

Abstract
Malaria is one of the most significant causes of childhood mortality but disease control efforts are threatened by resistance of the Plasmodium parasite to current therapies. Continued progress in combating malaria requires development of new, easy to administer drug combinations with broad ranging activity against all manifestations of the disease. DSM265, a triazolopyrimidine-based inhibitor of the pyrimidine biosynthetic enzyme dihydroorotate dehydrogenase (DHODH), is the first DHODH inhibitor to reach clinical development for treatment of malaria. We describe studies profiling the biological activity, pharmacological and pharmacokinetic properties, and safety of DSM265, which supported its advancement to human trials. DSM265 is highly selective towards DHODH of the malaria parasite Plasmodium, efficacious against both blood and liver stages of P. falciparum, and active against drug-resistant parasite isolates. Favorable pharmacokinetic properties of DSM265 are predicted to provide therapeutic concentrations for more than 8 days after a single oral dose in the range of 200–400 mg. DSM265 was well tolerated in repeat dose and cardiovascular safety studies in mice and dogs, was not mutagenic, and was inactive against panels of human enzymes/receptors. The excellent safety profile, blood and liver-stage activity, and predicted long human half-life position DSM265 as a new potential drug combination partner for either single-dose treatment or once weekly chemoprevention. DSM265 has advantages over current treatment options that are dosed daily or are inactive on the parasite liver-stage
Introduction
Malaria remains a significant global health challenge and despite effective drugs and insect control programs, still kills up to 580,000 people each year, the majority being children under 5 years old living in sub-Saharan Africa (1–3). No other parasitic infection has had such a broad ranging impact on human health. Its persistence has influenced the evolution of the human genome as highlighted by genetic polymorphisms that have arisen by conferring protection against severe malaria (1). Today, three billion people are at risk of malaria in 97 countries, leading to 200 million cases annually caused primarily by P. falciparum and P. vivax. Pregnant women and non-immune naive hosts are also particularly susceptible to severe disease, which is associated with complications such as severe anemia and/or sequestration of parasitized red blood cells into key organs, including brain (1). Lack of a fully protective vaccine, the constant threat of drug and insecticide resistance, and poverty all contribute to the difficult task of malaria control and elimination. In areas of intense malaria transmission, children are highly susceptible but adults develop protective immunity, and while harboring parasites, are largely asymptomatic. This reservoir of asymptomatic carriers contributes to transmission and further complicates the goal of malaria eradication (4).
Drug therapy provides the most effective option for treatment and prevention of malaria and a broad portfolio of clinically effective drugs has been developed and used throughout the modern history of the disease (1, 5). The propensity of the parasite to develop resistance to many of these agents (e.g. chloroquine and pyrimethamine) has hindered treatment and control. There is only a single drug for treatment of P. vivax latent liver forms (primaquine) and it is countraindicated in glucose-6-phosphate dehydrogenase (G6PD)-deficient patients (6), while chemopreventive medicines either have concerning side effects (mefloquine), or are expensive and require daily dosing (atovoquone-proguanil) (1, 5). The effectiveness of the artemisinin-based combination therapies (ACTs) for malaria treatment, combined with widespread use of insecticide-treated bed nets, has been credited with reduction in malaria deaths over the past decade. However, resistance to the artemisinins, manifested as delayed parasite clearance and linked to K13-propeller protein polymorphisms, has emerged in South East Asia and is threatening to derail malaria control efforts (7).
Over the past decade, a robust effort in antimalarial drug discovery has generated a broad portfolio of new drug candidates (5, 8, 9). To reduce the potential for emergence of resistance, new treatments are being developed as combination therapies (10). Candidate molecules with a broad spectrum of activities including treatment of the blood-stage infection, chemoprevention via activity on liver-stages, and transmission-blocking activity are required to contribute to the eradication agenda (11). Also key to this effort is improving patient compliance, thus compounds with pharmacokinetic properties supporting use in single dose combination treatments or once weekly chemoprevention are being prioritized for development.
Extensive and rapid replication of parasite DNA is required to propagate Plasmodium species in both liver and blood stages of infection (1). As a consequence, several clinically used antimalarial drugs target pyrimidine nucleotide biosynthesis, including the dihydrofolate reductase inhibitors pyrimethamine and P218 (9), and the cytochrome bc1 inhibitor atovoquone (12). Plasmodium species lack pyrimidine salvage enzymes, and unlike humans, rely entirely on the de novo pathway to acquire pyrimidines for DNA and RNA synthesis. A key step in this pathway is catalyzed by dihydroorotate dehydrogenase (DHODH) (13). We previously identified P. falciparum DHODH (PfDHODH) inhibitors from the triazolopyrimidine structural class with potent antimalarial activity (13). The series was optimized using an X-ray structure-guided medicinal chemistry program leading to the identification of potent DHODH inhibitors with good pharmacokinetic properties and in vivo efficacy in the P. falciparum SCID mouse model (14, 15). Herein, we describe the preclinical development of one of these analogs, DSM265 (Fig. 1A), the first DHODH inhibitor to advance to human clinical trials for the treatment of malaria.
Fig. 1. Chemical and protein bound inhibitor structures.

(A) Chemical structures of DSM265 (415 Da), DSM430 (430 Da) and DSM450 (431 Da). (B) X-ray structure of the inhibitor binding-site of PfDHODH bound to DSM265 (PDB 5BOO). DSM265 (pink); protein, orotate (Oro) and flavin (FMN) (carbons in teal); oxygen (red); nitrogen (blue); sulfur (yellow); phosphate (orange); fluorine (light blue).
Results
X-ray structure of DSM265 bound to PfDHODH
DHODH is a flavin (FMN)-dependent mitochondrial enzyme that catalyzes oxidation of dihydroorotate to orotic acid in a two-step reaction that requires Coenzyme Q (CoQ) for reoxidation of FMN (13). The crystal structure of DSM265 bound to PfDHODH was solved for two different crystal forms to 2.25 Å (form I) and 2.8 Å (form II) resolution, respectively (Table S1 and Fig. S1). The inhibitor binding-site is displayed for crystal form II because it had a more uniform DSM265-ligand density. The higher resolution structure determination of crystal form I allowed better quality refinement of the form II protein structure coordinates. DHODH is composed of a core β/α-barrel with an N-terminal α-helix that interacts with the mitochondrial membrane allowing binding of CoQ. The DSM265 inhibitor-binding pocket sits between FMN and the N-terminal α-helix and is also thought to be the binding-site for CoQ. This pocket is primarily hydrophobic and only two hydrogen bonds are formed between DSM265 and the protein via Arg-265 and His-185 (Fig. 1B). The amino acid composition of the binding-site is highly variable between the Plasmodium and human enzymes (Figs. 1B and S2) and this property is thought to underlie the strong selectivity for the parasite enzyme over human DHODH (13).
Activity and species selectivity of DSM265 and analogs against DHODH
DSM265 is a potent inhibitor of the Plasmodium enzymes PfDHODH and P. vivax DHODH (PvDHODH) with excellent selectivity versus human DHODH (14). To assess species selectivity towards other mammalian or parasite enzymes we evaluated DSM265 inhibitory activity towards DHODHs from species that would potentially be used in either toxicity or efficacy testing (Table 1). Given that the main impurity (DSM430) and the primary in vivo metabolite (DSM450) of DSM265 (Fig. 1A) (described below) would both be present following in vivo dosing, both compounds were also profiled for species selectivity. DSM265 inhibitory activity versus mammalian DHODHs showed significant differences. Like the human enzyme, rabbit, pig and monkey DHODH were not significantly inhibited [50% inhibitor concentration (IC50) >41 μg/ml]. In contrast, dog DHODH (IC50 = 10 μg/ml), and to a greater extent the mouse and rat enzymes (IC50 ~ 1 μg/ml for each), were sensitive to DSM265 (Table 1 and Fig. 2A). Comparison of the amino acid sequence in the inhibitor binding site shows that the rodent DHODH binding sites have diverged from human DHODH at 4 positions (M111L, F62V in rat and mouse, T360I in rat and T63I in mouse), whereas dog DHODH differs at only one (F62V) (Fig. S2). The remaining mammalian enzymes have conserved binding sites with human DHODH. DHODHs from the human malaria parasites, P. falciparum and P. vivax, and the simian parasite, P. cynomolgi, were inhibited by DSM265 with similar potency (Table 1). The IC50 for P. vivax DHODH was 2-fold higher than for PfDHODH while for P. cynomolgi it was 2-fold lower. In contrast, DSM265 had poor activity against DHODH from rodent Plasmodium species (P. berghei or P. yoelii) (14) and thus efficacy models using these species have not been useful for profiling DSM265.
Table 1.
Comparative kinetic analysis of DSM265 and derivatives on P. falciparum and mammalian DHODH.
| Compound | DSM265 | DSM430) | DSM450 |
|---|---|---|---|
|
| |||
| DHODH | IC50 (μg/mL | ||
| P. falciparum | 0.010 (0.0080 – 0.013) | 0.010 (0.0079 – 0.014) | 0.032 (0.026 – 0.040) |
| P. vivax | 0.020 (0.017 – 0.024) | ND | 0.11 (0.081 – 0.15) |
| P. cynomolgi | 0.0050 (0.0042 – 0.0058) | ND | ND |
| Human | ~ 41 | 3.7 (2.6 – 4.8) | >43 |
| Mouse | 1.1 (1.2 – 1.5) | 0.10 (0.082 – 0.12) | 4.5 (3.1 – 6.9) |
| Rat | 0.82 (0.63 – 1.1) | 0.082 (0.069 – 0.099) | 1.6 (1.2 – 2.0) |
| Dog | 11 (8.8 – 12) | 0.56 (0.48 – 0.65) | 25 (21 – 29) |
| Rabbit | > 41 | ND | >43 |
| Pig | > 41 | ND | ND |
| Monkey | > 41 | ND | ND |
Data represent the mean of 3 technical replicates with 95% confidence intervals for the fit shown in parentheses. Primary data are provided in Table S19. ND, not determined.
Fig. 2. In vitro activity of DSM265 and its analogs on DHODH and P. falciparum parasites.

(A) DHODH inhibition. IC50 values are reported in Table 1. Error bars show the standard error of the mean (SEM) for 3 technical replicates per concentration. Each fitted IC50 was obtained from 30 – 33 data points per fit. (B) In vitro P. falciparum 3D7 growth inhibition. Fitted EC50’s were 0.0018 (0.0011 – 0.0028), 0.079 (0.042 – 0.15) and 0.00020 (0.00011 – 0.00056) μg/mL for DSM265, DSM450 and DSM430, respectively, with the 95% confidence intervals in parenthesis (3 technical replicates per concentration and 18 – 24 data points per fit, the plot shows mean ± SEM). (C) Effects on P. falciparum 3D7 blood-stage phenotype and development. Cells were treated with drug at 10xEC50 and parasites were evaluated by microscopy 48 h after drug addition (minimally 200 cells were counted per condition). Atovaquone (Ato), artemisinin (Art), pyrimethamine (Pyr) were used as controls. Images of representative parasites from the counted stages are displayed (R, rings; YT, young trophozoite; MT, mature trophozoite; S, schizont; P, pyknotic). (D) In vitro killing curves for treatment of P. falciparum 3D7 blood-stage parasites. Data for 10x and 100xEC50 (where EC50=0.0046 μg/mL) (4 technical replicates showing the mean and standard deviation (SD)) are displayed for DSM265. Data for Art, Ato, Pyr and chloroquine (Chl) were previously reported (33). (E) Comparative DSM265 kill rates calculated from Fig. 2D. Parasite Reduction Ratio (PRR), the log number of parasites killed per asexual life cycle (48 h), Lag Phase, time before parasite killing begins, and parasite clearance time (PCT), time to achieve 99.9% parasite kill. Primary data are provided in Table S19.
DSM430 and DSM450 also inhibit PfDHODH but their activity against P. falciparum parasites and their species selectivity profiles are substantially different (Table 1). The metabolite DSM450 was 3-fold less potent than DSM265 as a PfDHODH inhibitor, however it was 43-fold less active against P. falciparum 3D7 parasites [effective concentration (EC50) = 0.079 μg/mL for DSM450 versus 0.0018 μg/mL for DSM265 (Fig. 2B)], thus it is not expected to contribute to in vivo parasite killing. The potency of the impurity DSM430 was comparable to that of DSM265 against PfDHODH, but it was10-fold more potent on the tested mammalian enzymes (Table 1). DSM430 parasite activity exceeded that of DSM265 (Fig. 2B; P. falciparum 3D7 EC50 = 0.0002 μg/mL). Given the lack of species selectivity of DSM430, concentrations of this impurity will need to be carefully controlled in clinical batches of DSM265. The reduced selectivity of DSM430 relative to the mammalian enzymes mirrors that of analogs containing para CF3-aniline when combined with meta-fluorines (16).
Antimalarial activities of DSM265: life-cycle profiling and kill rate
DSM265 activity was profiled against the full range of parasite life-cycle stages using strategies recently optimized with standard antimalarial compounds (17). In vitro blood-stage activity was evaluated for 9 strains of P. falciparum, including chloroquine-resistant and pyrimethamine-resistant parasites. DSM265 was equally effective (EC50 ranging from 0.001–0.004 μg/mL) against all tested strains (Table S2). Notably, these EC50 values are lower than previous values determined in media containing 10% human serum (14), which is a reflection of differential DSM265 protein binding in different media (Table 2). The effects of DSM265 treatment (48 h) on blood-stage parasites were evaluated by microscopy in comparison to standard antimalarial agents (Fig. 2C). DSM265 arrested parasites in the young trophozoite-stage showing a profile similar to atovaquone. In contrast, pyrimethamine-treated parasites arrested as mature trophozoites, and artemisinin-treated parasites were pyknotic.
Table 2.
DSM265 activity against P. falciparum parasites at different stages in the life cycle.
| Assay | MPC or EC50 (μg/mL) | Fraction unbound in assay media | a Unbound MPC or EC50 (μg/mL) |
|---|---|---|---|
| Blood-stage: P. falciparum 3D7 in vitro MPC (kill assay) | 0.014 a | 0.17 b | 0.0020 |
| SCID mouse in vivo MPC (plasma) | 1.1c | 0.001d | 0.0011 |
| Blood-stage: P. falciparum 3D7 in vitro growth assay | 0.0046 a | 0.17b | 0.00078 |
| Liver-stage: P. falciparum – schizont | 0.0057 | 0.215e | 0.0012 |
| P. falciparum early stage gametocytes stage III – IV; strain NF54 | > 0.42 | 0.004f | >0.0017 |
| Gametocyte assay P. falciparum stages IV–V | Inactive > 16 | ND | ND |
| P. falciparum dual gamete formation assay | Inactive > 42 | ND | ND |
| P. falciparum stage V oocyst | 38% at 1.0 | 0.001d | 38% at 0.001 |
Data represent EC50 values unless otherwise stated to be the MPC.
data taken from Fig. 2D;
0.5% Albumax II in RPMI-1640;
blood to plasma ratio in the humanized SCID mouse of 0.5 was used to convert blood concentrations to plasma concentrations;
human plasma;
10% fetal calf serum in DMEM or similar medium;
10% human serum in RPMI 1640.
ND=not determined. See Table S9 for protein binding data in serum and various media.
In vitro DSM265 kill rates were determined for P. falciparum 3D7 parasites over a concentration range of 1× (0.0046 μg/ml) to 100xEC50 (Figs. 2D, 2E and S3). DSM265 displayed maximum antimalarial effects at 3xEC50 and above, requiring a 24–48 h lag-phase prior to parasite killing. At 1xEC50 DSM265 parasite killing was further delayed (96 h lag-phase) (Fig. S3). Thus 3xEC50 (0.014 μg/mL, Table 2) represents the minimum concentration required to achieve the maximum kill rate (minimum parasiticidal concentration or MPC) in vitro. The DSM265 kill rate was similar to atovaquone, but significantly slower than observed for artemisinin and chloroquine.
DSM265 liver-stage activity was evaluated in vitro against P. falciparum and P. cynomolgi liver-stage parasites. In the P. falciparum liver-stage assay, DSM265 was unable to block liver cell invasion, and small exoerythrocytic forms (EEFs) were observed after treatment except at the highest dose (4.2 μg/mL); the EEF count was reduced by treatment with primaquine (Fig. 3A,B). However, DSM265 blocked EEF growth to the schizont stage with an EC50 of 0.0057 μg/mL (assessed by the diameter of the EEF), a value 30-fold more potent than obtained for primaquine (EC50 primaquine = 0.15 μg/mL) (Fig. 3C–E and Table 2). The unbound EC50 for P. falciparum liver-stage large forms was similar to the unbound P. falciparum blood-stage EC50 (Table 2), suggesting that DSM265 will be effective for the elimination of both blood and liver replicative forms. In the P. cynomolgi liver-stage model, DSM265 was also active against the liver-stage large form (multinucleated schizont) (EC50 = 0.13 μg/mL) while it had poor activity for against small forms representative of the non-dividing hypnozoite, a surrogate for the dormant forms in P. vivax infections (Fig. S4).
Fig. 3. Activity of DSM265 against P. falciparum liver stages.

(A) Inhibitory effect on hepatocyte infection rate. Number of exoerythrocytic forms (EEF)/10,000 hepatocytes quantitated at different concentrations of DSM265 relative to the DMSO control for three technical replicates using the same human donor, error bars show the SD for the replicates. Primary data are provided in Table S20. (B) Dose response curve showing the effect of DSM265 and primaquine (PQ) on infection rate. PQ EC50 = 0.20 (0.059 – 0.71) μg/mL and DSM265 EC50 = 7.0 (4.3 – 11) μg/mL. Errors are SEM for n=3. C) Inhibitory effect on EEF diameter for n = 20 – 22 EEFs. (D) Dose response curve showing the effect on EEF size. PQ EC50 = 0.15 (0.11 – 0.20) μg/mL and for DSM265 EC50 = 0.0057 (0.0048 – 0.0069) μg/mL. Error bars show the SD for n = 20 – 22. (E) Representative images of parasites day 3 post-infection treated with DMSO or DSM265 (0.42 μg/mL). Statistical significance was determined by ANOVA (*** p = 0.0002, **** p <0.0001; for comparison of drug-treated to DMSO controls, Panels A and C). Historical data for PQ (Panel B and D) and Ato (Panel A and C) using the same donor are shown.
Activity of DSM265 on P. falciparum sexual and mosquito stages was also evaluated. It had no activity on early- or late-stage gametocytes and was inactive in the P. falciparum dual gamete formation assay (Table 2). DSM265 inhibited formation of multinucleated oocysts with an approximate unbound EC50 (38% inhibition) that was within 3-fold of the unbound EC50 against the blood-stage (Table 2). At the infection intensity observed in this study (22 oocysts per mosquito), reduced oocyst density did not translate into decreased numbers of infected mosquitoes. However, oocyst density in the field is much lower (1–3 oocysts/mosquito) raising the possibility that there could be some transmission blocking activity under these conditions.
In vivo parasite killing activity: pharmacokinetic/pharmacodynamic (PK/PD) relationships
In order to define the MPC required for parasite killing in vivo, we undertook a detailed analysis of the oral dose-response profile in the P. falciparum SCID mouse 4-day dosing model. Because the half-life of DSM265 in mice is much shorter (2–4 h) than the predicted human half-life (described below), we dosed DSM265 twice daily (b.i.d.) to better mimic the sustained plasma exposure expected in humans. DSM265 had potent in vivo antimalarial activity with an effective dose (ED90) of 3 mg/kg/day (1.5 mg/kg b.i.d., Fig. 4, Table S3, Fig. S5), comparing favorably with chloroquine and mefloquine (Table S3B). The maximum rate of parasite killing occurred at and above a dose of 13 mg/kg/day (6.4 mg/kg b.i.d.) (Fig. 4A). Consistent with in vitro data (Fig. 2), a relatively steep dose response relationship was observed in vivo. The maximum blood concentrations (Cmax) and area under the blood concentration vs. time profiles (Day 1 area under the curve (AUC)0–10 h) were approximately dose proportional (Fig. 4B and Table S3A). The DSM265 blood concentration at 23 h after the last 13 mg/kg/day dose (0.53 μg/mL, Table S3A) was used as an estimate of the MPC, which corresponds to an unbound plasma concentration of 0.0011 μg/mL. This value shows good agreement with the unbound in vitro MPC (0.002 μg/mL) (Table 2). The ED90 in this SCID mouse study was 2.6-fold lower than in a preliminary study in SCID mice dosed once daily, which was limited by a small dose range and insufficient PK data to define the MPC (14).
Fig. 4. In vivo efficacy of DSM265 in the P.falciparum-infected SCID mouse.
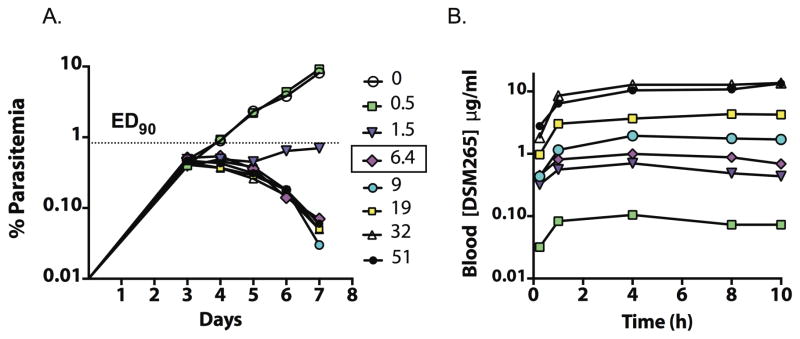
DSM265 was dosed as the tosylate salt b.i.d. for 4 days starting day 3 post infection. (A) Blood parasitemia versus days post infection for administered doses (mg/kg/12 h). The ED90 (1.5 mg/kg/12 h) and the MPC (dose = 6.4 mg/kg/12 h) were determined 24 h after the last dose. The parasite detection limit was 0.01%. (B) DSM265 blood concentrations (μg/mL) over 10 h after the first dose for doses as indicated. A single data point was collected per dose level with the exception of the no drug control where n = 3 biological replicates were obtained. Primary data are provided in Tables S3C and S3D.
Propensity of P. falciparum parasites to become resistant to DSM265
P. falciparum Dd2 and K1 parasites are multidrug-resistant strains commonly used to evaluate the propensity of parasites to become resistant to drugs in vitro (18). Dd2 parasites were selected under continuous DSM265 pressure at 3xEC50 over a range of parasite inocula to evaluate the minimum inoculum for resistance (MIR), the lowest number of parasites necessary to generate a resistant mutant, using atovaquone as a comparator. The MIR for DSM265 averaged 2×106 (ranged 2×105–2×107) (Tables S4A,C) over two independent studies while for atovaquone, the MIR was 2×107 (Tables S4A,D). The EC50 for DSM265-resistant Dd2 parasites increased 3–26-fold (Tables 3 and S5) over wild-type parasites compared to a 10-fold shift observed for atovaquone (Table S4D). The effect of DSM265 concentrations on propensity for resistance was also studied: drug concentrations of 33xEC50 were required to suppress emergence of atovaquone-resistant parasites compared to only 8xEC50 for DSM265 for a similar inoculum (2×107 parasites) (Table S4A and S4B). Additional selections at higher DSM265 concentrations (5 – 8xEC50) were also performed at a high starting inoculum (2×109) (Table S4C). Clonal lines from these selections showed EC50 shifts of ~30-fold (Table S5). Selection of DSM265-resistant parasites was significantly more difficult with the K1 strain (MIR of 2×108) (Table S4E). Resistant parasites were not obtained from selections with the HB3 strain at any DSM265 concentration, whereas resistant HB3 parasites could be obtained for selections with atovaquone (Table S4F). DSM265-resistant isolates derived from Dd2 and K1 remained fully sensitive to atovaquone and artemisinin (Table S5).
Table 3.
Evaluation of in vitro-generated DSM265 Dd2 resistant parasites
| Clone # | Inoculum | DSM265 selection concentration (μg/mL) | DSM265 Dd2 EC50 (μg/mL) | Gene copy # | DHODH mutation | PfDHODH IC50 (μg/mL) |
|---|---|---|---|---|---|---|
| Dd2/wt | N/A | N/A | 0.0022 | 1.0 | N/A | 0.0096 |
| PR.Cl1 | 2×107 | 0.0083 | 0.0083 [3.8] | 2.7 | none | N/A |
| PR.Cl2 | 2×107 | 0.0083 | 0.0099 [4.5] | 2.3 | none | N/A |
| DF.R10 Clb | 2×106 | 0.0095 | 0.058 [26] | 1.1 | G181C | 0.120 [12] |
Fold-change in square brackets. PfDHODH data are IC50 values determined on the recombinant wild-type (wt) or G181C mutant P. falciparum enzymes. All data were collected with a minimum of three technical replicates and then used for a global fit. Error analysis is provided in Table S5.
The resistance mechanism was evaluated for clonal Dd2 lines selected at 3xEC50. Both amplification of the DHODH gene and acquisition of a point mutation (G181C) were observed (Table 3 and S5), consistent with DHODH being the primary target of parasite killing. The G181C mutation maps to the DSM265 binding-site where this residue makes close contacts with the -CF2CH3 group (Fig. 1). Introduction of the G181C mutation into recombinant PfDHODH showed that the IC50 was 13-fold higher than for the wild-type enzyme, which correlates with the 26-fold shift in parasite EC50. Steady-state kinetic analysis of G181C PfDHODH showed that kcat was reduced by 2-fold compared to wild-type, while the Km for CoQd and L-dihydroorotate (DHO) were unchanged (Table S6).
Drug synergy studies to identify possible combination partners
To determine whether DSM265 has synergistic or antagonistic activity with possible partner compounds, interactions between DSM265 and other reported antimalarials were evaluated using the fixed-ratio isobologram method (Table S7). Compounds tested included several artemisinin derivatives, the synthetic ozonide OZ439, chloroquine, piperaquine, pyronaridine, mefloquine, lumefantrine, atovaquone, proguanil, and the dihydrofolate reductase inhibitors P218 and pyrimethamine. DSM265 activity was additive with all tested compounds and no evidence of synergistic or antagonistic activity was found.
Physicochemical properties, in vitro ADME and metabolite identification
Progression of DSM265 toward clinical development required detailed studies to define properties related to absorption, distribution, metabolism, and excretion (ADME). DSM265 free base was shown to be chemically stable in the solid state following storage at 30°/65% relative humidity for up to 24 months, but degradation of both the DSM265 free base and tosylate salt were observed when solutions were subjected to simulated sunlight necessitating that solutions be protected from light (Table S8A). DSM265 free base had poor aqueous solubility under simulated gastric conditions (6.8 μg/mL), and in fasted- and fed-state simulated intestinal fluids (FaSSIF and FeSSIF, 5.1 and 27.6 μg/mL, respectively, Table S8B). Across Caco-2 cell monolayers, DSM265 had high apical to basolateral permeability (Papp > 30 × 10−6 cm/s), with minimal evidence of efflux suggesting that it is likely to be well absorbed in vivo. The P-glycoprotein (Pgp) and breast cancer resistance protein (BCRP) inhibitors verapamil and fumitremorgin C, respectively, had no effect on DSM265 permeability, indicating that it is not a substrate for these transporters. High permeability integrated with low aqueous solubility suggested that DSM265 is a BCS Class II compound.
Plasma protein binding of DSM265 was highest in human plasma (99.9%), with slightly lower values in mouse and dog plasma (99.7 and 99.4%, respectively), and the lowest binding in rat plasma (97.7%) (Table S9). Due to the high plasma protein binding, DSM265 had a blood to plasma partitioning ratio of 0.5–0.7 across species (Table S9).
In hepatic microsomes and cryopreserved hepatocytes, DSM265 exhibited minimal degradation across species consistent with low in vivo hepatic clearance (Table S9). In vitro studies with individual human cytochrome P450 (CYP) enzymes (Supersomes™) indicated a low extent of metabolism in the presence of CYP2C8 (0.032 μL/min/pmol CYP) and CYP2C19 (0.039 μL/min/pmol CYP), while no measurable loss was observed with the other tested CYP isoforms (Table S9).
DSM265 metabolite identification and profiling was conducted using pooled plasma samples obtained from single dose pharmacokinetic studies in animals. Five metabolites were identified (Tables S10, S11 and Fig. S6) with metabolism primarily occurring via mono-oxidation to form the circulating metabolite M4 [DSM450, identity confirmed by chemical synthesis (Fig. 1)], followed by glucuronidation to give M2 and M3, and dehydration to form M5. Based on mass spectrometry peak area ratios, M4 (DSM450) was estimated to be the main plasma metabolite in all species, representing 4–27% of the parent AUC with other metabolites being minor.
In vivo pharmacokinetic (PK) properties and formulation studies
To support toxicological studies and human dose predictions and to test formulation performance, a series of PK studies were conducted in rodents, dogs and monkeys. Consistent with prior data in rodents (14), the DSM265 PK profile in monkeys and dogs after intravenous dosing was characterized by very low plasma clearance, moderate to high volume of distribution, and a long half-life (10 – 45 h) (Table S12). Oral PK studies in dogs suggested that increases in AUC were dose proportional at lower doses (3 and 30 mg/kg) but less so at higher doses (Table S13 and S14). The oral bioavailability of DSM265 was moderate in dogs (~20%) however there was evidence of enterohepatic recirculation, with secondary peaks noted following feeding (Fig. 5). Increases in Cmax and AUC were observed when DSM265 was administered orally to dogs with food (Fig. 5 and Table S14).
Fig. 5. Oral pharmacokinetic profiles for DSM265 in dogs.

Pharmacokinetic behavior of DSM265 was evaluated after a single oral dose (10 mg/kg) of either free base as a suspension in 0.5% carboxymethyl cellulose, 0.4% Tween 80 or SDD formulation administered as a suspension in Methocel A4M, in either the fasted or fed state. Data represent the mean ± SD for 6 dogs. Primary data are provided in Table S21.
Salt selection studies identified the tosylate salt based on improved solubility in water (3-fold). It showed improved exposure in mice, but not in rats and dogs (Tables S13 and S14). Efforts to increase the dissolution and absorption profile of crystalline DSM265 included particle size reduction (nanomilling) and preparation of amorphous spray dried dispersions (SDD, containing 25% DSM265 load), with the latter increasing solubility in simulated intestinal fluids to approximately 100 μg/mL. Following oral dosing to dogs, increases in Cmax and AUC were obtained with the nanomilled free base, or with the SDD formulation (Fig. 5 and Table S14). The SDD formulation all but eliminated the food effect in dogs (Table S14) and was selected for further development. The amorphous properties of the SDD formulation were stable for at least up to 9 months at 40°C/75% relative humidity.
Human pharmacokinetic predictions
Human PK parameters were estimated using a physiologically-based pharmacokinetic (PBPK) model inputting experimental values for physicochemical properties, permeability, protein binding and erythrocyte partitioning. Since the in vitro intrinsic clearance determined in liver microsomes and hepatocytes underestimated the observed in vivo clearance in all species tested, allometric scaling of the unbound in vivo clearance in preclinical species was used to estimate a human clearance value of 0.25 L/h. The PBPK model predicted a human volume of distribution of 46 L and a half-life of approximately 130 h. Human plasma concentration versus time profiles were simulated for different dose levels (Figure S7) and suggested that a single dose in the range of 200–400 mg would likely maintain plasma concentrations above the plasma MPC (1–2 μg/mL) for at least 8 days.
In vitro safety assessment
Safety of DSM265 was assessed in a series of standard in vitro assays (Table S15). DSM265 showed minimal activity in a standard panel of 106 human receptors and ion channels (IC50 values > 4.2 μg/mL for the full panel) performed by Cerep Inc. (Poitiers, France) and when tested against a panel of human kinases (University of Dundee, Scotland). In human liver microsomes in vitro, DSM265 caused no measurable inhibition of any of the key CYP enzymes (IC50 values >10 μg/mL) (Table S15) and did not induce CYP enzymes in human hepatocytes (Table S15).
Genotoxicity
DSM265 and SF5-aniline (synthetic precursor) were tested in a conventional 5-strain bacterial reverse mutation assay (Ames test) for mutagenic activity (Table S15). There was no increase in the number of revertant colonies for either compound and both were judged to be non-mutagenic. Genotoxicity was also studied in an oral bone marrow micronucleus test in male CD1 mice (Table S15). There was no increase in micronucleated polychromatic erythrocytes in this study, thus combined with the negative result in the Ames test it was concluded that DSM265 was not genotoxic.
Evaluation of cardiac electrophysiological risk
DSM265 was tested for activity against the human ether-a-go-go related gene (hERG) K+ channel in a standard patch clamp assay. Inhibition of this channel has been associated with QT prolongation and arrhythmias (19). DSM265 showed some inhibition of the hERG channel (IC50 = 0.66 – 2.9 μg/mL; Table S15). Notably, the predicted unbound maximum plasma concentration (Cmax < 0.01 μg/mL) of DSM265 in humans (Fig. S7) is 70–320-fold lower than the IC50 for hERG channel inhibition, suggesting a very low likelihood of an interaction with the channel in humans at the expected therapeutic dose. To further assess its potential cardiac risk, DSM265 was tested in a rabbit cardiac ventricular wedge assay; no Torsade de Pointes (TdP)-type arrhythmias were observed (tested up to 5 μg/mL). DSM265 did not cause any significant prolongation of the QT and Tp-e interval at 1 or 0.5 Hz and there was no change in the ratio of Tp-e/QT interval (Fig. S8). Tp-e duration and the ratio of Tp-e/QT interval reflect dispersion of repolarization and increases in these parameters are thought to elevate the risk of TdP arrhythmia (19, 20). As a final confirmation, cardiac safety was evaluated by telemetry in vivo in conscious dogs at DSM265 oral doses of 30 and 300 mg/kg (plasma concentrations 24 h after the high dose were 1.3 μg/mL) (Table S16). All ECGs were within normal limits and there were no other adverse effects on cardiac rate, rhythm or blood pressure.
Exploratory toxicology studies
Safety of DSM265 was evaluated in rats, mice and dogs in both single-dose and repeat-dose oral toxicity studies (Table S16). In the repeat-dose studies, mice and rats were dosed daily however dogs were dosed every other day as the long half-life was expected to lead to significant accumulation. Dogs were dosed in the fed state to increase exposure. In the 7-day repeat dose study in rats the maximum tolerated dose (MTD) was 50 mg/kg/day for both males and females. Rats showed weight loss, signs of dehydration (e.g. increased blood urea nitrogen and hematocrit), decreases in white blood cell counts and gastrointestinal tract damage that was dose dependent. As this was an early stage exploratory study, toxicokinetic (TK) data were not collected. We reasoned that the significant sensitivity of rat DHODH to DSM265 (IC50 = 0.82 μg/mL) combined with the lower plasma protein binding and higher unbound concentrations relative to other species (Table S9) resulted in the rat being atypically sensitive to DSM265 making it a poor model to study potential DSM265 toxicity in humans. It was therefore decided to evaluate the toxicological effects of DSM265 in mice and dogs as both species had plasma protein binding levels that were similar to humans, and dog DHODH was significantly less sensitive to DSM265 inhibition than the rat enzyme.
DSM265 was well tolerated at all dose levels in mice (25 – 200 mg/kg/day q.d. for 7 days) and dogs (30 – 480 mg/kg every other day for 10 days) and there were no mortalities in either species. In mice, there was no observation of clinical effects or noted changes in pathology. In dogs receiving the highest dose, there were instances of vomiting and reduced food consumption. A slight increase in bilirubin was also observed but this was not accompanied by changes in any other liver function markers. Based on these data, the MTD in both species was considered to have exceeded the highest dose (MTD >200 mg/kg in mice and >480 mg/kg in dogs). TK analysis confirmed that plasma exposure in both species increased with increasing dose (Tables S17–S18). There was no evidence of DSM265 accumulation over the 7-day study in mice but despite the alternate day dose regimen in dogs, significant accumulation was still apparent.
The average plasma concentration within a dosing interval (Cav) has been suggested to be a useful parameter for evaluation of safety margins in toxicology studies (21). Plasma Cav values at the highest dose tested in the mouse (Cav = 11.7 μg/mL, Day 7) and dog (Cav = 16.4 μg/mL, Day 1) were 10 and 15-fold higher, respectively, than the MPC for maximum parasite clearance in the SCID mouse efficacy study (Table 2). Full TK data were not collected in the dog after the final dose so Cav could not be calculated, but given the noted accumulation the margin in dogs is likely to be ~2-fold higher than that calculated from day 1 data.
Evaluation of safety in G6PD-deficient mice
Because of the prevalence of G6PD-deficient patients in malaria endemic regions (1), DSM265 was tested for evidence of hemolytic toxicity in NOD-SCID mice engrafted with blood type A- G6PD-deficient human red blood cells. DSM265 was not hemolytic in this model, suggesting that it will be safe for dosing in G6PD-deficient patients (Fig. S9). In contrast, treatment with primaquine led to a substantial loss in human RBCs, an increase in mouse reticulocytes and resulted in a substantial increase in spleen weight.
Discussion
The triazolopyrimidine-based inhibitor DSM265 represents a new class of antimalarial agent and is the first compound targeting plasmodial DHODH to reach clinical development. DSM265 fits a unique niche in the worldwide antimalarial portfolio, displaying both blood and liver-stage activity and a sufficiently long predicted human half-life to support either single dose treatment or once weekly dosing for chemoprevention, giving it considerable advantage over current agents that are dosed daily or lack liver-stage activity. Herein, we have described a comprehensive set of assays profiling biological activity, pharmacokinetic properties and safety, all of which support the advancement of DSM265 to clinical trials to evaluate safety and efficacy in humans.
The activities of DSM265 across the Plasmodium life cycle are consistent with its mechanism of action in blocking formation of nucleotides required for RNA and DNA synthesis. DSM265 inhibited formation of blood and liver-stage parasites with similar potency, arresting growth prior to formation of the multinucleated schizont stage in both cases. Modest activity against formation of the multinucleated insect stage (oocyst) was also observed, suggesting that DSM265 could exert some transmission blocking activity, particularly if dosed with a gametocytocidal agent. DSM265 blocked growth earlier in the parasite life-cycle than pyrimethamine, likely due to the fact that DSM265 acts upstream of DHFR inhibitors in the pyrimidine biosynthetic pathway. The earlier growth arrest of DSM265-treated parasites may be a consequence of RNA depletion, as growth arrest appears to precede the burst of DNA biosynthesis required for schizont formation.
DHODH is a long duration compound with a predicted human half-life of more than 100 h and with the potential to be useful for either treatment or prevention of malaria. Minimal metabolism by hepatic CYP enzymes and very high plasma protein binding both likely contribute to the sustained exposure after oral dosing. For P. falciparum, a single dose of 200 – 400 mg is predicted to maintain plasma concentrations above the MPC for at least 8 days. DSM265 is one of only a handful of compounds with potent activity on liver-stage parasites distinguishing it from many other compounds in the malaria drug portfolio (9, 17). Indeed, of the Phase II clinical candidates the only other agent with liver-stage activity is KAF156 (22) and it has an observed human half-life that is shorter than predicted for DSM265 (23). Recently a new preclinical candidate targeting P. falciparum translation elongation factor was reported to have blood and liver stage activity as well as transmission blocking activity, but this compound, while promising, is still in early stage preclinical development (24) and unlike DSM265, has not yet advanced to human clinical trials. While compounds lacking liver-stage activity have been used for chemoprevention (e.g. chloroquine and mefloquine), liver-stage activity would be expected to boost prophylactic potential, particularly for compounds like DSM265 that show a lag phase before blood-stage killing begins. While DSM265 blocks formation of the liver-stage schizont, it apparently lacks activity against hypnozoites (based on the P. cynomolgi data). Thus, DSM265 would not be useful for radical cure of P. vivax. DSM265’s activity profile is most similar to atovaquone and other cytochrome bc1 inhibitors (e.g. ELQ-300) (25), which also block uridine nucleotide biosynthesis. However, notably, a long-duration compound such as DSM265 has potential advantages for use in chemoprevention compared to atovaquone, which is dosed daily.
Careful monitoring for development of resistance against clinically used antimalarials is clearly of paramount importance given the history of drugs lost due to widespread resistance. The multidrug-resistant P. falciparum Dd2 strain developed resistance to DSM265 relatively easily, with a profile similar to atovaquone at low drug concentrations. However, at higher concentrations, or in other strains, it was significantly more difficult to select for DSM265-resistant parasites, suggesting that overall it may be harder for resistance to DSM265 to emerge compared to atovaquone. Published data on chloroquine and KAF156 suggest a lower propensity to develop resistance than DSM265 (MIR of 108 in Dd2 using the same 3xEC50 protocol) (22, 26). Similar data are not available for the ATP4 inhibitor (KAE609) in clinical development, although its target was identified through sequencing of resistant parasites (27). However, an MIR as low as 107 against P. falciparum 3D7 was recently reported for another ATP4 inhibitor (SJ733) (28). It is not known how these in vitro data will translate to the clinical situation, but clearly our findings point to the need to closely monitor for the emergence of DSM265-resistant parasites during clinical development.
Mechanistic studies indicated that DSM265 resistance was linked to increased DHODH gene copy number or to point mutations in the inhibitor binding pocket, providing easy markers to monitor resistance in the field. Both mechanisms appear to be common routes for the parasite to develop resistance after in vitro selections with DHODH inhibitors. Selections with a related triazolopyrimidine DSM1 also led to gene amplification (29), while point mutations in the inhibitor binding site were observed after selections using several unrelated chemical series in addition to another triazolopyrimidine DSM74, although the G181C mutation that we observed in parasites resistant to DSM265 was not identified in that study (30). If resistant parasites emerge readily in early clinical studies, DSM265, like atovaquone, might be best positioned for chemoprevention. Long duration compounds with potential for preventative treatment are particularly needed in areas of seasonal malaria transmission (1), and DSM265 appears particularly well suited to this application. Given the historical precedence for resistance, even for drugs lacking a specific protein target (e.g. chloroquine), it is clear that a combination strategy is the only mechanism to prolong the usefulness of new clinical agents against malaria (10). DSM265 will be formulated in combination with another antimalarial compound to mitigate this risk. We found through drug combination tests that DSM265 antimalarial activity was additive with a wide selection of potential partner compounds, thus these compounds remain viable partners for DSM265.
DSM265 showed excellent safety in short term multiple-dose studies in mice and dogs, but it was less well tolerated in rats. Significant species differences were identified both in the inhibitory activity of DSM265 towards DHODH from various mammalian species and in plasma protein binding. These data suggested that higher unbound concentrations coupled with significant DHODH inhibition led to greater toxicity in rats. This hypothesis is supported by the observation that only in rats were the toxicological findings suggestive of a direct effect on the myeloid elements of the bone marrow, similar to those reported for the immunosuppressive drug leflunomide, a clinically utilized DHODH inhibitor (31, 32). As human DHODH is much less sensitive to DSM265 inhibition than the rodent and dog enzymes, similar toxicological effects are not expected to occur in humans. While there was evidence of low micromolar activity against the hERG channel in vitro, the unbound concentration in vivo will likely be too low for an interaction with the channel to occur. Cardiovascular studies in dogs showed no evidence of any effect on the ECG or any evidence that DSM265 has the potential to cause cardiac arrhythmias.
The discovery and development of DSM265 is a rare example of a target-based strategy leading to identification of a new antimalarial, as most compounds in the pipeline have been identified by phenotypic screens (5). The advantage of the target-based approach is that it provides a clear path to development both by simplifying lead optimization and by allowing us to understand the toxicological findings in the rat, and to move beyond them. Thus, knowledge of the target has been essential to the successful advancement of DSM265. The preclinical development studies described herein, combined with a favorable toxicity profile, led to selection of DSM265 as a clinical development candidate and to advancement into Phase I human studies. While the studies described herein are expected to be very predictive of a desirable outcome in humans, they are limited by their restriction to in vitro analysis and the use of animal models. The full potential of DSM265 as a drug to treat malaria will not be fully defined until data from human safety and efficacy studies is collected and reported. In particular, whether the human half-life and dose prediction accurately reflect what is observed in humans, whether the predicted efficacy is observed against the range of Plasmodium species present in the field, and to what extent drug resistance is observed when the compound is used to treat human patients, are open questions. Finally, the choice of whether DSM265 should be advanced for treatment or chemoprevention will depend on both progress made with other compounds in the pipeline and on the ongoing safety and efficacy evaluation of DSM265 in humans.
Materials and Methods
Study Design
The program objectives were to characterize the efficacy, pharmacokinetic properties and safety of DSM265 to support its preclinical development and enable the compound to advance to human clinical trials. Efficacy was evaluated at both the enzyme target level and against the parasite using in vitro and in vivo models of Plasmodium infection. Enzyme inhibitor activity was evaluated against a range of different Plasmodium and mammalian species to demonstrate activity and to show selectivity relative to both human and other mammalian species that would be used in toxicological models. Studies of Plasmodium growth or infection were designed to evaluate efficacy across the full range of life cycle stages, to determine how fast the compound kills parasites, to obtain preliminary data on possible drug resistance mechanisms based on in vitro selections and to define the PK/PD relationships. Drug-like properties were evaluated using a series of in vitro ADME studies, and pharmacokinetic analysis was performed across multiple species. Finally both in vitro and in vivo toxicological profiling was undertaken to assess safety.
Animal welfare
Animal experiments were approved by the institutional animal care and use committees (IACUC) for each of the experimental sites. All studies were conducted according to the appropriate legislation and institutional policies on the care and use of animals.
DSM265 synthesis, impurities and metabolite synthesis
Synthesis of DSM265
DSM265 (2-(1,1-difluoroethyl)-5-methyl-[N-[4-(pentafluorosulfanyl)phenyl]1,2,4]triazolo[1,5-a]pyrimidin-7-amine) was prepared as previously described (14) to 14 kilogram scale (99.2% purity) by Wuxi App Tec (Shanghai, China) (Scheme S1). The major impurity (about 0.6%) in this GMP batch was DSM430 (Fig. 1), which derives from an impurity in the SF5-aniline raw material.
Synthesis of DSM430
DSM430 (2-(1,1-difluoroethyl)-N-(3-fluoro-4-(pentafluoro-l6-sulfanyl)phenyl)-5-methyl-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine) was synthesized using the same basic scheme as for DSM265 (Scheme S1) to 99% purity. Experimental data are as follows: MS 430.1; NMR (D6-DMSO) shows peaks at 2.15(3H, t), 2.51 (3H, s), 6.95 (1H, s), 7.5–7.6 (2H, m) and 8.05 (1H, m). 19F NMR (CDCl3) shows peaks at −104 ppm (aromatic F), −90.5 (CF3) and −68.8 ppm (SF5).
Synthesis of DSM450
DSM450 (2-(1,1-difluoroethyl)-7-((4-(pentafluoro-l6-sulfanyl)phenyl)amino)-[1,2,4]triazolo[1,5-a]pyrimidin-5-yl)methanol) was identified as the major in vivo metabolite of DSM265 and was synthesized to 99% purity by Syngene (Scheme S2). Experimental data are as follows: (m/z) 432.0 (M+) 1H NMR (400 MHz, DMSO-d6): δ 10.79 (s, 1H), 8.03 (d, J = 8.8 Hz, 2H), 7.69 (d, J = 8.6 Hz, 2H), 6.90 (s, 1H), 5.70 (t, J = 5.84 Hz, 3H), 4.53 (d, J = 5.92Hz, 2H), 2.13 (t, 19.1Hz 3H).
Nomenclature
We use the nomenclature EC50 (effective concentration) for concentration response curves on parasites, while we use IC50 (inhibitory concentration) for enzyme data as this distinction helps clarify whether the data are cell-based or enzyme-based.
DHODH purification and assays
DHODHs were expressed as recombinant proteins in E. coli and purified as previously described (14, 16). New expression constructs are described in supplemental methods. Steady-state kinetic analysis was performed using a 2,5-dichloroindophenol (DCIP)-based spectrophotometric method as described (14). Enzyme and substrate concentrations were: DHODH (E = 5–10 nM), substrates (0.2 mM L-dihydroorotate and 0.02 mM CoQd). DSM265 stock solutions (100x) were made in DMSO. Data were fitted to the log[I] vs response (three parameters) equation or for compounds where the IC50 >10 μM to the standard IC50 equation (Y=1/(1+X/(IC50))) in Graphpad Prism. Error represents the 95% confidence interval of the fit.
X-ray structure determination
The X-ray structure of PfDHODH in complex with DSM265 was solved as described in supplemental methods. The structure was displayed with PyMol (Pymol Molecular Graphics System (2000) DeLano Scientific, San Carlos, CA).
In vitro parasite efficacy assays
Blood-stage assays
P. falciparum cells were propagated in RPMI-1640 containing 0.5% Albumax-II (14). EC50 determinations for P. falciparum 3D7 cells and drug-resistant parasites were performed using the SYBR-green 72 h growth assay (16). Phenotypic assessment of blood-stage 3D7 parasites before and after treatment of DSM265 in comparison to control compounds was conducted at 10xIC50. Initial parasitemia was 1% (92% rings) at a hematocrit of 2%. Parasites were stained with Giemsa after drug exposure (48 h) and scored by microscopy with minimally 200 cells per condition. Parasite kill curves were assessed on P. falciparum 3D7 cells by incubating parasites with drug for the indicated time followed by washing and replating in drug free media to assess the number of remaining viable parasites as described (33). EC50 values reported for this study are for a 48 h growth assay.
Liver-stage assays
P. falciparum NF54 liver-stage assays were performed in media containing 10% FCS serum as described (34). Statistical significance was determined by ordinary one-way ANOVA with Dunnett’s multiple comparison using Graphpad Prism.
Sexual stage assays
Assays were performed as described in supplemental methods.
Curve fitting
For EC50 determinations data were fitted to the log(inhibitor) versus response –variable slope (four parameter) model in Graphpad Prism and error represents the 95% confidence interval of the fit.
Selection of DSM265-resistant parasites in vitro
Asynchronous cultures of cloned Plasmodium falciparum Dd2, K1 and HB3 parasite lines grown in culture medium containing 0.5% Albumax cell were used in continuous challenge experiments (29, 35). Cultures were maintained under DSM265 selection pressure for a minimum of 60 days. Gene copy number was determined as described (29).
In vivo parasite efficacy assays
SCID mouse efficacy study
NOD-scid IL-2Rγnull (NSG) mice (Jackson Laboratory, USA) (23–36 g) engrafted with human erythrocytes (36) were infected with 20 × 106 P. falciparum Pf3D70087/N9 by intravenous injection. DSM265 tosylate salt or chloroquine were administered orally in vehicle (saline solution for chloroquine; 0.5% w/v sodium carboxymethylcellulose, 0.5% v/v benzyl alcohol, 0.4% v/v Tween 80 in water for DSM265). DSM265 was administered b.i.d. at target doses ranging from 0.5 to 75 mg/kg (free base equivalent) starting on day 3 post infection. Formulation concentrations were measured to obtain actual doses administered. Parasitemia was monitored by flow cytometry. Blood DSM265 levels were measured by LC/MS/MS as described (14). Human biological samples were sourced ethically and their research use was in accordance with the terms of the informed consents.
Pharmacokinetic studies, physiochemical and in vitro ADME studies
Pharmacokinetic studies were conducted in female Swiss outbred mice, male Sprague Dawley rats, male beagle dogs, and female macaque monkeys. DSM265 was administered IV and orally in a range of formulations. Blood samples were collected into tubes containing anticoagulant, plasma was separated by centrifugation, and DSM265 and metabolite concentrations were determined by LC/MS. Physiochemical and in vitro ADME study methods are described in the supplemental methods.
Prediction of human pharmacokinetic parameters and human dose
Human clearance was estimated by allometric scaling of the unbound in vivo clearance in mice, rats, dogs, and monkeys. Distributional processes were estimated using a physiologically-based pharmacokinetic model (Gastroplus™, Simulations Plus Inc.), inputting the estimated human clearance, the measured Log DpH 7.4, apparent solubility from the amorphous SDD formulation, Caco-2 permeability, plasma protein binding, and blood to plasma ratio. Human plasma concentration versus time profiles were simulated for different dose levels to estimate the dose range that would provide concentrations above the MPC (1–2 μg/mL) for a period of more than 8 days.
Cyp inhibition and induction
CYP inhibition studies were performed using a substrate specific approach in human liver microsomes. Methods for this assay and for CYP induction assays are described in supplemental methods.
Rabbit wedge
The rabbit ventricular wedge assay was performed as described (20) (supplemental methods).
Hemolytic toxicity assay
Testing for hemolytic toxicity in G6PD-deficient human red blood cells was performed in NOD-SCID mice engrafted with blood from a G6PD A- donor (37).
Genotoxicity
Ames testing
DSM265 and the SF5-aniline (synthetic precursor) were examined for mutagenic activity in the Salmonella typhimurium-E. coli/microsome plate incorporation assay (supplemental methods). In vivo genotoxicity study. The in vivo micronuclei study was performed in male CD-1 mice under GLP conditions (supplemental methods)
Toxicology studies in rats, mice and dogs
Exploratory toxicological studies were performed in rats, mice and dogs (full details provided in the supplemental methods). Animals were monitored daily for clinical changes. Body weight and food consumption were also monitored. Hematology, clinical chemistry and histopathology were performed by standard methods. Dog cardiac studies were performed by standard methods.
Statistical Analysis
Statistical analyses were performed using GraphPad Prism 6.0 (GraphPad Inc). Ordinary one-way ANOVA with Dunnett’s multiple comparison or 1-way ANOVA with Bonferroni’s post test-were used to compare three or more groups. For enzyme and parasite IC50 or EC50 determination, data were fitted to the specified model as described in the methods using nonlinear least squares analysis and the mean and 95% confidence intervals are reported. Minimally, triplicate data were collected for each compound concentration and error bars on graphs represent the standard error of the mean (SEM). Pharmacokinetic data were collected typically for 2–6 animals and standard deviation is provided for data sets containing n>2. Details specific to each study are provided in the figure legends.
Supplementary Material
Fig. S1. (Fo-Fc) map for DSM265:PfDHODH binding-site
Fig. S2. DHODH sequence alignment
Fig. S3. In vitro parasite killing curves
Fig. S4. Activity of DSM265 against Plasmodium cynomolgi large (liver schizonts) and small (hypnozoites) forms
Fig. S5. The effect of DSM265 treatment on P. falciparum Pf3D70087/N9 in vivo
Fig. S6A. Proposed biotransformation pathways of DSM265 in plasma of mice, rabbits, monkeys and dogs.
Fig. S6B. Plasma concentrations of DSM265 and DSM450 (hydroxy metabolite).
Fig. S7. Simulated human plasma profiles using a physiologically-based pharmacokinetic model (Gastroplus™)
Fig. S8. Effect on ECG in the rabbit cardiac ventricular wedge assay
Fig. S9. Evaluation of the effects of DSM265 on G6PD deficient human RBCs engrafted into a NOD-SCID mouse
Table S1. PfDHODH-DSM265 X-ray diffraction data and refinement statistics
Table S2. In Vitro antimalarial activity of DSM265
Table S3A. Blood pharmacokinetic data for DSM265 in SCID mice
Table S3B. SCID mouse in vivo antimalarial activity
Table S3C. SCID mouse parasitemia levels
Table S3D. SCID mouse pharmacokinetic individual time point data
Table S4A. Selections for DSM265 resistant parasites in P. falciparum Dd2: Rathod lab
Table S4B. Selections for Atovaquone resistant parasites in P. falciparum Dd2: Rathod lab
Table S4C. Selections for DSM265 resistant parasites in P. falciparum Dd2: Fidock Lab
Table S4D. Selections for Atovaquone resistant parasites in P. falciparum Dd2: Fidock Lab
Table S4E. Selections for DSM265 resistant parasites in P. falciparum K1: Fidock Lab
Table S4F. Selections for DSM265 and Atovaquone P. falciparum HB3: Rathod Lab
Table S5. Summary of DSM265 resistant clones: analysis of parasites in whole cell assays
Table S6. Kinetic analysis of PfDHODH mutants
Table S7. Drug combination analysis
Table S8A. Stability data for DSM265 free base and tosylate salt
Table S8B. Solubility data for DSM265 free base
Table S9. In vitro ADME data for DSM265
Table S10. In vivo metabolite identification
Table S11. Relative plasma exposures of DSM265 metabolites in mice, rabbits, monkeys and dogs
Table S12. DSM265 plasma pharmacokinetics after a single intravenous dose in mice, rats, dogs and monkey
Table S13. DSM265 plasma pharmacokinetics after a single oral dose of DSM265 in mice, rats, dogs and monkeys
Table S14. Effect of salt form, formulation and food on the DSM265 plasma pharmacokinetics after oral dosing in beagle dogs
Table S15. Safety pharmacology
Table S16. Exploratory toxicology studies (non-GLP) in rodents and dogs
Table S17A. Toxicokinetic parameters on Day 1 and Day 7 in a mouse 7 day toxicology study
Table S17B. Individual mouse plasma concentrations 7 day toxicology study 25 mg/kg
Table S17C. Individual mouse plasma concentrations 7 day toxicology study 75 mg/kg
Table S17D. Individual mouse plasma concentrations 7 day toxicology study 200 mg/kg
Table S18A. Toxicokinetic data from a 10 day toxicology study in male beagle dogs
Table S18B. Toxicokinetic parameters day 1 of toxicology study in male beagle dogs
Table. S19. Primary data supporting Fig. 2
Table. S20. Primary data supporting Fig. 3A,B
Table. S21. Primary data supporting Fig. 5
Summary.
Malaria kills 0.6 million people annually yet current malaria drugs are no longer fully effective because the parasite that causes malaria is becoming resistant to these agents. We have identified a new drug that kills both drug sensitive and drug resistant malaria parasites by targeting the ability of the parasite to synthesize the nucleotide precursors required for synthesis of DNA and RNA. This drug kills parasites in both the blood and liver and is sufficiently long acting that it is expected to cure malaria after a single dose or to be effective if dosed weekly for chemoprevention.
Acknowledgments
The authors thank Dr. Leonard D. Shultz and The Jackson Laboratory for providing access to non-obese diabetic scid IL2Rγc null mice through their collaboration with GSK Tres Cantos Medicines Development Campus. We are also indebted to Dr. Tim Wells for thoroughly reviewing the manuscript. Results shown in this report are derived from work performed at Argonne National Laboratory, Structural Biology Center at the Advanced Photon Source. Argonne is operated by UChicago Argonne, LLC, for the U.S. Department of Energy, Office of Biological and Environmental Research under contract DE-AC02-06CH11357.
Funding: This work was supported by funds from the United States National Institutes of Health grants, U01AI075594 (to IB, MAP, PKR and SAC) and R01AI103947 (to MAP and PKR) and from Medicines for Malaria Venture (MMV). MAP acknowledges the support of the Welch Foundation (I-1257). MAP holds the Beatrice and Miguel Elias Distinguished Chair in Biomedical Science and the Carolyn R. Bacon Professorship in Medical Science and Education. BPRC (AZ and CK) and Columbia University (CN and DAF) were supported by funding from the Medicines for Malaria Venture. BBRC was also supported by a translational research grant (WT078285) from the Wellcome Trust.
Footnotes
Author contributions: MAP and SAC wrote the paper and contributed to overall study design and data interpretation; MAP and FEM designed, performed and or analyzed studies on enzyme inhibition; XD and DRT performed the protein X-ray crystallography; SAC, KLW, KM, YL designed, supervised and or analyzed ADME and PK studies; PKR, SK, TL, JNB performed or supervised chemical synthesis; JW, JWN, PKR, MAP, CN, DAF designed, performed or analyzed studies on in vitro drug resistance; SNB, SM, SW, MLM, FJGB, LMSA, MSM, MBJD, SFB, IA, AMZ, CK, RS, KD, VMA, SD, MD, RS, AR, IB, XD, BC, DL, JNB, MAP designed, performed and or analyzed in vitro and in vivo parasite efficacy studies; JH, JL, YC, AS performed, designed or interpreted rabbit wedge studies; JL, AD, KM, JM, TR designed and supervised toxicologic studies; JG, LI, ER, RR performed or supervised in vivo toxicologic studies in rats and dogs; AD, KM, JM, MJR interpreted the results from toxicologic studies.
Competing interests: MAP, SAC, PKR and JNB hold a pending patent covering DSM265 and related compounds (Antimalarial agents that are inhibitors of dihydroorotate dehydrogenase – WO2011/041304). TL and AD are paid consultants to MMV. SB is a paid consultant to Hepregen. RS and KD hold stock in TropIQ.
Data Deposition: The X-ray structures of DSM265 bound to PfDHODH have been deposited in the PDB database [form I (4RX0) and form II (5BOO)] and the DNA sequence of mini-pig DHODH has been deposited to Genbank (KR108306).
This manuscript has been accepted for publication in Science Translational Medicine. This version has not undergone final editing. Please refer to the complete version of record at www.sciencetranslationalmedicine.org/. The manuscript may not be reproduced or used in any manner that does not fall within the fair use provisions of the Copyright Act without the prior, written permission of AAAS.
References
- 1.White NJ, Pukrittayakamee S, Hien TT, Faiz MA, Mokuolu OA, Dondorp AM. Malaria. Lancet. 2014;383:723–735. doi: 10.1016/S0140-6736(13)60024-0. [DOI] [PubMed] [Google Scholar]
- 2.Miller LH, Ackerman HC, Su XZ, Wellems TE. Malaria biology and disease pathogenesis: insights for new treatments. Nat Med. 2013;19:156–167. doi: 10.1038/nm.3073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.WHO. World Malaria Report. 2014 http://www.who.int/malaria/publications/world_malaria_report_2014/en.
- 4.Cotter C, Sturrock HJ, Hsiang MS, Liu J, Phillips AA, Hwang J, Gueye CS, Fullman N, Gosling RD, Feachem RG. The changing epidemiology of malaria elimination: new strategies for new challenges. Lancet. 2013;382:900–911. doi: 10.1016/S0140-6736(13)60310-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Burrows JN, Burlot E, Campo B, Cherbuin S, Jeanneret S, Leroy D, Spangenberg T, Waterson D, Wells TN, Willis P. Antimalarial drug discovery - the path towards eradication. Parasitology. 2014;141:128–139. doi: 10.1017/S0031182013000826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Price RN, Nosten F. Single-dose radical cure of Plasmodium vivax: a step closer. Lancet. 2014;383:1020–1021. doi: 10.1016/S0140-6736(13)62672-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Menard D, Ariey F. Towards real-time monitoring of artemisinin resistance. Lancet Infect Dis. 2015;15:367–368. doi: 10.1016/S1473-3099(15)70046-0. [DOI] [PubMed] [Google Scholar]
- 8.Anthony MP, Burrows JN, Duparc S, Moehrle JJ, Wells TN. The global pipeline of new medicines for the control and elimination of malaria. Malaria journal. 2012;11:316. doi: 10.1186/1475-2875-11-316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yuthavong Y, Tarnchompoo B, Vilaivan T, Chitnumsub P, Kamchonwongpaisan S, Charman SA, McLennan DN, White KL, Vivas L, Bongard E, Thongphanchang C, Taweechai S, Vanichtanankul J, Rattanajak R, Arwon U, Fantauzzi P, Yuvaniyama J, Charman WN, Matthews D. Malarial dihydrofolate reductase as a paradigm for drug development against a resistance-compromised target. Proc Natl Acad Sci U S A. 2012;109:16823–16828. doi: 10.1073/pnas.1204556109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Duparc S, Lanza C, Ubben D, Borghini-Fuhrer I, Kellam L. Optimal dose finding for novel antimalarial combination therapy. Trop Med Int Health. 2012;17:409–413. doi: 10.1111/j.1365-3156.2012.02963.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Burrows JN, Hooft van Huijsduijnen R, Mohrle JJ, Oeuvray C, Wells TN. Designing the next generation of medicines for malaria control and eradication. Malar J. 2013;12:187. doi: 10.1186/1475-2875-12-187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stocks PA, Barton V, Antoine T, Biagini GA, Ward SA, O’Neill PM. Novel inhibitors of the Plasmodium falciparum electron transport chain. Parasitology. 2014;141:50–65. doi: 10.1017/S0031182013001571. [DOI] [PubMed] [Google Scholar]
- 13.Phillips MA, Rathod PK. Plasmodium dihydroorotate dehydrogenase: a promising target for novel anti-malarial chemotherapy. Infect Disord Drug Targets. 2010;10:226–239. doi: 10.2174/187152610791163336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Coteron JM, Marco M, Esquivias J, Deng X, White KL, White J, Koltun M, El Mazouni F, Kokkonda S, Katneni K, Bhamidipati R, Shackleford DM, Angulo-Barturen I, Ferrer SB, Jimenez-Diaz MB, Gamo FJ, Goldsmith EJ, Charman WN, Bathurst I, Floyd D, Matthews D, Burrows JN, Rathod PK, Charman SA, Phillips MA. Structure-guided lead optimization of triazolopyrimidine-ring substituents identifies potent Plasmodium falciparum dihydroorotate dehydrogenase inhibitors with clinical candidate potential. J Med Chem. 2011;54:5540–5561. doi: 10.1021/jm200592f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gujjar R, El Mazouni F, White KL, White J, Creason S, Shackleford DM, Deng X, Charman WN, Bathurst I, Burrows J, Floyd DM, Matthews D, Buckner FS, Charman SA, Phillips MA, Rathod PK. Lead-optimization of aryl and aralkyl amine based triazolopyrimidine inhibitors of Plasmodium falciparum dihydroorotate dehydrogenase with anti-malarial activity in mice. J Med Chem. 2011;54:3935–3949. doi: 10.1021/jm200265b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Deng X, Kokkonda S, El Mazouni F, White J, Burrows JN, Kaminsky W, Charman SA, Matthews D, Rathod PK, Phillips MA. Fluorine modulates species selectivity in the triazolopyrimidine class of Plasmodium falciparum dihydroorotate dehydrogenase inhibitors. J Med Chem. 2014;57:5381–5394. doi: 10.1021/jm500481t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Delves M, Plouffe D, Scheurer C, Meister S, Wittlin S, Winzeler EA, Sinden RE, Leroy D. The activities of current antimalarial drugs on the life cycle stages of Plasmodium: a comparative study with human and rodent parasites. PLoS medicine. 2012;9:e1001169. doi: 10.1371/journal.pmed.1001169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ding XC, Ubben D, Wells TN. A framework for assessing the risk of resistance for anti-malarials in development. Malar J. 2012;11:292. doi: 10.1186/1475-2875-11-292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kannankeril PJ, Roden DM. When should QT be measured? Summer solstice or Christmas Eve? Heart Rhythm. 2007;4:282–283. doi: 10.1016/j.hrthm.2006.11.027. [DOI] [PubMed] [Google Scholar]
- 20.Liu T, Brown BS, Wu Y, Antzelevitch C, Kowey PR, Yan GX. Blinded validation of the isolated arterially perfused rabbit ventricular wedge in preclinical assessment of drug-induced proarrhythmias. Heart Rhythm. 2006;3:948–956. doi: 10.1016/j.hrthm.2006.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Smith DA, Morgan P, Vogel WM, Walker DK. The use of C(av) rather than AUC in safety assessment. Regul Toxicol Pharmacol. 2010;57:70–73. doi: 10.1016/j.yrtph.2010.01.001. [DOI] [PubMed] [Google Scholar]
- 22.Kuhen KL, Chatterjee AK, Rottmann M, Gagaring K, Borboa R, Buenviaje J, Chen Z, Francek C, Wu T, Nagle A, Barnes SW, Plouffe D, Lee MC, Fidock DA, Graumans W, van de Vegte-Bolmer M, van Gemert GJ, Wirjanata G, Sebayang B, Marfurt J, Russell B, Suwanarusk R, Price RN, Nosten F, Tungtaeng A, Gettayacamin M, Sattabongkot J, Taylor J, Walker JR, Tully D, Patra KP, Flannery EL, Vinetz JM, Renia L, Sauerwein RW, Winzeler EA, Glynne RJ, Diagana TT. KAF156 is an antimalarial clinical candidate with potential for use in prophylaxis, treatment, and prevention of disease transmission. Antimicrob Agents Chemother. 2014;58:5060–5067. doi: 10.1128/AAC.02727-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Leong FJ, Zhao R, Zeng S, Magnusson B, Diagana TT, Pertel P. A first-in-human randomized, double-blind, placebo-controlled, single- and multiple-ascending oral dose study of novel Imidazolopiperazine KAF156 to assess its safety, tolerability, and pharmacokinetics in healthy adult volunteers. Antimicrob Agents Chemother. 2014;58:6437–6443. doi: 10.1128/AAC.03478-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Baragana B, Hallyburton I, Lee MC, Norcross NR, Grimaldi R, Otto TD, Proto WR, Blagborough AM, Meister S, Wirjanata G, Ruecker A, Upton LM, Abraham TS, Almeida MJ, Pradhan A, Porzelle A, Martinez MS, Bolscher JM, Woodland A, Norval S, Zuccotto F, Thomas J, Simeons F, Stojanovski L, Osuna-Cabello M, Brock PM, Churcher TS, Sala KA, Zakutansky SE, Jimenez-Diaz MB, Sanz LM, Riley J, Basak R, Campbell M, Avery VM, Sauerwein RW, Dechering KJ, Noviyanti R, Campo B, Frearson JA, Angulo-Barturen I, Ferrer-Bazaga S, Gamo FJ, Wyatt PG, Leroy D, Siegl P, Delves MJ, Kyle DE, Wittlin S, Marfurt J, Price RN, Sinden RE, Winzeler EA, Charman SA, Bebrevska L, Gray DW, Campbell S, Fairlamb AH, Willis PA, Rayner JC, Fidock DA, Read KD, Gilbert IH. A novel multiple-stage antimalarial agent that inhibits protein synthesis. Nature. 2015;522:315–320. doi: 10.1038/nature14451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nilsen A, LaCrue AN, White KL, Forquer IP, Cross RM, Marfurt J, Mather MW, Delves MJ, Shackleford DM, Saenz FE, Morrisey JM, Steuten J, Mutka T, Li Y, Wirjanata G, Ryan E, Duffy S, Kelly JX, Sebayang BF, Zeeman AM, Noviyanti R, Sinden RE, Kocken CH, Price RN, Avery VM, Angulo-Barturen I, Jimenez-Diaz MB, Ferrer S, Herreros E, Sanz LM, Gamo FJ, Bathurst I, Burrows JN, Siegl P, Guy RK, Winter RW, Vaidya AB, Charman SA, Kyle DE, Manetsch R, Riscoe MK. Quinolone-3-diarylethers: a new class of antimalarial drug. Sci Transl Med. 2013;5:177ra137. doi: 10.1126/scitranslmed.3005029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cooper RA, Ferdig MT, Su XZ, Ursos LM, Mu J, Nomura T, Fujioka H, Fidock DA, Roepe PD, Wellems TE. Alternative mutations at position 76 of the vacuolar transmembrane protein PfCRT are associated with chloroquine resistance and unique stereospecific quinine and quinidine responses in Plasmodium falciparum. Mol Pharmacol. 2002;61:35–42. doi: 10.1124/mol.61.1.35. [DOI] [PubMed] [Google Scholar]
- 27.White NJ, Pukrittayakamee S, Phyo AP, Rueangweerayut R, Nosten F, Jittamala P, Jeeyapant A, Jain JP, Lefevre G, Li R, Magnusson B, Diagana TT, Leong FJ. Spiroindolone KAE609 for falciparum and vivax malaria. N Engl J Med. 2014;371:403–410. doi: 10.1056/NEJMoa1315860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jimenez-Diaz MB, Ebert D, Salinas Y, Pradhan A, Lehane AM, Myrand-Lapierre ME, O’Loughlin KG, Shackleford DM, Justino de Almeida M, Carrillo AK, Clark JA, Dennis AS, Diep J, Deng X, Duffy S, Endsley AN, Fedewa G, Guiguemde WA, Gomez MG, Holbrook G, Horst J, Kim CC, Liu J, Lee MC, Matheny A, Martinez MS, Miller G, Rodriguez-Alejandre A, Sanz L, Sigal M, Spillman NJ, Stein PD, Wang Z, Zhu F, Waterson D, Knapp S, Shelat A, Avery VM, Fidock DA, Gamo FJ, Charman SA, Mirsalis JC, Ma H, Ferrer S, Kirk K, Angulo-Barturen I, Kyle DE, DeRisi JL, Floyd DM, Guy RK. (+)-SJ733, a clinical candidate for malaria that acts through ATP4 to induce rapid host-mediated clearance of Plasmodium. Proc Natl Acad Sci U S A. 2014;111:E5455–5462. doi: 10.1073/pnas.1414221111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Guler JL, Freeman DL, Ahyong V, Patrapuvich R, White J, Gujjar R, Phillips MA, DeRisi J, Rathod PK. Asexual populations of the human malaria parasite, Plasmodium falciparum, use a two-step genomic strategy to acquire accurate, beneficial DNA amplifications. PLoS Pathog. 2013;9:e1003375. doi: 10.1371/journal.ppat.1003375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ross LS, Javier Gamo F, Lafuente-Monasterio MJ, Singh OM, Rowland P, Wiegand RC, Wirth DF. In vitro resistance selections for Plasmodium falciparum dihydroorotate dehydrogenase inhibitors give mutants with multiple point mutations in the drug-binding site and altered growth. J Biol Chem. 2014;289:17980–17995. doi: 10.1074/jbc.M114.558353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Keen HI, Conaghan PG, Tett SE. Safety evaluation of leflunomide in rheumatoid arthritis. Expert Opin Drug Saf. 2013;12:581–588. doi: 10.1517/14740338.2013.798299. [DOI] [PubMed] [Google Scholar]
- 32.FDA. Leflunomide FDA Drug Approval Package: NDA 20905. 1998 http://www.accessdata.fda.gov/drugsatfda_docs/nda/98/20905_arava.cfm.
- 33.Sanz LM, Crespo B, De-Cozar C, Ding XC, Llergo JL, Burrows JN, Garcia-Bustos JF, Gamo FJ. P. falciparum in vitro killing rates allow to discriminate between different antimalarial mode-of-action. PloS one. 2012;7:e30949. doi: 10.1371/journal.pone.0030949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.March S, Ng S, Velmurugan S, Galstian A, Shan J, Logan DJ, Carpenter AE, Thomas D, Sim BK, Mota MM, Hoffman SL, Bhatia SN. A microscale human liver platform that supports the hepatic stages of Plasmodium falciparum and vivax. Cell Host Microbe. 2013;14:104–115. doi: 10.1016/j.chom.2013.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ekland EH, Schneider J, Fidock DA. Identifying apicoplast-targeting antimalarials using high-throughput compatible approaches. Faseb J. 2011;25:3583–3593. doi: 10.1096/fj.11-187401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jimenez-Diaz MB, Mulet T, Viera S, Gomez V, Garuti H, Ibanez J, Alvarez-Doval A, Shultz LD, Martinez A, Gargallo-Viola D, Angulo-Barturen I. Improved murine model of malaria using Plasmodium falciparum competent strains and non-myelodepleted NOD-scid IL2Rgammanull mice engrafted with human erythrocytes. Antimicrob Agents Chemother. 2009;53:4533–4536. doi: 10.1128/AAC.00519-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rochford R, Ohrt C, Baresel PC, Campo B, Sampath A, Magill AJ, Tekwani BL, Walker LA. Humanized mouse model of glucose 6-phosphate dehydrogenase deficiency for in vivo assessment of hemolytic toxicity. Proc Natl Acad Sci U S A. 2013;110:17486–17491. doi: 10.1073/pnas.1310402110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Baldwin J, Michnoff CH, Malmquist NA, White J, Roth MG, Rathod PK, Phillips MA. High-throughput screening for potent and selective inhibitors of Plasmodium falciparum dihydroorotate dehydrogenase. J Biol Chem. 2005;280:21847–21853. doi: 10.1074/jbc.M501100200. [DOI] [PubMed] [Google Scholar]
- 39.Malmquist NA, Gujjar R, Rathod PK, Phillips MA. Analysis of Flavin Oxidation and Electron-Transfer Inhibition in Plasmodium falciparum Dihydroorotate Dehydrogenase. Biochemistry. 2008;47:2466–2475. doi: 10.1021/bi702218c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Phillips MA, Gujjar R, Malmquist NA, White J, El Mazouni F, Baldwin J, Rathod PK. Triazolopyrimidine-based dihydroorotate dehydrogenase inhibitors with potent and selective activity against the malaria parasite, Plasmodium falciparum. J Med Chem. 2008;51:3649–3653. doi: 10.1021/jm8001026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Deng X, Gujjar R, El Mazouni F, Kaminsky W, Malmquist NA, Goldsmith EJ, Rathod PK, Phillips MA. Structural plasticity of malaria dihydroorotate dehydrogenase allows selective binding of diverse chemical scaffolds. J Biol Chem. 2009;284:26999–27009. doi: 10.1074/jbc.M109.028589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 43.Evans PR. An introduction to data reduction: space-group determination, scaling and intensity statistics. Acta Crystallogr D Biol Crystallogr. 2011;67:282–292. doi: 10.1107/S090744491003982X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.McCoy AJ. Solving structures of protein complexes by molecular replacement with Phaser. Acta Crystallogr D Biol Crystallogr. 2007;63:32–41. doi: 10.1107/S0907444906045975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D Biol Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dembele L, Gego A, Zeeman AM, Franetich JF, Silvie O, Rametti A, Le Grand R, Dereuddre-Bosquet N, Sauerwein R, van Gemert GJ, Vaillant JC, Thomas AW, Snounou G, Kocken CH, Mazier D. Towards an in vitro model of Plasmodium hypnozoites suitable for drug discovery. PloS one. 2011;6:e18162. doi: 10.1371/journal.pone.0018162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zeeman AM, van Amsterdam SM, McNamara CW, Voorberg-van der Wel A, Klooster EJ, van den Berg A, Remarque EJ, Plouffe DM, van Gemert GJ, Luty A, Sauerwein R, Gagaring K, Borboa R, Chen Z, Kuhen K, Glynne RJ, Chatterjee AK, Nagle A, Roland J, Winzeler EA, Leroy D, Campo B, Diagana TT, Yeung BK, Thomas AW, Kocken CH. KAI407, a potent non-8-aminoquinoline compound that kills Plasmodium cynomolgi early dormant liver stage parasites in vitro. Antimicrob Agents Chemother. 2014;58:1586–1595. doi: 10.1128/AAC.01927-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.D’Alessandro S, Silvestrini F, Dechering K, Corbett Y, Parapini S, Timmerman M, Galastri L, Basilico N, Sauerwein R, Alano P, Taramelli D. A Plasmodium falciparum screening assay for anti-gametocyte drugs based on parasite lactate dehydrogenase detection. J Antimicrob Chemother. 2013;68:2048–2058. doi: 10.1093/jac/dkt165. [DOI] [PubMed] [Google Scholar]
- 50.Duffy S, Avery VM. Identification of inhibitors of Plasmodium falciparum gametocyte development. Malar J. 2013;12:408. doi: 10.1186/1475-2875-12-408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Delves MJ, Ramakrishnan C, Blagborough AM, Leroy D, Wells TN, Sinden RE. A high-throughput assay for the identification of malarial transmission-blocking drugs and vaccines. Int J Parasitol. 2012;42:999–1006. doi: 10.1016/j.ijpara.2012.08.009. [DOI] [PubMed] [Google Scholar]
- 52.Delves MJ, Ruecker A, Straschil U, Lelievre J, Marques S, Lopez-Barragan MJ, Herreros E, Sinden RE. Male and female Plasmodium falciparum mature gametocytes show different responses to antimalarial drugs. Antimicrob Agents Chemother. 2013;57:3268–3274. doi: 10.1128/AAC.00325-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Snyder C, Chollet J, Santo-Tomas J, Scheurer C, Wittlin S. In vitro and in vivo interaction of synthetic peroxide RBx11160 (OZ277) with piperaquine in Plasmodium models. Exp Parasitol. 2007;115:296–300. doi: 10.1016/j.exppara.2006.09.016. [DOI] [PubMed] [Google Scholar]
- 54.Fivelman QL, Adagu IS, Warhurst DC. Modified fixed-ratio isobologram method for studying in vitro interactions between atovaquone and proguanil or dihydroartemisinin against drug-resistant strains of Plasmodium falciparum. Antimicrob Agents Chemother. 2004;48:4097–4102. doi: 10.1128/AAC.48.11.4097-4102.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Schleiferbock S, Scheurer C, Ihara M, Itoh I, Bathurst I, Burrows JN, Fantauzzi P, Lotharius J, Charman SA, Morizzi J, Shackleford DM, White KL, Brun R, Wittlin S. In vitro and in vivo characterization of the antimalarial lead compound SSJ-183 in Plasmodium models. Drug Des Devel Ther. 2013;7:1377–1384. doi: 10.2147/DDDT.S51298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jantratid E, Janssen N, Reppas C, Dressman JB. Dissolution media simulating conditions in the proximal human gastrointestinal tract: an update. Pharm Res. 2008;25:1663–1676. doi: 10.1007/s11095-008-9569-4. [DOI] [PubMed] [Google Scholar]
- 57.Roden DM, Kannankeril P, Darbar D. On the relationship among QT interval, atrial fibrillation, and torsade de pointes. Europace. 2007;9(Suppl 4):iv1–3. doi: 10.1093/europace/eum165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lu HR, Vlaminckx E, Van de Water A, Rohrbacher J, Hermans A, Gallacher DJ. In-vitro experimental models for the risk assessment of antibiotic-induced QT prolongation. Eur J Pharmacol. 2007;577:222–232. doi: 10.1016/j.ejphar.2007.07.070. [DOI] [PubMed] [Google Scholar]
- 59.Mortelmans K, Zeiger E. The Ames Salmonella/microsome mutagenicity assay. Mutat Res. 2000;455:29–60. doi: 10.1016/s0027-5107(00)00064-6. [DOI] [PubMed] [Google Scholar]
- 60.Mortelmans K, Riccio ES. The bacterial tryptophan reverse mutation assay with Escherichia coli WP2. Mutat Res. 2000;455:61–69. doi: 10.1016/s0027-5107(00)00076-2. [DOI] [PubMed] [Google Scholar]
- 61.Deferme S, Mols R, Van Driessche W, Augustijns P. Apricot extract inhibits the P-gp-mediated efflux of talinolol. J Pharm Sci. 2002;91:2539–2548. doi: 10.1002/jps.10262. [DOI] [PubMed] [Google Scholar]
- 62.Mao Q, Unadkat JD. Role of the breast cancer resistance protein (ABCG2) in drug transport. Aaps J. 2005;7:E118–133. doi: 10.1208/aapsj070112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhao YH, Le J, Abraham MH, Hersey A, Eddershaw PJ, Luscombe CN, Butina D, Beck G, Sherborne B, Cooper I, Platts JA. Evaluation of human intestinal absorption data and subsequent derivation of a quantitative structure-activity relationship (QSAR) with the Abraham descriptors. J Pharm Sci. 2001;90:749–784. doi: 10.1002/jps.1031. [DOI] [PubMed] [Google Scholar]
- 64.Chen VB, Arendall WB, 3rd, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr. 2010;66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Vennerstrom JL, Arbe-Barnes S, Brun R, Charman SA, Chiu FC, Chollet J, Dong Y, Dorn A, Hunziker D, Matile H, McIntosh K, Padmanilayam M, Santo Tomas J, Scheurer C, Scorneaux B, Tang Y, Urwyler H, Wittlin S, Charman WN. Identification of an antimalarial synthetic trioxolane drug development candidate. Nature. 2004;430:900–904. doi: 10.1038/nature02779. [DOI] [PubMed] [Google Scholar]
- 66.Brent RL. The history of the editorship of Teratology during the period from July 1, 1976 to January 1, 1993. Teratology. 2001;63:100–105. doi: 10.1002/1096-9926(200102)63:2<100::AID-TERA1016>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. (Fo-Fc) map for DSM265:PfDHODH binding-site
Fig. S2. DHODH sequence alignment
Fig. S3. In vitro parasite killing curves
Fig. S4. Activity of DSM265 against Plasmodium cynomolgi large (liver schizonts) and small (hypnozoites) forms
Fig. S5. The effect of DSM265 treatment on P. falciparum Pf3D70087/N9 in vivo
Fig. S6A. Proposed biotransformation pathways of DSM265 in plasma of mice, rabbits, monkeys and dogs.
Fig. S6B. Plasma concentrations of DSM265 and DSM450 (hydroxy metabolite).
Fig. S7. Simulated human plasma profiles using a physiologically-based pharmacokinetic model (Gastroplus™)
Fig. S8. Effect on ECG in the rabbit cardiac ventricular wedge assay
Fig. S9. Evaluation of the effects of DSM265 on G6PD deficient human RBCs engrafted into a NOD-SCID mouse
Table S1. PfDHODH-DSM265 X-ray diffraction data and refinement statistics
Table S2. In Vitro antimalarial activity of DSM265
Table S3A. Blood pharmacokinetic data for DSM265 in SCID mice
Table S3B. SCID mouse in vivo antimalarial activity
Table S3C. SCID mouse parasitemia levels
Table S3D. SCID mouse pharmacokinetic individual time point data
Table S4A. Selections for DSM265 resistant parasites in P. falciparum Dd2: Rathod lab
Table S4B. Selections for Atovaquone resistant parasites in P. falciparum Dd2: Rathod lab
Table S4C. Selections for DSM265 resistant parasites in P. falciparum Dd2: Fidock Lab
Table S4D. Selections for Atovaquone resistant parasites in P. falciparum Dd2: Fidock Lab
Table S4E. Selections for DSM265 resistant parasites in P. falciparum K1: Fidock Lab
Table S4F. Selections for DSM265 and Atovaquone P. falciparum HB3: Rathod Lab
Table S5. Summary of DSM265 resistant clones: analysis of parasites in whole cell assays
Table S6. Kinetic analysis of PfDHODH mutants
Table S7. Drug combination analysis
Table S8A. Stability data for DSM265 free base and tosylate salt
Table S8B. Solubility data for DSM265 free base
Table S9. In vitro ADME data for DSM265
Table S10. In vivo metabolite identification
Table S11. Relative plasma exposures of DSM265 metabolites in mice, rabbits, monkeys and dogs
Table S12. DSM265 plasma pharmacokinetics after a single intravenous dose in mice, rats, dogs and monkey
Table S13. DSM265 plasma pharmacokinetics after a single oral dose of DSM265 in mice, rats, dogs and monkeys
Table S14. Effect of salt form, formulation and food on the DSM265 plasma pharmacokinetics after oral dosing in beagle dogs
Table S15. Safety pharmacology
Table S16. Exploratory toxicology studies (non-GLP) in rodents and dogs
Table S17A. Toxicokinetic parameters on Day 1 and Day 7 in a mouse 7 day toxicology study
Table S17B. Individual mouse plasma concentrations 7 day toxicology study 25 mg/kg
Table S17C. Individual mouse plasma concentrations 7 day toxicology study 75 mg/kg
Table S17D. Individual mouse plasma concentrations 7 day toxicology study 200 mg/kg
Table S18A. Toxicokinetic data from a 10 day toxicology study in male beagle dogs
Table S18B. Toxicokinetic parameters day 1 of toxicology study in male beagle dogs
Table. S19. Primary data supporting Fig. 2
Table. S20. Primary data supporting Fig. 3A,B
Table. S21. Primary data supporting Fig. 5