PubMed] [Google Scholar]
- 93.Wilson DM, III, Seidman MM. A novel link to base excision repair? Trends Biochem Sci. 2010;35:247–52. doi: 10.1016/j.tibs.2010.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Gravel S, Chapman JR, Magill C, Jackson SP. DNA helicases Sgs1 and BLM promote DNA double-strand break resection. Genes Dev. 2008;22:2767–72. doi: 10.1101/gad.503108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Nimonkar AV, Ozsoy AZ, Genschel J, Modrich P, Kowalczykowski SC. Human exonuclease 1 and BLM helicase interact to resect DNA and initiate DNA repair. Proc Natl Acad Sci USA. 2008;105:16906–11. doi: 10.1073/pnas.0809380105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Suhasini AN, Rawtani NA, Wu Y, et al. Interaction between the helicases genetically linked to Fanconi anemia group J and Bloom’s syndrome. EMBO J. 2011;30:692–705. doi: 10.1038/emboj.2010.362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Litman R, Peng M, Jin Z, et al. BACH1 is critical for homologous recombination and appears to be the Fanconi anemia gene product FANCJ. Cancer Cell. 2005;8:255–65. doi: 10.1016/j.ccr.2005.08.004. [DOI] [PubMed] [Google Scholar]
- 98.Suhasini AN, Brosh RM., Jr Fanconi anemia and Bloom’s syndrome crosstalk through FANCJ-BLM helicase interaction. Trends Genet. 2012;28:7–13. doi: 10.1016/j.tig.2011.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Rudolf J, Makrantoni V, Ingledew WJ, Stark MJ, White MF. The DNA repair helicases XPD and FancJ have essential Iron-Sulfur domains. Mol Cell. 2006;23:801–8. doi: 10.1016/j.molcel.2006.07.019. [DOI] [PubMed] [Google Scholar]
- 100.Fan L, Fuss JO, Cheng QJ, et al. XPD helicase structures and activities: insights into the cancer and aging phenotypes from XPD mutations. Cell. 2008;133:789–800. doi: 10.1016/j.cell.2008.04.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Liu H, Rudolf J, Johnson KA, et al. Structure of the DNA repair helicase XPD. Cell. 2008;133:801–12. doi: 10.1016/j.cell.2008.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Wolski SC, Kuper J, Hanzelmann P, et al. Crystal structure of the FeS cluster-containing nucleotide excision repair helicase XPD. PLoS Biol. 2008;6:e149. doi: 10.1371/journal.pbio.0060149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Pugh RA, Honda M, Leesley H, et al. The iron-containing domain is essential in Rad3 helicases for coupling of ATP hydrolysis to DNA translocation and for targeting the helicase to the single-stranded DNA-double-stranded DNA junction. J Biol Chem. 2008;283:1732–43. doi: 10.1074/jbc.M707064200. [DOI] [PubMed] [Google Scholar]
- 104.Kuper J, Wolski SC, Michels G, Kisker C. Functional and structural studies of the nucleotide excision repair helicase XPD suggest a polarity for DNA translocation. EMBO J. 2011;31:494–502. doi: 10.1038/emboj.2011.374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Pugh RA, Wu CG, Spies M. Regulation of translocation polarity by helicase domain 1 in SF2B helicases. EMBO J. 2011;31:503–14. doi: 10.1038/emboj.2011.412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Wu Y, Brosh RM., Jr DNA helicase and helicase-nuclease enzymes with a conserved iron-sulfur cluster. Nucleic Acids Res. 2012;40(10):4247–60. doi: 10.1093/nar/gks039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Aggarwal M, Brosh RM., Jr Hitting the bull’s eye: novel directed cancer therapy through helicase-targeted synthetic lethality. J Cell Biochem. 2009;106:758–63. doi: 10.1002/jcb.22048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Aggarwal M, Sommers JA, Shoemaker RH, Brosh RM., Jr Inhibition of helicase activity by a small molecule impairs Werner syndrome helicase (WRN) function in the cellular response to DNA damage or replication stress. Proc Natl Acad Sci USA. 2011;108:1525–30. doi: 10.1073/pnas.1006423108. [DOI] [PMC free article] [PubMed] [Google Scholar]