

Abstract
Both human and rodent females are more susceptible to developing alcoholic liver disease following chronic ethanol (EtOH) ingestion. However, little is known about the relative effects of acute EtOH exposure on hepatotoxicity in female versus male mice. The nuclear receptor pregnane X receptor (PXR; NR1I2) is a broad-specificity sensor with species-specific responses to toxic agents. To examine the effects of the human PXR on acute EtOH toxicity, the responses of male and female PXR-humanized (hPXR) transgenic mice administered oral binge EtOH (4.5 g/kg) were analyzed. Basal differences were observed between hPXR males and females in which females expressed higher levels of two principal enzymes responsible for EtOH metabolism, alcohol dehydrogenase 1 and aldehyde dehydrogenase 2, and two key mediators of hepatocyte replication and repair, cyclin D1 and proliferating cell nuclear antigen. EtOH ingestion upregulated hepatic estrogen receptor α, cyclin D1, and CYP2E1 in both genders, but differentially altered lipid and EtOH metabolism. Consistent with higher basal levels of EtOH-metabolizing enzymes, blood EtOH was more rapidly cleared in hPXR females. These factors combined to provide greater protection against EtOH-induced liver injury in female hPXR mice, as revealed by markers for liver damage, lipid peroxidation, and endoplasmic reticulum stress. These results indicate that female hPXR mice are less susceptible to acute binge EtOH-induced hepatotoxicity than their male counterparts, due at least in part to the relative suppression of cellular stress and enhanced expression of enzymes involved in both EtOH metabolism and hepatocyte proliferation and repair in hPXR females.
Introduction
Gender is a major factor that impacts susceptibility to alcoholic liver disease (ALD); epidemiologic studies suggest that, for any given level of alcohol consumption, women have a higher likelihood of developing liver cirrhosis than men (Pares et al., 1986). Additionally, liver injury progresses faster in women with alcoholic hepatitis who stop or reduce drinking (Pares et al., 1986). Several theories have been proposed to explain this disparity based on gender differences in ethanol (EtOH) pharmacokinetics, estrogen levels, and alcohol elimination rates (Sato et al., 2001).
Ethanol is metabolized predominantly in the liver by enzymes located in different subcellular compartments of the hepatocyte (Zakhari and Li, 2007). Alcohol dehydrogenase (ADH), a cytosolic NAD+-dependent enzyme, is the main enzyme that catalyzes the conversion of EtOH to acetaldehyde, a potent toxicant that accounts for most of the toxic effects of EtOH (Zakhari and Li, 2007). Acetaldehyde produced from EtOH is further converted to the nontoxic acetate by a mitochondrial aldehyde dehydrogenase (ALDH or Aldh) (Zakhari and Li, 2007). In addition to ADH, the endoplasmic reticulum enzyme CYP2E1, which is induced by EtOH, can metabolize EtOH to acetaldehyde at high alcohol doses (Zakhari and Li, 2007). Catalase, located in the peroxisomes, is also capable of oxidizing EtOH to acetaldehyde (Zakhari and Li, 2007).
The effect of gender on ADH and ALDH activity is controversial. Pharmacokinetic studies revealed that women have lower levels of gastric and hepatic ADH, resulting in increased blood alcohol levels and EtOH toxicity (Frezza et al., 1990; Chrostek et al., 2003). However, Maly and Sasse (1991) found that hepatic ADH activity is enhanced in women. Although only minor variations in ALDH activity between men and women have been reported, ALDH activity is higher in male hamsters than in females (Maly and Sasse, 1991; Lee et al., 2001; Chrostek et al., 2003).
We have previously shown that nuclear receptors, a class of intracellular transcription factors activated by ligands, are involved in the pathogenesis of ALD (Gyamfi et al., 2006, 2008). Notably, the nuclear hormone receptor pregnane X receptor (PXR; NR1I2), expressed primarily in the liver and intestine, was originally characterized as a xenobiotic receptor important for defense against toxic agents and for eliminating drugs and other xenobiotics (Kliewer et al., 2002). Interestingly, PXR activation induces hepatic triglyceride accumulation, a characteristic feature of ALD, suggesting the possibility that PXR is associated with ALD pathogenesis (Zhou et al., 2006). However, it is not known whether PXR is involved in the gender dimorphism of EtOH hepatotoxicity.
Due to differences between the mouse and human ligand binding domain sequences, species-specific responses to ligand activation of human and mouse PXR have been reported (Lehmann et al., 1998). Thus, some chemical ligands, such as rifampicin and rifaximin, that activate human PXR usually have little effect on the mouse form of this receptor and vice versa (Ma et al., 2007a,b). To this end, PXR-humanized (hPXR) transgenic mice were developed to provide a more valid in vivo model of human xenobiotic responses (Ma et al., 2007a). Interestingly, recent reports indicate that transgenic mice expressing the human PXR gene are more prone to high-fat diet–induced hyperglycemia in both genders compared with their respective wild-type counterparts (Spruiell et al., 2014a,b). Further, high-fat diet–fed hPXR females express lower basal protein levels of both hepatic and white adipose tissue estrogen receptor α (ERα) (Spruiell et al., 2014a). Given that higher estrogen levels and/or signaling in women have been implicated in EtOH-induced liver injury, we reasoned that the human PXR gene may play a role in sex differences in EtOH-induced liver injury (Eagon, 2010).
Most rodent studies investigating sex-specific differences in EtOH hepatotoxicity have used the chronic EtOH ingestion model (Kono et al., 2000; Nanji et al., 2001; Colantoni et al., 2002; Ronis et al., 2004). In comparison, relatively few studies have investigated the effects of acute EtOH exposure on hepatotoxicity in male and female mice (Wagnerberger et al., 2013). Although some studies have addressed gender dimorphisms in EtOH hepatotoxicity using rodents, extrapolation of the results to humans can be difficult. In this study, binge EtOH was administered to male and female hPXR mice, revealing that the basal protein expression levels of both ADH1 and ALDH2 are higher in the liver of female hPXR mice. As a result, female hPXR mice eliminate EtOH more effectively than their male counterparts, thereby mitigating acute EtOH hepatotoxicity.
Materials and Methods
Animal Care and Treatment.
Breeding pairs of male and female hPXR mice on a C57BL/6 background were transferred from a colony housed at the National Cancer Institute (National Institutes of Health, Bethesda, MD) (Ma et al., 2007a). The hPXR mice were generated by bacterial artificial chromosome transgenesis, in which the transgene contains the complete human PXR gene and the 5′- and 3′-flanking sequences, and then bred with Pxr-null mice to produce hPXR mice carrying a C57BL/6 genetic background (Ma et al., 2007a). Human PXR was coexpressed with CYP3A in hPXR mice in the liver, duodenum, jejunum, and ileum, matching the gene expression pattern in humans and “humanizing” mouse liver and intestine with respect to PXR (Ma et al., 2007a). Treatment with PXR ligands revealed a clear species difference between wild-type and hPXR mice in their response to xenobiotics, suggesting that this bacterial artificial chromosome–transgenic hPXR mouse model is useful for investigating human PXR function in vivo (Ma et al., 2007a). Mice (3–5 mice/cage) were housed in polycarbonate cages on racks directly vented via the facility’s exhaust system at 22°C with a 12-hour light/dark cycle at the Animal Resources Complex at North Carolina Central University (Durham, NC). Both male and female age-matched (10–12 weeks of age) hPXR mice were randomly separated into two groups (n = 8–9 for each group) and were gavaged with three doses of 4.5 g/kg 50% (v/v) EtOH or saline solution every 12 hours at 9:00 AM, 9:00 PM, 9:00 AM the next day, as previously reported (Kirpich et al., 2012; Wang et al., 2013a). Four hours after the final dose at 1:00 pm, mice were anesthetized with isoflurane and killed. Sections of liver were rapidly dissected, weighed, snap-frozen in liquid nitrogen, and kept at –80°C. Blood samples collected by cardiac puncture from anesthetized mice were centrifuged at 3000 rpm for 15 minutes to collect serum and stored at −80°C to determine EtOH concentration, liver enzymes, triglycerides, and nonesterified fatty acid (NEFA) concentrations. All procedures conducted in accordance with the National Institutes of Health Guidelines for the Care and Use of Laboratory Animals were approved by the North Carolina Central University Institutional Animal Care and Use Committee.
H&E Staining of Liver Sections.
Liver slices were fixed in 10% formalin/phosphate-buffered saline, and then stained with H&E for histologic examination.
Serum Alanine Aminotransferase, Aspartate Aminotransferase, Triglyceride, and NEFA Measurements.
Serum was processed from blood and stored at −80°C. Serum alanine aminotransferase (ALT) and aspartate aminotransferase (AST) activity was determined using the Cholestech LDX analyzer (Cholestech Corporation, Hayward, CA) as reported previously (Spruiell et al., 2014a,b). Serum triglyceride and NEFA levels were quantified using Triglyceride and NEFA-HR (2) test kits (Wako Pure Chemical Industries, Richmond, VA).
Determination of Serum Alcohol Concentration.
Blood samples were collected after three doses of binge EtOH administration and centrifuged at 3000 rpm for 15 minutes to collect serum for blood EtOH concentration (BEC). In a separate study, male and female hPXR mice were administered a single dose of 50% (v/v) EtOH (4.5 g/kg) by gastric intubation after overnight starvation. Mice (n = 4–5) were sacrificed 1, 2, 4, 6, and 8 hours after EtOH administration, and blood samples were collected to prepare serum. The EtOH L3K assay kit for quantitative measurement of EtOH concentration (Sekisui Diagnostics P.E.I. Inc, Charlottetown, PEI, Canada) was used according to the manufacturer’s instructions as reported previously (Perides et al., 2005). The reaction is based on the enzymatic conversion of EtOH by ADH to acetaldehyde and NADH. EtOH concentration in the serum was quantified as the rate of increase in NADH absorbance due to the reduction of NAD+ at 340 nm.
Hepatic Triglyceride and Cholesterol Levels.
Total liver lipids were extracted from 100 mg of liver homogenate using methanol and chloroform as previously described (Gyamfi et al., 2008). Hepatic triglyceride and cholesterol levels were quantified using Triglyceride and Cholesterol test kits (Wako Pure Chemical Industries).
Measurements of Lipid Peroxidation in Liver Tissues.
The extent of lipid peroxidation (LPO) in liver homogenates was quantitatively determined by measuring the concentration of the thiobarbituric acid–reactive product, malondialdehyde (MDA), using the thiobarbituric acid–reactive substances kit (ZeptoMetrix, Buffalo, NY) as we previously reported (Tanaka et al., 2008). Protein contents in liver homogenates were determined by the BCA protein assay kit (Thermo Scientific, Rockford, IL).
Preparation of Liver Extracts for Western Blot Analyses.
Frozen livers were homogenized at 4°C, and Western blot analysis was performed on extracts as described previously (Gyamfi et al., 2006). Protein content in liver homogenates were determined by the BCA protein assay kit (Thermo Scientific). Liver homogenate (40 μg/lane) was mixed in Laemmli loading buffer containing β-mercaptoethanol, boiled for 5 minutes, separated on 10 or 15% SDS-PAGE gels, and transferred to a polyvinylidene difluoride membrane. The membranes were probed with one or more of the following antibodies according to the manufacturers’ recommendations: anti-CYP2E1 and anti-catalase (Abcam, Cambridge, MA); anti-ERα, anti-CYP3A, anti-GRP78, anti-ALDH2, and anti-ADH1 (Santa Cruz Biotechnology, Santa Cruz, CA); anti–phospho-p38 mitogen-activated protein kinase (p-p38 MAPK; Thr180/Tyr182), anti–p38 MAPK, anti–phospho-p44/42 MAPK [i.e., extracellular regulated kinases 1/2 (ERK1/2), p-ERK1/2; Thr202/Tyr204], anti-ERK1/2, anti–caspase 12, anti–cyclin D1, and anti–phosphorylated-elF2α (Ser51; Cell Signaling Technology, Boston, MA); or anti–proliferating cell nuclear antigen (PCNA; Sigma-Aldrich, St. Louis, MO). Blots were then incubated with the appropriate peroxidase-conjugated anti-rabbit IgG secondary antibodies (Santa Cruz Biotechnology) diluted in Tris-buffered saline/Tween 20 plus 1% milk for 60 minutes at room temperature. Following initial probing, blots were stripped and reprobed with anti–α-tubulin antibody (Cell Signaling Technology). Proteins were visualized using enhanced chemiluminescence, and band intensity was quantified using ImageJ software (National Institutes of Health).
Quantification of mRNA Levels Using Real-Time Polymerase Chain Reaction.
Total RNA was isolated from frozen liver tissues using the Trizol reagent according to the manufacturer’s protocol (Invitrogen, Carlsbard, CA). Total RNA (5 µg) was reverse transcribed into cDNA with random hexamer primers using a Tetro cDNA Synthesis Kit (Bioline, Taunton, MA) as we previously described (Spruiell et al., 2014b). The cDNA was then diluted 20-fold with water and subjected to real-time quantitative polymerase chain reaction (PCR) by the SensiFast SYBR Hi-ROX Kit (Bioline) to quantify the mRNA levels of peroxisome proliferator–activated receptor α (PPARα), PPARγ, sterol regulatory element-binding protein-1c (SREBP-1c), CPT1 (carnitine palmitoyltransferase 1), ACOX-1 (acyl-CoA Oxidase 1), L-FABP-1 (liver-type fatty acid-binding protein 1), microsomal triglyceride transfer protein (MTTP), fatty acid translocase (CD36), ACC-1α (acetyl-CoA carboxylase 1α), FAS (fatty acid synthase), SCD1 (stearoyl-CoA desaturase-1), and phosphatidylethanolamine N-methyltransferase (PEMT). The primers (Table 1) for mRNA encoding PPARα, PPARγ, SREBP-1c, CPT1, ACOX-1, L-FABP, MTTP, CD36, ACC-1α, FAS, SCD1, PEMT, and GAPDH (glyceraldehyde 3-phosphate dehydrogenase) were designed using Primer Express 2.0 (Applied Biosystems, Foster City, CA). Furthermore, the following proprietary TaqMan Gene Expression Assays for isoforms of Adh and Aldh2 were purchased from Applied Biosystems/Life Technologies (Grand Island, NY) and used for real-time quantitative PCR: Adh1 (class I Adh, no. 00507711_m1), Adh4 (class II Adh, no. 00478838_m1), Adh5 (class III Adh, no. 00475804_g1), Aldh2 (no. 00477463_m1), and Gapdh (housekeeping gene; no. 99999915_g1). The amplification reactions were carried out in the ABI 7900HT Fast Real-Time PCR System (Applied Biosystems) as previously described (Gyamfi et al., 2008). Results were presented as levels of expression relative to that of controls after normalizing with Gapdh mRNA using the comparative CT method.
TABLE 1.
Sequences of primers used for real-time quantitative PCR
| Name | Sequence | Accession No. |
|---|---|---|
| PPARα | ||
| Sense | GATTCAGAAGAAGAACCGGAACA | NM011144 |
| Antisense | TGCTTTTTCAGATCTTGGCATTC | |
| PPARγ | ||
| Sense | CCCAATGGTTGCTGATTACAAA | NM011146 |
| Antisense | GAGGGAGTTAGAAGGTTCTTCATGA | |
| SREBP-1c | ||
| Sense | CATGCCATGGGCAAGTACAC | NM011480 |
| Antisense | TGTTGCCATGGAGATAGCATCT | |
| CPT1 | ||
| Sense | CGATCATCATGACTATGCGCTACT | NM013495 |
| Antisense | GCCGTGCTCTGCAAACATC | |
| ACOX-1 | ||
| Sense | TTTGTTGTCCCTATCCGTGAGA | NM 015729 |
| Antisense | GCCGATATCCCCAACAGTGA | |
| L-FABP | ||
| Sense | TGCATGAAGGGAAGAAAATCAAA | NM017399 |
| Antisense | CCCCCAGGGTGAACTCATT | |
| MTTP | ||
| Sense | CCGCTGTGCTTGCAGAAGA | NM008642 |
| Antisense | TTTGACACTATTTTTCCTGCTATGGT | |
| CD36 | ||
| Sense | TCCAGCCAATGCCTTTGC | NM007643 |
| Antisense | TGGAGATTACTTTTTCAGTGCAGAA | |
| ACC-1α | ||
| Sense | ATGTCCGCACTGACTGTAACCA | NM133360 |
| Antisense | TGCTCCGCACAGATTCTTCA | |
| FAS | ||
| Sense | CCCGGAGTCGCTTGAGTATATT | NM007988 |
| Antisense | GGACCGAGTAATGCCATTCAG | |
| SCD1 | ||
| Sense | CGTTCCAGA ATGACGTGTACGA | NM009127 |
| Antisense | AGGGTCGGCGTGTGTTTC | |
| PEMT | ||
| Sense | TCTGCATCCTGCTTTTGAACA | NM008819 |
| Antisense | TGGGCTGGCTCATCATAGC | |
| GAPDH | ||
| Sense | TGTGTCCGTCGTGGATCTGA | NM001001303 |
| Antisense | CCTGCTTCACCACCTTCTTGA |
ACC-1α, acetyl-CoA carboxylase 1α; ACOX-1, acyl-CoA oxidase 1; CPT1, carnitine palmitoyltransferase 1; FAS, fatty acid synthase; L-FABP, liver-type fatty acid-binding protein; SCD1, stearoyl-CoA desaturase-1.
Statistical Analysis.
Data are presented as means ± S.E.M. (n = 8–9). Statistical analysis was performed using one-way analysis of variance followed by Tukey’s HSD post-hoc test. A P value of <0.05 was considered statistically significant. Statistical analyses were performed using IBM SPSS Statistics 20 software (Armonk, NY).
Results
EtOH Induces Lipid Accumulation in Both Male and Female hPXR Mice.
Body and liver weights were significantly higher in male hPXR mice than female hPXR mice; however, binge EtOH ingestion had no effect on body weight, liver weight, or ratios of liver to body weight of either sex (Fig. 1, A–C). H&E staining revealed that hepatic lipid droplets in the control-fed mice of both genders were less abundant than their EtOH-fed counterparts (Fig. 2A). Furthermore, EtOH-induced accumulation of lipid droplets correlated with increased hepatic triglyceride levels in both male and female hPXR mice (Fig. 2, A and B). However, EtOH treatment did not significantly impact hepatic cholesterol levels in either male or female hPXR mice (Fig. 2C). Furthermore, neither serum triglycerides nor NEFA levels differed between genders or treatments (Fig. 2, D and E).
Fig. 1.
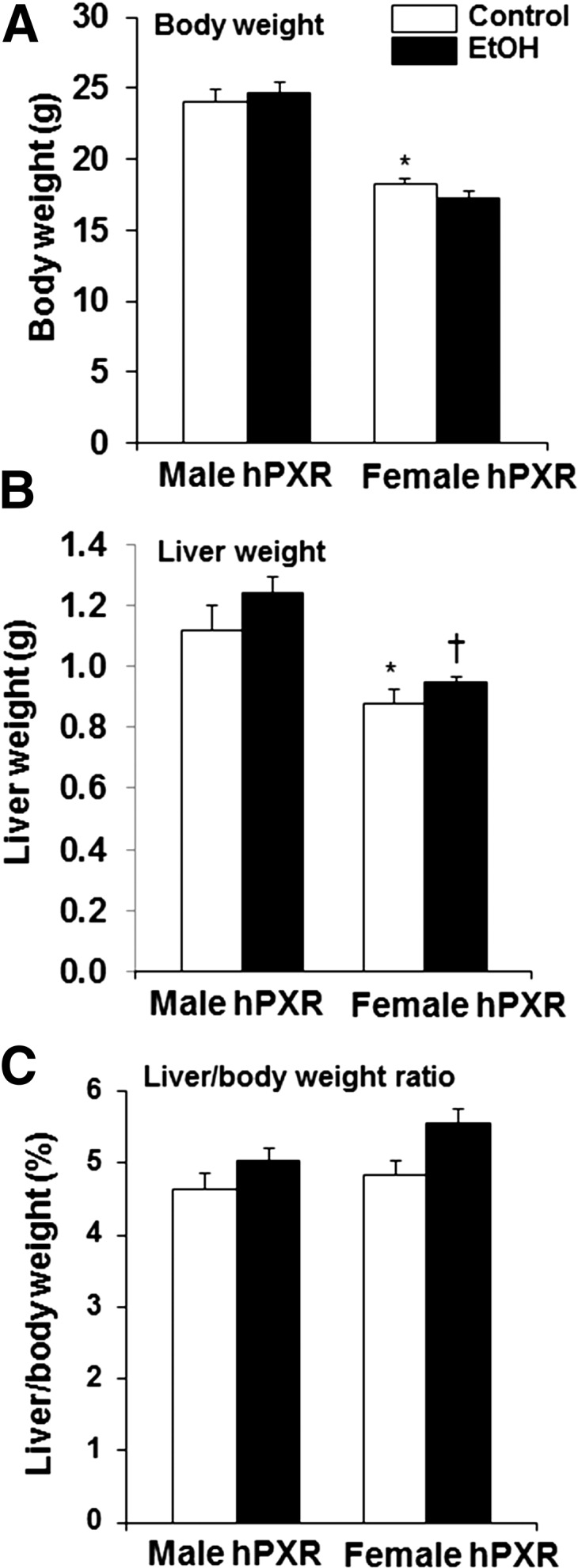
Body weight, liver weight, and liver-to-body-weight ratio in male and female hPXR mice. Male and female hPXR mice were orally administered saline (control; □) or EtOH (4.5 g/kg; ▪) every 12 hours for a total of three doses and killed 4 hours after the final dose. Body weight (A), liver weight (B), and liver-to-body-weight ratios (C) were determined. Data represent the mean ± S.E.M. (n = 8–9). *P < 0.05 between male and female mice treated with saline (control); †P < 0.05 between male and female mice treated with EtOH.
Fig. 2.

Characterization of hepatic and serum lipids in control and binge EtOH-fed male and female hPXR mice. Male and female hPXR mice were orally administered saline (control, □) or EtOH (4.5 g/kg; ▪) every 12 hours for a total of three doses and were killed 4 hours after the final dose. H&E staining (original magnification, 400×) (A), hepatic triglyceride (B), hepatic cholesterol (C), serum triglyceride (D), and serum NEFA levels (E) were assayed as described in Materials and Methods. Steatosis (indicated by arrows) in both male and female hPXR mice fed EtOH. Data represent the mean ± S.E.M. (n = 5–6). #P < 0.05 between male and female mice treated with saline (control) and EtOH.
Ethanol Ingestion Upregulates Hepatic ERα and Its Target Gene Cyclin D1 in Both Male and Female hPXR Mice.
In humans and in rodents, EtOH ingestion was found to alter circulating sex steroid levels and increase hepatic ER expression in males (Colantoni et al., 2002). Furthermore, a link between 1) gender-dependent recovery from liver injury and 2) upregulation of cell cycle genes, including the ERα target gene cyclin D1 and the cell proliferation marker PCNA, was observed (Balasenthil et al., 2004; Wang et al., 2013b). Furthermore, MAPKs have been implicated in EtOH-induced cyclin D1 expression, cell cycle inhibition, and cell survival (Stepniak et al., 2006). To investigate the effect of sex steroid levels in male and female hPXR mice on gender differences in ALD, we used Western blot analysis to determine hepatic levels of ERα, cyclin D1, PCNA, and two MAPK family members, ERK and p38, and their activated phosphorylated forms. As expected, the basal hepatic protein levels of ERα were higher in control female hPXR mice compared with their male counterparts (Fig. 3A). EtOH ingestion induced hepatic expression of ERα protein in male hPXR mice (Fig. 3A). Hepatic ERα protein levels were also enhanced 3.5-fold in female hPXR mice by EtOH ingestion (Fig. 3A). Similarly, basal hepatic protein levels of the ERα target gene cyclin D1 (Fig. 3B) and PCNA (Fig. 3C) involved in liver regeneration were significantly higher (2.5- and 2.1-fold, respectively) in female hPXR mice than in hPXR males (Fig. 3, B and C) (Balasenthil et al., 2004). EtOH significantly increased cyclin D1 protein levels in both male (2.2-fold) and female (1.3-fold) hPXR mice (Fig. 3, B and C). Cyclin D1 levels were subject to modest upregulation by binge EtOH treatment in males and females, whereas PCNA levels were not (Fig. 3, B and C). EtOH did not significantly alter hepatic ERK1/2 and p38 protein expression (Fig. 3, D and E). However, EtOH ingestion inhibited ERK1/2 activation in the livers of male and female hPXR mice by 71 and 53%, respectively, compared with their untreated controls (Fig. 3D). Furthermore, EtOH also suppressed p38 activation in both male hPXR (by 81%) and female hPXR mice by 69% (Fig. 3E).
Fig. 3.

Immunoblot analysis of hepatic ERα, cyclin D1, PCNA, and mitogen-activated protein kinases ERK1/2 and p38 MAPK in male and female hPXR mice. Male and female hPXR mice were orally administered saline (control; □) or EtOH (4.5 g/kg; ▪) every 12 hours for a total of three doses and were killed 4 hours after the final dose. Western blots of liver homogenate (40 μg/lane) probed with antibodies to ERα (A), cyclin D1 (B), PCNA (C), ERK1/2 (D), or p38 (E). Bands were quantified and normalized to α-tubulin. Data represent the mean ± S.E.M. of two independent experiments of 3–4 mice/group. *P < 0.05 between male and female mice treated with saline (control); #P < 0.05 between male and female mice treated with saline (control) and EtOH; †P < 0.05 between male and female mice fed EtOH.
Ethanol Effect on Hepatic Lipid Synthesis, Uptake, and Oxidation Genes Is More Pronounced in Female hPXR Mice than in Males.
There are several regulators of hepatic lipid synthesis, metabolism, oxidation, storage, and transport. PPARs are ligand-activated transcription factors that primarily regulate genes involved in lipid metabolism; PPAR isoforms vary in fatty livers (Memon et al., 2000). SREBP-1c, a transcription factor known to play an important role in de novo fatty acid and triglyceride synthesis, may also contribute to lipid accumulation in the liver (Horton et al., 2002). PEMT is the only enzyme in liver that converts phosphatidylethanolamine into phosphatidylcholine, a process that is required for very-low-density lipoprotein (VLDL) assembly and lipid export from hepatocytes (Vance, 2014). Furthermore, PEMT deficiency or inhibition promotes steatosis and liver damage (Vance, 2014). Because acute EtOH exposure resulted in increased lipid accumulation in both genders (Fig. 2, A and B), changes in the expression of these regulatory factors and their target genes were explored. Basal hepatic Pparα mRNA levels were comparable between control male and female hPXR mice (Fig. 4A). EtOH decreased Pparα mRNA levels in both male and female hPXR mice; however, the decrease was not statistically significant compared with their respective controls (Fig. 4A). Similarly, basal Cpt1 and Acox-1 mRNA levels did not vary between the two hPXR genders, and their mRNA levels were not different after EtOH treatment (Fig. 4A). Although constitutive L-fabp-1 mRNA levels did not vary between the two hPXR genders and EtOH did not have any significant effects on L-fabp-1 mRNA levels in male hPXR mice, EtOH induced a 1.5-fold increase in L-fabp-1 mRNA levels in female hPXR mice (Fig. 4A). Similarly, constitutive Mttp mRNA levels did not differ between male and female hPXR mice, whereas EtOH significantly induced the Mttp gene 1.7-fold in female hPXR mice only (Fig. 4A). The basal levels of Srebp-1c mRNA and SREBP-1c target genes Acc-1α, Fas, and Scd1 mRNAs were comparable between male and female hPXR mice (Fig. 4B). However, EtOH administration diminished Srebp-1c mRNA levels by 60% in male hPXR mice only (Fig. 4B). EtOH had no significant effect on Acc-1α mRNA in males, but increased Acc-1α mRNA levels 1.6-fold in female hPXR mice (Fig. 4B). Fas mRNA was the only gene to be upregulated by EtOH in male hPXR mice (1.6-fold), but this increase did not reach statistical significance and merely matched basal Fas mRNA levels in control hPXR females (Fig. 4B). In contrast, EtOH significantly increased Fas mRNA levels (1.9-fold) in female hPXR mice (Fig. 4B). Unexpectedly, EtOH ingestion significantly decreased Scd1 mRNA levels (64%) in female hPXR mice, but not in male hPXR mice (Fig. 4B). Basal hepatic Pparγ mRNA levels did not vary between male and female hPXR mice treated with saline, even though basal mRNA levels of the Pparγ downstream target Cd36 were 2.7-fold higher in female hPXR mice compared with male hPXR mice (Fig. 4C) (Tontonoz et al., 1998). Although EtOH ingestion did not significantly affect Pparγ and Cd36 mRNA levels in male hPXR mice, it induced an increase in hepatic Pparγ and Cd36 expression by 1.5- and 1.8-fold, respectively, in hPXR females (Fig. 4C). In contrast, hepatic Pemt mRNA levels did not vary significantly between male and female hPXR control mice and between control and EtOH-treated hPXR females (Fig. 4D). Pemt gene expression was suppressed by EtOH (47%) in male hPXR mice.
Fig. 4.

Gene expression of hPXR mouse hepatic enzymes involved in lipid metabolism. Male and female hPXR mice were orally administered saline (control; □) or EtOH (4.5 g/kg; ▪) every 12 hours for a total of three doses and killed 4 hours after the final dose. Total hepatic mRNA levels of Pparα, carnitine palmitoyltransferase 1 (CPT1), acyl Coenzyme A oxidase 1 (ACOX-1), liver fatty acid binding protein (L-FABP), microsomal triglyceride transfer protein (MTTP) (A), sterol regulatory element binding protein 1c (SREBP-1c), acetyl-CoA carboxylase 1 (ACC1), fatty acid synthase (FAS), stearoyl-CoA desaturase 1 (SCD1) (B), Pparγ, fatty acid translocase (FAT/CD36) (C), and phosphatidylethanolamine N-methyltransferase (PEMT) (D) were quantified by the SensiFast SYBR Hi-ROX Kit (Bioline) as described in Materials and Methods. Results were presented as levels of expression relative to that of controls after normalizing with glyceraldehyde 3-phosphate dehydrogenase (GAPDH) mRNA using the comparative CT method. Data represent the mean ± S.E.M. (n = 5–6). *P < 0.05 between male and female mice treated with saline (control); #P < 0.05 between male and female mice treated with saline (control) and EtOH; †P < 0.05 between male and female mice fed EtOH.
Ethanol-Induced Hepatotoxicity Is Greater in Male hPXR Mice.
Serum ALT and AST activities were measured as indices of hepatocyte/organ injury. EtOH increased both ALT and AST levels in male hPXR mice by 1.8- and 2.1-fold, respectively (Fig. 5, A and B), and in female mice by 1.6- and 1.5-fold, respectively (Fig. 5, A and B). However, the increases in ALT and AST activity in females were not statistically significant, suggesting that EtOH-induced hepatotoxicity is more severe in male hPXR mice (Fig. 5, A and B). LPO, an early biochemical feature of EtOH toxicity and a major indicator of oxidative stress, was quantified by measuring the thiobarbituric acid–reactive product, MDA. Levels of MDA were significantly increased by EtOH only in the livers of male hPXR (7.3-fold) mice (Fig. 5C). Moreover, EtOH-induced increases in MDA levels were significantly higher in hPXR males compared with EtOH-fed hPXR females (Fig. 5C). Expression of hepatic endoplasmic reticulum stress markers phospho-elF2α and GRP78 was upregulated 11.1- and 1.4-fold, respectively, in EtOH-treated versus control males (Fig. 5, D and E). Phospho-elF2α protein levels were elevated in the livers of control-fed female hPXR mice by 2.6-fold compared with their male counterparts (Fig. 5D). Intriguingly, EtOH exposure elevated hepatic phospho-elF2α protein levels (4.3-fold) but not GRP78 in hPXR females (Fig. 5, D and E). Hepatic protein levels of active caspase 12, the endoplasmic reticulum stress-specific caspase, was increased only in EtOH-fed hPXR males (2.5-fold), and this increase was higher than in EtOH-fed female hPXR mice (Fig. 5F).
Fig. 5.

Markers of hepatotoxicity in control and binge EtOH-treated male and female hPXR mice. Male and female hPXR mice were orally administered saline (control; □) or EtOH (4.5 g/kg; ▪) every 12 hours for a total of three doses and were killed 4 hours after the final dose. Serum ALT (A) and AST (B) levels were determined as described in Materials and Methods. The extent of lipid peroxidation in liver tissues was quantified by measuring the thiobarbituric acid–reactive product, MDA (C). Western blots of liver homogenate (40 μg/lane) were probed with antibodies to phospho-elF2α (D), GRP78 (E), and caspase 12 (F). Bands were quantified and normalized to α-tubulin. Data represent the mean ± S.E.M. of two independent experiments from 3–4 mice/group. #P < 0.05 between male and female mice treated with saline (control) and EtOH; †P < 0.05 between male and female mice fed EtOH.
Basal ADH and ALDH2 Protein, but Not Gene Expression, Is Greater in Female hPXR Mice.
Alcohol metabolism is considered a major factor in alcohol-related liver damage (Zakhari and Li, 2007; Ronis et al., 2010). The hepatotoxicity data (Fig. 5, A and B) prompted further examination of ADH, ALDH2, catalase, CYP2E1, and CYP3A enzymes known to be involved in EtOH metabolism in male and female hPXR mice (Zakhari and Li, 2007). The basal hepatic Adh1 (class 1 Adh) mRNA levels did not vary significantly between genders or treatments (Fig. 6A). However, constitutive Adh4 (class II Adh) mRNA levels tended to be lower in female hPXR mice (59%) compared with control male hPXR mice, but the difference was not statistically significant (P = 0.052) (Fig. 6B). Although EtOH did not have any significant effects on Adh4 mRNA levels in female hPXR mice, it significantly decreased the levels in male hPXR mice (54%) compared with control males (Fig. 6B). The hepatic Adh5 (class III Adh) mRNA levels did not vary significantly between male and female hPXR control mice (Fig. 6C). Although EtOH treatment did not alter hepatic Adh5 mRNA levels in female hPXR mice, it tended to inhibit Adh5 gene expression in male hPXR mice (Fig. 6C). In contrast, the basal mRNA levels of Aldh2 were somewhat (33%) but not significantly higher in female hPXR mice compared with hPXR males (Fig. 6D). EtOH treatment significantly decreased Aldh2 mRNA levels only in female hPXR mice (Fig. 6D). Unexpectedly, immunoblot analysis indicated a dramatic increase in basal hepatic ADH1 protein in female hPXR (4.3-fold) compared with male hPXR mice (Fig. 7A), even though mRNA levels did not change significantly (Fig. 6A). EtOH ingestion did not affect hepatic ADH1 protein expression in male hPXR mice; however, it inhibited ADH1 protein levels somewhat in female hPXR mice compared with control hPXR females (Fig. 7A). Furthermore, after EtOH treatment, hepatic ADH1 protein levels in female hPXR mice were significantly higher than in EtOH-fed males (Fig. 7A). Similarly, whereas the mRNA levels did not vary, similar to ADH1, the basal hepatic ALDH2 protein levels were significantly higher in female hPXR mice (3.0-fold) than in males (Fig. 7B). Also, as with ADH1, ALDH2 protein levels were not altered by EtOH treatment in male hPXR mice (Fig. 7B). In contrast, EtOH treatment inhibited ALDH2 protein levels by 37% in female hPXR mice compared with controls, consistent with the EtOH inhibition of mRNA expression of this gene (Figs. 6D and 7B). Even so, ALDH2 protein levels in EtOH-treated females remained about 1.5-fold higher than in EtOH-fed males (Fig. 7B). By contrast, basal hepatic catalase protein levels did not vary between male and female hPXR mice or between saline and EtOH treatments (Fig. 7C). CYP2E1 (Fig. 7D) and CYP3A11 (Fig. 7E) protein expression in the liver did not differ between control male and female mice. However, CYP2E1, but not CYP3A11, protein expression was upregulated (2.0-fold) by EtOH treatment in both male and female hPXR mice (Fig. 7D).
Fig. 6.

Gene expression of hPXR transgenic mouse hepatic enzymes involved in EtOH metabolism. Male and female hPXR mice were orally administered saline (control; □) or EtOH (4.5 g/kg; ▪) every 12 hours for a total of three doses and were killed 4 hours after the final dose. Total hepatic RNA was isolated and cDNA prepared using Tetro cDNA Synthesis Kit (Bioline). (A) Adh1, (B) Adh4, (C) Adh5, and (D) Aldh2, were determined using proprietary Taqman Gene Expression Assays purchased from Applied Biosystems/Life Technologies as described in Materials and Methods. Data represent the mean ± S.E.M. (n = 5–6). #P < 0.05 between male and female mice treated with saline (control) and EtOH.
Fig. 7.

Immunoblot analysis of hepatic EtOH-metabolizing enzymes in hPXR mice. Male and female hPXR mice were orally administered saline (control; □) or EtOH (4.5 g/kg; ▪) every 12 hours for a total of three doses and were killed 4 hours after the final dose. Western blots of liver homogenate (40 μg/lane) were probed with antibodies to ADH1 (A), ALDH2 (B), catalase (C), CYP2E1 (D), or CYP3A11 (E). Bands were quantified and normalized to α-tubulin. Data represent the mean ± S.E.M. of two independent experiments of 3–4 mice/group. *P < 0.05 between male and female mice treated with saline (control); #P < 0.05 between male and female mice treated with saline (control) and EtOH; †P < 0.05 between male and female mice fed EtOH.
Female hPXR Mice Eliminate Blood EtOH More Efficiently than Males.
To determine the impact on EtOH metabolism of elevated levels of ADH1 and ALDH2 protein expression observed in female hPXR mice (Fig. 7, A and B), residual concentrations of EtOH were measured after administration of three doses of binge EtOH. Elevated levels of alcohol-metabolizing enzymes in female hPXR mice resulted in lower BEC in EtOH-fed females compared with EtOH-fed hPXR males (Fig. 8A). Gender differences in EtOH oxidation in the gut, or “first-pass” metabolism by gastric ADH, may explain the sex-related differences in alcohol bioavailability and toxicity (DiPadova et al., 1987). Fasting eliminates first-pass metabolism and diminishes the contribution of gastric ADH to EtOH oxidation (DiPadova et al., 1987). Therefore, to study the impact of the high hepatic ADH1 protein levels in female hPXR mice, EtOH elimination rates were determined in fasting hPXR mice after a single dose of EtOH by gastric intubation. BECs were measured at various time points. Although BECs remained high in both male and female hPXR mice up to 4 hours after EtOH challenge, female hPXR mice were able to clear blood EtOH more efficiently than males by 6 hours, and by 8 hours BEC was virtually eliminated in females (Fig. 8B).
Fig. 8.

BEC and EtOH elimination rates in male and female mice. (A) Male and female hPXR mice were orally administered saline (control; □) or EtOH (4.5 g/kg; ▪) every 12 hours for a total of three doses and were killed 4 hours after the final dose. Serum was assayed for BEC as described in Materials and Methods. (B) Male and female hPXR mice were orally administered a single dose of 4.5 g/kg EtOH (50% v/v) by gastric intubation after overnight starvation. Mice were sacrificed 1, 2, 4, 6, and 8 hours after EtOH administration, and BEC was determined for each time point. Data represent the mean ± S.E.M. (n = 4–6). #P < 0.05 between male and female mice treated with saline (control) and EtOH; †P < 0.05 between male and female mice fed EtOH.
Discussion
Despite numerous studies investigating chronic EtOH responses in rodents, sex differences in acute EtOH hepatotoxicity have not been thoroughly investigated. In this study, binge EtOH was administered to male and female hPXR mice, and sex differences in the gene and protein expression of enzymes associated with EtOH and lipid metabolism, EtOH clearance, hepatotoxicity hepatocyte proliferation, and endoplasmic reticulum stress were examined. Surprisingly, female hPXR mice were less susceptible to acute binge EtOH-induced liver injury than their male counterparts. Once EtOH is absorbed and distributed, sex differences could occur in hepatic EtOH metabolism, leading to liver damage (Zakhari and Li, 2007; Ronis et al., 2010).
We first determined the basal gene and protein expression levels of ADH1, the major enzyme responsible for EtOH catabolism, and the ADH4 and ADH5 isoforms active at high EtOH concentrations (Zakhari and Li, 2007). In the current study, basal Adh1, Adh4, and Adh5 mRNA levels did not differ between male and female hPXR mice. EtOH significantly downregulated the expression of Adh4 mRNA in male hPXR mice only. Contrary to gene expression, basal hepatic protein expression of ADH1 was higher in female hPXR mice than males. These findings are consistent with previous reports showing that hepatic ADH regulation is post-transcriptional, and that ADH protein expression increases without a corresponding increase in Adh mRNA levels (Tussey and Felder, 1989; Mezey et al., 2005; Gyamfi et al., 2006). The current findings are also consistent with several reports that hepatic ADH is more highly expressed in female rodents and in women (Maly and Sasse, 1991; Harada et al., 1998; Aasmoe and Aarbakke, 1999; Kishimoto et al., 2002).
Variations in EtOH-metabolizing enzyme expression and activity influence not only EtOH metabolism but also EtOH clearance and EtOH-induced liver injury (Gyamfi et al., 2006, 2008; Zakhari and Li, 2007). Besides ADH1, the basal hepatic protein levels of ALDH2 were also upregulated in female hPXR mice, suggesting that this metabolic pathway converting EtOH to acetate via acetaldehyde is more efficient in female hPXR animals. Correspondingly, measurements of BEC, the EtOH concentration in the blood after metabolism of consumed EtOH in hPXR males and females treated with or without binge EtOH, were lower in females relative to males. Consistent with this, pharmacological activation of ALDH2 reversed alcoholic steatosis and apoptosis through accelerating acetaldehyde clearance, suggesting that higher ALDH2 levels in hPXR females may provide enhanced protection against alcohol hepatotoxicity (Zhong et al., 2015).
EtOH metabolism and elimination may vary depending on gender, age, body weight, stomach content, and medication use (Jones and Sternebring, 1992). Gastric ADH activity is higher in males than in females, likely resulting in differences in first-pass EtOH metabolism and BEC (Frezza et al., 1990). With evidence that shows fasting increases EtOH absorption and eliminates the contribution of gastric ADH to BEC, a fasting model was used to focus exclusively on the role of hepatic ADH and other EtOH-detoxifying enzymes in the liver (DiPadova et al., 1987). Although peak BEC levels were similar between fasting male and female hPXR mice after a single EtOH dose, the rate of EtOH elimination was enhanced in female hPXR mice compared with males. Similarly, serum EtOH levels were lower in female C3H/HeNCrj mice after acute EtOH administration, with females exhibiting enhanced ADH and ALDH activity (Kishimoto et al., 2002). The phenomenon that women metabolize EtOH more rapidly than men has also been observed consistently in human subjects (Mishra et al., 1989; Jones, 2010). The similarity in EtOH elimination between our hPXR mice and humans suggests that hPXR mice may be an appropriate model to further explore the effect of gender, EtOH concentration, and hepatic metabolic capacity on EtOH catabolism.
Estrogen and its receptors impact the gender differences we observed in EtOH metabolism in hPXR mice, either directly or indirectly. First, ADH is hormonally, nutritionally, and developmentally regulated (Boleda et al., 1992; Rao et al., 1997). The emergence of gender differences in ADH activity coincides with sexual development: a relative increase in ADH activity in females is not detected until the sixth week of age, the age at which rodents reach sexual maturity, and remains elevated thereafter (Rao et al., 1997). Second, estrogen increases hepatic ADH activity, whereas testosterone decreases it (Rachamin et al., 1980; Harada et al., 1998). Similar to ADH, estrogen also increases mitochondrial ALDH activity, consistent with the sexual dimorphisms in both ADH and ALDH expression seen in hPXR females in the current study (Kishimoto et al., 2002). Third, in humans and in Sprague-Dawley rats, EtOH ingestion leads to increased ER expression in males, not females (Colantoni et al., 2002). Curiously, we observed an increase in hepatic ERα protein levels not only in EtOH-fed male hPXR mice but also in their female counterparts, unlike the rat model in which ERα levels in females did not increase significantly upon chronic EtOH exposure (Colantoni et al., 2002). Taken together, our data suggest that a functional relationship exists between the human PXR and ERα. Further indirect evidence gleaned from the literature supports this possibility. PXR expression is enhanced in ERα-negative breast cancer cells (Dotzlaw et al., 1999), and expression of both PXR and ERα is upregulated in an in vitro reporter system by the organochlorine insecticide chlordecone (Lee et al., 2008).
Earlier studies indicate that feminization of the liver protects males, but not females, against EtOH-induced liver injury (Colantoni et al., 2002). In contrast, we found a significant increase in levels of two markers of liver damage, ALT and AST, in EtOH-fed hPXR males, whereas females showed only a modest (statistically insignificant) increase in serum ALT and AST levels. Thus, male hPXR mice appear to be more susceptible to acute EtOH-induced hepatotoxicity than hPXR females, contrary to previous findings that female mice were more prone to acute EtOH-induced steatosis (Wagnerberger et al., 2013). However, similar to our hepatotoxicity results, serum ALT levels were significantly increased only in male C57BL/6J mice, not females, after acute EtOH (6 g/kg) administration (Wagnerberger et al., 2013). CYP2E1 activity is induced after EtOH exposure and has been implicated as the source of oxidative stress, which further exacerbates liver injury (Zakhari and Li, 2007). In the current study, CYP2E1 was induced about 2-fold by EtOH in livers of both male and female hPXR mice; however, LPO, a marker of oxidative stress, was significantly increased in EtOH-fed hPXR males, but not in hPXR females. Consistent with an increase in the serum levels of liver damage markers ALT and AST in EtOH-fed male hPXR mice, we found that endoplasmic reticulum stress markers caspase 12, GRP78, and phospho-elF2α protein levels were higher in male hPXR mice than in females (Dara et al., 2011).
Compensatory tissue repair also influences the final outcome of hepatotoxicity (Chanda and Mehendale, 1996). Cyclin D1 signals hepatocyte commitment to division and liver regeneration (Apte et al., 2004). p38 MAPK regulates cyclin D1 expression (Stepniak et al., 2006). In the current study, basal levels and EtOH-induced upregulation of regenerative cyclin D1 were elevated in female hPXR mice. However, although cyclin D1 was upregulated by EtOH in male hPXR mice, the cell proliferation marker PCNA was not, even though hepatic PCNA protein levels were elevated in control and EtOH-treated hPXR females, consistent with the possibility of increased DNA repair after liver damage as reported by others (Essers et al., 2005). It is possible that the suppression of p38 and ERK activation by EtOH in both male and female hPXR mice was involved in cyclin D1 upregulation seen in the current study, as previously reported (Stepniak et al., 2006).
A surprising observation of the current study was that, although EtOH-induced steatosis and triglyceride accumulation were observed in both genders, the effect of EtOH on hepatic genes involved in lipid metabolism was strongly influenced by gender. Importantly, mRNA levels of L-fabp-1 (involved in the uptake of fatty acids and fatty acid oxidation) and Mttp (involved in the assembly and secretion of VLDL from hepatocytes) were induced by EtOH in hPXR females only (Desvergne et al., 1998; Letteron et al., 2003). Moreover, EtOH also induced genes involved in lipid synthesis and uptake, including Acc-1α, Fas, Pparγ, and Cd36, but did not affect levels of Srebp-1c in female hPXR mice. Although male hPXR mice were resistant to EtOH-induced modulation of genes involved in fatty acid synthesis, uptake, and oxidation, they were more prone to inhibition of the phosphatidylcholine biosynthesis enzyme PEMT, which may lead to decreased VLDL formation and steatosis, as seen in the hPXR males. Considered together, the current results suggest different mechanisms for EtOH-induced steatosis in male and female hPXR mice; whereas EtOH-induced inhibition of Pemt gene expression is the major mechanism for steatosis in males, upregulation of fatty acid synthesis and uptake genes may account for steatosis in females. Our data provide evidence for the contribution of the human PXR gene to sexual dimorphism of lipid metabolism in response to EtOH ingestion. However, the detailed mechanisms remain to be determined.
In conclusion, the present results demonstrate gender differences in EtOH-induced expression of lipid metabolic genes, EtOH elimination, and hepatotoxicity. Surprisingly, female hPXR mice are less susceptible to acute EtOH-induced liver injury compared with their male counterparts and are more efficient in reducing BEC, at least in part because of increased expression of 1) ADH1 and ALDH2, involved in metabolism of EtOH; 2) cyclin D1 and PCNA, important for repair of injured hepatocytes; and 3) suppression of EtOH-induced activation of LPO and endoplasmic reticulum stress. To our knowledge, this is the first report of sexual dimorphism in ADH1 and ALDH2 protein expression in hPXR mice.
Abbreviations
- ADH
alcohol dehydrogenase
- ALD
alcoholic liver disease
- ALDH
aldehyde dehydrogenase
- ALT
alanine aminotransferase
- AST
aspartate aminotransferase
- BEC
blood ethanol concentration
- ER
estrogen receptor
- ERK
extracellular regulated kinase
- EtOH
ethanol
- hPXR
PXR-humanized
- LPO
lipid peroxidation
- MAPK
mitogen-activated protein kinase
- MDA
malondialdehyde
- MTTP
microsomal triglyceride transfer protein
- NEFA
nonesterified fatty acid
- p38 MAPK
phospho-p38 mitogen-activated protein kinase
- PCNA
proliferating cell nuclear antigen
- PCR
polymerase chain reaction
- PEMT
phosphatidylethanolamine N-methyltransferase
- PPAR
peroxisome proliferator–activated receptor
- PXR
pregnane X receptor
- VLDL
very-low-density lipoprotein
Authorship Contributions
Participated in research design: M. Gyamfi, Gonzalez.
Conducted experiments: Spruiell, A. Gyamfi, M. Gyamfi.
Contributed new reagents or analytic tools: M. Gyamfi, Richardson, Gonzalez.
Performed data analysis: M. Gyamfi, Yeyeodu.
Wrote or contributed to the writing of the manuscript: M. Gyamfi, Yeyeodu, Richardson, Gonzalez.
Footnotes
This work was supported by the National Institutes of Health National Institute on Alcohol Abuse and Alcoholism [Grant AA019765]; the National Institutes of Health National Institute on Minority Health and Health Disparities [Grant MD000175]; and the National Institutes of Health National Cancer Institute [Grant CA92077]. This research was also supported in part by the Intramural Research Program of the National Institutes of Health [National Cancer Institute].
References
- Aasmoe L, Aarbakke J. (1999) Sex-dependent induction of alcohol dehydrogenase activity in rats. Biochem Pharmacol 57:1067–1072. [DOI] [PubMed] [Google Scholar]
- Apte UM, McRee R, Ramaiah SK. (2004) Hepatocyte proliferation is the possible mechanism for the transient decrease in liver injury during steatosis stage of alcoholic liver disease. Toxicol Pathol 32:567–576. [DOI] [PubMed] [Google Scholar]
- Balasenthil S, Barnes CJ, Rayala SK, Kumar R. (2004) Estrogen receptor activation at serine 305 is sufficient to upregulate cyclin D1 in breast cancer cells. FEBS Lett 567:243–247. [DOI] [PubMed] [Google Scholar]
- Boleda MD, Farrés J, Guerri C, Parés X. (1992) Alcohol dehydrogenase isoenzymes in rat development. Effect of maternal ethanol consumption. Biochem Pharmacol 43:1555–1561. [DOI] [PubMed] [Google Scholar]
- Chanda S, Mehendale HM. (1996) Hepatic cell division and tissue repair: a key to survival after liver injury. Mol Med Today 2:82–89. [DOI] [PubMed] [Google Scholar]
- Chrostek L, Jelski W, Szmitkowski M, Puchalski Z. (2003) Gender-related differences in hepatic activity of alcohol dehydrogenase isoenzymes and aldehyde dehydrogenase in humans. J Clin Lab Anal 17:93–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colantoni A, Emanuele MA, Kovacs EJ, Villa E, Van Thiel DH. (2002) Hepatic estrogen receptors and alcohol intake. Mol Cell Endocrinol 193:101–104. [DOI] [PubMed] [Google Scholar]
- Dara L, Ji C, Kaplowitz N. (2011) The contribution of endoplasmic reticulum stress to liver diseases. Hepatology 53:1752–1763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desvergne B, IJpenberg A, Devchand PR, Wahli W. (1998) The peroxisome proliferator-activated receptors at the cross-road of diet and hormonal signalling. J Steroid Biochem Mol Biol 65:65–74. [DOI] [PubMed] [Google Scholar]
- DiPadova C, Worner TM, Julkunen RJ, Lieber CS. (1987) Effects of fasting and chronic alcohol consumption on the first-pass metabolism of ethanol. Gastroenterology 92:1169–1173. [DOI] [PubMed] [Google Scholar]
- Dotzlaw H, Leygue E, Watson P, Murphy LC. (1999) The human orphan receptor PXR messenger RNA is expressed in both normal and neoplastic breast tissue. Clin Cancer Res 5:2103–2107. [PubMed] [Google Scholar]
- Eagon PK. (2010) Alcoholic liver injury: influence of gender and hormones. World J Gastroenterol 16:1377–1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Essers J, Theil AF, Baldeyron C, van Cappellen WA, Houtsmuller AB, Kanaar R, Vermeulen W. (2005) Nuclear dynamics of PCNA in DNA replication and repair. Mol Cell Biol 25:9350–9359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frezza M, di Padova C, Pozzato G, Terpin M, Baraona E, Lieber CS. (1990) High blood alcohol levels in women. The role of decreased gastric alcohol dehydrogenase activity and first-pass metabolism. N Engl J Med 322:95–99. [DOI] [PubMed] [Google Scholar]
- Gyamfi MA, He L, French SW, Damjanov I, Wan YJ. (2008) Hepatocyte retinoid X receptor alpha-dependent regulation of lipid homeostasis and inflammatory cytokine expression contributes to alcohol-induced liver injury. J Pharmacol Exp Ther 324:443–453. [DOI] [PubMed] [Google Scholar]
- Gyamfi MA, Kocsis MG, He L, Dai G, Mendy AJ, Wan YJ. (2006) The role of retinoid X receptor alpha in regulating alcohol metabolism. J Pharmacol Exp Ther 319:360–368. [DOI] [PubMed] [Google Scholar]
- Harada S, Tachiyashiki K, Imaizumi K. (1998) Effect of sex hormones on rat liver cytosolic alcohol dehydrogenase activity. J Nutr Sci Vitaminol (Tokyo) 44:625–639. [DOI] [PubMed] [Google Scholar]
- Horton JD, Goldstein JL, Brown MS. (2002) SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J Clin Invest 109:1125–1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones AW. (2010) Evidence-based survey of the elimination rates of ethanol from blood with applications in forensic casework. Forensic Sci Int 200:1–20. [DOI] [PubMed] [Google Scholar]
- Jones AW, Sternebring B. (1992) Kinetics of ethanol and methanol in alcoholics during detoxification. Alcohol Alcohol 27:641–647. [PubMed] [Google Scholar]
- Kirpich I, Ghare S, Zhang J, Gobejishvili L, Kharebava G, Barve SJ, Barker D, Moghe A, McClain CJ, Barve S. (2012) Binge alcohol-induced microvesicular liver steatosis and injury are associated with down-regulation of hepatic Hdac 1, 7, 9, 10, 11 and up-regulation of Hdac 3. Alcohol Clin Exp Res 36:1578–1586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishimoto R, Ogishi Y, Ueda M, Matsusaki M, Amako K, Goda K, Park SS. (2002) Gender-related differences in mouse hepatic ethanol metabolism. J Nutr Sci Vitaminol (Tokyo) 48:216–224. [DOI] [PubMed] [Google Scholar]
- Kliewer SA, Goodwin B, Willson TM. (2002) The nuclear pregnane X receptor: a key regulator of xenobiotic metabolism. Endocr Rev 23:687–702. [DOI] [PubMed] [Google Scholar]
- Kono H, Wheeler MD, Rusyn I, Lin M, Seabra V, Rivera CA, Bradford BU, Forman DT, Thurman RG. (2000) Gender differences in early alcohol-induced liver injury: role of CD14, NF-kappaB, and TNF-alpha. Am J Physiol Gastrointest Liver Physiol 278:G652–G661. [DOI] [PubMed] [Google Scholar]
- Lee J, Scheri RC, Zhang Y, Curtis LR. (2008) Chlordecone, a mixed pregnane X receptor (PXR) and estrogen receptor alpha (ERalpha) agonist, alters cholesterol homeostasis and lipoprotein metabolism in C57BL/6 mice. Toxicol Appl Pharmacol 233:193–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SF, Chen ZY, Fong WP. (2001) Gender difference in enzymes related with alcohol consumption in hamster, an avid consumer of alcohol. Comparative biochemistry and physiology. Toxicology & pharmacology. CBP 129:285–293. [DOI] [PubMed] [Google Scholar]
- Lehmann JM, McKee DD, Watson MA, Willson TM, Moore JT, Kliewer SA. (1998) The human orphan nuclear receptor PXR is activated by compounds that regulate CYP3A4 gene expression and cause drug interactions. J Clin Invest 102:1016–1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lettéron P, Sutton A, Mansouri A, Fromenty B, Pessayre D. (2003) Inhibition of microsomal triglyceride transfer protein: another mechanism for drug-induced steatosis in mice. Hepatology 38:133–140. [DOI] [PubMed] [Google Scholar]
- Ma X, Shah Y, Cheung C, Guo GL, Feigenbaum L, Krausz KW, Idle JR, Gonzalez FJ. (2007a) The PREgnane X receptor gene-humanized mouse: a model for investigating drug-drug interactions mediated by cytochromes P450 3A. Drug Metab Dispos 35:194–200. [DOI] [PubMed] [Google Scholar]
- Ma X, Shah YM, Guo GL, Wang T, Krausz KW, Idle JR, Gonzalez FJ. (2007b) Rifaximin is a gut-specific human pregnane X receptor activator. J Pharmacol Exp Ther 322:391–398. [DOI] [PubMed] [Google Scholar]
- Maly IP, Sasse D. (1991) Intraacinar profiles of alcohol dehydrogenase and aldehyde dehydrogenase activities in human liver. Gastroenterology 101:1716–1723. [DOI] [PubMed] [Google Scholar]
- Memon RA, Tecott LH, Nonogaki K, Beigneux A, Moser AH, Grunfeld C, Feingold KR. (2000) Up-regulation of peroxisome proliferator-activated receptors (PPAR-alpha) and PPAR-gamma messenger ribonucleic acid expression in the liver in murine obesity: troglitazone induces expression of PPAR-gamma-responsive adipose tissue-specific genes in the liver of obese diabetic mice. Endocrinology 141:4021–4031. [DOI] [PubMed] [Google Scholar]
- Mezey E, Rennie-Tankersley L, Potter JJ. (2005) Effect of leptin on liver alcohol dehydrogenase. Biochem Biophys Res Commun 337:1324–1329. [DOI] [PubMed] [Google Scholar]
- Mishra L, Sharma S, Potter JJ, Mezey E. (1989) More rapid elimination of alcohol in women as compared to their male siblings. Alcohol Clin Exp Res 13:752–754. [DOI] [PubMed] [Google Scholar]
- Nanji AA, Jokelainen K, Fotouhinia M, Rahemtulla A, Thomas P, Tipoe GL, Su GL, Dannenberg AJ. (2001) Increased severity of alcoholic liver injury in female rats: role of oxidative stress, endotoxin, and chemokines. Am J Physiol Gastrointest Liver Physiol 281:G1348–G1356. [DOI] [PubMed] [Google Scholar]
- Parés A, Caballería J, Bruguera M, Torres M, Rodés J. (1986) Histological course of alcoholic hepatitis. Influence of abstinence, sex and extent of hepatic damage. J Hepatol 2:33–42. [DOI] [PubMed] [Google Scholar]
- Perides G, Tao X, West N, Sharma A, Steer ML. (2005) A mouse model of ethanol dependent pancreatic fibrosis. Gut 54:1461–1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rachamin G, MacDonald JA, Wahid S, Clapp JJ, Khanna JM, Israel Y. (1980) Modulation of alcohol dehydrogenase and ethanol metabolism by sex hormones in the spontaneously hypertensive rat. Effect of chronic ethanol administration. Biochem J 186:483–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao UN, Aravindakshan M, Satyanarayan V, Chauhan PS. (1997) Genotype- and gender-dependent hepatic alcohol dehydrogenase (ADH) activity in developing mice. Alcohol 14:527–531. [DOI] [PubMed] [Google Scholar]
- Ronis MJ, Korourian S, Blackburn ML, Badeaux J, Badger TM. (2010) The role of ethanol metabolism in development of alcoholic steatohepatitis in the rat. Alcohol 44:157–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronis MJ, Korourian S, Yoon S, Ingelman-Sundberg M, Albano E, Lindros KO, Badger TM. (2004) Lack of sexual dimorphism in alcohol-induced liver damage (ALD) in rats treated chronically with ethanol-containing low carbohydrate diets: The role of ethanol metabolism and endotoxin. Life Sci 75:469–483. [DOI] [PubMed] [Google Scholar]
- Sato N, Lindros KO, Baraona E, Ikejima K, Mezey E, Järveläinen HA, Ramchandani VA. (2001) Sex difference in alcohol-related organ injury. Alcohol Clin Exp Res 25(5, Suppl ISBRA):40S–45S. [DOI] [PubMed] [Google Scholar]
- Spruiell K, Jones DZ, Cullen JM, Awumey EM, Gonzalez FJ, Gyamfi MA. (2014a) Role of human pregnane X receptor in high fat diet-induced obesity in pre-menopausal female mice. Biochem Pharmacol 89:399–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spruiell K, Richardson RM, Cullen JM, Awumey EM, Gonzalez FJ, Gyamfi MA. (2014b) Role of pregnane X receptor in obesity and glucose homeostasis in male mice. J Biol Chem 289:3244–3261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stepniak E, Ricci R, Eferl R, Sumara G, Sumara I, Rath M, Hui L, Wagner EF. (2006) c-Jun/AP-1 controls liver regeneration by repressing p53/p21 and p38 MAPK activity. Genes Dev 20:2306–2314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka Y, Aleksunes LM, Yeager RL, Gyamfi MA, Esterly N, Guo GL, Klaassen CD. (2008) NF-E2-related factor 2 inhibits lipid accumulation and oxidative stress in mice fed a high-fat diet. J Pharmacol Exp Ther 325:655–664. [DOI] [PubMed] [Google Scholar]
- Tontonoz P, Nagy L, Alvarez JG, Thomazy VA, Evans RM. (1998) PPARgamma promotes monocyte/macrophage differentiation and uptake of oxidized LDL. Cell 93:241–252. [DOI] [PubMed] [Google Scholar]
- Tussey L, Felder MR. (1989) Tissue-specific genetic variation in the level of mouse alcohol dehydrogenase is controlled transcriptionally in kidney and posttranscriptionally in liver. Proc Natl Acad Sci USA 86:5903–5907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vance DE. (2014) Phospholipid methylation in mammals: from biochemistry to physiological function. Biochim Biophys Acta 1838:1477–1487. [DOI] [PubMed] [Google Scholar]
- Wagnerberger S, Fiederlein L, Kanuri G, Stahl C, Millonig G, Mueller S, Bischoff SC, Bergheim I. (2013) Sex-specific differences in the development of acute alcohol-induced liver steatosis in mice. Alcohol Alcohol 48:648–656. [DOI] [PubMed] [Google Scholar]
- Wang T, Yang P, Zhan Y, Xia L, Hua Z, Zhang J. (2013a) Deletion of circadian gene Per1 alleviates acute ethanol-induced hepatotoxicity in mice. Toxicology 314:193–201. [DOI] [PubMed] [Google Scholar]
- Wang Y, Ye F, Ke Q, Wu Q, Yang R, Bu H. (2013b) Gender-dependent histone deacetylases injury may contribute to differences in liver recovery rates of male and female mice. Transplant Proc 45:463–473. [DOI] [PubMed] [Google Scholar]
- Zakhari S, Li TK. (2007) Determinants of alcohol use and abuse: Impact of quantity and frequency patterns on liver disease. Hepatology 46:2032–2039. [DOI] [PubMed] [Google Scholar]
- Zhong W, Zhang W, Li Q, Xie G, Sun Q, Sun X, Tan X, Sun X, Jia W, Zhou Z. (2015) Pharmacological activation of aldehyde dehydrogenase 2 by Alda-1 reverses alcohol-induced hepatic steatosis and cell death in mice. J Hepatol 62:1375–1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Zhai Y, Mu Y, Gong H, Uppal H, Toma D, Ren S, Evans RM, Xie W. (2006) A novel pregnane X receptor-mediated and sterol regulatory element-binding protein-independent lipogenic pathway. J Biol Chem 281:15013–15020. [DOI] [PMC free article] [PubMed] [Google Scholar]