

Abstract
There is discrepancy regarding the duration of reperfusion required using 2,3,5-triphenyl-2H-tetrazolium chloride (TTC) staining to assess myocardial infarction in an isolated, perfused heart model. Several investigators prefer long-term reperfusion (120 minutes) to determine myocardial injury, while others have used a shorter duration (30-40 minutes). We investigated whether oxygen surrounding the myocardium during ischemia plays a critical role in the installation of myocardial infarction during reperfusion. Mice hearts were perfused with a Langendorff apparatus using Krebs Henseleit (KH) buffer oxygenated with 95% O2 plus 5% CO2 at 37°C. Hearts were either immersed in KH or suspended in air during 18 minutes of global ischemia in a normothermic, water-jacketed chamber. Hearts then were reperfused for 40, 60, or 90 minutes. We found that hearts immersed in KH had decreased recovery of function and increased myocardial infarct size, reaching a steady-state level after 40 minutes of reperfusion. In contrast, hearts suspended in air approached steady-state after 90 minutes of reperfusion. Thus, mitochondrial reactive oxygen species (ROS) production was much lower in air-maintained hearts than in KH-immersed hearts. To investigate whether an increase in oxygen surrounding the myocardium during ischemia might cause further damage, we bubbled the KH solution with nitrogen (KH+N2) rather than oxygen (KH+O2). With this alteration, recovery of cardiac function was improved and myocardial infarct size and mitochondrial ROS production were reduced compared with hearts immersed in KH+O2. In conclusion, short-term (40 minutes) reperfusion is sufficient to reach steady-state myocardial infarct size when hearts are immersed in physiologic solution during ischemia; however, a longer duration of reperfusion (90 minutes) is required if hearts are suspended in air. Thus, oxygen surrounding the heart during ischemia determines the extent of myocardium injury during reperfusion.
Keywords: Oxygen, reperfusion injury, ischemic conservation, heart function, myocardial infarct size
Introduction
A model using isolated perfused heart subjected to ischemia and reperfusion is an adequate alternative to the experimental model utilizing in vivo left anterior descending artery occlusion, with the isolated perfused heart allowing rigorous control of parameters that is not readily achievable by an in vivo model. The initial perfusion systems for the isolated mammalian heart were developed more than a century ago by H. Newell Martin and Oscar Langendorff. In the working mode, the isolated perfused heart was established in the 1960s. Consequently, during the past three decades, the isolated perfused heart system has been extensively used as an experimental model, despite the disadvantages of the lack of the normal humoral background and extrinsic neuronal regulation or the multiple conditions needed for a better isolated heart preparation. Sutherland stated that, although the isolated mouse heart has been used for many years in the Langendorff perfusion mode [1,2] and working heart mode [3,4], many characteristics remain undefined [5,6]. For example, there is no agreement on the reperfusion duration necessary to assess myocardial infarction with use of the 2,3,5-triphenyl-2H-tetrazolium chloride (TTC) staining method. Using isolated perfused rat or mouse heart, several groups have performed long-term reperfusion (120 minutes) to determine myocardial infarct size by the TTC staining method [7-9]. However, with application of the same method, other authors have preferred short-term reperfusion (30-40 minutes) to assess myocardial infarct size [10-13]. Moreover, with the same duration of ischemia, some investigators have used long-term reperfusion, while others have preferred short-term reperfusion [14,15]. In fact, Ruan and co-workers have subjected hearts to 30 minutes of global ischemia followed by 120 minutes of reperfusion to reveal a reduction in myocardial infarct size, determined by TTC staining, in PTEN (CKO) mice hearts compared to wild-type mice hearts [14]. In contrast, Zhao and co-workers have subjected isolated hearts to 30 minutes of ischemia followed by only 30 minutes of reperfusion to assess myocardial infarct size with TTC staining [15]. Specific reasons for the choice of reperfusion duration are not well established in studies. However, some investigators have concerns about assessing myocardial infarction by the TTC staining method after short-term reperfusion (30-60 minutes). Many studies have been rejected for publication because of the use of short-term reperfusion to assess myocardial infarction.
In this study, we investigated the optimal duration of reperfusion required to assess myocardial infarction by the TTC staining method as well as the role of oxygen surrounding the myocardium during ischemic conservation while installation of the myocardial injury is performed. To this end, hearts were reperfused for different times following the same duration of global normothermic ischemia. Hearts were either suspended in air or immersed in a normothermic physiologic solution (Krebs Henseleit [KH] buffer) during ischemia. To define the specific role of oxygen surrounding the myocardium during injury installation, during the reperfusion, KH was bubbled with nitrogen (KH+N2) instead of oxygen (KH+O2) during ischemia. We found that when hearts were immersed in KH during ischemia, recovery of function was much lower and myocardial infarct size was greater, reaching a steady-state level before 40 minutes of reperfusion. In contrast, hearts suspended in air had conserved basal function and a smaller myocardial infarct size approaching steady-state after 90 minutes of reperfusion. Consistent with the decreased myocardial injury, mitochondrial reactive oxygen species (ROS) production by stimulating complex I was much lower in air-maintained than in KH-immersed hearts. We also found that when hearts were immersed in KH+N2, recovery of cardiac function was improved and myocardial infarct size as well as ROS production were reduced as compared to hearts immersed in KH+O2. These results suggest an important role of oxygen surrounding the myocardium during ischemia in the installation of myocardium injury during reperfusion.
Materials and methods
Animals
Adult male wild-type (C57BL/6NCrl) mice, 8-12 weeks old, were used. Protocols were approved by the University of Texas Health Science Center at San Antonio Institutional Animal Care and Use Committee. The investigation conformed to the Guide for the Care and Use of Laboratory Animals, published by the US National Institute of Health (NIH Publication No. 85-23, revised 1996).
Langendorff preparation
The technique of isolated perfused mouse heart preparation has been recently described [16]. Mice were anesthetized by intraperitoneal injection of pentobarbital (60 mg/kg), and heparin (200 IU/kg) was injected to prevent blood coagulation. Once the animal was completely anesthetized, a transabdominal incision was performed and the diaphragm was cut to expose the thoracic cavity. The heart was then removed and immediately immersed and function was arrested in cold (4°C) KH buffer solution (mM): glucose 11.1, NaCl 118, KCl 4.7, MgSO4 1.2, KH2PO4 1.2, NaHCO3 25.0, and CaCl2 2 at pH 7.4. The aorta was rapidly cannulated and the heart was retrograde perfused at a constant rate (3 ml/l) in the Langendorff mode using KH solution. The buffer was bubbled with 95% O2 plus 5% CO2 at 37°C.
Experimental protocol design
Figure 1 illustrates the dispositive used to perfuse and conserve the heart during the ischemia/reperfusion cycle. Isolated mice hearts were assigned to one of the experimental groups presented in Figure 3A. All of the hearts underwent 18 min of global ischemia after 20 minutes of basal perfusion, followed by different times of reperfusion (Rep) (40 minutes for Rep-40, 60 minutes for Rep-60, or 90 minutes for Rep-90). Global normothermic ischemia was induced by clamping the aorta. During ischemic conservation hearts were either suspended in an air-jacketed chamber at 36.5°C or immersed in KH+O2 or KH+O2 in the water-jacketed chamber at the same temperature as shown in Figure 1.
Figure 1.
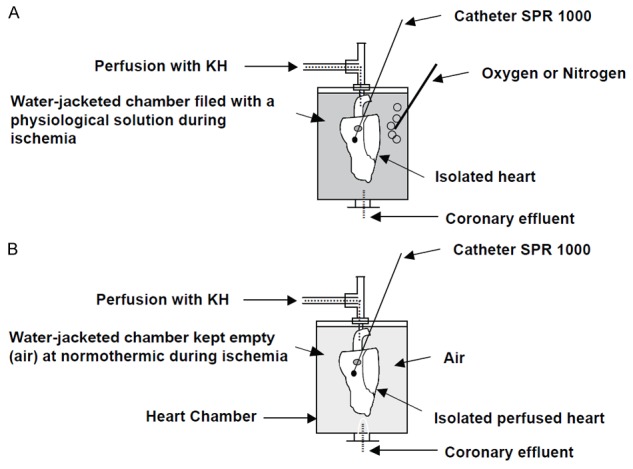
Illustration of the experimental dispositive (Langendorff) used to perfuse and conserve the heart during ischemia reperfusion. Note that the heart is maintained in the water-jacketed chamber during perfusion as well as reperfusion and the ischemic conservation period, and the temperature of perfusion is maintained constant at 36.5°C by a heater circulating pump. A. The heart is kept immersed in a physiological solution bublled either with oxygen or with nitrogen; B. The heart is kept suspended in air the normothermic chamber.
Figure 3.
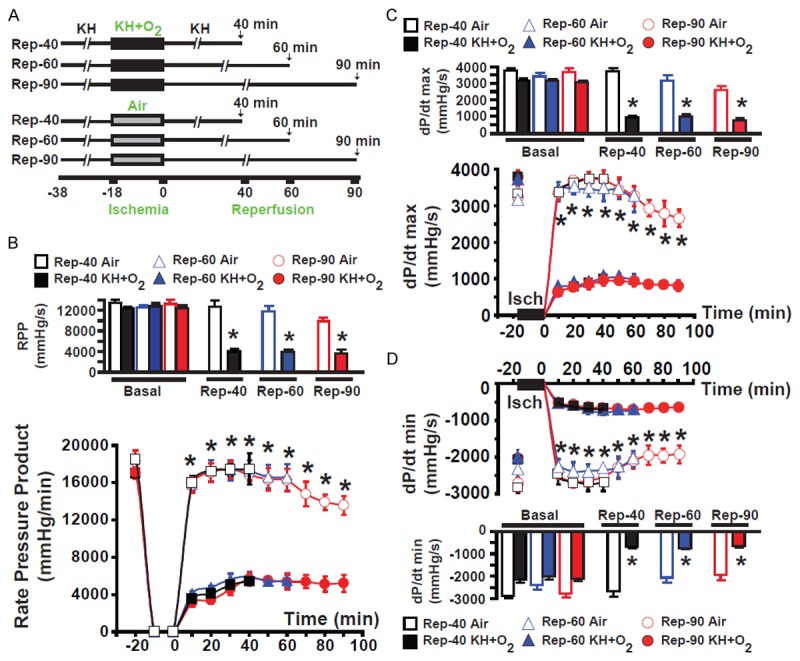
Graphs showing improved heart function in hearts suspended in air compared to hearts immersed in KH+O2 during normothermic ischemia. (A) The experimental procedure used to perfuse hearts in different groups. (B) The mean values of the rate pressure product (RPP) of hearts in different groups subjected to 18 minutes of global normothermic ischemia conserved either by immersion in KH or suspended in air and reperfused for 40 minutes (Rep-40) (black) or 60 minutes (Rep-60) (blue), of dP/dt max (C) and -dP/dt min (D). There were no significant differences throughout reperfusion between any group of hearts immersed in KH during ischemia. Similar results were obtained with hearts suspended in air during ischemia. However, recovery of cardiac function was much greater in hearts suspended in air than in hearts immersed in KH during the ischemic period. Values are expressed as mean ± SEM; *P<0.05 hearts immersed in KH+O2 versus hearts maintained in air during ischemia (n = 5-10/group).
Heart function measurements during ischemia and reperfusion
Left ventricular systolic pressure (LVSP), left ventricular end-diastolic pressure (LVEDP), and heart rate (HR) were recorded with a pressure transducer (Millar catheter) directly inserted into the left ventricle (LV). The LV-developed pressure (LVDP = LVSP - LVEDP), rate pressure product (RPP = LVDP x HR), maximum rate of rise of the LV contraction velocity (dP/dt max), and maximum isovolumetric rate of relaxation (-dP/dt min) were directly calculated using Chart software (LabChart 5.5).
Myocardial infarct size
Myocardial infarction was assessed at the end of the reperfusion by measurement of myocardial infarct size using 2,3,5-triphenyltetrazolium chloride (TTC) staining. At the end of reperfusion, each heart was cut into four transverse slices, parallel to the atrioventricular groove. After removal of right ventricular tissue, heart slices were weighed and incubated for 10 minutes in 1% TTC in Tris-HCl (pH 7.4) at 37°C followed by fixation with 4% paraformaldehyde. This procedure differentiates the infarcted (white) from viable (red) myocardial tissue. The slices were photographed using digital microscopic imaging. Extent of the area of necrosis was quantified by computerized planimetry with Adobe Photoshop. The total area of necrosis was calculated and expressed as the percentage of total LV area.
Preparation of isolated mitochondria
Mitochondria were isolated after only 10 minutes of reperfusion because at that time no infarction was present in the myocardium. All of the procedures were carried out at 4°C as previously described [17]. Myocardial sections (approximately 0.15-0.22 g) were placed in isolation buffer A (mM): 70 sucrose, 210 mannitol, 1 EDTA, and 50 Tris-HCl, pH 7.4. The tissue was finely minced with scissors and homogenized in the same buffer A (0.1 g of tissue/ml of buffer) using Kontes and Potter-Elvehjem tissue grinders. The homogenate was centrifuged at 1,300 xg for 3 minutes, and the supernatant was filtered through cheesecloth and centrifuged at 10,000 xg for 10 minutes. The resultant supernatant was discarded, and the pellet was gently washed 3 times with 500 µl of buffer B (in mM): 150 sucrose, 50 KCl, 2 KH2PO4, 5 succinic acid, and 20 Tris/HCl, pH 7.4. The pellet was then resuspended in 50 µl of the same buffer B. Mitochondrial protein concentration was assayed using the Bradford method. Protein was adjusted to a final concentration of 25 mg/ml.
Mitochondrial H2O2 measurement
Mitochondrial ROS generation was measured spectrophotometrically (560-nm excitation and 590-nm emission) in 125 µg/ml of mitochondrial protein incubated in a solution containing: 20 mM Tris, 250 mM sucrose, 1 mM EGTA, 1 mM EDTA, and 0.15% bovine serum albumin adjusted to pH 7.4 at 25°C with continuous stirring. ROS was measured with the H2O2-sensitive dye amplex red (10 µM) according to the manufacturer’s instructions (Nitrogen). H2O2 levels were measured from a calibration curve obtained from the fluorescence emission intensity as a function of H2O2 concentration. The sodium salt of glutamate/malate (3 mM) was used to activate complex I of the mitochondrial electron transfer chain.
Statistical analysis
Error bars indicate the standard errors of the mean (± SEM) for a minimum of three independent hearts (n ≥ 3). For cardiac infarct size and mitochondrial calcium retention capacity (CRC), means were compared between groups using one-way analysis of variance (ANOVA). For the ex vivo functional studies, mean profiles over time were compared across groups using repeated-measure ANOVA methods. Under the ANOVA model, the significance of pairwise mean comparisons was determined using the Tukey studentized range criterion. SPSS, version 13.0 (SPSS Inc, Chicago, IL), was used to carry out the computations. Because all outcomes were continuous, results were summarized with means ± SEMs. P < 0.05 was considered statistically significant.
Results
Heart function recovery
To investigate the role of ischemic conservation in the myocardial injury installation during reperfusion, cardiac function was recorded in all groups throughout the experiment, as shown in the protocol (Figure 3A). Figure 2 presents examples of recording of mice hearts subjected to ischemia/reperfusion in an ex vivo model using the Langendorff system. Figure 2 presents typical recordings of hearts reperfused for different durations (40, 60, and 90 minutes) following 18 minutes of global normothermic ischemia. Hearts were either maintained in KH+O2 (Figure 2A) or suspended in air in the air-jacketed chamber. We selected an ischemic period of 18 minutes that, in the ex vivo mouse model, typically shows ~50% of the infarct size in control conditions (Figure 4D). There was no significant difference in cardiac function between all groups before ischemia, as the RPP, dP/dt max, and dP/dt min were similar (Figure 3B-D). All other functional parameters are presented in Table 1. The post-ischemic LV function recovery was significantly decreased from baseline in all groups of hearts immersed in KH+O2 when compared with respective hearts suspended in air during ischemia with RPP: 5401±320 vs. 17465±1669 mmHg/min; dP/dt max: 940±115 vs. 3744±183 mmHg/s, and dP/dt min: -701±69 vs. -2688±229 mmHg/s in Rep-40; RPP: 5464±645 vs. 16557±1405 mmHg/min, dP/dt max: 976±145 vs. 3172±342 mmHg/s, and dP/dt min: -730±56 vs. -2047±225 mmHg/s in Rep-60; and RPP: 5195±917 vs. 13558±992 mmHg/min, dP/dt max: 805±149 vs. 2657±240 mmHg/s, and dP/dt min: -639±82 vs. -1920±243 mmHg/s in Rep-90 (p < 0.05). Mean values were obtained at the end of reperfusion in all groups. These data indicate that the recovery of heart function is better preserved when hearts are suspended in air during the ischemic period. However, we found that recovery of cardiac function was nearly identical in all groups of hearts immersed in KH+O2 during ischemic conservation (RPP: 5401±320 mmHg/min for Rep-40, (RPP: 5464±645 mmHg/min for Rep-60, and RPP: 5195±917 mmHg/min for Rep-90) (Figure 3A-C, see also other functional parameters presented in Table 1). Similarly, hearts suspended in air developed the same cardiac function recovery parameters (RPP: 17465±1669 mmHg/min for Rep-40; RPP: 16557±1405 mmHg/min for Rep-60; and RPP: 14558±992 mmHg/min for Rep-90). These results suggest that the increased duration of reperfusion from 40 to 90 minutes does not affect the recovery of heart function in either condition. In summary, recovery of heart function is much lower (steady-state at less than 40 minutes of reperfusion) when hearts are immersed in the KH buffer compared with the recovery in hearts suspended in air during ischemia (steady-state after 90 minutes of reperfusion).
Figure 2.
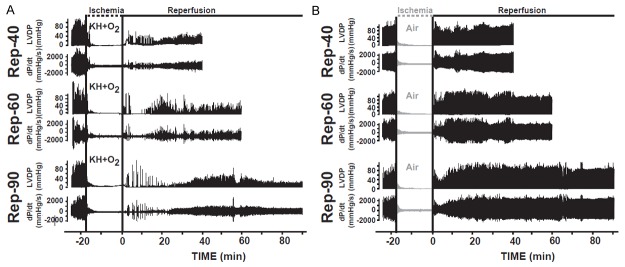
Heart function recovery was better conserved in hearts suspended in air compared to hearts immersed in KH+O2 during normothermic ischemic period. A. Recordings of ventricular-developed pressure (LVDP) and velocity of contraction and relaxation (dP/dt) in hearts immersed in KH+O2 during ischemic conservation and reperfused for 40 minutes (Rep-40), for 60 minutes (Rep-60), and for 90 minutes (Rep-90). B. Examples of heart function recordings showing the left ventricular-developed pressure (LVDP) and the maximum velocity of contraction and relaxation (dP/dt) of hearts suspended in air during the ischemic conservation period and reperfused for 40 minutes (Rep-40), 60 minutes (Rep-60), and 90 minutes (Rep-90). Note that in all groups, values before ischemia were similar, whereas after the ischemic period heart function recovery was greatly conserved in hearts suspended in air compared to the groups immersed in KH during ischemia.
Figure 4.
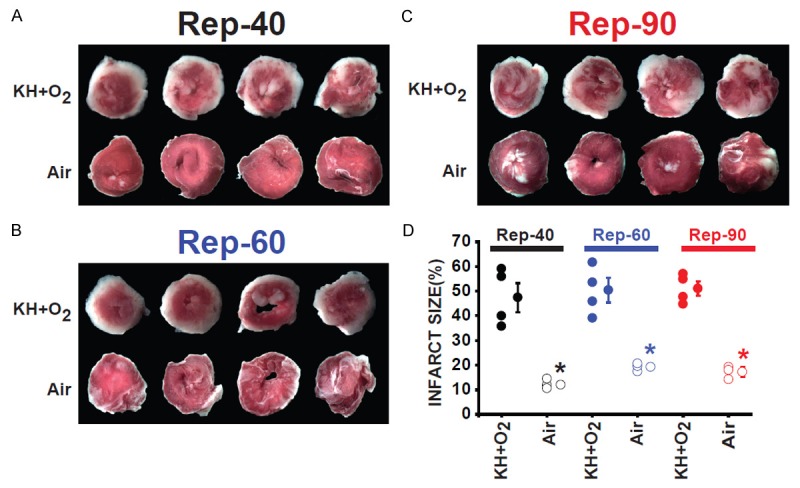
Myocardial infarct size was much smaller in hearts suspended in air compared to hearts immersed in KH+O2 during the normothermic ischemic period at the end of the reperfusion. Images of four slices of different regions (four transverse slices parallel to the atrioventricular groove) from the same heart in Rep-40 (A), Rep-60 (B), and Rep-90 (C) groups of hearts immersed in KH compared with hearts suspended in air during the ischemic period and reperfused for the same duration. Hearts were stained with TTC, showing infarcted regions in white and viable healthy regions in red. (D) Bar graph showing the individual percentage of myocardial infarct size in Rep-40, Rep-60, and Rep-90 in both conditions of ischemic conservation. Infarct size did not change significantly after the different times of reperfusion between the three groups immersed in KH as well as the groups suspended in air during the ischemic insult. However, the myocardial infarct size was much smaller in the groups of hearts suspended in air when compared to hearts immersed in KH during the ischemic period. Values mean ± SEM.; *P<0.05 hearts immersed in KH+O2 versus hearts maintained in air during ischemia (n = 4-5/group).
Table 1.
All heart functional recovery parameters
| Time (min) | Ischemic conservation | Groups | LVSP (mmHg) | LVEDP (mmHg) | LVDP (mmHg) | HR (beats·min-1) |
|
| ||||||
| KH+O2 | Rep-40 | 99±4 | 3±1 | 96±4 | 167±5 | |
| Air | Rep-40 | 115±7 | 1±1 | 115±6 | 162±10 | |
| Basal | KH+O2 | Rep-60 | 97±5 | 2±1 | 95±5 | 172±21 |
| KH+N2 | Rep-60 | 111±10 | 2±2 | 109±9 | 162±9 | |
| Perfusion | Air | Rep-60 | 112±7 | 2±1 | 111±7 | 158±7 |
| KH+O2 | Rep-90 | 101±5 | 4±1 | 97±6 | 165±43 | |
| Air | Rep-90 | 109±5 | 1±0.7 | 108±6 | 169±9 | |
|
| ||||||
| Reperfusion | ||||||
|
| ||||||
| KH+O2 | Rep-40 | 41±10* | 16±10 | 25±2* | 168±17 | |
| Air | Rep-40 | 112±5 | 1±1 | 112±4 | 155±6 | |
| KH+O2 | Rep-60 | 37±4 | 8±3 | 29±2 | 165±5 | |
| 20 min | Air | Rep-60 | 115±15 | 1±1 | 115±15 | 150±5 |
| KH+N2 | Rep-60 | 53±5+ | 3±2 | 50±5+ | 160±12 | |
| KH+O2 | Rep-90 | 44±16* | 20±15 | 24±3* | 153±21 | |
| Air | Rep-90 | 111±6 | 1±1 | 111±5 | 161±8 | |
| KH+O2 | Rep-40 | 43±6* | 13±7 | 30±3* | 165±5 | |
| Air | Rep-40 | 116±13 | 1±1 | 115±24 | 156±19 | |
| KH+O2 | Rep-60 | 43±3* | 7±2 | 36±2* | 168±5 | |
| 40 min | Air | Rep-60 | 114±13 | 1±1 | 113±13 | 158±10 |
| KH+N2 | Rep-60 | 68±8+ | 3±2 | 65±7+ | 174±5 | |
| KH+O2 | Rep-90 | 43±10* | 12±10 | 31±3* | 167±8 | |
| Air | Rep-90 | 108±7 | 1±0.64 | 108±7 | 160±8 | |
| KH+O2 | Rep-60 | 45±6* | 11±5 | 33±4 | 165±6 | |
| Air | Rep-60 | 94±11 | 1±0.4 | 94±11 | 180±9 | |
| 60 min | KH+N2 | Rep-60 | 74±8+ | 2±2 | 72±7+ | 162±16 |
| KH+O2 | Rep-90 | 52±11* | 19±11 | 33±7* | 170±9 | |
| Air | Rep-90 | 94±9 | 1±0.34 | 94±9 | 176±9 | |
| 90 min | KH+O2 | Rep-90 | 53±16* | 23±15 | 30±5* | 162±3 |
| Air | Rep-90 | 79±8 | 1±0.33 | 178±8 | 161±8 | |
All heart function recovery parameters were better preserved when hearts were suspended in air or immersed in nitrogen-KH solution (KH+N2) compared to hearts immersed in oxygenated-KH solution (KH+O2) during the normothermic ischemic period. Time course of cardiac function recovery parameters, including left ventricular systolic pressure (LVSP); left ventricular end-diastolic pressure (LVEDP); left ventricular developed pressure (LVDP) and heart rate (HR) of the three groups: 1) hearts immersed in KH+O2, 2) hearts immersed in KH+N2, and 3) in hearts suspended in air. Note that hearts immersed in KH+O2 and hearts suspended in air where subjected to three different reperfusion durations (Rep-40, Rep-60, and Rep-90), while hearts immersed in KH+N2 received only 60 minutes of reperfusion. Values are mean ± SEM.;
P<0.05 hearts immersed in KH+O2 versus hearts maintained in air;
P<0.05 hearts immersed in KH+O2 versus hearts immersed in KH+N2 (n = 4-10/group).
Myocardial infarct size
There was no myocardial infarct in non-ischemic groups after 20 minutes of initial perfusion between all four groups. Figure 4A-C shows typical heart cross sections at the end of different reperfusion durations after TTC staining. The red area represents the viable region, and the white area is the infarct size. The myocardial infarct size in groups of hearts immersed in KH+O2 (Rep-40, Rep-60, and Rep-90) was similar but significantly increased compared with hearts suspended in air during the ischemic period (47±5% vs. 12±1%, 50±5% vs. 17±2%, and 51±5% vs. 17±2%, respectively; p < 0.001) (Figure 4D). These data suggest that when hearts are immersed in KH+O2 during ischemia, the maximum injury occurs before 40 minutes of reperfusion. However, a much longer time of reperfusion is required when hearts are maintained in air during ischemia.
Reduction of oxygen during ischemia induces cardioprotection
We found that recovery of function was better preserved (Figures 2 and 3) and myocardial infarct size was less (Figure 4) in hearts suspended in air in the normothermic water-jacketed chamber during ischemia compared with hearts immersed in KH+O2. These data suggest that an increase in oxygen surrounding hearts immersed in KH+O2 during ischemia (100% compared to 21% in air) might be responsible for the greater injury observed. To test this hypothesis, we reduced the level of oxygen surrounding the hearts during ischemic conservation by using KH+N2. We found that, in this condition, the recovery of heart function was significantly improved in the KH+N2 group compared with the KH+O2 group (RPP: 11221±701 vs. 5464±645 mmHg/min, dP/dt max: 2808±173 vs. 976±145 mmHg/s, dP/dt min: -1915±207 vs. -730±56 mmHg/s; p < 0.05), (Figure 5A-C). Table 1 shows that all cardiac function parameters recorded were improved in hearts immersed in KH+N2 as compared to hearts immersed in KH+O2 (Figure 6A-C and Table 1). Furthermore, myocardial infarct size was reduced in hearts immersed in KH+N2 as compared to those immersed in KH+O2 (17±7% vs. 51±5%, respectively). Interestingly, myocardial infarct size in hearts immersed in KH+N2 was similar to the size in hearts maintained in air during ischemia (17±2%; p < 0.05; n = 4-5/group) (Figure 5D, 5E). These results suggest that the reduction in oxygen concentration surrounding the heart during ischemic conservation induces the decrease in myocardial injury and the improvement of the recovery of heart function. This finding also indicates that oxygen surrounding the heart during ischemia is one of the key factors that controls the installation of myocardial injury during reperfusion.
Figure 5.
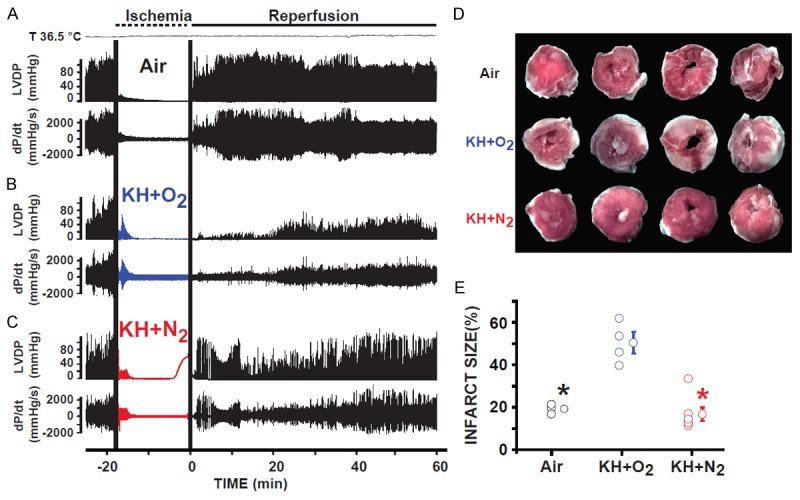
Hearts immersed in KH+N2 had improved recovery of function and reduced myocardial infarct size when compared to hearts immersed in (KH+O2 during the normothermic ischemic period. Recordings of ventricular-developed pressure (LVDP) and velocity of contraction and relaxation (dP/dt) in hearts suspended in air (A), immersed in KH+N2 (B), and immersed in KH+O2 (C) during the ischemic period and reperfused for 60 minutes. Note that the recovery of cardiac function was much greater in the groups of hearts immersed in KH+N2 than in hearts immersed in KH+O2 during the ischemic period. (D), Images of 4 slices of different regions from the same heart suspended in air, immersed in KH+N2, or immersed in KH+O2 during the ischemic period; hearts were obtained at the end of 60 minutes of reperfusion. (E) Bar graph showing the individual percentage of myocardial infarct size in the three conditions of ischemic conservation. The myocardial infarct size was much smaller in the groups of hearts suspended in air than in hearts immersed in KH+O2 during the ischemic period. The myocardial infarct size significantly decreased when hearts were immersed in KH+N2 when compared to hearts immersed in KH+O2. Values are expressed as mean ± SEM.; *P<0.05 hearts immersed in KH+O2 versus hearts maintained in air during ischemia; +P<0.05 hearts immersed in KH+O2 versus hearts immersed in KH+N2 during ischemia (n = 4-5/group).
Figure 6.
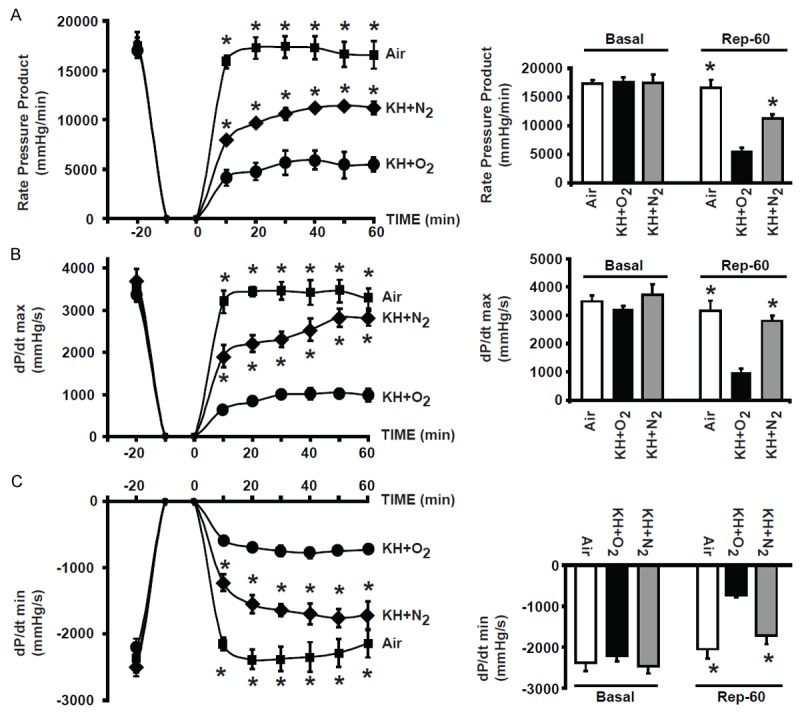
Graphs showing improved recovery of function in hearts immersed in KH+N2 compared to hearts immersed in KH+O2 during the normothermic ischemic period. Graph representing the mean values of the rate pressure product (RPP) (A), of dP/dt max (B) and -dP/dt min (C) in hearts suspended in air, immersed in KH+N2, or immersed in KH+O2 during the ischemic period, and followed by 60 minutes of reperfusion. Note that the cardiac function recovery parameters were much greater in the groups of hearts suspended in air than in hearts immersed in KH+O2 during the ischemic period. Recovery of heart function was significantly improved when hearts were immersed in KH+N2 compared to hearts immersed in KH+O2. Values are expressed as mean ± SEM; *P<0.05 hearts immersed in KH+O2 versus hearts maintained in air during ischemia; +P<0.05 hearts immersed in KH+O2 versus hearts immersed in KH+N2 during ischemia (n = 5-10/group).
Mitochondrial ROS production
With use of cell labeling methods, it has been demonstrated that an increase in mitochondrial ROS production is relevant in many pathological situations, such as myocardial infarction, diabetes mellitus, and cancer, and participates in necrosis and apoptosis associated with ischemia/reperfusion injury [18,19]. Because we found that oxygen surrounding the heart during ischemia played an important role in the installation of myocardial injury during reperfusion, we investigated whether the increase in myocardial injury observed in hearts immersed in KH+O2 might be associated with the increase in mitochondrial ROS production during reperfusion. We found that mitochondrial ROS produced by stimulating complex I of the electron transfer chain with glutamate/malate was much higher in the mitochondria of hearts immersed in KH+O2 compared to those immersed in KH+N2, as well as the mitochondria of hearts suspended in air (Figure 7A). In fact, the ROS production level in mitochondria isolated from the hearts immersed in KH+O2 was 272±13 pmol./min/mg of mitochondrial protein. This level of ROS production was much higher than that of mitochondrial protein produced by mitochondria isolated from hearts immersed in KH+N2 (187±15 pmol./min/mg), and of mitochondrial protein in mitochondria isolated from hearts maintained in air (192±18 pmol./min/mg), p < 0.05 (Figure 7A, 7B). This result suggests that the increase in mitochondrial ROS production may be responsible for the increase in myocardial injury during reperfusion in hearts immersed KH+O2.
Figure 7.
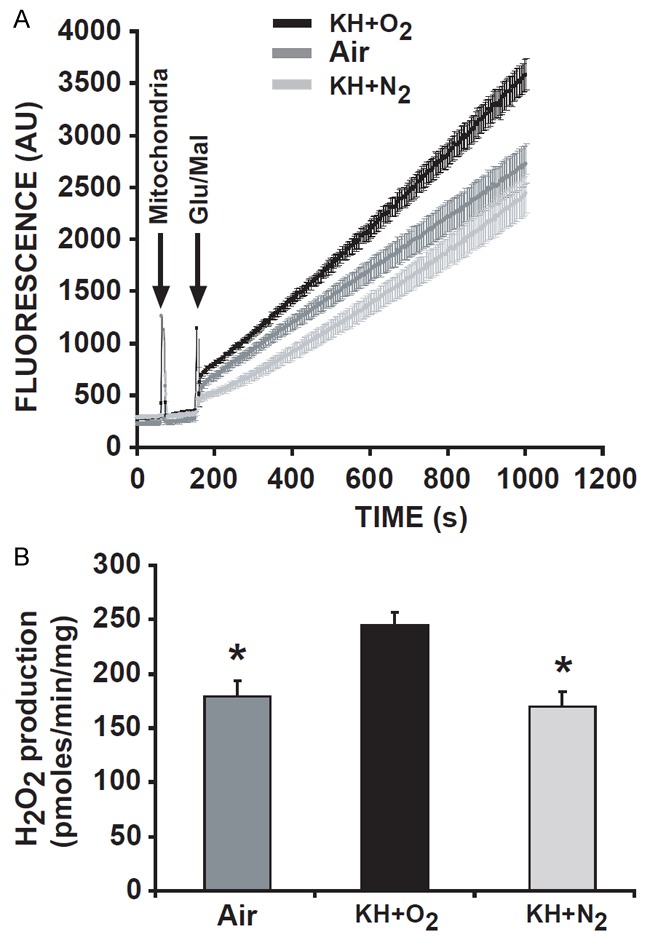
Mitochondrial ROS production was much lower in hearts suspended in air and immersed in KH+N2 than in hearts immersed in KH+O2 during the ischemic period. Graph representing the mean values of the rate of ROS production in mitochondria isolated from hearts suspended in air, immersed in KH+N2, or immersed in KH+O2 during the ischemic period (A), and the bar graph corresponding to H2O2 production in all groups (B). Note that mitochondrial ROS production was much lower in groups of hearts suspended in air than in hearts immersed in KH+O2 during the ischemic period. In addition, mitochondrial ROS production was significantly reduced when hearts were immersed in KH+N2 compared to hearts immersed in KH+O2. Values are expressed as mean ± SEM; *P<0.05 hearts immersed in KH+O2 versus hearts maintained in air during ischemia; +P<0.05 hearts immersed in KH+O2 versus hearts immersed in KH+N2 during ischemia (n = 9/group).
Discussion
In this study, we found that hearts immersed in a physiologic solution that is bubbled with oxygen during ischemic conservation developed much lower recovery of cardiac function and larger myocardial infarct size as compared to hearts immersed in the same solution but bubbled with nitrogen or those suspended in air. We also found that hearts suspended in air required a greater duration of reperfusion compared to hearts immersed in a physiologic solution. Heart function recovery and myocardial infarct size can be assessed by TTC staining with a shorter time of reperfusion in immersed hearts. Our finding suggests that oxygen surrounding the heart during ischemia has an important role in the installation of myocardial injury during reperfusion.
The duration of reperfusion needed to determine the degree the myocardial infarct by TTC staining after a normothermic ischemia insult is subject to discrepancy. For the same isolated perfused mouse heart model, some investigators have chosen to use two durations (120 or 180 minutes) of reperfusion, while others have preferred to reperfuse for only 30 or 40 minutes [7,10,11,14,15]. Sicard and co-workers, using TTC staining in the isolated perfused mouse heart, reperfused hearts for 120 minutes after 30 minutes of global normothermic ischemia. With that method, SB203580 eliminated the reduction in myocardial infarct size caused by preconditioning [7]. Using the same experimental protocol with TTC staining, Ruan and co-workers revealed a significant reduction in myocardial infarct size in PTEN (CKO) mice hearts when compared with controls [14]. However, Zhao and co-workers have subjected isolated mice hearts to only 30 minutes of reperfusion after 30 minutes of ischemia (the same duration used by Ruan and colleagues) to assess myocardial infarct size after TTC staining [15]. Furthermore, Jin and co-workers used a protocol consisting of 20 minutes of global ischemia followed by only 30 minutes of reperfusion to observe the reduction of myocardial infarct size induced by ischemic preconditioning eliminated by N,N-dimethyl sphingosine (DMS) [10].
In this study, we determined myocardial infarct size and recovery of heart function with different durations of reperfusion. Using the isolated perfused mice heart model, we compared the myocardial infarct size at the end of 40, 60, and 90 minutes of reperfusion after 18 minutes of global normothermic ischemia of hearts immersed in KH+O2 solution. In these conditions, we found that myocardial infarct size and the heart function recorded were stable after 40 minutes of reperfusion. This result indicates that when hearts are immersed in a normothermic physiologic solution, the installation of the maximum level of myocardial infarct size assessable by TTC staining occurs before 40 minutes of reperfusion. In fact, Jin and co-workers have measured 45±4% of myocardial infarct size after 20 minutes of ischemia followed by only 30 minutes of reperfusion in isolated mouse heart [10]. These observations are similar to our finding of 47±5% of myocardial infarct size when hearts were immersed in a solution during ischemia followed by 40 minutes of reperfusion. Short-term reperfusion to determine myocardial infarction is supported by other investigators. Wang and co-workers have subjected mice hearts to 30 minutes of ischemia followed by 30 minutes of reperfusion in a Langendorff-perfused heart model to assess myocardial damage [13]. Several groups have observed that myocardial infarct size assessed at the end of 60 minutes of reperfusion was similar to the size at 120 minutes in isolated perfused rat hearts subjected to 40 minutes of ischemia [20,21].
The rationale for use of a specific duration of reperfusion is not mentioned in most articles. However, the main factor in the determination of the reperfusion duration in an ischemia/reperfusion protocol is undoubtedly related to the manner in which hearts are maintained during the ischemia insult. When hearts are suspended in air in the normothermic water-jacketed chamber, as represented in Figure 1, the temperature inside the myocardium will not rapidly reach 37°C. Consequently, such myocardium kept in mild hypothermia will require a much longer time of reperfusion (120 minutes) for installation of myocardial infarction to be determined by TTC staining. However, when hearts are immersed in a normothermic physiologic solution in the water-jacketed chamber, the temperature inside the myocardium will rapidly reach normothermia (37°C) during ischemic conservation, probably due to a much higher convection of the heat from liquid to myocardium. In this case, hearts do not require a much longer reperfusion period for installation of myocardial injury. A short reperfusion period of 30-40 minutes is sufficient to result in a maximum myocardial infarct size assessable by TTC staining.
We found that when hearts were conserved by immersion in KH+O2 during ischemia, the reperfusion duration needed to reach the steady-state level of myocardial infarction is very short, while a much longer duration is required for hearts suspended in air in the normothermic water-jacketed chamber. The present study validates the use of short-term reperfusion (~40 minutes) to assess myocardial infarction by TTC staining in a perfused isolated heart model.
Because ROS production is an important factor involved in cell death after ischemia/reperfusion, we investigated whether oxygen transfer from the milieu of ischemic conservation to the myocardium might cause an increase in mitochondrial ROS production and play a key role in myocardial infarction installation during reperfusion. To this end, we reduced the O2 concentration present in the physiologic solution during the ischemic conservation by bubbling the KH buffer with nitrogen instead of oxygen. Figure 5 indicates that hearts maintained in KH+N2 during ischemia have improved recovery of cardiac function and decreased myocardial infarct size when compared with hearts maintained in KH+O2 (Figure 5B-E). This result suggests that oxygen surrounding the myocardium during ischemic conservation plays a key role in the installation of myocardial injury during reperfusion. The adverse effect of higher oxygen concentration in the myocardium during reperfusion can be explained by its facility to increase oxidative stress, resulting in an increase in ROS production. In fact, we found that mitochondrial ROS production measured in isolated mitochondria from hearts immersed in KH+O2 during the ischemic insult was much higher than in mitochondria that were isolated from hearts immersed in KH+N2 and hearts suspended in air (Figure 6) during ischemia. The increase in mitochondrial ROS production in hearts immersed in KH+O2 during ischemic insult might be explained by a higher concentration of oxygen compared within the concentration in air (~21% O2), as well as, by a much higher diffusion of oxygen across the endocardium-solution interface than the endocardium-air interface. The resultant increase in oxygen concentration inside the myocardium triggers a cascade of oxidative reactions during reperfusion that causes an increase in cellular oxidative stress responsible for the increase in ROS production and a decrease in ATP production in the post-ischemic myocardium. The increase in ROS production associated with mitochondrial calcium overload are two known key factors that promote the opening of the mitochondrial permeability transition pore leading to cell death by apoptosis and necrosis. This process may explain why hearts immersed in KH+O2 need a short time of reperfusion to reach the maximum of myocardial infarction installation that can be determined by TTC staining. In conclusion, oxygen surrounding the heart during ischemic conservation determines the installation of myocardial injury during reperfusion.
Acknowledgements
Supported by Bopassa’s AHA fellowship 09POST2190008, Voelcker fund (Bopassa).
Disclosure of conflict of interest
None.
References
- 1.Galinanes M, Hearse DJ. Assessment of ischemic injury and protective interventions: the Langendorff versus the working rat heart preparation. Can J Cardiol. 1990;6:83–91. [PubMed] [Google Scholar]
- 2.Hearse DJ, Yamamoto F, Shattock MJ. Calcium antagonists and hypothermia: the temperature dependency of the negative inotropic and anti-ischemic properties of verapamil in the isolated rat heart. Circulation. 1984;70:I54–I64. [PubMed] [Google Scholar]
- 3.Grupp IL, Subramaniam A, Hewett TE, Robbins J, Grupp G. Comparison of normal, hypodynamic, and hyperdynamic mouse hearts using isolated work-performing heart preparations. Am J Physiol. 1993;265:H1401–H1410. doi: 10.1152/ajpheart.1993.265.4.H1401. [DOI] [PubMed] [Google Scholar]
- 4.Hearse DJ, Mullins AJ. An isolated perfused working guinea-pig heart preparation. J Physiol. 1972;223:4P–5P. [PubMed] [Google Scholar]
- 5.Sutherland FJ, Shattock MJ, Baker KE, Hearse DJ. Mouse isolated perfused heart: characteristics and cautions. Clin Exp Pharmacol Physiol. 2003;30:867–78. doi: 10.1046/j.1440-1681.2003.03925.x. [DOI] [PubMed] [Google Scholar]
- 6.Sutherland FJ, Hearse DJ. The isolated blood and perfusion fluid perfused heart. Pharmacol Res. 2000;41:613–27. doi: 10.1006/phrs.1999.0653. [DOI] [PubMed] [Google Scholar]
- 7.Sicard P, Clark JE, Jacquet S, Mohammadi S, Arthur JS, O’Keefe SJ, Marber MS. The activation of p38 alpha, and not p38 beta, mitogen-activated protein kinase is required for ischemic preconditioning. J Mol Cell Cardiol. 2010;48:1324–8. doi: 10.1016/j.yjmcc.2010.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sun J, Murphy E. Calcium sensing receptor: a sensor and mediator of ischemic preconditioning in the hearts. Am J Physiol Heart Circ Physiol. 2010;299:H1309–17. doi: 10.1152/ajpheart.00373.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bak I, Czompa A, Juhasz B, Lekli I, Tosaki A. Reduction of reperfusion-induced ventricular fibrillation and infarct size via heme oxygenase-1 overexpression in isolated mouse hearts*. J Cell Mol Med. 2010;14:2268–72. doi: 10.1111/j.1582-4934.2010.01142.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jin ZQ, Goetzl EJ, Karliner JS. Sphingosine kinase activation mediates ischemic preconditioning in murine heart. Circulation. 2004;110:1980–9. doi: 10.1161/01.CIR.0000143632.06471.93. [DOI] [PubMed] [Google Scholar]
- 11.Tokudome S, Sano M, Shinmura K, Matsuhashi T, Morizane S, Moriyama H, Tamaki K, Hayashida K, Nakanishi H, Yoshikawa N, Shimizu N, Endo J, Katayama T, Murata M, Yuasa S, Kaneda R, Tomita K, Eguchi N, Urade Y, Asano K, Utsunomiya Y, Suzuki T, Taguchi R, Tanaka H, Fukuda K. Glucocorticoid protects rodent hearts from ischemia/reperfusion injury by activating lipocalin-type prostaglandin D synthase-derived PGD2 biosynthesis. J Clin Invest. 2009;119:1477–88. doi: 10.1172/JCI37413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Siddall HK, Warrell CE, Yellon DM, Mocanu MM. Ischemia-reperfusion injury and cardioprotection: investigating PTEN, the phosphatase that negatively regulates PI3K, using a congenital model of PTEN haploinsufficiency. Basic Res Cardiol. 2008;103:560–8. doi: 10.1007/s00395-008-0735-y. [DOI] [PubMed] [Google Scholar]
- 13.Wang QD, Swardh A, Sjoquist PO. Relationship between ischaemic time and ischaemia/reperfusion injury in isolated Langendorff-perfused mouse hearts. Acta Physiol Scand. 2001;171:123–8. doi: 10.1046/j.1365-201x.2001.00788.x. [DOI] [PubMed] [Google Scholar]
- 14.Ruan H, Li J, Ren S, Gao J, Li G, Kim R, Wu H, Wang Y. Inducible and cardiac specific PTEN inactivation protects ischemia/reperfusion injury. J Mol Cell Cardiol. 2009;46:193–200. doi: 10.1016/j.yjmcc.2008.10.021. [DOI] [PubMed] [Google Scholar]
- 15.Zhao TC, Zhang LX, Cheng G, Liu JT. gp-91 mediates histone deacetylase inhibition-induced cardioprotection. Biochim Biophys Acta. 2010;1803:872–80. doi: 10.1016/j.bbamcr.2010.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bopassa JC, Eghbali M, Toro L, Stefani E. A novel estrogen receptor GPER inhibits mitochondria permeability transition pore opening and protects the heart against ischemia-reperfusion injury. Am J Physiol Heart Circ Physiol. 2010;298:H16–H23. doi: 10.1152/ajpheart.00588.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bopassa JC, Ferrera R, Gateau-Roesch O, Couture-Lepetit E, Ovize M. PI 3-kinase regulates the mitochondrial transition pore in controlled reperfusion and postconditioning. Cardiovasc Res. 2006;69:178–85. doi: 10.1016/j.cardiores.2005.07.014. [DOI] [PubMed] [Google Scholar]
- 18.Chen Q, Moghaddas S, Hoppel CL, Lesnefsky EJ. Ischemic defects in the electron transport chain increase the production of reactive oxygen species from isolated rat heart mitochondria. Am J Physiol Cell Physiol. 2008;294:C460–C466. doi: 10.1152/ajpcell.00211.2007. [DOI] [PubMed] [Google Scholar]
- 19.Droge W. Free radicals in the physiological control of cell function. Physiol Rev. 2002;82:47–95. doi: 10.1152/physrev.00018.2001. [DOI] [PubMed] [Google Scholar]
- 20.Ferrera R, Benhabbouche S, Bopassa JC, Li B, Ovize M. One hour reperfusion is enough to assess function and infarct size with TTC staining in Langendorff rat model. Cardiovasc Drugs Ther. 2009;23:327–31. doi: 10.1007/s10557-009-6176-5. [DOI] [PubMed] [Google Scholar]
- 21.Kanno S, Lee PC, Zhang Y, Ho C, Griffith BP, Shears LL, Billiar TR. Attenuation of myocardial ischemia/reperfusion injury by superinduction of inducible nitric oxide synthase. Circulation. 2000;101:2742–8. doi: 10.1161/01.cir.101.23.2742. [DOI] [PubMed] [Google Scholar]