

Abstract
Highly active antiretroviral therapy prolongs the life of HIV-infected individuals, but it requires lifelong treatment and results in cumulative toxicities and viral-escape mutants. Gene therapy offers the promise of preventing progressive HIV infection by sustained interference with viral replication in the absence of chronic chemotherapy. Gene-targeting strategies are being developed with RNA-based agents, such as ribozymes, antisense, RNA aptamers and small interfering RNA, and protein-based agents, such as the mutant HIV Rev protein M10, fusion inhibitors and zinc-finger nucleases. Recent advances in T-cell–based strategies include gene-modified HIV-resistant T cells, lentiviral gene delivery, CD8+ T cells, T bodies and engineered T-cell receptors. HIV-resistant hematopoietic stem cells have the potential to protect all cell types susceptible to HIV infection. The emergence of viral resistance can be addressed by therapies that use combinations of genetic agents and that inhibit both viral and host targets. Many of these strategies are being tested in ongoing and planned clinical trials.
Controlling HIV infection continues to be a major challenge in both underdeveloped and developed nations. Although the drug cocktails used in highly active antiretroviral therapy (HAART) have markedly changed the profile of progression to AIDS in HIV-infected individuals, they are not without significant problems and drawbacks. Pharmacokinetic differences between individuals result in many drug-related toxicities, leading to problems of nonadherence, although the increase in side effects is due in part to the improved lifespan brought by the very success of antiretroviral therapies. There is a need for personalized dosing regimens and combinations and for continued therapeutic monitoring of the drugs themselves. Drug failures for those on HAART continue to occur as a consequence of viral resistance and other complications arising from a lifelong regimen of chemotherapy. In addition, treatment guidelines traditionally have not recommended initiating therapy in the early stages of infection, despite the risks associated with loss of immunological function, increased likelihood of transmission and development of a larger pool of viral subspecies that serve as a reservoir for potential resistance. However, there has been a recent shift toward starting retroviral therapy when the CD4 count is in the range of 300 × 106 to 350 × 106/liter (Office of AIDS Research Advisory Council guidelines, http://AIDSinfo.nih.gov)1,2.
The importance of developing new antiretroviral drugs cannot be overstated. However, that HAART is lifelong and may be associated with cumulative toxicities underscores the need for new approaches. Given the increasing knowledge of mechanisms that allow control of HIV infection2, several investigators are focusing their attention on gene therapy, either as a stand-alone approach or as an adjuvant to pharmacological regimens. Several million HIV-infected individuals live in settings where there is sufficient infrastructure to support such an approach with current technology. Gene-based approaches present conundrums and trade-offs analogous to those of conventional drugs. One consideration is the issue of viral versus cellular targets. RNA antivirals can be designed with high specificity, and HIV-1 products are the preferred target (Fig. 1). However, viral escape is a major problem that will confound even gene therapy approaches. Cellular targets are far less prone to mutational escape, but the side effects of downregulating cellular targets for the long term are unknown. This article reviews some of the genetic approaches that have been used in gene therapy clinical trials for HIV-1 treatment as well as approaches that are about to be tested. We also discuss the virtues and problems associated with T-cell therapies versus hematopoietic stem (HS) cell therapies for the treatment of HIV-1 infection in the era of HAART. The review is not meant to be exhaustive but should provide an overview of the possibilities for treating HIV-1 infection using gene therapy either as a stand-alone approach or in conjunction with HAART.
Figure 1.
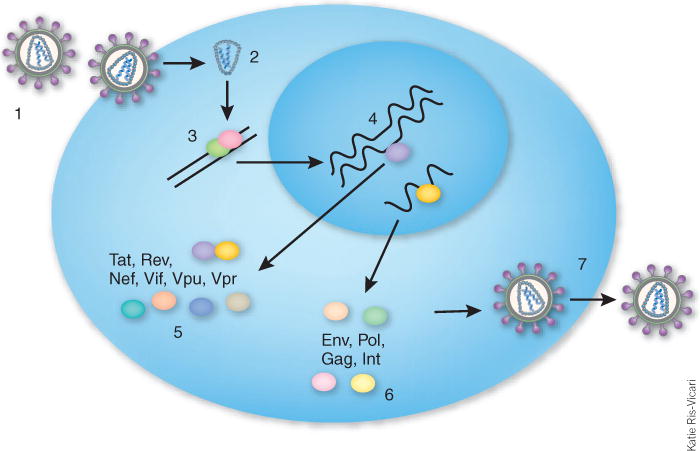
HIV life cycle. (1) HIV binds to CD4 and co-receptors CCR5 and CXCR4 and is internalized. (2) Uncoating of virus. (3) Reverse transcription. (4) Integration into host chromosomal DNA. (5) Expression of early viral proteins from multiply spliced mRNAs. (6) Expression of late mRNAs encoding the structural proteins Env, Gag, Pol and integrase. (7) Packaging of unspliced genomic RNA and release of viral particles.
Targeting HIV genes and their products
Over the past 15 years several different anti–HIV-1 gene therapy approaches have been tested in hematopoietic cells. These approaches can be classified into two categories (Fig. 2): (i) RNA-based agents (including antisense, ribozymes, aptamers and RNA interference (RNAi)); and (ii) protein-based agents (including dominant-negative proteins, intrabodies, intrakines, fusion inhibitors and zinc-finger nucleases).
Figure 2.
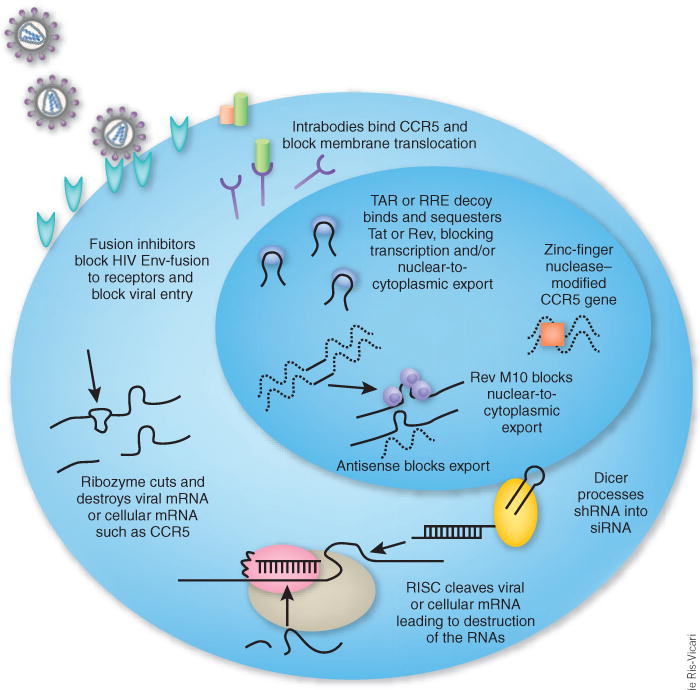
Inhibitory agents used in HIV hematopoietic cell gene therapy trials.
RNA-based inhibitory agents
Ribozymes are antisense RNAs that enzymatically cleave targeted mRNAs. Since the first demonstration that ribozymes can inhibit HIV replication3, hundreds of publications have demonstrated related ribozyme-based strategies for the treatment of HIV infection. Three separate clinical trials have used ribozymes targeting HIV genes, including tat, rev and the viral U5 region. The ribozymes were expressed either from the retroviral long terminal repeats (LTRs) as long, capped, polyadenylated transcripts from the retroviral LTR promoter4–6 or as a discrete, chimeric polymerase III (Pol III) tRNA-ribozyme transcript7. Two of the trials involved retroviral vector delivery of the ribozyme genes into autologous hematopoietic progenitor cells isolated from HIV-1–infected individuals. After retroviral transduction the cells were reinfused into the patients either without bone marrow conditioning, or in one case, with bone marrow conditioning to treat AIDS-related lymphomas4–6. The third trial used autologous peripheral blood mononuclear cells isolated from HIV-infected individuals that were transduced with a retroviral vector expressing a single hairpin-type ribozyme7. Although these trials have not shown significant anti-HIV efficacy, they demonstrated that it is safe to mobilize stem cells or to collect peripheral blood mononuclear cells from persons with HIV, genetically modify the cells with retroviral-ribozyme vectors and reinfuse them into patients. The level of marking achieved in these studies was very low, however, so they may not support the safety of current HS cell gene transfer protocols, where much higher levels of gene transfer are likely to be observed.
Short and long antisense RNA transgenes that simply pair with HIV transcripts to form nonfunctional duplexes have also proven to be effective in blocking HIV replication in hematopoietic cells. The first demonstration of this principle came from studies using adeno-associated virus to deliver a short anti–U5-region antisense RNA8. More recently, a clinical trial using an HIV LTR–expressed anti-env antisense has been reported9. Although the actual mechanism by which these antisense transcripts inhibit HIV replication is not clear, it may involve triggering extensive adenosine deamination of the HIV-antisense duplex, resulting in nuclear retention of transcripts or the generation of multiple viral-disabling mutations10.
Another group of RNA molecules, RNA aptamers, have been evolved in vitro to bind targeted ligands with high affinity11–13. Although aptamers against HIV show promise, thus far there have been no clinical trials using anti-HIV aptamers. One potential problem is that aptamers selected in vitro may not form the required tertiary structure in cells to effectively bind target proteins. On the other hand, expressed RNA decoys based on HIV TAR (trans-activating response region) and RRE (Rev responsive element) are amenable to gene therapy, and one of us (D.B.K.) has tested in the clinic an expressed RRE decoy that binds and sequesters Rev14. RNAi is a regulatory mechanism of most eukaryotic cells that uses small double-stranded RNA molecules as triggers to direct homology-dependent control of gene activity15. Known as small interfering RNAs (siRNAs), these ~21- to 22–base pair (bp) double-stranded RNA molecules have characteristic two-nucleotide 3′ overhangs that allow them to be recognized by the host RNAi enzymatic machinery, leading to homology-dependent degradation of the target mRNA (Fig. 2). RNAi triggers can be produced by expressing short hairpin (shRNA) precursors that partly resemble endogenous microRNA precursors, allowing them to be exported to the cytoplasm and processed by the RNAi machinery. Expressing short hairpin precursors encoding siRNAs targeting viral or cellular sequences can be readily accomplished from the backbone of viral vectors used in gene therapy.
HIV-1 was one of the first infectious agents targeted by RNAi as a result of the virus’ well-understood life cycle and pattern of gene expression. Virtually all the HIV-encoded RNAs—including tat, rev, gag, pol, nef, vif, env, vpr and the LTR—are susceptible to RNAi downregulation in cell lines16–20. A substantial challenge for clinical applications of RNAi triggers is the high viral mutation rate of HIV, which generates mutants that escape being targeted21–24. One approach to avoid this problem is to target cellular transcripts that encode functions required for HIV-1 entry and replication. To this end, cellular cofactors such as nuclear factor kappa B (NF-κB), the HIV receptor CD4 and the co-receptors CCR5 (C-C motif receptor 5) and CXCR4 (C-X-C motif receptor 4) have all been downregulated, thereby blocking viral replication or entry18,19,25–28.
Enthusiasm for targeting CD4 is diminished by genetic studies indicating that CD4 disruption causes substantial immunodeficiency. In contrast, the macrophage-tropic CCR5 co-receptor holds particular promise as a target. Disruption of CCR5 is compatible with immune function, in that individuals homozygous for the 32-bp deletion mutant of CCR5 receptor (delta32-CCR5) are more resistant to R5 strains of HIV than individuals who express the wild-type receptor29–31. Several major pharmaceutical companies have initiated programs to develop small molecules or antibodies to block the binding of HIV to CCR5, and one such drug, maraviroc (Selzentry), developed by Pfizer, has been recently approved. CXCR4 is essential for homing of HS cells to bone marrow and subsequent T-cell differentiation32–34, and targeting this receptor may not therefore be a viable approach. At the same time, targeting the CCR5 co-receptor alone may be insufficient, in that HIV-1 switches to CXCR4 tropism during the course of AIDS, sometimes creating a more virulent infection35. Such considerations suggest that downregulation of both viral and host targets should be considered in any RNAi strategy against HIV. Targeting of other host genes involved in the viral life cycle (for example, the LEDGF/p75 protein, which facilitates HIV-1 integration) may also prove beneficial.
As described above, siRNAs can be produced from shRNA precursors (Fig. 2) expressed from retroviral or lentiviral vector backbones by transcription from either Pol III or Pol II promoters. Because the transcription units are short in both cases, shRNAs can readily be multiplexed in various combinations. Using multiple shRNAs to target separate conserved sites in HIV—akin to the HAART approach—should prevent cross-resistance among different RNAi effectors or among RNAi effectors and conventional pharmaceuticals. Multiple RNAi effectors would thus have the advantage of limiting escape and targeting a range of sequences as is found in different viral genotypes or quasi species36,37. Viruses that escape the antiviral effects of RNAi can be reinhibited by targeting different sequences. Thus, a multiple inhibitory approach should aim to target distinct genomic regions of HIV-1 or, alternatively, target host-derived factors that contribute to viral replication.
A potential drawback of using multiple shRNAs is that expressed hairpins and the siRNAs processed from them can compete with endogenous microRNAs for nuclear-to-cytoplasmic export and incorporation into the RNA silencing machinery. The expression levels of shRNAs can be a critical determinant of whether they are toxic, so caution is necessary in using expressed shRNAs for gene therapy28,38. The toxicity of an shRNA targeting CCR5 in primary blood mononuclear cells has been shown to depend on its absolute expression level and is alleviated by damping expression28.
Rather than relying solely upon RNAi for anti-HIV therapy, a potent combinatorial approach is to mix an shRNA with other antiviral genes. For example, one of our groups (J.J.R.) has co-expressed from a single vector backbone an anti–tat/rev shRNA, a nucleolar localizing TAR decoy and an anti-CCR5 ribozyme39. This triple combination vector has recently been approved by the US Food and Drug Administration (FDA; Rockville, MD) for use in a clinical trial of autologous peripheral blood stem cell transplantation in AIDS/lymphoma patients at the City of Hope Medical Center in Duarte, California and is a combined effort between City of Hope and Benitec, Inc. The first patients are currently undergoing eligibility screening for entry into this trial. A somewhat different combination used an shRNA with a dominant-negative Rev M10 protein in a co-expression system; this may represent a future direction for Tat-regulated expression of antiviral transgenes40.
Protein-based inhibitors
Similar to the RNA-based inhibitors of HIV, proteins can be directed to inhibit either cellular or viral targets. The majority of the protein inhibitors have been expressed from the viral vector LTRs, but in several instances they were produced from strong constitutive promoters inserted within the bodies of the viral vectors. The first protein used in an HIV gene therapy trial is a mutant form of the HIV Rev protein called M10 (ref. 41). Rev M10 is believed to work by blocking the export of singly spliced and unspliced HIV RNA from the nucleus to the cytoplasm, thereby preventing packaging and subsequent transmission. This mutant protein is one of the most potent inhibitors of HIV replication. Intracellular antibodies and intrakines have also proven to be very potent inhibitors of HIV replication42–46. These proteins work by binding to viral or cellular target proteins, most often resulting in targeting of the proteins to the proteasome for degradation. Of all these approaches, thus far only the M10 dominant-negative protein has been tested in human clinical trials41.
A new entrant in the pool of protein-based agents is fusion inhibitors, which bind to HIV gp41 at the cell surface and block viral entry47,48. As with the other protein-based inhibitors, these entry-blocking proteins can be expressed constitutively from the backbone of retroviral or lentiviral vectors, making them suitable for use in gene therapy.
A different protein-based approach uses zinc-finger nuclease (ZFN) fusion proteins49. ZFN proteins can be engineered to bind with exquisite selectivity to specific sequence motifs in the genome, and the associated nuclease cleaves the targeted DNA. When these double-stranded breaks are repaired, high-frequency deletions and insertions are introduced at the site of cleavage. The CCR5 gene is a target for ex vivo gene therapy of HS cells. Disruption of the coding sequence of this gene will generate nonfunctional CCR5 mutants, rendering the cells resistant to CCR5-tropic HIV. The challenge with this approach is to transiently introduce the ZFN protein or a genetic transcription unit into primary hematopoietic cells (stable expression of the ZFN may cause genotoxicity)50. The goal is to have a single hit of mutagenesis and eliminate the nuclease from the cells after that hit. The efficiency of ZFN-mediated gene modification must be high to achieve bi-allelic CCR5 gene knockout. Despite these challenges, this is a particularly exciting approach in that the ex vivo–modified cells should have a selective growth advantage in HIV-infected individuals. Furthermore, the recent approval of CCR5 inhibitor maraviroc for advanced HIV infection increases enthusiasm for strategies that target this co-receptor. That said, the emergence of dual-tropic or CXCR4-tropic virus would abrogate this advantage. In addition, potential genotoxicities of ZFNs, from chromosomal breakage, such as translocations, or effects of ‘off-target’ DNA cleavage, must also be determined.
T-cell gene therapy
As the role of T cells in adaptive cell-mediated immune responses against viral agents becomes better understood, opportunities are increasing for their co-option for anti-HIV treatments.
Advances in T-cell biology
Advances in the understanding of T-cell biology coupled with the advances in genetic engineering described above have led to several new adoptive transfer strategies that are now poised for translation into clinical trials. Over the past decade, significant advances have been made in the manipulation and growth of T cells ex vivo. In particular, the discovery that the anergy induced by stimulation of T cells with CD3 alone could be overcome through costimulation of the CD3 and CD28 receptors permitted large-scale amplification of T cells51–53. Furthermore, CD28 costimulation induces a state of resistance ad interim to HIV infection by CCR5-tropic virus in CD4+ cells53. The feasibility of T-cell processing to produce sufficient doses of cellular product from HIV-infected individuals has been demonstrated. Early trials raised safety concerns about administration of CD4+ cells to those infected with HIV54; however, the viral load of HIV-infected individuals is not increased by adoptively transferred CD4+ T cells produced using present processing, expansion and infusion technologies55. Although the T-cell–based HIV gene therapy trials thus far have reported no or modest effects on viral load, they have established an encouraging body of data supporting safety, a selective advantage of gene-modified HIV-resistant T cells in vivo and the ability of gene-modified CD4+ T cells to persist long term. T-cell therapy may also prove to be a fertile testing and validation ground for subsequent stem cell–based clinical trials, which take longer to reach endpoints.
T-cell subsets
CD4+ T cells exist in several distinct stages of differentiation. Naive CD4+ T cells undergo unique developmental programs after antigen activation, generating effector memory T cells (TEM) and long-lived central memory T cells (TCM). The TCM cells, being the least differentiated of the antigen-stimulated T cells, retain the developmental options of naive T cells, including their capacity for marked clonal expansion and self-renewal56. In adoptive transfer experiments, TCM cells show superior therapeutic effects compared with TEM cells on a per-cell basis57. Thus, the long-term survival of subsets of T cells has increased enthusiasm for T-cell–based gene therapy trials. Genetically modified T cells persist for more than a decade in children with adenosine deaminase deficiency58.
Adoptive immunotherapy strategies
AIDS is a disorder of the immune system that is caused by collapse of immunity driven primarily by depletion of CD4+ T cells. Therefore, prevention of AIDS onset by protection of the CD4+ T-cell compartment by genetic modification is an attractive hypothesis. Possible in vivo mechanisms of action of gene-modified T cells that may lead to clinical benefit include: (i) selective outgrowth of HIV-resistant cells to a tipping point where overall HIV replication is thwarted59,60; (ii) generation of an expanding HIV-resistant T-cell population through spread of conditionally replicating HIV vectors61; (iii) protection and/or boosting of critical HIV-specific immunity by HIV-resistant helper cells. A combination of these approaches may be required for success.
Preventing viral entry
Modeling studies suggest that blocking of an early step in the HIV life cycle will be important to confer a selective advantage to vector-modified cells in vivo and hence to allow outgrowth of HIV-resistant cells in the patient60. In 2003, the new anti-retroviral drug enfuvirtide (Fuzeon; Roche), commonly known as T20, was adopted into clinical practice62. T20 blocks HIV entry by inhibiting the conformational changes needed for fusion of the viral envelope with the cellular membrane. In a genetic approach, von Laer and colleagues47 have developed a retroviral vector (M87o) that encodes the membrane-anchored antiviral peptide C46, which contains T20 sequences and is derived from the second heptad repeat of the HIV-1 envelope glycoprotein gp41. A pilot clinical trial was carried out by van Lunzen et al.63 in 10 patients with late-stage HIV/AIDS and HAART failure, who received an infusion of CD4+ T cells transduced with the retroviral M87o vector. The approach was shown to be safe, although viral loads were not affected, despite a significant rise in CD4+ T-cell counts. Gene marking was detected throughout the 1-year followup. The M87o payload has also been inserted in a lentiviral vector and was effective in preclinical studies48. Single-chain antibodies that bind gp120 were tested in CD4+ cells and found effective in preclinical studies64. Although resistance has not yet been documented in the gene therapy setting, the emergence of resistance after T20 treatment suggests that it may be important to use anti-HIV surface peptides in combination with other surface inhibitors or other modalities to interfere with the virus’ replication cycle65.
The CCR5 and CXCR4 co-receptors have been targeted in T cells using ribozymes, RNAi, intrakines, single-chain antibodies (intrabodies) and ZFNs66,67. As a variation on this, trans-dominant mutant variants of CCR5 also interfere with HIV infection of CD4+ T cells68. Lentiviral vectors expressing a single-chain antibody against CCR5 in primary CD4+ T cells disrupt CCR5 cell surface expression and provide protection from R5-tropic viral isolates69. Single-chain antibodies targeted to CXCR4 and cyclin T1 inhibit the replication of various HIV strains70,71. Further testing is required, however, to show that targeting such cellular factors in primary lymphoid cells will not result in immunodeficiency or toxicity.
Expression of rhesus tripartite motif 5α (TRIM5α) protein, which binds to the HIV capsid and interferes with the uncoating process, strongly protects human cells from productive HIV-1 infection72. The human version of TRIM5α is not efficient at blocking HIV, presumably because the capsid protein has evolved to reduce the interaction73. Changing one residue in human TRM5α confers substantial resistance to infection by HIV-1 in human cells, mimicking the rhesus phenotype74. Thus, gene therapy using this gene may not be immunogenic, because only minor modifications to human TRIM5α are sufficient to augment innate HIV-1 resistance by increasing affinity for the HIV capsid, which could result in efficient destruction of the viral particle.
Early genetic antivirals in T cells
The first proof that genetic antivirals can protect cells from HIV in vivo was in a clinical competitive repopulation experiment: CD4+ T lymphocytes were genetically modified to express either the trans-dominant-negative protein Rev-M10 or a marking vector with no antiviral payload, and a mixture of both was infused41,75. Autologous CD4+ T cells were modified with gold microparticles or by a murine leukemia virus (MLV) vector expressing the dominant-negative protein. Analysis of engraftment showed that the transduced cells containing the antiviral gene but not the control-transduced cells had a selective advantage in individuals chronically infected with HIV. Cells expressing Rev-M10 were detectable for an average of 6 months compared with 3 weeks for control cells.
More recently, Morgan et al.76 have reported long-term engraftment of T cells engineered to express an antisense TAR element or Rev M10. Robust antiviral effects were documented, particularly in patients with high viral loads. Furthermore, Macpherson et al.77 have reported persistent engraftment of T cells for longer than 4 years after treatment of syngeneic CD4+ T cells with an MLV vector expressing an anti-tat ribozyme. This study was similar to that of reference 75 in that cells were transduced either with an empty vector or a vector expressing an anti–HIV-1 payload. But in contrast to references 75 and 76, no selective advantage of the HIV-resistant CD4+ cells was observed. However, a companion study testing this vector in CD34+ cells did observe a selective survival advantage for CD4+ cells derived from CD34+ cells78.
Lentiviral vectors in the clinic
Numerous antisense targets have been tested in preclinical studies targeting both coding and noncoding regions of the HIV-1 genome, and many effectively inhibit HIV-1 replication79–81.The first clinical trial to use lentiviral vectors was recently reported9. The vector expressed a long antisense against the HIV-1 envelope gene in autologous CD4+ T cells. Vector delivery was efficient, with an average of one to two vector copies per cell, and engraftment and persistence were prolonged, with ongoing detection of gene-modified T cells for more than 1 year in two of the subjects. The magnitude of engraftment ranged from ~0.1% to 4% of CD4+ cells 90 d after infusion of genetically modified CD4+ cells. Transient vector mobilization was observed, most likely because cis-acting sequences remained intact (Box 1). Analyses of vector integration sites in blood cells revealed a preference for gene-rich regions typical of lentivirus82. Follow-up over 3 years has not detected any adverse clinical effects. Notably, there has been no evidence of insertional mutagenesis83,84. A second phase 1/2 trial is under way to evaluate the therapy using structured treatment interruption, and a follow-up phase 2 repeat-dosing exploratory trial is in progress. Many other groups have developed lentiviral vectors with various payloads that confer antiviral effects in T cells. The vectors designed by the group of J.R.R. are described above39,85. Another group designed an HIV-1 LTR-specific translational inhibitor that seems promising in preclinical studies86.
Box 1. Mobilizing vectors, defective interference and conditional replication: friend or foe?
Most viral vectors are engineered to be nonreplicating. However, in some circumstances it may be advantageous for vectors to mobilize and spread their anti-HIV sequences throughout the T-cell population and other HIV reservoirs in the body. Naturally occurring and engineered defective interfering viruses have been described that consist of mutated or deleted pathogenic viruses that replicate and compete for packaging into virions at the expense of infectious helper virus130,131. Conditionally replicating HIV vectors contain none of the trans elements necessary for viral packaging and instead carry an antiviral gene that inhibits any of numerous wild-type HIV-1 functions132. At the cellular level, conditional replication has the potential to convert viral-producing cells into latently infected cells by competing for factors that are required for HIV replication. If a cell carrying an integrated copy of a conditionally replicating HIV vector becomes infected with wild-type virus, the antiviral vector payload acts to limit the production of HIV. A model describing the potential for conditionally replicating anti-HIV vectors to overcome wild-type infection in vivo has been described59. Conditional replication that was self-limiting has occurred during a T-cell gene therapy trial9; however, the long-term safety of this approach remains to be demonstrated.
Targeting CD8+ T cells
Substantial data exist to indicate that CD8+ T cells can affect the outcome and viral load in HIV-1 infection. Naturally occurring gag-specific cytotoxic T-lymphocyte (CTL) responses are inversely associated with viremia87. Adoptive therapy with natural CTL clones for cytomegalovirus and Epstein-Barr virus infection is effective in immunosuppressed individuals88,89. In contrast, adoptive CTL therapy for HIV/AIDS, though demonstrating safety and promising engraftment and trafficking of cells to sites of viral replication, has not been clinically effective90. Mathematical modeling suggests that adoptive transfer of CTLs should augment HIV-1 immunity and control viral replication, but only when the replicative capacity of the genetically modified CTLs is preserved and functional CD4+ T cells are present59,91. Thus, an attractive strategy is the use of genetically enhanced CTLs to facilitate immune-mediated control of viral replication. Ultimately, a two-pronged approach of CD4+ T-cell protection and CTL augmentation therapy might be optimal.
T-body approaches
Studies initiated in the early 1990s examined the potential of engineering HIV-specific CTLs using the CD4 extracellular domain or a gp41-specific antibody coupled to the ζ signaling chain of the CD3 T-cell receptor (TCR)92,93, generating antibody-based chimeric proteins expressed in T cells known as ‘T bodies’. These preclinical studies showed that redirected CD8+ T cells respond by interleukin-2 secretion upon binding to HIV-1 and have robust CTL activity against HIV-1 in vitro equal to that of natural CTLs. The CD4-CD3ζ approach has since been translated to the clinic94–96. Analysis of rectal mucosal biopsy specimens and of peripheral T cells showed lymphoid tissue trafficking and stable engraftment of modified cells. In one study the CD4-CD3ζ transgene was detected in 1–3% of blood mononuclear cells at 8 weeks and at 0.1% frequency 1 year after infusion95. A randomized phase 2 study of the CD4-CD3ζ vector in 40 patients (20 treated and 20 control patients) confirmed that T-cell infusions resulted in elevated CD4+ T-cell counts and stable persistence of vector-modified cells96. This trial also showed modest antiviral effects (P < 0.07) on the viral reservoir in well-controlled patients and established the feasibility of multicenter phase 2 trials with genetically modified T cells. Together with another natural CD4+ T-cell adoptive transfer trial not discussed here55, these trials provide substantial data demonstrating the safety of multiple infusions of gene-modified autologous T cells in HIV-infected individuals.
Engineered TCRs
The failure of most patients to control HIV-1 replication is related to acquired CTL dysfunction and TCR repertoire contraction97,98. In preclinical studies, Cooper and colleagues99 isolated a human leukocyte antigen (HLA)-A3–restricted p17 gag-specific TCR from a donor infected with HIV-1. Using a retroviral vector they expressed this TCR in CD8+ T-cell clones and showed that the clones killed HIV-infected cells. Advances in vector design with TCRs now permit clinical testing of approaches to convert polyclonal T cells into redirected potent CTLs, a strategy that has shown promise in cancer patients99. In that trial, retroviral gene transfer of redirected TCRs for MART-1 in CD8+ CTLs was found to be safe in melanoma patients.
Improving the affinity of natural TCRs could be beneficial in HIV. This concept is supported by a recent study in which a T-cell line engineered to contain a high-affinity TCR (Kd = 10 nM) responded to significantly lower peptide concentrations than cells expressing the parental TCR100. Several approaches are under investigation, such as improving the intrinsic avidity of the TCR or improving functional avidity by enhancing signal transduction downstream of the TCR101. In principle, T cells engineered to express high-avidity or high-affinity TCRs could be produced in large numbers and used to kill infected T cells at an earlier point in the viral life cycle, when fewer peptide–major histocompatibility complexes (MHC) are available to be targeted at the cell surface. In addition, such TCRs could limit the generation of escape mutants. A general limitation of this approach for humans is that each TCR is specific for a given peptide–MHC complex, such that each vector would be useful only for individuals with shared MHC alleles and HIV-1 infections that retain and express the targeted epitope. Another technical issue with the redirected TCR approach is the potential for off-target effects due to mispairing of modified TCR chains with endogenous TCR chains. Several approaches to direct the pairing and induce ‘allelic exclusion’ of natural TCR genes have been described, including introduction of an artificial disulfide bond, which is reported to increase surface expression and pairing efficiency102.
Opportunities and future directions of T-cell gene therapy
In contrast to the challenges of evaluating the efficacy of stem-cell gene therapy, an attractive feature of T-cell approaches is that it is straightforward to determine therapeutic effects. Brief analytical treatment interruptions, if carefully performed, are safe and can provide definitive information on the antiviral efficacy of the vector by measuring changes in viral load or CD4+ cell counts over time after the interruption. Because the correlates of immune protection are largely unknown, and although anti–HIV-1 effects can be assessed in vitro, the best way to test the functionality of the antiviral response is to discontinue therapy and investigate the ability of the engineered host responses to control viral replication and protect CD4+ cells. Long-term structured treatment interruptions may increase the risk of HIV progression and are discouraged.
The availability of preclinical models in which to optimize vectors is essential. Studies in nonhuman primates are costly, and host restriction factors may preclude testing of lentiviral vectors in these models. Rather than re-engineering vectors into viruses that are permissive to nonhuman primates, it is preferable to test candidate vectors in human cells. Improved humanized mouse models of HIV-1 infection103,104 may be useful for preclinical vector testing.
In T-cell gene therapy, preservation of the replicative life span of memory T cells is probably vital for long-term antiviral effects and immune protection. Given that HIV-1 infection induces changes consistent with accelerated aging of the immune system105, regenerative medicine approaches might be used to restore lymphocyte function in individuals with advanced HIV/AIDS. Genetic engineering of T cells to restore CD28 expression, enhance cytokines that promote T-cell survival and restore eroded telomeres may rejuvenate T cells (reviewed by C.H.J. in ref.106). Finally, knowledge of gene expression patterns associated with the acquisition of T-cell memory56,107 might be used to reprogram HIV-1–specific T cells to have TCM qualities, such as long life spans.
HS cell therapies
HS cells represent an attractive cell target for gene therapy for HIV-1. Because HS cells produce all the cells involved in HIV-1 pathogenesis (CD4+ T cells, macrophages, dendritic cells and microglia), genetic modification of these cells could protect the entire spectrum of susceptible cells. HS cells may function for years and could therefore serve as an enduring source of HIV-1–resistant cells, including cells generated by de novo lymphopoiesis to replenish central and mucosal lymphoid organs.
HS cells present in bone marrow, peripheral blood (after mobilization from the marrow by administration of granulocyte colony-stimulating factor for 3–5 d) or umbilical cord blood can be isolated and enriched based upon their expression of the CD34+ protein. CD34+ cells are typically cultured ex vivo for 2–4 d in a mixture of recombinant cytokines (for example, c-kit ligand and flt-3 ligand) to stimulate the cells to proliferate while they are exposed to gene transfer vectors. No reliable methods for expanding HS cells in vitro have yet been identified; instead, the culture of HS cells leads to progressive loss of stem-cell capacity. Thus, genetic modification methods that require minimal ex vivo manipulation are most likely to preserve HS cells that can engraft and differentiate into T cells and other blood cells after reinfusion.
Because HS cells proliferate extensively once they begin to contribute to blood cell production, any introduced genetic modification must be permanent so that it will be passed on to the progeny cells. Most efforts to add anti–HIV-1 genes have used gamma-retroviral vectors derived from the Moloney MLV, which covalently integrates the gene into the cellular chromosome108. More recently, lentiviral vectors derived from HIV-1 are being investigated for HS cell gene therapy, with several clinical trials under development109,110. Lentiviral vectors have the potential to transduce a greater percentage of HS cells with a shorter ex vivo culture duration than gamma-retroviral vectors108. Although there is a theoretical potential for recombination between the minimal HIV-1–derived sequences present in the lentiviral vector backbone and a person’s wild-type HIV-1, it is difficult to imagine a recombinant that would be more pathogenic than the wild-type virus. Some anti–HIV-1 genes may interfere with the production of HIV-1–based lentiviral vectors, which may limit their use in the clinic if sufficient titers cannot be achieved.
In several preclinical studies, human (or rhesus) CD34+ cells were modified with anti-HIV genes, differentiated to produce monocytic cells or T lymphocytes in vitro and then challenged with HIV-1 infection to assess the conferred inhibition of HIV-1 replication6,111–114. Although viral replication was effectively impaired, the major limitation of these in vitro studies is that they used relatively mature progenitor cells rather than true HS cells, which lead to long-term lymphopoiesis after transplant. A few studies in immune-deficient mouse xenograft models have shown that mature T cells produced from transduced human CD34+ cells were relatively resistant to HIV-1 infection115–117. Surprisingly, there have been no reported studies in nonhuman primate models transplanted with autologous CD34+ cells transduced with anti-HIV genes and challenged in vivo with simian immunodeficiency virus, to test this approach in the most relevant preclinical models. The Chen group118 has recently shown that HS cells transduced with a lentivirus to express an siRNA against CCR5 have stable long-term reduction of CCR5 in nonhuman primates, with resistance to simian immunodeficiency virus infection after ex vivo growth118.
The results of clinical trials for HIV-1 using HS cells have been modest14,78,119,120. The numbers of peripheral blood cells containing the introduced gene have been low or undetectable in most subjects after the first few months, indicating low engraftment of gene-modified HS cells. One child had a late reappearance of CD4+ T cells containing an anti–HIV-1 gene during a period of noncompliance with HAART and an HIV-1 resurgence, suggesting a relative selective survival advantage of gene-protected cells121. On the basis of these findings of low numbers of gene-marked cells, current efforts are directed at increasing the gene transduction of HS cells using lentiviral vectors instead of gamma-retroviral vectors.
In recent clinical trials of gene therapy for genetic diseases of blood cells, the administration of chemotherapy agents intended to ablate some of the endogenous bone marrow (for example, busulfan or melphalan) before reinfusing the ex vivo gene-modified HS cells has significantly increased the fraction of gene-modified cells in circulation122,123. Except for X-linked severe combined immune deficiency, where the selective advantage of genetically normal lymphoid progenitors is very strong124, it will probably be necessary to use cytoreductive conditioning for HS cell gene therapy to produce a significant percentage of gene-modified cells. These agents add to the risks of the procedure, both from potential short-term toxicity and from potential adverse effects on residual immunity. However, the experience in the setting of genetic disease has shown that dosages of these agents that are well tolerated clinically do considerably increase the amount of engrafting of gene-modified HS cells.
Safety concerns were raised by the major adverse effect of insertional oncogenesis that occurred in infants with X-linked severe combined immune deficiency undergoing gene therapy125. Particularly high-risk features in those cases may have included the specific transgene, γC, encoding a cytokine receptor protein, which may provide a subtle proliferative signal to cells, the relatively high number of bone marrow CD34+ cells that were present in the infants’ marrow and the massive lymphoid expansion that occurred upon engraftment of the corrected cells, possibly aided by the highly supportive thymic microenvironment present during infancy. In contrast, in HIV-1 gene therapy the genes themselves are not expected to confer any autonomous proliferative capacity on cells, the content of bone marrow stem cells may be lower in older individuals with HIV-1 infection, and prolonged antiretroviral therapy and the thymic function may be greatly diminished. Thus, the same factors that currently limit the efficacy of gene therapy for HIV-1 using HS cells may also limit the risks.
Genetically modified HS cells may show complex patterns of proliferation, with some clones proliferating early but then becoming exhausted, whereas others may be quiescent for some months and then proliferate to produce blood cells126. De novo production of peripheral blood T cells after bone marrow transplant with CD34+ cells in persons with X-linked severe combined immune deficiency who lack endogenous lymphocytes takes at least 3 months, with migration and differentiation of marrow-derived lymphoid progenitors in the thymus being a limiting step. CD34+ populations include a subset of common lymphoid progenitors (CLPs) that are restricted in their differentiation capacity to production of only lymphoid, and not myeloid, cells127. CLPs may be responsible for the initial wave of lymphopoiesis after transplant of CD34+ cells, and transduced CLPs may give rise to the first wave of protected T cells after a few months. Although expansion of HS cells has not been convincingly achieved, expansion of human CLPs has been successful, and increased numbers of CLPs may shorten the lag of de novo lymphopoiesis. Gene-modified CLPs expanded in vitro may play a role in the rapid production of gene-protected T cells.
Conclusions
Many options are available for using gene therapy in the treatment of HIV infection, whether the transgene encodes an RNA-based or protein-based agent. The major issues facing the field are targeting specificity of the anti-HIV gene (maximal activity against HIV-1 and minimal cellular toxicity), averting viral resistance and potential antigenicity of the antiviral agents. Considering the continuing health and financial costs of the HIV-1 epidemic, it is prudent to continue to explore a variety of therapeutic strategies. Gene therapy has the potential to complement conventional antiretroviral therapies and to augment the effects of currently available vaccine technologies that fall short of desired efficacy.
Combining gene-modified CD4+ T cells and CD34+ HS cells may yield additive effects (Fig. 3). The transfused T cells would be present immediately but decline over time, whereas the CD34+ cells would produce T cells over subsequent months. If these T cells contributed to immunity or otherwise diminished HIV-1 replication, their more prolonged presence may be beneficial.
Figure 3.
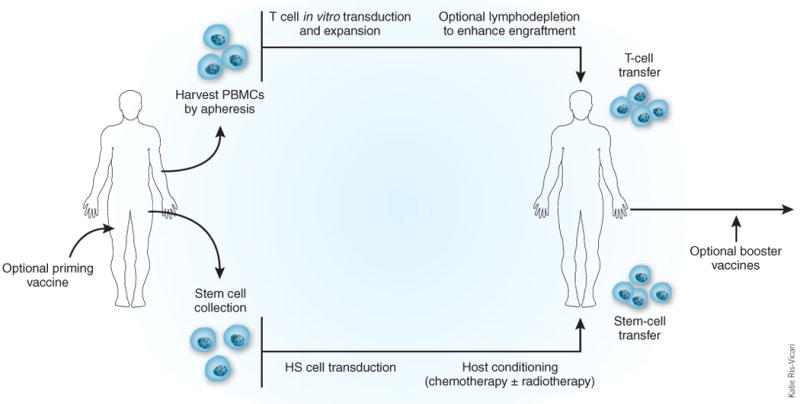
Adoptive immunotherapy strategies with gene-modified T cells and HS cells. Gene transfer approaches have tested engineered T cells and HS cells. Lymphodepletion enhances engraftment of both cell types. Other strategies under consideration include the use of common lymphoid progenitor cells (CLPs). PBMC, peripheral blood mononuclear cell.
Several clinical trials testing gene transfer strategies in T cells and HS cells have been reported or are in development (Table 1). Although progress and benefits from genetic therapies for HIV-1 have come more slowly than hoped, the same may be said for results of the far larger effort at developing an HIV-1 vaccine. Thus far, no gene transfer trials that test combination approaches have been carried out. In light of the success achieved with combined drugs in HAART, it makes the most sense to devise gene therapy schemes in which combinations of antiviral genes are co-expressed in target hematopoietic cells. Targeting combinations of CCR5 and viral genes also has a theoretical advantage over simply targeting cellular or viral genes. Given the repertoire of antiviral genes now available, this should be the goal of future gene therapy trials.
Table 1.
Completed and ongoing gene therapy trials for HIV
| Protocol description | Phase | Status | Payload | Cellular vehicle | Transfer vector | References |
|---|---|---|---|---|---|---|
| T cells | ||||||
| A randomized study of HIV-specific T-cell gene therapy in subjects with undetectable plasma viremia on combination antiretroviral therapy | 2 | Completed | CD4 receptor coupled with the CD3 signaling chain ζ | Autologous CD4+ and CD8+ T cells; (3) repeat doses | Murine retrovirus | 95 |
| Evaluation of safety, tolerability and persistence of escalating and repeat doses of genetically modified syngeneic CD8+ or CD4+/CD8+ cells | 1–2 | Completed | CD4 receptor coupled with the CD3 signaling chain ζ | Syngeneic CD8+ or CD4+/CD8+ cells; single or multiple doses | Murine retrovirus | 93 |
| Evaluation of safety, tolerability and tissue trafficking of a single dose of genetically modified autologous CD4+ and CD8+ cells. | 1–2 | Completed | CD4 receptor coupled with the CD3 signaling chain ζ | Autologous CD4+ and CD8+ cells; single dose | Murine retrovirus | 94 |
| Evaluation of safety and tolerability of a single infusion of autologous CD4+ T cells modified with a dominant-negative anti-HIV gene | 1–2 | Completed | Rev M10 | Autologous CD4+ cells; single dose | Gold particles / murine retrovirus | 41,74 |
| A marker study of therapeutically transduced CD4+ peripheral blood lymphocytes in HIV-discordant identical twins | 1 | Completed | Anti–HIV-1 tat ribozyme (Rz2) | Syngeneic CD4+ cells (twin study); single dose | Murine retrovirus | 76 |
| Evaluation of safety and tolerability of multiple infusions of syngeneic CD4+ lymphocytes modified with anti-HIV genes | 1 | Completed | Trans-dominant rev and/or trans-dominant rev with TAR antisense | Syngeneic CD4+ cells (twin study); two doses | Murine retrovirus | 75 |
| Evaluation of safety and tolerability of ribozyme gene therapy of HIV-1 infection | 1 | Completed | Anti–HIV-1 Rz to the U5 leader sequence | Autologous CD4+ cells; single dose | Murine retrovirus | 128 |
| Evaluation of safety and tolerability of a single dose of autologous T cells transduced with VRX496 in HIV-positive patient subjects | 1–2 | Completed | Anti–HIV-1 antisense against the envelope gene | Autologous CD4+ T cells; single dose | HIV-derived lentivirus, conditionally replicating | 9 |
| Evaluation of safety, tolerability and antiviral effects of autologous CD4+ T cells expressing the HIV fusion inhibitor M87 | 1 | Completed | gp41 fusion peptide inhibitor | Autologous CD4+ T cells | Murine retrovirus | 62 |
| An open-label, multicenter study to evaluate the safety, tolerability and biological activity of repeated doses of autologous T cells transduced with VRX496 in HIV-positive subjects | 2 | Ongoing | Anti HIV-1 antisense against the envelope gene | Autologous CD4+ T cells; (4 or 8) repeat doses | HIV-derived lentivirus, conditionally replicating | |
| An open-label, single-center study to evaluate the tolerability, trafficking and therapeutic effects of repeated doses of autologous T cells transduced with VRX496 in HIV-infected subjects | 1–2 | Ongoing | Anti HIV-1 antisense against the envelope gene | Autologous CD4+ T cells; (6) repeat doses | HIV-derived lentivirus, conditionally replicating | |
| HS cells | ||||||
| Nonmyeloablative conditioning followed by transplantation of genetically modified HLA-matched peripheral blood progenitor cells for hematological malignancies in persons with acquired immunodeficiency syndrome | 1 | Completed | Trans-dominant Rev | Autologous CD34+ cells isolated from mobilized peripheral blood | Murine retrovirus | 129 |
| Evaluation of retroviral-mediated transfer of a rev-responsive element decoy gene into CD34+ cells from the bone marrow of HIV-1–infected children | 1 | Completed | RRE decoy | Autologous CD34+ bone marrow cells | Murine retrovirus | 14 |
| Evaluation of safety, tolerability and persistence of transplantation with autologous bone marrow transduced with a retroviral vector expressing dominant-negative Rev or a control gene | 1 | Completed | Trans-dominant Rev | Autologous CD34+ bone marrow cells | Murine retrovirus | 120 |
| Evaluation of safety and tolerability of autologous CD34+ hematopoietic progenitor cells transduced with an anti-HIV ribozyme | 1 | Completed | Anti–HIV-1 tat ribozyme (Rz2) | Autologous CD34+ cells isolated from mobilized peripheral blood | Murine retrovirus | 77,119 |
| A randomized, double-blind, controlled trial to evaluate the safety and efficacy of autologous CD34+ hematopoietic progenitor cells transduced with placebo or an anti–HIV-1 ribozyme (OZ1) in patients with HIV-1 infection | 2 | Ongoing | Anti-HIV ribozyme OZ1 | Autologous CD34+ cells isolated from mobilized peripheral blood | Murine retrovirus | http://clinicaltrials.gov/show/NCT00074997 |
| A pilot study of safety and feasibility of stem-cell therapy for AIDS lymphoma using stem cells treated with a lentivirus vector encoding multiple anti-HIV RNAs | 1 | Ongoing | Triple combination vector co-expressing an anti tat/rev shRNA, a nucleolar localizing TAR decoy and an anti-CCR5 ribozyme in a single vector backbone | Autologous CD34+ HS cells | HIV-derived lentivirus | http://clinicaltrials.coh.org/specific_result.aspx?dise=0&category=1023&age=&protgroup=&phase=&gender=&keyword1=&frmexact |
Footnotes
COMPETING INTERESTS STATEMENT
The authors declare competing financial interests: details accompany the full-text HTML version of the paper at http://www.nature.com/naturebiotechnology/.
References
- 1.Deeks SG. Antiretroviral treatment of HIV infected adults. Br Med J. 2006;332:1489. doi: 10.1136/bmj.332.7556.1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Phillips AN, Gazzard BG, Clumeck N, Losso MH, Lundgren JD. When should antiretroviral therapy for HIV be started? Br Med J. 2007;334:76–78. doi: 10.1136/bmj.39064.406389.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sarver N, et al. Ribozymes as potential anti-HIV-1 therapeutic agents. Science. 1990;247:1222–1225. doi: 10.1126/science.2107573. [DOI] [PubMed] [Google Scholar]
- 4.Michienzi A, et al. RNA-mediated inhibition of HIV in a gene therapy setting. Ann NY Acad Sci. 2003;1002:63–71. doi: 10.1196/annals.1281.008. [DOI] [PubMed] [Google Scholar]
- 5.Ngok FK, et al. Clinical gene therapy research utilizing ribozymes: application to the treatment of HIV/AIDS. Methods Mol Biol. 2004;252:581–598. doi: 10.1385/1-59259-746-7:581. [DOI] [PubMed] [Google Scholar]
- 6.Bauer G, et al. Inhibition of human immunodeficiency virus-1 (HIV-1) replication after transduction of granulocyte colony-stimulating factor-mobilized CD34+ cells from HIV-1–infected donors using retroviral vectors containing anti–HIV-1 genes. Blood. 1997;89:2259–2267. [PubMed] [Google Scholar]
- 7.Leavitt MC, et al. Transfer of an anti–HIV-1 ribozyme gene into primary human lymphocytes. Hum Gene Ther. 1994;5:1115–1120. doi: 10.1089/hum.1994.5.9-1115. [DOI] [PubMed] [Google Scholar]
- 8.Chatterjee S, Johnson PR, Wong KK., Jr Dual-target inhibition of HIV-1 in vitro by means of an adeno-associated virus antisense vector. Science. 1992;258:1485–1488. doi: 10.1126/science.1359646. [DOI] [PubMed] [Google Scholar]
- 9.Levine BL, et al. Gene transfer in humans using a conditionally replicating lentiviral vector. Proc Natl Acad Sci USA. 2006;103:17372–17377. doi: 10.1073/pnas.0608138103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lu X, et al. Antisense-mediated inhibition of human immunodeficiency virus (HIV) replication by use of an HIV type 1–based vector results in severely attenuated mutants incapable of developing resistance. J Virol. 2004;78:7079–7088. doi: 10.1128/JVI.78.13.7079-7088.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Held DM, Kissel JD, Patterson JT, Nickens DG, Burke DH. HIV-1 inactivation by nucleic acid aptamers. Front Biosci. 2006;11:89–112. doi: 10.2741/1782. [DOI] [PubMed] [Google Scholar]
- 12.Joshi PJ, Fisher TS, Prasad VR. Anti-HIV inhibitors based on nucleic acids: emergence of aptamers as potent antivirals. Curr Drug Targets Infect Disord. 2003;3:383–400. doi: 10.2174/1568005033481060. [DOI] [PubMed] [Google Scholar]
- 13.Symensma TL, Giver L, Zapp M, Takle GB, Ellington AD. RNA aptamers selected to bind human immunodeficiency virus type 1 Rev in vitro are Rev responsive in vivo. J Virol. 1996;70:179–187. doi: 10.1128/jvi.70.1.179-187.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kohn DB, et al. A clinical trial of retroviral-mediated transfer of a rev-responsive element decoy gene into CD34+ cells from the bone marrow of human immunodeficiency virus-1–infected children. Blood. 1999;94:368–371. [PubMed] [Google Scholar]
- 15.Hannon GJ, Rossi JJ. Unlocking the potential of the human genome with RNA interference. Nature. 2004;431:371–378. doi: 10.1038/nature02870. [DOI] [PubMed] [Google Scholar]
- 16.Jacque JM, Triques K, Stevenson M. Modulation of HIV-1 replication by RNA interference. Nature. 2002;418:435–438. doi: 10.1038/nature00896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee NS, et al. Expression of small interfering RNAs targeted against HIV-1 rev transcripts in human cells. Nat Biotechnol. 2002;20:500–505. doi: 10.1038/nbt0502-500. [DOI] [PubMed] [Google Scholar]
- 18.Martinez MA, Clotet B, Este JA. RNA interference of HIV replication. Trends Immunol. 2002;23:559–561. doi: 10.1016/s1471-4906(02)02328-1. [DOI] [PubMed] [Google Scholar]
- 19.Novina CD, et al. siRNA-directed inhibition of HIV-1 infection. Nat Med. 2002;8:681–686. doi: 10.1038/nm725. [DOI] [PubMed] [Google Scholar]
- 20.Coburn GA, Cullen BR. Potent and specific inhibition of human immunodeficiency virus type 1 replication by RNA interference. J Virol. 2002;76:9225–9231. doi: 10.1128/JVI.76.18.9225-9231.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Boden D, Pusch O, Lee F, Tucker L, Ramratnam B. Human immunodeficiency virus type 1 escape from RNA interference. J Virol. 2003;77:11531–11535. doi: 10.1128/JVI.77.21.11531-11535.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Westerhout EM, Ooms M, Vink M, Das AT, Berkhout B. HIV-1 can escape from RNA interference by evolving an alternative structure in its RNA genome. Nucleic Acids Res. 2005;33:796–804. doi: 10.1093/nar/gki220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Das AT, et al. Human immunodeficiency virus type 1 escapes from RNA interference–mediated inhibition. J Virol. 2004;78:2601–2605. doi: 10.1128/JVI.78.5.2601-2605.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sabariegos R, Gimenez-Barcons M, Tapia N, Clotet B, Martinez MA. Sequence homology required by human immunodeficiency virus type 1 to escape from short interfering RNAs. J Virol. 2006;80:571–577. doi: 10.1128/JVI.80.2.571-577.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Anderson J, Akkina R. CXCR4 and CCR5 shRNA transgenic CD34+ cell derived macrophages are functionally normal and resist HIV-1 infection. Retrovirology. 2005;2:53. doi: 10.1186/1742-4690-2-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cordelier P, Morse B, Strayer DS. Targeting CCR5 with siRNAs: using recombinant SV40-derived vectors to protect macrophages and microglia from R5-tropic HIV. Oligonucleotides. 2003;13:281–294. doi: 10.1089/154545703322616961. [DOI] [PubMed] [Google Scholar]
- 27.Surabhi RM, Gaynor RB. RNA interference directed against viral and cellular targets inhibits human immunodeficiency virus type 1 replication. J Virol. 2002;76:12963–12973. doi: 10.1128/JVI.76.24.12963-12973.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.An DS, et al. Optimization and functional effects of stable short hairpin RNA expression in primary human lymphocytes via lentiviral vectors. Mol Ther. 2006;14:494–504. doi: 10.1016/j.ymthe.2006.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Garred P, et al. Dual effect of CCR5 delta 32 gene deletion in HIV-1–infected patients. Copenhagen AIDS Study Group. Lancet. 1997;349:1884. doi: 10.1016/s0140-6736(05)63874-3. [DOI] [PubMed] [Google Scholar]
- 30.Samson M, et al. Resistance to HIV-1 infection in Caucasian individuals bearing mutant alleles of the CCR-5 chemokine receptor gene. Nature. 1996;382:722–725. doi: 10.1038/382722a0. [DOI] [PubMed] [Google Scholar]
- 31.Huang Y, et al. The role of a mutant CCR5 allele in HIV-1 transmission and disease progression. Nat Med. 1996;2:1240–1243. doi: 10.1038/nm1196-1240. [DOI] [PubMed] [Google Scholar]
- 32.Lapidot T. Mechanism of human stem cell migration and repopulation of NOD/SCID and B2m null NOD/SCID mice. The role of SDF-1/CXCR4 interactions. Ann NY Acad Sci. 2001;938:83–95. doi: 10.1111/j.1749-6632.2001.tb03577.x. [DOI] [PubMed] [Google Scholar]
- 33.Lapidot T, Kollet O. The essential roles of the chemokine SDF-1 and its receptor CXCR4 in human stem cell homing and repopulation of transplanted immune-deficient NOD/SCID and NOD/SCID/B2m(null) mice. Leukemia. 2002;16:1992–2003. doi: 10.1038/sj.leu.2402684. [DOI] [PubMed] [Google Scholar]
- 34.Kahn J, et al. Overexpression of CXCR4 on human CD34+ progenitors increases their proliferation, migration, and NOD/SCID repopulation. Blood. 2004;103:2942–2949. doi: 10.1182/blood-2003-07-2607. [DOI] [PubMed] [Google Scholar]
- 35.Ariën KK, et al. Replicative fitness of CCR5-using and CXCR4-using human immunodeficiency virus type 1 biological clones. Virology. 2006;347:65–74. doi: 10.1016/j.virol.2005.11.045. [DOI] [PubMed] [Google Scholar]
- 36.Chang LJ, Liu X, He J. Lentiviral siRNAs targeting multiple highly conserved RNA sequences of human immunodeficiency virus type 1. Gene Ther. 2005;12:1133–1144. doi: 10.1038/sj.gt.3302509. [DOI] [PubMed] [Google Scholar]
- 37.Berkhout B, Haasnoot J. The interplay between virus infection and the cellular RNA interference machinery. FEBS Lett. 2006;580:896–902. doi: 10.1016/j.febslet.2006.02.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Grimm D, et al. Fatality in mice due to oversaturation of cellular microRNA/short hairpin RNA pathways. Nature. 2006;441:537–541. doi: 10.1038/nature04791. [DOI] [PubMed] [Google Scholar]
- 39.Li MJ, et al. Long-term inhibition of HIV-1 infection in primary hematopoietic cells by lentiviral vector delivery of a triple combination of anti-HIV shRNA, anti-CCR5 ribozyme, and a nucleolar-localizing TAR decoy. Mol Ther. 2005;12:900–909. doi: 10.1016/j.ymthe.2005.07.524. [DOI] [PubMed] [Google Scholar]
- 40.Unwalla HJ, et al. Novel Pol II fusion promoter directs human immunodeficiency virus type 1–inducible coexpression of a short hairpin RNA and protein. J Virol. 2006;80:1863–1873. doi: 10.1128/JVI.80.4.1863-1873.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Woffendin C, Ranga U, Yang Z, Xu L, Nabel GJ. Expression of a protective gene-prolongs survival of T cells in human immunodeficiency virus–infected patients. Proc Natl Acad Sci USA. 1996;93:2889–2894. doi: 10.1073/pnas.93.7.2889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lobato MN, Rabbitts TH. Intracellular antibodies as specific reagents for functional ablation: future therapeutic molecules. Curr Mol Med. 2004;4:519–528. doi: 10.2174/1566524043360384. [DOI] [PubMed] [Google Scholar]
- 43.Marasco WA, LaVecchio J, Winkler A. Human anti–HIV-1 tat sFv intrabodies for gene therapy of advanced HIV-1-infection and AIDS. J Immunol Methods. 1999;231:223–238. doi: 10.1016/s0022-1759(99)00159-3. [DOI] [PubMed] [Google Scholar]
- 44.Poluri A, van Maanen M, Sutton RE. Genetic therapy for HIV/AIDS. Expert Opin Biol Ther. 2003;3:951–963. doi: 10.1517/14712598.3.6.951. [DOI] [PubMed] [Google Scholar]
- 45.Schroers R, Davis CM, Wagner HJ, Chen SY. Lentiviral transduction of human T-lymphocytes with a RANTES intrakine inhibits human immunodeficiency virus type 1 infection. Gene Ther. 2002;9:889–897. doi: 10.1038/sj.gt.3301711. [DOI] [PubMed] [Google Scholar]
- 46.Yang AG, Bai X, Huang XF, Yao C, Chen S. Phenotypic knockout of HIV type 1 chemokine coreceptor CCR-5 by intrakines as potential therapeutic approach for HIV-1 infection. Proc Natl Acad Sci USA. 1997;94:11567–11572. doi: 10.1073/pnas.94.21.11567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Egelhofer M, et al. Inhibition of human immunodeficiency virus type 1 entry in cells expressing gp41-derived peptides. J Virol. 2004;78:568–575. doi: 10.1128/JVI.78.2.568-575.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Perez EE, Riley JL, Carroll RG, von Laer D, June CH. Suppression of HIV-1 infection in primary CD4 T cells transduced with a self-inactivating lentiviral vector encoding a membrane expressed gp41-derived fusion inhibitor. Clin Immunol. 2005;115:26–32. doi: 10.1016/j.clim.2005.02.019. [DOI] [PubMed] [Google Scholar]
- 49.Mani M, Kandavelou K, Dy FJ, Durai S, Chandrasegaran S. Design, engineering, and characterization of zinc finger nucleases. Biochem Biophys Res Commun. 2005;335:447–457. doi: 10.1016/j.bbrc.2005.07.089. [DOI] [PubMed] [Google Scholar]
- 50.Lombardo A, et al. Gene editing in human stem cells using zinc finger nucleases and integrase-defective lentiviral vector delivery. Nat Biotechnol. 2007;25:1298–1306. doi: 10.1038/nbt1353. [DOI] [PubMed] [Google Scholar]
- 51.Levine BL, et al. Antiviral effect and ex vivo CD4+ T cell proliferation in HIV-positive patients as a result of CD28 costimulation. Science. 1996;272:1939–1943. doi: 10.1126/science.272.5270.1939. [DOI] [PubMed] [Google Scholar]
- 52.Levine BL, et al. Effects of CD28 costimulation on long term proliferation of CD4+ T cells in the absence of exogenous feeder cells. J Immunol. 1997;159:5921–5930. [PubMed] [Google Scholar]
- 53.Carroll RG, et al. Differential regulation of HIV-1 fusion cofactor expression by CD28 costimulation of CD4+ T cells. Science. 1997;276:273–276. doi: 10.1126/science.276.5310.273. [DOI] [PubMed] [Google Scholar]
- 54.Bex F, et al. Syngeneic adoptive transfer of anti–human immunodeficiency virus-1 (HIV-1)–primed lymphocytes from a vaccinated HIV-seronegative individual to his HIV-1–infected identical twin. Blood. 1994;84:3317–3326. [PubMed] [Google Scholar]
- 55.Levine BL, et al. Adoptive transfer of costimulated CD4+ T cells induces expansion of peripheral T cells and decreased CCR5 expression in HIV infection. Nat Med. 2002;8:47–53. doi: 10.1038/nm0102-47. [DOI] [PubMed] [Google Scholar]
- 56.Fearon DT, Manders P, Wagner SD. Arrested differentiation, the self-renewing memory lymphocyte, and vaccination. Science. 2001;293:248–250. doi: 10.1126/science.1062589. [DOI] [PubMed] [Google Scholar]
- 57.Barber DL, Wherry EJ, Ahmed R. Cutting edge: rapid in vivo killing by memory CD8 T cells. J Immunol. 2003;171:27–31. doi: 10.4049/jimmunol.171.1.27. [DOI] [PubMed] [Google Scholar]
- 58.Muul LM, et al. Persistence and expression of the adenosine deaminase gene for 12 years and immune reaction to gene transfer components: long-term results of the first clinical gene therapy trial. Blood. 2003;101:2563–2569. doi: 10.1182/blood-2002-09-2800. [DOI] [PubMed] [Google Scholar]
- 59.Lund O, et al. Gene therapy of T helper cells in HIV infection: mathematical model of the criteria for clinical effect. Bull Math Biol. 1997;59:725–745. doi: 10.1007/BF02458427. [DOI] [PubMed] [Google Scholar]
- 60.von Laer D, Hasselmann S, Hasselmann K. Impact of gene-modified T cells on HIV infection dynamics. J Theor Biol. 2006;238:60–77. doi: 10.1016/j.jtbi.2005.05.005. [DOI] [PubMed] [Google Scholar]
- 61.Weinberger LS, Schaffer DV, Arkin AP. Theoretical design of a gene therapy to prevent AIDS but not human immunodeficiency virus type 1 infection. J Virol. 2003;77:10028–10036. doi: 10.1128/JVI.77.18.10028-10036.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lalezari JP, et al. A controlled phase II trial assessing three doses of enfuvirtide (T-20) in combination with abacavir, amprenavir, ritonavir and efavirenz in non-nucleoside reverse transcriptase inhibitor-naive HIV-infected adults. Antivir Ther. 2003;8:279–287. [PubMed] [Google Scholar]
- 63.van Lunzen J, et al. Transfer of autologous gene-modified T cells in HIV-infected patients with advanced immunodeficiency and drug-resistant virus. Mol Ther. 2007;15:1024–1033. doi: 10.1038/mt.sj.6300124. [DOI] [PubMed] [Google Scholar]
- 64.Masiero S, et al. T-cell engineering by a chimeric T-cell receptor with antibody-type specificity for the HIV-1 gp120. Gene Ther. 2005;12:299–310. doi: 10.1038/sj.gt.3302413. [DOI] [PubMed] [Google Scholar]
- 65.Briz V, Poveda E, Soriano V. HIV entry inhibitors: mechanisms of action and resistance pathways. J Antimicrob Chemother. 2006;57:619–627. doi: 10.1093/jac/dkl027. [DOI] [PubMed] [Google Scholar]
- 66.Strayer DS, et al. Current status of gene therapy strategies to treat HIV/AIDS. Mol Ther. 2005;11:823–842. doi: 10.1016/j.ymthe.2005.01.020. [DOI] [PubMed] [Google Scholar]
- 67.Swan CH, Torbett BE. Can gene delivery close the door to HIV-1 entry after escape? J Med Primatol. 2006;35:236–247. doi: 10.1111/j.1600-0684.2006.00172.x. [DOI] [PubMed] [Google Scholar]
- 68.Luis AJ, et al. Novel interfering bifunctional molecules against the CCR5 coreceptor are efficient inhibitors of HIV-1 infection. Mol Ther. 2003;8:475–484. doi: 10.1016/s1525-0016(03)00202-8. [DOI] [PubMed] [Google Scholar]
- 69.Swan CH, Buhler B, Tschan MP, Barbas CF, III, Torbett BE. T-cell protection and enrichment through lentiviral CCR5 intrabody gene delivery. Gene Ther. 2006;13:1480–1492. doi: 10.1038/sj.gt.3302801. [DOI] [PubMed] [Google Scholar]
- 70.Bai J, et al. Inhibition of Tat-mediated transactivation and HIV-1 replication by human anti-hCyclinT1 intrabodies. J Biol Chem. 2003;278:1433–1442. doi: 10.1074/jbc.M208297200. [DOI] [PubMed] [Google Scholar]
- 71.BouHamdan M, et al. Inhibition of HIV-1 infection by down-regulation of the CXCR4 co-receptor using an intracellular single chain variable fragment against CXCR4. Gene Ther. 2001;8:408–418. doi: 10.1038/sj.gt.3301411. [DOI] [PubMed] [Google Scholar]
- 72.Stremlau M, et al. The cytoplasmic body component TRIM5α restricts HIV-1 infection in Old World monkeys. Nature. 2004;427:848–853. doi: 10.1038/nature02343. [DOI] [PubMed] [Google Scholar]
- 73.Luban J, Cyclophilin A. TRIM5, and resistance to human immunodeficiency virus type 1 infection. J Virol. 2007;81:1054–1061. doi: 10.1128/JVI.01519-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Li Y, Li X, Stremlau M, Lee M, Sodroski J. Removal of arginine 332 allows human TRIM5α to bind human immunodeficiency virus capsids and to restrict infection. J Virol. 2006;80:6738–6744. doi: 10.1128/JVI.00270-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ranga U, et al. Retroviral delivery of an antiviral gene in HIV-infected individuals. Proc Natl Acad Sci USA. 1998;95:1201–1206. doi: 10.1073/pnas.95.3.1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Morgan RA, et al. Preferential survival of CD4+ T lymphocytes engineered with anti–human immunodeficiency virus (HIV) genes in HIV-infected individuals. Hum Gene Ther. 2005;16:1065–1074. doi: 10.1089/hum.2005.16.1065. [DOI] [PubMed] [Google Scholar]
- 77.Macpherson JL, et al. Long-term survival and concomitant gene expression of ribozyme-transduced CD4+ T-lymphocytes in HIV-infected patients. J Gene Med. 2005;7:552–564. doi: 10.1002/jgm.705. [DOI] [PubMed] [Google Scholar]
- 78.Amado RG, et al. Anti–human immunodeficiency virus hematopoietic progenitor cell–delivered ribozyme in a phase I study: myeloid and lymphoid reconstitution in human immunodeficiency virus type-1–infected patients. Hum Gene Ther. 2004;15:251–262. doi: 10.1089/104303404322886101. [DOI] [PubMed] [Google Scholar]
- 79.VandenDriessche T, et al. Inhibition of clinical human immunodeficiency virus (HIV) type 1 isolates in primary CD4+ T lymphocytes by retroviral vectors expressing anti-HIV genes. J Virol. 1995;69:4045–4052. doi: 10.1128/jvi.69.7.4045-4052.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Veres G, et al. Comparative analyses of intracellularly expressed antisense RNAs as inhibitors of human immunodeficiency virus type 1 replication. J Virol. 1998;72:1894–1901. doi: 10.1128/jvi.72.3.1894-1901.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Humeau LM, et al. Efficient lentiviral vector-mediated control of HIV-1 replication in CD4 lymphocytes from diverse HIV+ infected patients grouped according to CD4 count and viral load. Mol Ther. 2004;9:902–913. doi: 10.1016/j.ymthe.2004.03.005. [DOI] [PubMed] [Google Scholar]
- 82.Schroder AR, et al. HIV-1 integration in the human genome favors active genes and local hotspots. Cell. 2002;110:521–529. doi: 10.1016/s0092-8674(02)00864-4. [DOI] [PubMed] [Google Scholar]
- 83.Dave UP, Jenkins NA, Copeland NG. Gene therapy insertional mutagenesis insights. Science. 2004;303:333. doi: 10.1126/science.1091667. [DOI] [PubMed] [Google Scholar]
- 84.Sadelain M. Insertional oncogenesis in gene therapy: how much of a risk? Gene Ther. 2004;11:569–573. doi: 10.1038/sj.gt.3302243. [DOI] [PubMed] [Google Scholar]
- 85.Li MJ, et al. Inhibition of HIV-1 infection by lentiviral vectors expressing Pol III–promoted anti-HIV RNAs. Mol Ther. 2003;8:196–206. doi: 10.1016/s1525-0016(03)00165-5. [DOI] [PubMed] [Google Scholar]
- 86.Eberhardy SR, et al. Inhibition of human immunodeficiency virus type 1 replication with artificial transcription factors targeting the highly conserved primer-binding site. J Virol. 2006;80:2873–2883. doi: 10.1128/JVI.80.6.2873-2883.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kiepiela P, et al. CD8+ T-cell responses to different HIV proteins have discordant associations with viral load. Nat Med. 2007;13:46–53. doi: 10.1038/nm1520. [DOI] [PubMed] [Google Scholar]
- 88.Walter EA, et al. Reconstitution of cellular immunity against cytomegalovirus in recipients of allogeneic bone marrow by transfer of T-cell clones from the donor. N Engl J Med. 1995;333:1038–1044. doi: 10.1056/NEJM199510193331603. [DOI] [PubMed] [Google Scholar]
- 89.Heslop HE, et al. Long-term restoration of immunity against Epstein-Barr virus infection by adoptive transfer of gene-modified virus–specific T lymphocytes. Nat Med. 1996;2:551–555. doi: 10.1038/nm0596-551. [DOI] [PubMed] [Google Scholar]
- 90.Brodie SJ, et al. HIV-specific cytotoxic T lymphocytes traffic to lymph nodes and localize at sites of HIV replication and cell death. J Clin Invest. 2000;105:1407–1417. doi: 10.1172/JCI8707. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 91.Wodarz D, Nowak MA. Specific therapy regimes could lead to long-term immunological control of HIV. Proc Natl Acad Sci USA. 1999;96:14464–14469. doi: 10.1073/pnas.96.25.14464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Roberts MR, et al. Targeting of human immunodeficiency virus–infected cells by CD8+ T lymphocytes armed with universal T-cell receptors. Blood. 1994;84:2878–2889. [PubMed] [Google Scholar]
- 93.Yang OO, et al. Lysis of HIV-1–infected cells and inhibition of viral replication by universal receptor T cells. Proc Natl Acad Sci USA. 1997;94:11478–11483. doi: 10.1073/pnas.94.21.11478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Walker RE, et al. Long-term in vivo survival of receptor-modified syngeneic T cells in patients with human immunodeficiency virus infection. Blood. 2000;96:467–474. [PubMed] [Google Scholar]
- 95.Mitsuyasu RT, et al. Prolonged survival and tissue trafficking following adoptive transfer of CD4zeta gene-modified autologous CD4+ and CD8+ T cells in human immunodeficiency virus–infected subjects. Blood. 2000;96:785–793. [PubMed] [Google Scholar]
- 96.Deeks SG, et al. A phase II randomized study of HIV-specific T-cell gene therapy in subjects with undetectable plasma viremia on combination anti-retroviral therapy. Mol Ther. 2002;5:788–797. doi: 10.1006/mthe.2002.0611. [DOI] [PubMed] [Google Scholar]
- 97.Letvin NL, Walker BD. Immunopathogenesis and immunotherapy in AIDS virus infections. Nat Med. 2003;9:861–866. doi: 10.1038/nm0703-861. [DOI] [PubMed] [Google Scholar]
- 98.Cooper LJ, Kalos M, Lewinsohn DA, Riddell SR, Greenberg PD. Transfer of specificity for human immunodeficiency virus type 1 into primary human T lymphocytes by introduction of T-cell receptor genes. J Virol. 2000;74:8207–8212. doi: 10.1128/jvi.74.17.8207-8212.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Morgan RA, et al. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science. 2006;314:126–129. doi: 10.1126/science.1129003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Holler PD, Lim AR, Cho BK, Rund LA, Kranz DM. CD8− T cell transfectants that express a high affinity T cell receptor exhibit enhanced peptide-dependent activation. J Exp Med. 2001;194:1043–1052. doi: 10.1084/jem.194.8.1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Li QJ, et al. miR-181a is an intrinsic modulator of T cell sensitivity and selection. Cell. 2007;129:147–161. doi: 10.1016/j.cell.2007.03.008. [DOI] [PubMed] [Google Scholar]
- 102.Boulter JM, et al. Stable, soluble T-cell receptor molecules for crystallization and therapeutics. Protein Eng. 2003;16:707–711. doi: 10.1093/protein/gzg087. [DOI] [PubMed] [Google Scholar]
- 103.Shultz LD, Ishikawa F, Greiner DL. Humanized mice in translational biomedical research. Nat Rev Immunol. 2007;7:118–130. doi: 10.1038/nri2017. [DOI] [PubMed] [Google Scholar]
- 104.Berges BK, et al. HIV-1 infection and Cd4 T cell depletion in the humanized Rag2−/−gamma c−/− (RAG-hu) mouse model. Retrovirology. 2006;3:76. doi: 10.1186/1742-4690-3-76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.van Baarle D, Tsegaye A, Miedema F, Akbar A. Significance of senescence for virus-specific memory T cell responses: rapid ageing during chronic stimulation of the immune system. Immunol Lett. 2005;97:19–29. doi: 10.1016/j.imlet.2004.10.003. [DOI] [PubMed] [Google Scholar]
- 106.June CH. Principles of adoptive T cell cancer therapy. J Clin Invest. 2007;117:1204–1212. doi: 10.1172/JCI31446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Kaech SM, Hemby S, Kersh E, Ahmed R. Molecular and functional profiling of memory CD8 T cell differentiation. Cell. 2002;111:837–851. doi: 10.1016/s0092-8674(02)01139-x. [DOI] [PubMed] [Google Scholar]
- 108.Mazurier F, et al. Lentivector-mediated clonal tracking reveals intrinsic heterogeneity in the human hematopoietic stem cell compartment and culture-induced stem cell impairment. Blood. 2004;103:545–552. doi: 10.1182/blood-2003-05-1558. [DOI] [PubMed] [Google Scholar]
- 109.Check E. Pioneering HIV treatment would use interference and gene therapy. Nature. 2005;437:601. doi: 10.1038/437601b. [DOI] [PubMed] [Google Scholar]
- 110.Bahner I, et al. Lentiviral vector transduction of a dominant-negative Rev gene into human CD34+ hematopoietic progenitor cells potently inhibits human immunodeficiency virus-1 replication. Mol Ther. 2007;15:76–85. doi: 10.1038/sj.mt.6300025. [DOI] [PubMed] [Google Scholar]
- 111.Bahner I, Kearns K, Hao QL, Smogorzewska M, Kohn DB. Transduction of human CD34+ hematopoietic progenitor cells by a retroviral vector expressing an RRE decoy inhibits HIV-1 replication in the myelomonocytic cells produced in long-term culture. J Virol. 1996;70:4352–4360. doi: 10.1128/jvi.70.7.4352-4360.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Rosenzweig M, et al. Intracellular immunization of rhesus CD34+ hematopoietic progenitor cells with a hairpin ribozyme protects T cells and macrophages from simian immunodeficiency virus infection. Blood. 1997;90:4822–4831. [PubMed] [Google Scholar]
- 113.Davis BR, et al. Targeted transduction of CD34+ cells by transdominant negative Rev-expressing retrovirus yields partial anti-HIV protection of progeny macrophages. Hum Gene Ther. 1998;9:1197–1207. doi: 10.1089/hum.1998.9.8-1197. [DOI] [PubMed] [Google Scholar]
- 114.Braun SE, et al. Inhibition of simian/human immunodeficiency virus replication in CD4+ T cells derived from lentiviral-transduced CD34+ hematopoietic cells. Mol Ther. 2005;12:1157–1167. doi: 10.1016/j.ymthe.2005.07.698. [DOI] [PubMed] [Google Scholar]
- 115.Su L, et al. Hematopoietic stem cell–based gene therapy for acquired immunodeficiency syndrome: efficient transduction and expression of RevM10 in myeloid cells in vivo and in vitro. Blood. 1997;89:2283–2290. [PubMed] [Google Scholar]
- 116.Bai J, et al. RNA-based anti–HIV-1 gene therapeutic constructs in SCID-hu mouse model. Mol Ther. 2002;6:770–782. doi: 10.1006/mthe.2002.0800. [DOI] [PubMed] [Google Scholar]
- 117.Anderson J, et al. Safety and efficacy of a lentiviral vector containing three anti-HIV genes—CCR5 ribozyme, tat-rev siRNA, and TAR decoy—in SCID-hu mouse-derived T cells. Mol Ther. 2007;15:1182–1188. doi: 10.1038/sj.mt.6300157. [DOI] [PubMed] [Google Scholar]
- 118.An DS, et al. Stable reduction of CCR5 by RNAi through hematopoietic stem cell transplant in non-human primates. Proc Natl Acad Sci USA. 2007;104:13110–13115. doi: 10.1073/pnas.0705474104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Zaia JA, et al. Autologous stem cell transplantation using retrovirus-transduced peripheral blood progenitor cells in HIV-infected persons: comparison of gene marking post-engraftment with and without myeloablative therapy. Blood. 1999;94(Suppl 1):642a. [Google Scholar]
- 120.Amado RG, et al. A phase I trial of autologous CD34+ hematopoietic progenitor cells transduced with an anti-HIV ribozyme. Hum Gene Ther. 1999;10:2255–2270. doi: 10.1089/10430349950017239. [DOI] [PubMed] [Google Scholar]
- 121.Podsakoff GM, et al. Selective survival of peripheral blood lymphocytes in children with HIV-1 following delivery of an anti-HIV gene to bone marrow CD34+ cells. Mol Ther. 2005;12:77–86. doi: 10.1016/j.ymthe.2005.02.024. [DOI] [PubMed] [Google Scholar]
- 122.Aiuti A, et al. Correction of ADA-SCID by stem cell gene therapy combined with nonmyeloablative conditioning. Science. 2002;296:2410–2413. doi: 10.1126/science.1070104. [DOI] [PubMed] [Google Scholar]
- 123.Ott MG, et al. Correction of X-linked chronic granulomatous disease by gene therapy, augmented by insertional activation of MDS1–EVI1, PRDM16 or SETBP1. Nat Med. 2006;12:401–409. doi: 10.1038/nm1393. [DOI] [PubMed] [Google Scholar]
- 124.Cavazzana-Calvo M, et al. Gene therapy of human severe combined immunodeficiency (SCID)-X1 disease. Science. 2000;288:669–672. doi: 10.1126/science.288.5466.669. [DOI] [PubMed] [Google Scholar]
- 125.Hacein-Bey-Abina S, et al. LMO2-associated clonal T cell proliferation in two patients after gene therapy for SCID-X1. Science. 2003;302:415–419. doi: 10.1126/science.1088547. [DOI] [PubMed] [Google Scholar]
- 126.Shepherd BE, et al. Hematopoietic stem-cell behavior in nonhuman primates. Blood. 2007;110:1806–1813. doi: 10.1182/blood-2007-02-075382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Payne KJ, Crooks GM. Immune-cell lineage commitment: translation from mice to humans. Immunity. 2007;26:674–677. doi: 10.1016/j.immuni.2007.05.011. [DOI] [PubMed] [Google Scholar]
- 128.Wong-Staal F, Poeschla EM, Looney DJ. A controlled, phase 1 clinical trial to evaluate the safety and effects in HIV-1 infected humans of autologous lymphocytes transduced with a ribozyme that cleaves HIV-1 RNA. Hum Gene Ther. 1998;9:2407–2425. doi: 10.1089/hum.1998.9.16-2407. [DOI] [PubMed] [Google Scholar]
- 129.Kang EM, et al. Gene therapy–based treatment for HIV-positive patients with malignancies. J Hematother Stem Cell Res. 2002;11:809–816. doi: 10.1089/152581602760404612. [DOI] [PubMed] [Google Scholar]
- 130.Barrett AD, Dimmock NJ. Defective interfering viruses and infections of animals. Curr Top Microbiol Immunol. 1986;128:55–84. doi: 10.1007/978-3-642-71272-2_2. [DOI] [PubMed] [Google Scholar]
- 131.Roux L, Simon AE, Holland JJ. Effects of defective interfering viruses on virus replication and pathogenesis in vitro and in vivo. Adv Virus Res. 1991;40:181–211. doi: 10.1016/S0065-3527(08)60279-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Mautino MR, Morgan RA. Inhibition of HIV-1 replication by novel lentiviral vectors expressing transdominant Rev and HIV-1 env antisense. Gene Ther. 2002;9:421–431. doi: 10.1038/sj.gt.3301674. [DOI] [PubMed] [Google Scholar]