

Abstract
Poly (lactic-co-glycolic acid) (PLGA) copolymers have been extensively used in cancer research. PLGA can be chemically engineered for conjugation or encapsulation of drugs in a particle formulation. We reported that oseltamivir phosphate (OP) treatment of human pancreatic tumor-bearing mice disrupted the tumor vasculature with daily injections. Here, the controlled release of OP from a biodegradable PLGA cylinder (PLGA-OP) implanted at tumor site was investigated for its role in limiting tumor neovascularization, growth, and metastasis. PLGA-OP cylinders over 30 days in vitro indicated 20%–25% release profiles within 48 hours followed by a continuous metronomic low dose release of 30%–50% OP for an additional 16 days. All OP was released by day 30. Surgically implanted PLGA-OP containing 20 mg OP and blank PLGA cylinders at the tumor site of heterotopic xenografts of human pancreatic PANC1 tumors in RAGxCγ double mutant mice impeded tumor neovascularization, growth rate, and spread to the liver and lungs compared with the untreated cohort. Xenograft tumors from PLGA and PLGA-OP-treated cohorts expressed significant higher levels of human E-cadherin with concomitant reduced N-cadherin and host CD31+ endothelial cells compared with the untreated cohort. These results clearly indicate that OP delivered from PLGA cylinders surgically implanted at the site of the solid tumor show promise as an effective treatment therapy for cancer.
Keywords: pancreatic cancer, oseltamivir phosphate, PLGA, tumor neovascularization, metastasis
Introduction
Cancer therapy can involve multiple injections of a therapeutic agent over an extended period. The design of controlled release devices is partly in a response to the need for extended term pharmacological treatment. These devices ensure dosing within an appropriate therapeutic window, particularly if it is directed to the immediate area of the tumor.
The current research interest on targeting the tumor microenvironment has focused on anti-angiogenesis therapy, which involves drugs that either prevent the formation of new blood vessel supply to the tumor or impair existing blood vessels.1 The strategic approach of these anti-angiogenic drugs can involve either selective targeting of the tumor-associated vasculature, increasing the bioavailability of tumor endothelial cells to systemically-administered anti-angiogenic drugs, or using metronomic therapy to reduce systemic drug toxicity.2,3 To achieve these therapeutic outcomes, polymer technology has been used to facilitate more efficient targeting and killing of the tumor-associated vasculature. Several polymer parameters of interest include the size of the polymer particles that allows them to intrinsically home in to metastasized tumors through the enhanced permeability and retention effect, to evade the immune system, to improve the drug’s half-life, and to allow for potent selective targeting due to their high surface density.2,4–6 The enhanced permeability and retention effect is the property by which unique sizes of structures such as liposomes, nanoparticles (NPs), and macromolecular drugs tend to accumulate in tumor tissue much more so than they do in normal tissues.7 Thus, polymer delivery of novel drug designs for cancer treatment are emerging new paradigms targeting anti-angiogenesis.
We have recently reported that oseltamivir phosphate (OP) can prevent the tumor neovascularization, growth, and metastasis of human triple-negative breast8 and pancreatic9 cancer cells in heterotopic xenografts of these tumors in RAGxCγ double mutant mice. The findings also disclosed a novel signaling paradigm that regulates epidermal growth factor receptors (EGFRs).9,10 Indeed, over 90% of pancreatic cancer cases overly express mutations of the EGFR, which are the major contributing factors to the aggressive nature of pancreatic tumors.11 These mutations lead to a constitutive activation of EGFR, causing rapid, uncontrolled cellular growth. Chemotherapeutics designed to target EGFR have failed in the clinic because of the inability to synthesize drugs that can accommodate the full repertoire of EGFR mutations. This type of therapeutic approach significantly limits the possibility of highly targeted chemotherapies being implemented as part of a treatment regime.11–13 Alternatively, molecular-targeting therapies involving metronomic therapy to disrupt tumor neovascularization and to increase the pro-apoptotic effects of chemo-drugs in cancer have become promising approaches.
In the present study, the metronomic therapy of a slow-released OP encapsulated in a biodegradable poly (lactic-co-glycolic acid) (PLGA-OP) cylinder was designed, developed, characterized, and demonstrated in vitro, and then surgically implanted at the tumor site to examine its efficacy to impede tumor neovascularization, tumor growth, and metastasis in a mouse model of human pancreatic epithelioid carcinoma. PLGAs offer a delivery approach that can provide sustained, controlled slow release of the drug for up to several weeks.14 A PLGA-designed cylinder based on a delivery vehicle can be implanted at the tumor site without the need for repeated drug administration. Here, the data indicate that implanted blank PLGA and PLGA-OP cylinders at the tumor site were found to impede tumor neovascularization, growth, and metastasis in heterotopic xenografts of tumors growing in a mouse model of human pancreatic cancer.
Materials and methods
Reagents
Contents of pharmacy-based OP 75 mg capsules were reconstituted in 25 mL distilled water, vortexed for 3 minutes, and centrifuged at 1,100× g for 5 minutes to remove insoluble materials, as previously reported by us.15 Supernatant was transferred to a 50 mL vial, frozen at −80°C, lyophilized for 24–48 hours, and stored at −80°C. The stock-extracted OP solution had a concentration of 15 mg/mL. OP (~98% purity) was obtained from Hangzhou DayangChem Co, Ltd (Hangzhou City, People’s Republic of China). Cell culture Dulbecco’s Modified Eagle’s conditioned medium (1× DMEM) containing different concentrations of OP (200–800 μg/mL) was used for the in vitro and in vivo experiments.
SPAN 80 (sorbitol monooleate) was purchased from Sigma Aldrich (Oakville, ON, Canada). Glycerol was obtained from Rougier Pharma (Mirabel, QC, Canada). Purasorb 5002, poly (D,L-lactic-co-glycolic) acid (D,L; 50:50) with a mean molecular weight of 17 kDa was obtained from Purac Biomaterials (Lincolnshire, IL, USA).
Fabrication of PLGA-OP matrix cylinders
PLGA cylinders were fabricated by dissolving 80 mg PLGA in 400 μL acetone and adding 4–6 mg of Span 80 followed by 10 mg OP, 20 mg OP, or without OP as a blank PLGA. The suspension was vortexed in a glass 20 mL vial for 30 seconds, followed by sonication for 2 minutes until PLGA was visibly dissolved. The suspension was transferred using a 1 mL glass syringe from a height of 4 cm onto a smooth Teflon sheet to form a flat, circular disk. The solvent was left to evaporate in a fume hood for 1 hour, followed by refrigeration at 5°C overnight. The disk was then lifted from the Teflon sheet using a razor blade, and rolled onto a glycerol lubricated Precision Glide 18 gauge syringe tip, forming a cylinder. The fabricated PLGA cylinders were extracted from the syringe needle and stored at −80°C.
Cylinder characterization
PLGA-OP cylinder dimensions were measured using a digital micrometer. Scanning electron microscope (JEOL 840; Peabody, MA, USA) images were obtained on gold sputter-coated, microtone sections of the cylinders. Differential scanning calorimetry (Mettler-Toledo; Mississauga, ON, Canada) was conducted by placing 10 mg cylinder sections into hermetically sealed aluminum pans, cooled to −50°C and equilibrated for 1 minute. Samples were then heated to 75°C and cooled back to −50°C twice, first to refresh the polymers, and second to record a value for the glass transition temperature of the PLGA.
OP release kinetics from PLGA-OP cylinders
PLGA-OP cylinders were placed into 3 mL of 0.2 M sodium phosphate buffer (pH 7.4), mixed on an orbital shaker at 40 rpm and stored in an incubator at 37°C. Supernatants (2 mL) were extracted periodically and placed in the freezer at −20°C. At sampling points, the cylinder was rinsed twice with buffer solution before being resuspended in fresh 3 mL buffer. Release experiments were performed in triplicate to construct release kinetic profiles.
OP in supernatant samples was measured using a method developed by the Centers for Disease Control.16 In a 20 mL glass vial, 150 μL supernatant was combined with 250 μL of 1 mg/mL aqueous Congo red solution, 350 μL of 0.1 M potassium phthalate buffer (pH 4.2), and 3 mL ethyl acetate. Samples were vigorously hand-shaken for 1 minute, followed by centrifugation at 500× g for 2 minutes. Samples were left to settle for 2 hours to facilitate solvent-water partitioning. The ethyl acetate solvent phase was then carefully removed using a glass Pasteur pipette and spectrophotometrical readings were performed at 480 nm. Readings were compared to a calibration curve constructed from fresh OP solution standards.
Cell line
PANC1 (human pancreatic carcinoma, epithelial-like, ATCC® CRL-1469™) cell line was obtained from the American Type Culture Collection (Manassas, VA, USA). The cells were grown in a 5% CO2 incubator at 37°C in culture medium containing 1× DMEM (Gibco, Rockville, MD, USA) supplemented with 10% fetal calf serum (HyClone, Logan, UT, USA) and 5 μg/mL Plasmocin™ (InvivoGen, San Diego, CA, USA). When the cells reached ~80% confluence, they were passaged at least five times for use in the experiments.
Heterotopic xenograft mouse model of human pancreatic cancer
An immunodeficient mouse with a double mutation combining recombinase activating gene (RAG) and common cytokine receptor γ chain (Cγ) was used as the xenograft model, as previously reported by us.9 RAGxCγ double mutant mice on a NOD genetic background are completely alymphoid (deficient in T cells, B cells, and natural killer cells), show no spontaneous tumor formation, and exhibit normal hematopoietic parameters. These mice were generated by intercrossing and were maintained in specific pathogen-free isolators in the animal care facility at Queen’s University, Kingston, ON, Canada. A colony was established in the animal facility. All mice were kept under sterile conditions in micro-isolators or air-filtered cages, and were provided with autoclaved food and water. Mice were used when aged 6–8 weeks, following protocols approved by the animal care committee at Queen’s University.
Mice were injected subcutaneously into the right back flank, with 0.5×106 PANC1 cells. Tumor volume was measured twice a week using the formula ((width squared/2) × length) and permitted to grow to 35 days prior to implantation of PLGA and PLGA-OP cylinders.
Surgical implantation of PLGA-empty and PLGA-OP in mice
PLGA-empty and PLGA-OP cylinders were sterilized in 25 mL of 95% ethanol for 20 seconds, followed by 20 minutes per side ultraviolet (UV) exposure in a sterile biological hood prior to implantation.
Mice were anesthetized using a continuous flow of isoflurane and oxygen, and administered meloxicam subcutaneously prior to surgery. The cylinders were implanted subcutaneously approximately 1–2 cm from the tumor site, 35 days post-implantation of PANC1 cells when the tumor volume reached 100–120 mm3. Mouse weight and tumor volumes were recorded for an additional 50 days, after which animals were sacrificed and necropsied.
Statistical analysis
Graphing and statistical analysis were performed using GraphPad Prism. One-way ANOVA unpaired t-test comparisons were performed.
Results
Fabrication and characterization PLGA-OP cylinders
Poly (D,L-lactic-co-glycolic) acid cylinders loaded with OP at different doses were fabricated using a novel method as depicted in Figure 1A. There is no loss of OP during the formulation, and granular OP was uniformly distributed throughout the polymer matrix. The procedure involves drop-wise extrusion of the PLGA solution containing insoluble OP crystals dispersed by a surfactant. Extruded droplets form circular disks on a smooth Teflon surface, enabling rapid evaporation and thus removal of the solvent. The disks were readily removed from Teflon with a sharp blade, and sufficiently flexible to be wrapped around a lubricated syringe needle forming a cylinder. The advantage in the use of a syringe needle to form cylinders is that different needle diameters allow flexibility in the fabrication of cylinders of different diameters. PLGA was selected as matrix polymer for its sustained, controlled release properties resulting from bulk hydrolysis of the polymer.17 As well, various medical devices and pharmaceutical products formulated from PLGA have US Food and Drug Administration approval.18,19
Figure 1.


Therapeutic design of PLGA-OP cylindrical implants.
Notes: (A) Fabrication of PLGA-OP; (a, b) dissolved suspension of PLGA in acetone containing Span 80 with OP or without as a blank was transferred using a 1 mL glass syringe from a height of 4 cm onto a smooth Teflon sheet to form a flat, circular disk, in a fume hood for 1 hour followed by refrigeration at 5°C overnight; (c) PLGA-OP disk was lifted from the teflon sheet using a razor blade, and (d) rolled onto a glycerol lubricated precision glide 18 gauge syringe tip to form a cylinder; (e) fabricated PLGA-OP cylinders were extracted from the syringe needle and stored at −80°C; (f) surgical implantation of blank PLGA and PLGA-OP at tumor site (image showing PLGA-OP near necropsied live tumor at end point of experiment. (B) Photograph of PLGA-empty and PLGA-OP cylinders. (C) SEM micrographs of PLGA-OP cylinder; (a) micrograph of a single layer cross-section with hollow continuous center throughout the entire PLGA-OP structure, (b) top surface, (c) magnified porous surface structure, (d) magnified surface structure, and (e) magnified internal structure of PLGA-OP showing crystals of OP.
Abbreviations: OP, oseltamivir phosphate; PLGA, poly (lactic-co-glycolic acid); SEM, scanning electron microscope.
Different surfactants and solvents were tested to achieve a uniform distribution of OP crystals. The fabrication protocol as described provides an even distribution (Figure 1Ca) of OP crystals based on visual examination of the cylinders.
Cylinder diameters ranged from 2.4 to 3.0 mm and length was ~1 cm (Figure 1B). Both diameter and length were easily modified. Scanning electron microscope micro-graphs of cross-section, surface, and internal morphologies of PLGA-OP cylinders are presented in Figure 1C. Discrete OP crystals are visibly embedded within the polymer matrix in Figure 1Ce, as well as protruding from the top and bottom surface of the polymer film. The cylinder appears smooth and continuous as seen in Figure 1Cb. Figure 1Ca shows a cross-sectional view of the cylinder illustrating a hollow core of approximately 0.8 mm diameter, extending through the axis of the cylinder, which is comprised of 3–4 layers of polymer. The surface pores are visible in the magnified view in Figure 1Cc. Internally, Figure 1Cd shows a porous morphology that is highly variegated, along with cracks that extend into the core of the thin film polymer outer layer.
Differential scanning calorimetry shows the effect of OP loading on glass transition temperature (Tg) of the PLGA polymer making up the cylinders. Thermograms in Figure 2A reveal Tg of 5.8, 15.2, and 32.8°C with PLGA cylinders containing 0, 10, and 20 mg OP, respectively, and Tg of 32.7°C for native PLGA polymer. Tg values, while varied with drug loading, were all below body temperature, and therefore the glass transition.
Figure 2.
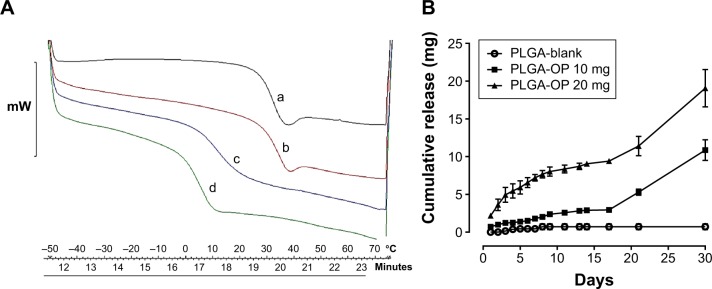
(A) A thermogram showing the glass transition temperatures of (a) PLGA polymer (32.7°C), PLGA loaded with (b) 20 mg (32.8°C), (c), 10 mg (15.2°C), and (d) 0 mg OP (5.8°C). (B) Cumulative release kinetics for PLGA blank, 10 mg and 20 mg OP loaded PLGA. All PLGA-OP samples exhibited a biphasic release profile.
Abbreviations: OP, oseltamivir phosphate; PLGA, poly (lactic-co-glycolic acid).
OP release kinetics from PLGA-OP cylinders
OP release from PLGA-OP cylinders was monitored over a 30-day period as depicted in Figure 2B. For the 20 mg PLGA-OP cylinder, 20%–25% of the total OP loading was released in the initial 48 hours. This initial release of OP was followed by a period of slow release for an additional 16 days. By day 17, 45% of the OP was released followed by a secondary release phase resulting in the remaining OP being released by day 30. An initial burst release was not observed with the 10 mg PLGA-OP, and about 30% of the OP loading was released by day 17. A secondary release phase was then observed with full release of the OP seen by day 30. Both formulations exhibited similar biphasic release profiles, which are typically observed with this polymer.20 It is interesting that the typical burst release of drug from PLGA microparticles and NP including that of large molecular weight protein-based drugs21 was not observed in this case for a small molecular weight hydrophilic drug from PLGA cylinders. Higher drug amount led to higher rate of release in the first 48 hours. It is generally reported that PLGA releases drug through bulk erosion;14 however, this could be facilitated by water permeation into the polymer at a higher rate, enabled by a doubling in the number of drug-loaded pockets within the polymer cylinder, clearly visible in Figure 1Ce. An examination of Figure 2B shows that after 30 days the cumulative amount of OP released from the cylinders equaled the initial amount of OP encapsulated within the cylinders, thus demonstrating complete retention of the OP during the fabrication of the cylinders. No signal was detected with blank PLGA cylinders during the course of the assay, indicating that the products of PLGA degradation did not affect the response of the assay.
PLGA-OP cylinders were releasing OP into 3 mL of supernatant volume, which is comparable to blood volume of a mouse. Supernatants were replaced at each sample point, which may represent the continuous filtering of blood and body fluids through circulation and normal replacement. Thus, the experiment was designed to best replicate OP release profiles, in vivo. The intent was that the initial burst release of OP from PLGA-OP cylinders over 48 hours would promote rapid induction of OP into the surrounding tissues including that of the tumor site followed by a continual infusion of released OP for a period of up to 30 days.
Metronomic therapy of slow-release OP from PLGA-OP impedes tumor vascularization, growth, and invasiveness in heterotopic xenografts of PANC1 tumors in RAGxCγ double mutant mice
Anti-tumorigenesis of OP released at the tumor site was investigated in the RAGxCγ double mutant mouse model of human PANC1 pancreatic cancer cells. The model lacks mature T and B cells, functional NK cells, and is deficient in cytokine signaling, leading to better engraftment of human cells than any other published mouse strain.9 As previously demonstrated for a pancreatic epithelial carcinoma cell line,9 the hypothesis is that metronomic therapy of slow-released OP from PLGA-OP impedes tumor neovascularization, growth, and spread in the heterotopic xenograft mouse model of human pancreatic cancer. PANC1 epithelioid carcinoma cells at 0.5×106 cells in 0.2 mL were implanted subcutaneously in the right back flank. Twice a week, each mouse was monitored for tumor volume, body weight, and body condition scoring. Loss of body weight was not observed in any of the animals, and body condition scoring was within the normal range. Sterile blank PLGA and PLGA-OP containing 20 mg OP (PLGA-OP 20 mg) cylinders were implanted subcutaneously approximately 1–2 cm from the tumor site, 35 days post-implantation of PANC1 cells, when the tumor volume reached 100–120 mm3.
Data presented in Figure 3A show that OP released from PLGA-OP 20 mg cylinders effectively inhibited tumor growth for approximately 30 days when implanted at day 35. Beyond this point, the tumor grew slightly, eventually reaching a volume similar to that of the blank PLGA cohorts by day 77. Tumor growth rate was substantial for the untreated control cohort. It is noteworthy that the 30-day period of tumor suppression in mice receiving PLGA-OP cylinders was consistent with the in vitro 30-day OP release period observed in Figure 2B.
Figure 3.

(A) PLGA-OP treatment of RAGxCγ double mutant mice bearing heterotopic xenografts of pancreatic PANC1 tumors. Cells at 0.5×106 in 0.2 mL were implanted subcutaneously in the right back flank of these mice. Twice a week following implantation of the PANC1 cancer cells, each mouse was monitored for tumor volume ((width squared/2) × length) at the site of implantation. Mice were surgically implanted with sterilized PLGA-empty (four mice) and PLGA-OP (four mice cylinders) containing 20 mg OP at day 35 post-implantation of cancer cells when the tumor volume reached approximately 50–120 mm3. (B) Graph of mean ± SEM of live necropsy tumor weight per mouse body weight. Statistical analysis using unpaired t-test was carried out using GraphPad Prism, and the results were compared with untreated control cohort.
Abbreviations: OP, oseltamivir phosphate; PLGA, poly (lactic-co-glycolic acid); SEM, standard error of the mean; RAG, recombinase activating gene.
The blank PLGA cohort exhibited a shrinkage of tumor growth post-day 65 until the end point of the experiment. To confirm these findings, tumor weight per mouse body weight was measured at necropsy. The data in Figure 3B indicated a significant reduction of tumor weight per body weight for both the blank PLGA and PLGA-OP 20 mg cohorts at day 86 compared with the untreated cohort (Figure 3B). The average ± standard deviation (SD) of body weight (g) for the PLGA-empty cohort was 35.7±0.6 g while for the PLGA-OP cohort was 34.5±0.7 g. Final tumor volume for the PLGA-OP cohort was 1% that of the untreated group, and also significantly less than that of the PLGA cohort at 6.5%.
If tumor volume is examined 30 days post-implantation, at the point where the OP was expected to be fully released, then data in Figure 3A indicate that the PLGA-OP cohort showed a tumor volume of 10% to that of the control cohort whereas the PLGA blank cohort in the absence of OP showed a tumor volume of 87% to that of the untreated cohort. Both comparisons, at 30 days post-implantation and at necropsy, demonstrate a significant reduction in tumor volume of the PLGA-OP cohort relative to the untreated group.
At necropsy, it was observed that the treatment of tumor-bearing mice with PLGA-OP cylinders at day 35 attenuated the aggressive tumor vascularization (Figure 4C) compared with the untreated control cohort (Figure 4A). There is also indication that PLGA cylinder implants on their own attenuate tumor vascularization and potentially, tumor growth. There is some evidence of residual PLGA cylinder in proximity to the tumor, as seen for example in the image of the tumor in Figure 4C.
Figure 4.

Notes: (A) Untreated control (three mice), (B) PLGA-empty (four mice), and (C) PLGA-OP (four mice) 20 mg treated cohorts. Necropsy tumors, HE staining of tumors, and paraffin-embedded tumor sections (5 μm) on glass slides were processed for immunohistochemistry using primary DyLight 488 conjugated rat monoclonal anti-mouse CD31+ (PECAM-1) antibody, primary anti-E-cadherin, and N-cadherin antibodies followed with polyclonal goat anti-rabbit Alexa Fluor® 488 secondary antibody and Entellan® rapid mounting media. Background control (not shown) sections were prepared without the primary antibodies and relative staining density was 2–4×105. The bar on the stained tissue sections represents 200 μm. Images are representative of at least five fields of view from two tumor sections. (D) Quantitative analysis was done by assessing the density of tumor staining corrected for background in each panel using Corel Photo Paint 8.0 software. Each symbol in the figure represents the mean ± SEM corrected density of tumor staining within the respective images. Statistical analysis using unpaired t-test was carried out using GraphPad Prism and results were compared with the untreated cohort.
Abbreviations: OP, oseltamivir phosphate; PLGA, poly (lactic-co-glycolic acid); SEM, standard error of the mean.
Paraffin-embedded tumors were immunostained for host endothelial cell marker CD31/PECAM-1, and tumor human E- and N-cadherin expressions. Immunostained images (Figure 4A–C) and results from image analysis (Figure 4D) appear to show an increased migration of host CD31+ cells to the tumors with the untreated control cohort, but significantly reduced CD31+ host cells for the blank PLGA and PLGA-OP treated cohorts. Significantly high levels of tumor human E-cadherin expressions were observed with PLGA and PLGA-OP cohorts, with concomitant reduced N-cadherin levels for the PLGA-OP compared with the untreated cohort (Figure 4D).
Because both the PLGA-OP and blank PLGA cylinders had an impact on tumor neovascularization and growth, the effect of blank PLGA on PANC1 cells in culture was examined for 15 days (Figure 5). There appears to be an effect on day 3, but less so with time, to the point of 15 days where there was no difference between PLGA and control. Blank PLGA cylinder had no effect on cell viability.
Figure 5.

Blank PLGA cylinders in culture with PANC1 cells. Morphology of viable cells at 3, 6, 10, and 15 days. Viability of cells was counted using 0.4% trypan blue solution and hemacytometer. Cell count was expressed as a percentage of control.
Abbreviation: PLGA, poly (lactic-co-glycolic acid).
Necropsy live livers (Figure 6A) and lungs (Figure 6B) were lacking visible tumor nodules for the blank PLGA and PLGA-OP treated cohorts compared with extensive visible tumors for the untreated controls. There were neither tumor nodules nor metastatic clusters of cancer cells in the liver or lung for the PLGA-OP treated cohort. Both blank PLGA and PLGA-OP treatments markedly attenuated the metastatic spread of PANC1 cancer cells to the lungs and liver (Figure 6A and B) compared with extensive evidence of metastatic spread in the untreated cohort. Lack of metastatic spread to the lung and liver following PLGA treatment may be due in part to a disrupted tumor vasculature development and reduced host CD31+ endothelial cell migration, or result from a decreased expression of the cell surface mesenchymal marker, N-cadherin, and an increased expression of the cell surface epithelial marker, E-cadherin as previously reported by us for pancreatic cancer.15
Figure 6.

Necropsy liver (A) and lung (B). RAGxCγ double mutant mice were implanted with 0.5×106 PANC1 pancreatic cancer cells subcutaneously on the rear flank and PLGA-empty and PLGA-OP 20 mg cylinders were surgically implanted at day 35 post-implantation when tumors reached 100–120 mm3. Paraffin-embedded tissue sections (5 μm) on glass slides were processed for HE staining for each mouse necropsied at end point of the study. Stained tissue sections were photographed using a Zeiss Imager M2 fluorescence microscope at 400× magnification. Images are representative of at least five fields of view from two tissue sections. Metastatic tissue clusters were microscopically counted per tissue sections (5 μm) and plotted in the graph.
Abbreviations: OP, oseltamivir phosphate; PLGA, poly (lactic-co-glycolic acid); RAG, recombinase activating gene.
Discussion
In the present study, we explored the metronomic controlled release of OP from an implantable biodegradable PLGA implant at the tumor site in preventing tumor development. The challenge in the delivery of OP from implantable biodegradable devices, similar to that of other low molecular weight hydrophilic drugs, is to ensure efficient entrapment of the hydrophilic OP in the formulation of the delivery vehicle without premature release of the drug. PLGA cylindrical implants loaded with OP were fabricated using a novel method where there is minimal loss of OP during the formulation, and OP is uniformly distributed throughout the polymer matrix. Other reports have used polymeric cylindrical implant for drug delivery applications.14,21–25 It is noteworthy that all of these studies with the exception of one did not present in vivo data, and the one that had in vivo data, did not have in vitro release profiles. It also appears that the methods used tend to be more focused, but not entirely, on hydrophobic drugs.
Many PLGA-based drug delivery systems involve NP.26 However, there are two major problems using PLGA-NP formulations. First, small molecular weight hydrophilic drugs are difficult to encapsulate in PLGA-NP resulting in poor loading, and second, high burst release of the drug is often observed. Even if PLGA-NP can be formed with high encapsulation efficiencies, resulting drug loading is generally as low as 1%, suggesting a NP content of 1 mg active drug per 100 mg NP. The second major problem is the high burst release, observed for most PLGA-based drug NP resulting in potential toxicity and reduced ability to reach the target tissue or cells.26 In addition, there are concerns around the issue of nanotoxicology, where interactions of the NP with biological systems may have contraindicative effects.27 The size and surface properties of the NP may also modify the behavior of the copolymer components in the host.
Here, we used a novel design to encapsulate water soluble, low molecular weight OP into PLGA cylinders. This method avoids the common problem areas described above for NPs, essentially achieving full encapsulation efficiency of a low molecular weight hydrophilic drug, and enabling extensive control of drug loading. The downside is that the cylinders are not injectable in the present form. In the PLGA-OP formulation, lyophilized OP was added in a fine crystalline particulate form to a solvent phase where it is insoluble, and thus stabilized. This fabrication method was built on our experience in loading crystalline insulin within PLGA microparticles, and subsequently into alginate sponge dressing.28,29 A stabilized and released insulin from PLGA microparticles was reported for up to 25 days, following a similar biphasic release pattern,30 as demonstrated here for OP. Thus, it appears that both low and high molecular weight therapeutics including hydrophilic compounds can follow a predictable release profile from the PLGA polymer, whether in spherical or cylindrical morphology. Hydrophilic drugs and particularly low molecular weight compounds are typically difficult to encapsulate in controlled release formulations involving biphasic formulation methods. This necessitated insulin and OP being entrapped in a solid crystalline form, avoiding the use of aqueous formulations for the applications described. The benefit of this fabrication process is essentially full encapsulation efficiency of the drug, stable preparations, and extended term, sustained release profiles.
For PLGA-OP cylinders, in vitro studies demonstrated controlled release of OP over a 30-day period for cylinders containing 10 and 20 mg OP. When 20 mg OP cylinders were surgically implanted near the tumor site, tumor growth was blocked over a 30-day period, corresponding to the period of OP release observed in vitro. Beyond 30 days post-implantation, tumor volume began to increase for 12 days, then stabilize to the end of the experiment. For the PLGA blank cylinders, there was a sharp increase in tumor volume, directly paralleling the no-treatment control in the 20 days post-implantation. After 30 days, tumor volumes of no-treatment controls continued to increase, while tumor volumes for the blank PLGA cohort remained stagnant for the remainder of the experiment to day 86, 52 days post-implantation.
The data in Figure 4 show distinct visual separation between tumor and cylinder in mice necropsied 50 days post-implantation. The bulk of erosion of PLGA cylinders could be expected to occur after 17–18 days post-plantation, based on the shift in release kinetics observed at this time point in vitro, as observed in Figure 2B. While it is clear that released OP is arresting tumor growth, it is apparent that blank PLGA is also to some extent impacting tumor neovascularization, growth, and metastasis, but to a much lesser extent than observed with PLGA-OP. Differences between PLGA blank and no-treatment controls became apparent at 30 days post-implantation, at the same point when PLGA-OP cylinders could be expected to have depleted their content of OP. The reasons for the effect of blank PLGA impacting tumor development in vivo are unclear. The data in Figure 5 indicate that blank PLGA cylinders have no direct effect on the cell viability of PANC1 tumor cells in culture. Based on these results, it is proposed that PLGA may have an effect on the tumor-associated vasculature. Evidence to support this hypothesis is shown in Figure 4B where tumor-associated vasculature is reduced or negligible, or diverted toward the PLGA polymer in one live tumor necropsy.
Earlier study by Mooney et al31 using fabricated hollow, tubular devices from porous films of various polymers of PLGA showed that after surgical implantation in the mesentery and omentum of Lewis rats, there was an initial acute inflammatory response to the devices within the first few days. Fibrin deposition and macrophage infiltration were noted within the pores of the devices by 3 days, and fibrovascular tissue formed in the pores of the devices and on the luminal surface over 7–14 days. Capillary ingrowth through the pores accompanied the fibrous tissue ingrowth. In our studies, it would appear that the blank PLGA cylinders may have competed with the tumor for vasculature development impeding tumor growth, although our cylinders were not fabricated with the porous structure as described in Mooney’s study.31 More recent studies on angiogenesis and the microstructure of the PLGA meshes have reported that the micropattern of the capillary network and vascular endothelial growth factor provided a synergistic effect on the micropatterned angiogenesis process.32 The progress of PLGA scaffolds with particular emphasis on fabrication approaches, mechanical properties, and degradation behaviors has been reviewed by Pan and Ding.33
Another explanation for PLGA-reduced tumor vasculature is that tumor microenvironment might be metabolically and acid stressed due to the metabolic accumulation of lactic and glycolic acids as degradation products. Using PLGA porous scaffolds for tissue engineering and regenerative medicine, Chang et al34 and Pan and Ding33 measured the local pH of a block copolymer hydrogel composed of poly(ethylene glycol) and PLGA after subcutaneous injection. The initial local pH dropped to 4, but after 10 hours, the PLGA microenvironment recovered to nearly neutral. The reports also disclosed that in the case of subcutaneous implanting of PLGA, the body fluid exchange was rapid to neutralize the initial acid-stressed microenvironment. Mooney et al31 also observed that glycolic and lactic acid monomers were only released in meaningful quantities from 50:50 PLGA polymer after substantial reduction in PLGA molecular weight, occurring after 20 days in vitro. A recent study on the safety evaluation of PLGA and/or poly(lactic-acid) microspheres through intravitreal injection in rabbits showed that the microspheres did not cause retinal histological changes or functional damage, and were biocompatible and safe for controlled drug delivery.35
The multiple biological activities of lactic acid in cancer with particular focus on tumor growth, angiogenesis, and metastasis have been extensively reviewed by Dhup et al.36 Glycolic acid, an alpha-hydroxy acid derived from fruit and milk sugars, was shown to reduce UV-induced skin tumor development with a concomitant decreased expression of the cell-cycle regulatory proteins such as proliferating cell nuclear antigen, cyclin D1, cyclin E, cdk2, and cdk4 and the signal mediators c-Jun NH2-terminal kinase, p38 kinase, and mitogen-activated protein kinase kinase.37 The report also disclosed that the inhibition of the activation of transcription factors AP-1 and NF-κB could contribute significantly to the inhibitory effect of glycolic acid on tumor development.
Due to the strong differences in tumor vasculature observed between the different test cohorts, paraffin-embedded tumors were immunostained for host endothelial cell marker CD31/PECAM-1 for angiogenesis. There was a migration of host CD31+ cells to the untreated tumors consistent with the bloody vasculature and extensive tumor growth observed in Figure 4, but reduced levels were observed for the blank PLGA and PLGA-OP treated cohorts, consistent with the absence of the bloody tumor vasculature, and considerable reduction in tumor growth. The necropsy tumors from the PLGA-OP and blank PLGA cylinder-treated cohorts revealed higher levels of human tumor E-cadherin compared with the untreated group, with concomitant reduced N-cadherin expression levels compared with the untreated group (Figure 4). Since the implanted PLGA and PLGA-OP had an inhibitory effect on tumor neovascularization and tumor E- and N-cadherin expressions, we also examined metastatic spread to the lung and liver. Necropsy liver (Figure 6A) and lung (Figure 6B) showed extensive visible tumor nodules for the untreated cohort. In contrast, there were few or no lung or liver, visible tumor nodules for the PLGA and PLGA-OP cohorts, respectively. HE staining of necropsy liver and lung sections from treated and untreated mice were examined to quantify the number of metastatic clusters per 5 μm sections. For the blank PLGA and PLGA-OP treated cohorts, a significant reduction in metastatic clusters in the liver and lung was observed relative to the untreated cohort. The lack of metastases observed with blank PLGA and further reduced with the PLGA-OP treatments may be due in part to a disruption of tumor vasculature (eg, reduced host CD31+ endothelial cell migration) or a decreased expression of N-cadherin with concomitant increase in E-cadherin expression.
The findings with the PLGA-OP cylinders impacting tumor neovascularization are consistent with our previous reports on molecular-targeting studies.8,38 We have also observed an abnormal and robust bloody tumor vasculature in xenografts of A2780 ovarian38 and MDA-MB-231 triple-negative8 tumors growing in RAGxCγ double mutant mice. When Snail shRNA A2780 ovarian cancer cells were injected subcutaneously, the xenografts of tumors showed no robust bloody tumor vascularization compared with A2780 and A2780 shRNA Slug tumors, and the mice were completely devoid of tumor growth and spread to the lungs.38 For triple-negative breast cancer, OP monotherapy ablated tumor neovascularization with a concomitant reduction in host CD31+ endothelial cells.8 These findings propose a novel organizational signaling platform connecting the Snail-MMP-9 signaling axis in amplifying the Neu1 sialidase and MMP-9 cross-talk in regulating EGFRs in promoting tumor neovascularization, growth, and invasiveness as previously reported by us.9,15 Molecular-targeting Neu1 with OP downregulated the Snail-MMP9 signaling axis. Other studies have similarly reported that curcumin (diarylheptanoid) encapsulated in PLGA microspheres downregulated CD31+ cells and vascular endothelial growth factor in nude mice bearing human breast MDA-MB-231 xenografts.39,40
Conclusion
The findings in this report clearly indicate that PLGA cylinders surgically implanted at the site of the solid tumor impede tumor neovascularization, growth, and metastasis in mouse model of human pancreatic carcinoma. The results from our studies provide proof-of-concept using PLGA-OP cylinder implantation at the tumor site, which shows promise for potential application in the treatment of pancreatic cancer.
It is our goal to move toward injectable implants providing similar sustained release profiles achieved for OP from PLGA cylinder implants.
Acknowledgments
This work was supported in part by grants to MR Szewczuk and RJ Neufeld from the Natural Sciences and Engineering Research Council of Canada and private sector cancer funding from the Josefowitz Family to MR Szewczuk. The cancer contents of this report are covered by a PCT international patent application number, PCT/CA2011/050690. We thank Lacey Brodhagen and Alexandra Morris, Veterinarian Technologists for their veterinarian expertise and assistance with the animal work.
Footnotes
Authors’ contributions
All authors contributed toward data analysis, drafting and revising the paper and agree to be accountable for all aspects of the work. JP Ellis, JAM Steele, and M Hrynyk fabricated and characterized PLGA cylinders; S Abdulkhalek generated and maintained cancer cell line and prepared for animal experiments; S Allison confirmed the release studies of OP and carried out in vitro study with PANC1 cells in presence of PLGA; F Haxho performed the immunohistochemistry and the immunostaining for host CD31+ cells in tumor tissues and the liver and lung metastatic tumor analyses; MR Szewczuk and RJ Neufeld supervised the research design and prepared the manuscript.
Disclosure
M Hrynyk was the recipient of the Natural Sciences and Engineering Research Council of Canada (NSERC) Graduate Scholarship (CGS-D3). S Abdulkhalek was a recipient of the RS McLaughlin scholarship, the Ontario Graduate scholarship, and the Canadian Institutes of Health Research Doctoral award (Frederick Banting and Charles Best Canada Graduate Scholarship). S Allison holds the Queen’s Graduate Award (QGA). F Haxho holds the QGA, Graduate Entrance Tuition Award (GETA), and the NSERC Alexander Graham Bell Canada Graduate Scholarship-Master’s (CGS M).
The authors report no conflicts of interest in this work.
References
- 1.Folkman J. Fundamental concepts of the angiogenic process. Curr Mol Med. 2003;3(7):643–651. doi: 10.2174/1566524033479465. [DOI] [PubMed] [Google Scholar]
- 2.Segal E, Satchi-Fainaro R. Design and development of polymer conjugates as anti-angiogenic agents. Adv Drug Deliv Rev. 2009;61(13):1159–1176. doi: 10.1016/j.addr.2009.06.005. [DOI] [PubMed] [Google Scholar]
- 3.Kerbel RS, Kamen BA. The anti-angiogenic basis of metronomic chemotherapy. Nat Rev Cancer. 2004;4(6):423–436. doi: 10.1038/nrc1369. [DOI] [PubMed] [Google Scholar]
- 4.Peer D, Karp JM, Hong S, Farokhzad OC, Margalit R, Langer R. Nanocarriers as an emerging platform for cancer therapy. Nat Nanotechnol. 2007;2(12):751–760. doi: 10.1038/nnano.2007.387. [DOI] [PubMed] [Google Scholar]
- 5.Davis ME, Chen ZG, Shin DM. Nanoparticle therapeutics: an emerging treatment modality for cancer. Nat Rev Drug Discov. 2008;7(9):771–782. doi: 10.1038/nrd2614. [DOI] [PubMed] [Google Scholar]
- 6.Suri SS, Fenniri H, Singh B. Nanotechnology-based drug delivery systems. J Occup Med Toxicol. 2007;2:16. doi: 10.1186/1745-6673-2-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Noguchi Y, Wu J, Duncan R, et al. Early phase tumor accumulation of macromolecules: a great difference in clearance rate between tumor and normal tissues. Jpn J Cancer Res. 1998;89(3):307–314. doi: 10.1111/j.1349-7006.1998.tb00563.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Haxho F, Allison S, Alghamdi F, et al. Oseltamivir phosphate monotherapy ablates tumor neovascularization, growth, and metastasis in mouse model of human triple-negative breast adenocarcinoma. Breast Cancer Targets Ther. 2014;6:191–203. doi: 10.2147/BCTT.S74663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gilmour AM, Abdulkhalek S, Cheng TS, et al. A novel epidermal growth factor receptor-signaling platform and its targeted translation in pancreatic cancer. Cell Signal. 2013;25(12):2587–2603. doi: 10.1016/j.cellsig.2013.08.008. [DOI] [PubMed] [Google Scholar]
- 10.Abdulkhalek S, Hrynyk M, Szewczuk MR. A novel G-protein-coupled receptor-signaling platform and its targeted translation in human disease. Res Rep Biochem. 2013;3:17–30. [Google Scholar]
- 11.Xiong HQ, Abbruzzese JL. Epidermal growth factor receptor-targeted therapy for pancreatic cancer. Semin Oncol. 2002;29(5 Suppl 14):31–37. doi: 10.1053/sonc.2002.35645. [DOI] [PubMed] [Google Scholar]
- 12.Safran H, Dipetrillo T, Iannitti D, et al. Gemcitabine, paclitaxel, and radiation for locally advanced pancreatic cancer: a Phase I trial. Int J Radiat Oncol Biol Phys. 2002;54(1):137–141. doi: 10.1016/s0360-3016(02)02902-4. [DOI] [PubMed] [Google Scholar]
- 13.Safran H, Rathore R. Paclitaxel as a radiation sensitizer for locally advanced pancreatic cancer. Crit Rev Oncol Hematol. 2002;43(1):57–62. doi: 10.1016/s1040-8428(01)00184-6. [DOI] [PubMed] [Google Scholar]
- 14.Desai KG, Mallery SR, Schwendeman SP. Effect of formulation parameters on 2-methoxyestradiol release from injectable cylindrical poly(DL-lactide-co-glycolide) implants. Eur J Pharm Biopharm. 2008;70(1):187–198. doi: 10.1016/j.ejpb.2008.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.O’Shea LK, Abdulkhalek S, Allison S, Neufeld RJ, Szewczuk MR. Therapeutic targeting of Neu1 sialidase with oseltamivir phosphate (Tamiflu(R)) disables cancer cell survival in human pancreatic cancer with acquired chemoresistance. Onco Targets Ther. 2014;7:117–134. doi: 10.2147/OTT.S55344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Green MD, Nettey H, Wirtz RA. Determination of oseltamivir quality by colorimetric and liquid chromatographic methods. Emerg Infect Dis. 2008;14(4):552–556. doi: 10.3201/eid1404.061199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Degim IT, Celebi N. Controlled delivery of peptides and proteins. Curr Pharm Des. 2007;13(1):99–117. doi: 10.2174/138161207779313795. [DOI] [PubMed] [Google Scholar]
- 18.Chaubul M. Polylactides/glycolides – excipients for injectable drug delivery and beyond. Drug Deliv Technol. 2002;2:34–36. [Google Scholar]
- 19.Lu JM, Wang X, Marin-Muller C, et al. Current advances in research and clinical applications of PLGA-based nanotechnology. Expert Rev Mol Diagn. 2009;9(4):325–341. doi: 10.1586/erm.09.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kreye F, Siepmann F, Siepmann J. Drug release mechanisms of compressed lipid implants. Int J Pharm. 2011;404(1–2):27–35. doi: 10.1016/j.ijpharm.2010.10.048. [DOI] [PubMed] [Google Scholar]
- 21.Ansary RH, Awang MB, Rahman MM. Biodegradable poly(D,L-lactic-co-glycolic acid)-based micro/nanoparticles for sustained release of protein drugs – a review. Trop J Pharm Res. 2014;13(7):1179–1190. [Google Scholar]
- 22.Arnold W, Senn P, Hennig M, et al. Novel slow- and fast-type drug release round-window microimplants for local drug application to the cochlea: an experimental study in guinea pigs. Audiol Neurootol. 2005;10(1):53–63. doi: 10.1159/000082575. [DOI] [PubMed] [Google Scholar]
- 23.Hou J, Li C, Cheng L, Guo S, Zhang Y, Tang T. Study on hydrophilic 5-fluorouracil release from hydrophobic poly(epsilon-caprolactone) cylindrical implants. Drug Dev Ind Pharm. 2011;37(9):1068–1075. doi: 10.3109/03639045.2011.559658. [DOI] [PubMed] [Google Scholar]
- 24.Hiremath J, Patel B, Aitha RK, Mura P, Palavalli GS. Characterization of cylindrical and strip-shaped tamoxifen citrate-loaded biodegradable implants. Asian J Pharm. 2011;5:209–214. [Google Scholar]
- 25.Hiremath JG, Devi VK. Paclitaxel loaded biodegradable poly(sebacic acid-co-ricinoleic acid) cylindrical implants for local delivery – in vitro characterization. Asian J Pharm. 2013;7:125–132. [Google Scholar]
- 26.Danhier F, Ansorena E, Silva JM, Coco R, Le Breton A, Preat V. PLGA-based nanoparticles: an overview of biomedical applications. J Control Release. 2012;161(2):505–522. doi: 10.1016/j.jconrel.2012.01.043. [DOI] [PubMed] [Google Scholar]
- 27.Carlin DJ. Nanotoxicology and nanotechnology: new findings from the NIEHS and Superfund Research Program scientific community. Rev Environ Health. 2014;29(1–2):105–107. doi: 10.1515/reveh-2014-0026. [DOI] [PubMed] [Google Scholar]
- 28.Hrynyk M, Martins-Green M, Barron AE, Neufeld RJ. Alginate-PEG sponge architecture and role in the design of insulin release dressings. Biomacromolecules. 2012;13(5):1478–1485. doi: 10.1021/bm300186k. [DOI] [PubMed] [Google Scholar]
- 29.Hrynyk M, Martins-Green M, Barron AE, Neufeld RJ. Sustained prolonged topical delivery of bioactive human insulin for potential treatment of cutaneous wounds. Int J Pharm. 2010;398(1–2):146–154. doi: 10.1016/j.ijpharm.2010.07.052. [DOI] [PubMed] [Google Scholar]
- 30.Ahmed AR, Bodmeier R. Preparation of preformed porous PLGA microparticles and antisense oligonucleotides loading. Eur J Pharm Biopharm. 2009;71(2):264–270. doi: 10.1016/j.ejpb.2008.09.007. [DOI] [PubMed] [Google Scholar]
- 31.Mooney DJ, Breuer C, McNamara K, Vacanti JP, Langer R. Fabricating tubular devices from polymers of lactic and glycolic acid for tissue engineering. Tissue Eng. 1995;1(2):107–118. doi: 10.1089/ten.1995.1.107. [DOI] [PubMed] [Google Scholar]
- 32.Oh HH, Lu H, Kawazoe N, Chen G. Micropatterned angiogenesis induced by poly(d,l-lactic-co-glycolic acid) mesh-structured scaffolds. J Bioact Compat Polym. 2012;27(2):97–106. [Google Scholar]
- 33.Pan Z, Ding J. Poly(lactide-co-glycolide) porous scaffolds for tissue engineering and regenerative medicine. Interface Focus. 2012;2(3):366–377. doi: 10.1098/rsfs.2011.0123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chang G, Ci T, Yu L, Ding J. Enhancement of the fraction of the active form of an antitumor drug topotecan via an injectable hydrogel. J Control Release. 2011;156(1):21–27. doi: 10.1016/j.jconrel.2011.07.008. [DOI] [PubMed] [Google Scholar]
- 35.Rong X, Yuan W, Lu Y, Mo X. Safety evaluation of poly(lactic-co-glycolic acid)/poly(lactic-acid) microspheres through intravitreal injection in rabbits. Int J Nanomed. 2014;9:3057–3068. doi: 10.2147/IJN.S64100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dhup S, Dadhich RK, Porporato PE, Sonveaux P. Multiple biological activities of lactic acid in cancer: influences on tumor growth, angiogenesis and metastasis. Curr Pharm Des. 2012;18(10):1319–1330. doi: 10.2174/138161212799504902. [DOI] [PubMed] [Google Scholar]
- 37.Hong JT, Kim EJ, Ahn KS, et al. Inhibitory effect of glycolic acid on ultraviolet-induced skin tumorigenesis in SKH-1 hairless mice and its mechanism of action. Mol Carcinog. 2001;31(3):152–160. doi: 10.1002/mc.1050. [DOI] [PubMed] [Google Scholar]
- 38.Abdulkhalek S, Geen O, Brodhagen L, et al. Transcriptional factor Snail controls tumor neovascularization, growth and metastasis in mouse model of human ovarian carcinoma. Clin Transl Med. 2014;3:28. doi: 10.1186/s40169-014-0028-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shahani K, Panyam J. Highly loaded, sustained-release microparticles of curcumin for chemoprevention. J Pharm Sci. 2011;100(7):2599–2609. doi: 10.1002/jps.22475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shahani K, Swaminathan SK, Freeman D, Blum A, Ma L, Panyam J. Injectable sustained release microparticles of curcumin: a new concept for cancer chemoprevention. Cancer Res. 2010;70(11):4443–4452. doi: 10.1158/0008-5472.CAN-09-4362. [DOI] [PMC free article] [PubMed] [Google Scholar]