

Abstract
Interstitial lung disease (ILD) is frequently associated with collagen disease. It is then designated as collagen vascular disease-associated ILD (CVD-ILD), and influences patients’ prognosis. The prognosis of acute-onset diffuse ILD (AoDILD) occurring in patients with collagen disease is quite poor. Here, we report our investigation of auto-antibody (Ab) profiles to determine whether they may be useful in diagnosing CVD-ILD or AoDILD in collagen disease. Auto-Ab profiles were analyzed using the Lambda Array Beads Multi-Analyte System, granulocyte immunofluorescence test, Proto-Array Human Protein Microarray, AlphaScreen assay, and glutathione S-transferase capture enzyme-linked immunosorbent assay in 34 patients with rheumatoid arthritis (RA) with or without CVD-ILD and in 15 patients with collagen disease with AoDILD. The average anti-major histocompatibility complex class I-related chain A (MICA) Ab levels were higher in RA patients with CVD-ILD than in those without (P = 0.0013). The ratio of the average anti-MICA Ab level to the average anti-human leukocyte antigen class I Ab level (ie, MICA/Class I) was significantly higher in RA patients with CVD-ILD compared with those without (P = 4.47 × 10−5). To the best of our knowledge, this is the first report of auto-Ab profiles in CVD-ILD. The MICA/Class I ratio could be a better marker for diagnosing CVD-ILD than KL-6 (Krebs von den lungen-6).
Keywords: auto-antibody profile, anti-MICA antibody, collagen disease, interstitial lung disease, biological marker
Introduction
Interstitial lung disease (ILD) is characterized by interstitial inflammation of the lung and is frequently associated with collagen diseases including rheumatoid arthritis (RA), systemic lupus erythematosus, systemic sclerosis (SSc), polymyositis/dermatomyositis (PM/DM), and polyarteritis nodosa. ILD associated with collagen disease is then designated as collagen vascular disease-associated ILD (CVD-ILD). The presence of CVD-ILD is one of the major manifestations of collagen disease that influences prognosis.1,2 Acute-onset diffuse ILD (AoDILD) occurs in patients with collagen disease with or without underlying CVD-ILD.3,4 AoDILD includes acute exacerbation of ILD, drug-induced ILD, and Pneumocystis pneumonia. The prognosis of AoDILD is quite poor. Thus, biomarkers for the early detection of CVD-ILD and AoDILD are urgently needed.
Krebs von den lungen-6 (KL-6) and surfactant protein-D (SP-D) are currently used as serum surrogate markers for ILD screening. However, these markers have low sensitivity for the discrimination of CVD-ILD.5,6 It was also previously reported that some auto-antibodies (Abs) in the sera of patients with collagen disease are good markers for ILD screening. RA patients with high rheumatoid factor levels have a higher risk of ILD, while there was an association of high levels of anti-cyclic citrullinated peptide Abs and ILD in RA.7 Anti-CADM-140 (clinically amyopathic dermatomyositis-140)/MDA5 (melanoma differentiation-associated gene 5)/IFIH1 (interferon-induced helicase C domain-containing protein 1) Abs are detected in sera of patients with dermatomyositis without clinical findings of myositis and are accepted as good markers for AoDILD in dermatomyositis.8 ILD is also frequently associated with the presence of anti-aminoacyl-tRNA synthetase Abs in PM/DM patients.9 However, neither anti-CADM-140/MDA5/IFIH1 Abs nor anti-aminoacyl-tRNA synthetase Abs were present in sera from patients with collagen diseases other than PM/DM.10,11
Transfusion-related acute lung injury (TRALI) is as an acute respiratory distress syndrome associated with blood transfusion; some of its causes are thought to be anti-human leukocyte antigen (HLA) or anti-granulocyte Abs.12 However, it is still unknown how these Abs cause the injury only in lung in TRALI. Thus, Abs recognizing self-antigens may be associated with CVD-ILD or AoDILD. Together, these extensive studies suggest that the detection of auto-Ab markers using auto-Ab profiling may provide a useful indicator for the presence of CVD-ILD and AoDILD. Hence, we investigated the auto-Ab profiles of CVD-ILD and AoDILD in patients with collagen disease.
Materials and Methods
Patients
Thirty-four Japanese patients with RA were recruited at Sagamihara Hospital. ILD was diagnosed from computed tomography findings. Images were reviewed by two physicians specializing in CVD-ILD, and categorized from A to Z, according to the Sagamihara Criteria.13 RA cases in categories A–D were diagnosed as “RA with ILD [ILD(+)RA]” and those in G and H were diagnosed as “RA without ILD [ILD(−)RA]”. RA cases in categories A or H were included in this study.
Twenty-five patients with collagen disease [mean age ± standard deviation (SD): 65.9 ± 10.8 years; 11 men) were admitted to Sagamihara Hospital between 2001 and 2010, because of AoDILD requiring corticosteroid pulse therapy, as previously reported.4 AoDILD was defined as acute onset and progression within a month, the presence of clinical symptoms (fever, dry cough, or dyspnea), hypoxia, and computed tomography findings of ILD.3,4 Patients with evidence of apparent bacterial infection or heart disease were excluded. They were classified according to the American College of Rheumatology criteria for RA,14 SSc,15 and Bohan’s criteria for PM/DM.16 Diagnoses of the patients included 20 RA, 2 SSc, and 3 PM/DM. None of these 20 RA patients was included in the aforementioned 34 RA patients with or without ILD.
Ethical statements
This study was reviewed and approved by the Sagamihara Hospital Research Ethics Committee. Written informed consent was obtained from all study participants except those already deceased before starting this study. The serum samples collected before this study began were anonymized in a manner preventing any link with the patients identification, and their analysis was approved on that condition by the Sagamihara Hospital Research Ethics Committee. This study was conducted in accordance with the principles expressed in the Declaration of Helsinki.
Sera
Sera from the 34 RA patients with or without ILD were collected, and these individual sera were analyzed for anti-HLA Ab profiles.
Sera from the 15 collagen disease patients with AoDILD were collected on admission and in the stable state, at least three months before admission. Two samples were collected from each patient. These individual sera were analyzed for anti-HLA Ab profiles. The sera from these patients either with AoDILD or in the stable state were combined; the two pooled sera at these two states were screened for the auto-Ab profiling to be described in the following sections (Fig. 1).
Figure 1.

Auto-Ab identification methods used in this study. (A) Sera from the 34 rheumatoid arthritis (RA) patients with or without ILD were analyzed for anti-HLA Ab profiles. (b) Sera from the 15 collagen disease patients with acute-onset diffuse ILD (AoDILD) were collected on admission and in the stable state. Two samples were collected from each patient. These individual sera were analyzed for anti-HLA Ab profiles or granulocyte immunofluorescence test (GIFT). (C) The sera from these patients either with AoDILD or in the stable state were combined; the two pooled sera at these two states were screened for the auto-Ab profiling in ProtoArray and AlphaScreen systems. (D) These individual sera were analyzed by glutathione S-transferase (GST) capture enzyme-linked immunosorbent assay (ELISA). Analyses of individual sera are written in black and those of pooled sera in red.
Rheumatoid factor and anti-citrullinated peptide antibody were detected using N-latex RF kit (Siemens Healthcare Diagnostics, Germany) and Mesacup-2 test CCP (Medical & Biological Laboratories, Japan), respectively. KL-6 and SP-D were detected using a Picolumi KL-6 Electrochemiluminescence immunoassay system (EIDIA Co., Ltd, Japan) and SP-D kit “Yamasa” EIA II (Yamasa Corporation, Japan), respectively.
Anti-HLA Ab analysis
Anti-HLA class I, class II, and major histocompatibility complex class I-related chain A (MICA) Ab levels were analyzed using the Lambda Array Beads Multi-Analyte System (LABScreen Mixed Beads Lot#016, One Lambda, Supplementary Table S1), according to the manufacturer’s protocol.17 Briefly, 20 µL of serum was incubated with the beads for 30 minutes at room temperature in the dark. The beads were washed three times with 1 × wash buffer, and 100 µL of phycoerythrin-conjugated goat anti-human IgG Abs (diluted 1:100 in 1 × wash buffer, One Lambda) was added. After a 30-minute incubation step at room temperature in the dark, the beads were washed twice and analyzed using a Luminex 100 system (Luminex). The obtained data were analyzed by HLA Fusion 2.0 (One Lambda). Trimmed means, which remove the lower and upper 5% of the extreme fluorescence intensity, were calibrated by the subtraction of the negative control signal.
Granulocyte immunofluorescence test (GIFT)
GIFT was performed as previously described, with some modifications.17,18 Briefly, 6 mL blood samples from healthy individuals (C-1~6) were treated with ethylenediaminetetraacetic acid disodium salt (Wako Pure Chemical Industries, Japan), overlaid on 5 mL Lymphosepar I density gradient (Immuno-Biological Laboratories Company, Japan), and centrifuged at 2000 g for 10 minutes. After isolation of the granulocyte layer, erythrocytes were lysed in 10 mL of 0.19 M NH4Cl lysis buffer at 37°C for 15 minutes. The cells were washed and resuspended in 1% paraformaldehyde (MERCK) in phosphate-buffered saline (PBS) and incubated for 10 minutes at room temperature. After three washes, 5 × 105 cells were incubated with 50 µL sera [1:4 diluted in 2% horse serum (Cedarlane) in 0.1% NaN3−PBS] in 96-well U-bottomed plates for 30 minutes at 37°C, followed by three washing steps and incubation with 50 µL fluorescein isothiocyanate-conjugated goat F(ab′)2 anti-human IgG Fc Abs (1:100, Jackson ImmunoResearch Laboratories) for 30 minutes at 4°C. After two more washing steps, anti-granulocyte Abs were detected by flow cytometric analysis using an FC 500 MPL (Beckman-Coulter). The mode fluorescence intensity (MFI) of the cells gated for granulocytes in the scattergram was taken for data analysis.
Auto-Ab profiling
Immune response biomarker profiling was performed with ProtoArray Human Protein Micro-array v5.0 (Invitrogen) for detection of auto-Abs in pooled sera from 15 patients with collagen disease either with AoDILD or in the stable state, according to the manufacturer’s protocol.19 The two pooled sera at AoDILD or the stable states were screened for the auto-Ab profiling. The array contains 9,483 probes for 7,783 human proteins. After blocking and washing, the array was incubated with the pooled sera diluted 1:500 in 5 mL of washing buffer at 4°C for 1.5 hours. After five washes, the array was incubated with Alexa Fluor 647 goat anti-human IgG Abs (0.9 µg/mL) and Alexa Fluor 647 anti-V5 Ab. After another five washes, data were acquired with a microarray scanner GenePix 4000B (Molecular Devices), quantified using GenePix Pro 5.1 (Molecular Devices), and finally analyzed by ProtoArray Prospector 5.20 (Invitrogen). The obtained signals were calibrated by the subtraction of negative control signals. Calibrated signals were normalized for comparison between arrays using the Z-factor, which is a metric that takes into account the signal dynamic range and the variation associated with the control and sample features [Z = 1 − (3σs + 3σc−)/|µs − µc−|, where σs is the signal sample SD for the protein features, σc− is the signal sample SD for the negative control features, µs is the mean signal for the protein features, and µc− is the mean signal for the negative control features).20 A Z-factor >0.5 indicates a signal greater than twofold above the noise. A protein with a Z-factor >0.4 was considered as the auto-antigen reacting with the auto-Abs in the sera. The data obtained with Protoarray were deposited in the public repository, Gene Expression Omnibus at the National Center for Biotechnology Information, and are accessible by accession number GSE59687.
The AlphaScreen assay was also performed for the detection of auto-Abs in pooled sera from patients either with AoDILD or in the stable state, according to the manufacturer’s protocol (PerkinElmer Life and Analytical Sciences).21 Antigens were synthesized using cell-free protein synthesis systems prepared from wheat embryos.22,23 The assay contains 2,426 probes for 2,307 human proteins. AlphaScreen reactions were carried out in 25 µL of reaction volume in 384-well plates. For the antigen–auto-Ab reaction, the translation mixture expressing the biotinylated protein was mixed with the pooled sera diluted 1:730 in 10 µL of reaction buffer and incubated at 26°C for 30 minutes. Subsequently, 10 µL of streptavidin-coated donor beads and protein A-conjugated acceptor beads (PerkinElmer Life and Analytical Sciences) were added to a final concentration of 20 µg/mL and incubated at 26°C for 1 hour in the dark. Fluorescence emission was measured with the EnVision plate reader (PerkinElmer Life and Analytical Sciences), and the data were analyzed using the AlphaScreen detection program. The cutoff level for auto-Ab positivity was designated as the mean + 3SD of negative control signals.
Glutathione S-transferase capture enzyme-linked immunosorbent assay (ELISA)
Potential auto-antigens were selected for further glutathione S-transferase (GST) capture ELISA analysis of auto-Ab target proteins in individual patient serum, based on the AlphaScreen data. The ratios of the AlphaScreen signals between stable and AoDILD states for these antigens were higher than 1.2 or lower than 0.8, and the differences were larger than 30 or smaller than −50.
A GST capture ELISA system was employed for the detection of auto-Abs in each individual serum.24 Briefly, ELISA plates precoated with 20 µg/mL glutathione-casein were blocked by 0.2% casein, followed by incubation with eight selected GST-fused human recombinant proteins [ATPBD1C, AYTL1, C12orf32 (RHNO1), CD44, CD55, IL13RA2, MUC20, SYT1] synthesized using the wheat germ cell-free protein production system.22,23 The coated ELISA plates were incubated with individual sera diluted 1:300 in 100 µL. Bound auto-Abs were detected by alkaline phosphatase-conjugated goat anti-human IgG-Fc Abs (Sigma-Aldrich). The alkaline phosphatase activity was determined by colorimetric assay for p-nitrophenyl phosphate substrate conversion by reading the absorption of each well at 405 and 510 nm for reference using a 96-well microplate reader. All GST capture ELISAs were carried out in triplicate.
Statistical analysis
Differences in patient characteristics were analyzed by Mann–Whitney’s U-test or Fisher’s exact test using 2 × 2 contingency tables. Mann–Whitney’s U-test was performed for the comparison of Ab assay results between ILD(+)RA and ILD(−)RA. Logistic regression analysis was conducted to develop an ILDIndex for ILD in RA from KL-6 values and MICA/Class I ratios. Wilcoxon signed-rank test was performed for the comparison of Ab assay results between AoDILD and in the stable state. The sample size needed on this study with the power of 80% to detect association was estimated with Java Applets for Power and Sample Size (http://www.stat.uiowa.edu/~rlenth/Power.).25 Statistical significance was defined as P < 0.05.
Results
Characteristics of the RA patients
Characteristics of the ILD(+)RA and ILD(−)RA groups are described in Table 1. KL-6 was greatly elevated in the ILD(+)RA group compared with the ILD(−)RA group. There were no differences in the mean age, percentage of males, disease duration, Steinbrocker stage,26 smoking habit, positivity for rheumatoid factor, anti-citrullinated peptide Ab, or SP-D between the two groups.
Table 1.
Characteristics of the RA patients.
| ILD(+)RA | ILD(−)RA | P | ||
|---|---|---|---|---|
| Number | 17 | 17 | ||
| Age | Years | 67.0 (9.5) | 65.8 (7.2) | *0.8092 |
| Males | n (%) | 8 (47.1) | 2 (11.8) | 0.0570 |
| Disease duration | Years | 12.4 (7.7) | 18.7 (11.3) | *0.0755 |
| Steinbrocker stage III and IV | n (%) | 9 (52.9) | 13 (76.5) | 0.2818 |
| Smoker or past smoker | n (%) | 7 (41.2) | 3 (17.6) | 0.2451 |
| Rheumatoid factor-positive | n (%) | 15 (88.2) | 15 (88.2) | 1.0000 |
| Anti-citrullinated peptide Ab-positive | n (%) | 14 (82.4) | 13 (76.5) | 1.0000 |
| KL-6 | U/mL | 605.4 (418.4) | 258.9 (131.1) | *0.0005 |
| SP-D | ng/mL | 91.4 (84.4) | 45.7 (36.5) | *0.1165 |
Notes: Average values of each group are shown. Standard deviations or percentages are shown in parenthesis. Differences were tested by Mann-Whitney’s U test or Fisher’s exact test using 2 × 2 contingency tables.
Mann–Whitney’s U-test was employed.
Abbreviations: RA, rheumatoid arthritis; ILD(+)RA, RA with ILD; ILD(−)RA, RA without ILD; Ab, antibody; KL-6, Krebs von den lungen-6; SP-D, surfactant protein-D.
Anti-HLA Ab profiles of RA patients with or without ILD
Anti-HLA Ab profiles in RA patients were analyzed in ILD(+)RA and ILD(−)RA groups (Table 2). Anti-HLA class I (beads# 003, 004, 005, 009, 011, 014, 016, 017, 046, 049, 077, and 078, Supplementary Table S1), anti-class II (beads# 050, 053, 056, 057, and 059), and anti-MICA (beads# 074 and 075) Abs were compared.
Table 2.
Anti-HLA Ab profiles of the RA patients with or without ILD.
| BEADS# | NC | ILD(+)RA | ILD(−)RA | P | |
|---|---|---|---|---|---|
| Class I | 003 | 18.1 | 101.6 (92.7) | 205.3 (574.8) | 0.3796 |
| Class I | 004 | 23.6 | 83.5 (63.0) | 228.3 (422.1) | 0.5240 |
| Class I | 005 | 24.6 | 107.9 (79.3) | 276.4 (856.9) | 0.4384 |
| Class I | 009 | 25.9 | 118.7 (131.6) | 263.0 (751.2) | 0.3613 |
| Class I | 011 | 69.2 | 145.7 (69.9) | 381.9 (1086.8) | 0.4183 |
| Class I | 014 | 29.6 | 113.2 (79.1) | 251.4 (584.6) | 0.5698 |
| Class I | 016 | 34.4 | 119.9 (79.6) | 104.4 (147.4) | 0.1965 |
| Class I | 017 | 16.4 | 70.9 (39.3) | 121.6 (167.5) | 0.6668 |
| Class I | 046 | 26.2 | 94.3 (67.0) | 301.8 (964.6) | 0.3613 |
| Class I | 049 | 30.8 | 124.6 (143.5) | 197.9 (478.5) | 0.3987 |
| Class I | 077 | 18.0 | 89.7 (70.3) | 269.3 (855.1) | 0. 4181 |
| Class I | 078 | 21.0 | 65.9 (66.7) | 182.1 (574.1) | 0.2931 |
| Class II | 050 | 20.3 | 86.8 (99.8) | 176.0 (536.1) | 0.1339 |
| Class II | 053 | 102.9 | 283.3 (305.1) | 278.2 (353.4) | 0.7697 |
| Class II | 056 | 44.0 | 187.5 (239.4) | 188.7 (337.1) | 0.2779 |
| Class II | 057 | 50.1 | 164.8 (159.6) | 484.3 (1635.2) | 0.1630 |
| Class II | 059 | 28.3 | 163.0 (245.4) | 196.4 (504.5) | 0.1339 |
| MICA | 074 | 27.7 | 139.2 (280.6) | 30.5 (19.8) | 0.0014 |
| MICA | 075 | 19.5 | 348.7 (1060.5) | 69.2 (157.4) | 0.0026 |
| Average | |||||
| Class I | 28.2 | 103.0 (70.7) | 231.9 (610.0) | 0.6419 | |
| Class II | 49.1 | 177.1 (194.4) | 264.7 (665.6) | 0.2486 | |
| Class I, Class II | 34.3 | 124.8 (85.3) | 241.6 (470.4) | 0.5934 | |
| MICA | 23.6 | 244.0 (669.3) | 49.8 (80.0) | 0.0013 | |
| Ratio of average | |||||
| MICA/Class I | 0.8 | 1.7 (2.9) | 0.4 (0.3) | 4.47 × 10−5 | |
| MICA/Class II | 0.5 | 1.7 (3.7) | 0.7 (1.7) | 0.0040 | |
| MICA/(Class I, Class II) | 0.7 | 1.6 (3.1) | 0.3 (0.3) | 0.0001 | |
Notes: Average values of each group are shown. Standard deviations are shown in parenthesis. Differences were tested by Mann–Whitney’s U-test. Specific antigens coated on the fluorescence beads are described in Supplementary Table S1.
Abbreviations: HLA, human leukocyte antigen; RA, rheumatoid arthritis; ILD, interstitial lung disease; ILD(+)RA, RA with ILD; ILD(–)RA, RA without ILD; NC, negative control; Ab: antibody.
Anti-HLA class I and class II Ab levels in ILD(+)RA tended to be lower than in the ILD(−)RA group, but these differences did not reach statistical significance. However, anti-MICA Ab levels were significantly elevated in the ILD(+)RA group. Mean Ab levels were calculated so that they reflected the levels of auto-Abs for common domains of HLA molecules by reducing the effects of allele-specific allo-Abs. The averages of anti-HLA class I and class II Ab levels in ILD(+)RA tended to be lower than in the ILD(−)RA group. In contrast, the average of anti-MICA Ab levels was significantly higher in the ILD(+)RA group. The ratio of the average anti-MICA Ab levels to the average anti-HLA class I Ab levels (MICA/Class I) was markedly higher in the ILD(+)RA group (Table 2, Fig. 2A). The sample size estimated on this analysis with the power of 80% to detect association is 50 each. Similarly, the ratio of anti-MICA Ab levels to anti-HLA class II Ab levels and of anti-MICA Ab levels to anti-HLA Ab levels were also higher in the ILD(+)RA group. Thus, anti-MICA Ab levels in the ILD(+)RA group are higher than in the ILD(−)RA group.
Figure 2.

Evaluation of the ratio of the average anti-major histocompatibility complex class I-related chain A (MICA) Ab levels to the average anti-human leukocyte antigen (HLA) class I Ab levels (MICA/Class I), as a marker for interstitial lung disease (ILD) in rheumatoid arthritis (RA) patients. (A) Distribution of the MICA/Class I ratio. Horizontal bars denote the means. The horizontal dotted line represents an optimized cut-off level (MICA/Class I = 0.770, with specificity and sensitivity of 0.882 and 0.824, respectively). ILD(+)RA: RA with ILD, ILD(−)RA: RA without ILD. (b) The receiver operating characteristic (ROC) curve using the MICA/Class I ratio (solid line) and KL-6 (dotted line) as markers for ILD in RA. The area under the curve (AUC) value of the ROC curve for MICA/Class I is 0.912 and that for KL-6 is 0.853. (C) Distribution of KL-6. Horizontal bars denote the means. The horizontal dotted line represents an optimized cut-off level (KL-6 = 296, with specificity and sensitivity of 0.824 and 0.875, respectively).
The area under the curve (AUC) value of the receiver operating characteristic (ROC) curve for the MICA/Class I ratio was 0.912 (Fig. 2B). Specificities and sensitivities of this ratio were estimated from the ROC curve conditional on the highest Youden’s index. The optimized cut-off level was 0.770 for ILD in RA with relatively high sensitivity (0.824) and specificity (0.882). On the other hand, the AUC of the KL-6 ROC curve in this study was 0.853. The optimized cut-off level of KL-6 was 296 with a sensitivity of 0.875 and specificity of 0.824 (Fig. 2C). These data suggest that the MICA/Class I ratio could perform better than KL-6 as a screening marker for ILD in RA.
ILDIndex was generated from KL-6 values and MICA/Class I ratios and evaluated as a marker for ILD in RA; ILDIndex = exp[4.28 × 10−4 × (KL-6) + 0.576 × (MICA/Class I)]/[1 + exp{4.28 × 10−4 × (KL-6) + 0.576 × (MICA/Class I)}]. ILDIndex was markedly higher in the ILD(+)RA group [Figure 3A, P = 6.71 × 10−6, mean ± SD = 0.714 ± 0.089 in ILD(+)RA, 0.586 ± 0.047 in ILD(−)RA]. The AUC value of the ROC curve for the ILDIndex was 0.960 (Fig. 3B). Specificities and sensitivities of ILDIndex were estimated from the ROC curve conditional on the highest Youden’s index. The optimized cut-off level was 0.652 for ILD in RA with relatively high sensitivity (1.000) and specificity (0.882).
Figure 3.
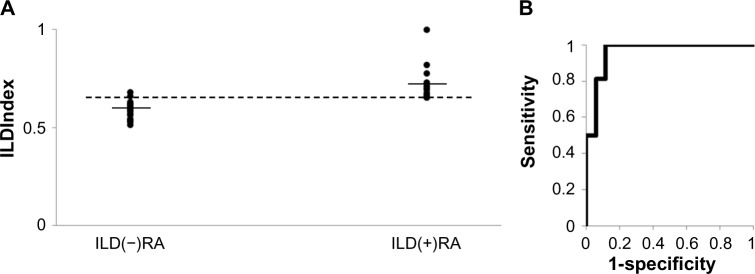
Evaluation of ILDIndex as a marker for interstitial lung disease (ILD) in rheumatoid arthritis (RA) patients. (A) Distribution of the ILDIndex. Horizontal bars denote the means. The horizontal dotted line represents an optimized cut-off level (ILDIndex = 0.652, with specificity and sensitivity of 0.882 and 1.000, respectively). ILD(+)RA: RA with ILD, ILD(−)RA: RA without ILD. (b) The receiver operating characteristic (ROC) curve using the ILDIndex as markers for ILD in RA. The area under the curve (AUC) value of the ROC curve for ILDIndex is 0.960.
Anti-HLA Ab profiles of patients with collagen disease in stable and AoDILD states
Next, the anti-HLA Ab profiles between the stable and the AoDILD states of each patient with collagen disease were compared. Characteristics of the 15 collagen disease patients with AoDILD were as previously described; albumin and sodium ion concentrations were decreased and KL-6 was increased in AoDILD.6 Anti-HLA class I and class II, and anti-MICA Abs were compared in both states (Table 3). Anti-HLA class I levels for several beads (003, 009, 011, 014, 017, 046, 049, 077, and 078) in AoDILD were lower than in the stable state. Anti-HLA class II and anti-MICA Ab levels were comparable in AoDILD and stable states. Mean anti-HLA class I Ab levels in AoDILD were also lower than in the stable state. The sample size estimated on this analysis with the power of 80% to detect association is 108 each. The averages of anti-HLA class II and anti-MICA Ab between AoDILD and stable states were comparable. Thus, the anti-HLA Ab profiles were different between AoDILD and stable states.
Table 3.
Anti-HLA Ab profiles of patients with collagen disease in the stable and AoDILD states.
| BEADS# | NC | STABLE | AODILD | P | |
|---|---|---|---|---|---|
| Class I | 003 | 0.0 | 231.7 (192.6) | 164.6 (147.9) | 0.0038 |
| Class I | 004 | 10.7 | 208.8 (208.0) | 169.3 (174.6) | 0.1578 |
| Class I | 005 | 23.8 | 211.4 (200.9) | 170.2 (200.0) | 0.1155 |
| Class I | 009 | 21.3 | 276.6 (244.6) | 193.4 (173.8) | 0.0066 |
| Class I | 011 | 54.4 | 272.1 (180.9) | 207.2 (98.3) | 0.0324 |
| Class I | 014 | 69.7 | 273.1 (263.7) | 207.1 (232.3) | 0.0214 |
| Class I | 016 | 24.1 | 230.4 (252.0) | 183.5 (231.5) | 0.0980 |
| Class I | 017 | 1.1 | 229.1 (273.0) | 175.0 (239.4) | 0.0422 |
| Class I | 046 | 33.0 | 228.5 (194.3) | 172.2 (163.1) | 0.0264 |
| Class I | 049 | 13.1 | 298.5 (409.5) | 243.1 (383.6) | 0.0422 |
| Class I | 077 | 13.5 | 192.2 (234.5) | 152.2 (229.1) | 0.0480 |
| Class I | 078 | 1.8 | 129.5 (123.5) | 89.2 (71.7) | 0.0230 |
| Class II | 050 | 0.0 | 160.3 (201.0) | 113.7 (150.4) | 0.0736 |
| Class II | 053 | 100.1 | 503.0 (709.8) | 554.3 (848.1) | 0.4118 |
| Class II | 056 | 18.2 | 314.5 (532.4) | 317.7 (491.0) | 0.2418 |
| Class II | 057 | 46.6 | 311.6 (388.3) | 233.1 (290.3) | 0.0653 |
| Class II | 059 | 5.0 | 233.9 (351.1) | 250.8 (353.4) | 0.5449 |
| MICA | 074 | 38.6 | 233.6 (173.3) | 183.7 (169.5) | 0.0980 |
| MICA | 075 | 12.7 | 198.3 (413.2) | 252.5 (598.3) | 0.0780 |
| Average | |||||
| Class I | 22.2 | 231.8 (214.2) | 177.2 (185.6) | 0.0186 | |
| Class II | 34.0 | 304.7 (405.2) | 293.9 (412.6) | 0.1658 | |
| MICA | 25.7 | 215.9 (241.8) | 218.1 (318.7) | 0.1658 | |
Notes: Average values of each group are shown. Standard deviations are shown in parenthesis. Differences were tested by Wilcoxon signed-rank test. Specific antigens coated on the fluorescence beads are described in Supplementary Table S1.
Abbreviations: HLA, human leukocyte antigen; AoDILD, acute-onset diffuse interstitial lung disease; NC, negative control; Ab, antibody.
GIFT of the patients with collagen disease in stable and AoDILD states
Anti-granulocyte Ab levels were compared by GIFT in the AoDILD and stable states of the 5 of the 15 collagen disease patients with AoDILD as described in the Materials and Methods section (Table 4). No marked change of anti-granulocyte Ab levels was detected in patients #1, #3, and #4 with RA or PM/DM. Anti-granulocyte Ab levels for C-1, C-2, C-3, and C-4 were decreased in the AoDILD state of patient #2 with RA. The extremely high level of anti-granulocyte Ab for C-1 in the stable state had disappeared in the AoDILD state of patient #5 with RA.
Table 4.
GIFT of the collagen disease patients in the stable and AoDILD states.
| PATIENT# | GRANULOCYTES FROM HEALTHY INDIVIDUALS (C-1–6) | ||||||
|---|---|---|---|---|---|---|---|
| UNDERLYING DISEASE | STATE | C-1 | C-2 | C-3 | C-4 | ||
| 1 | RA | Stable | 30.7 | 26.4 | 32.7 | 28.9 | |
| AoDILD | 31.3 | 23.3 | 32.7 | 29.4 | |||
| 2 | RA | Stable | 44.8 | 42.1 | 51.8 | 38.8 | |
| AoDILD | 35.5 | 27.1 | 37.8 | 31.9 | |||
| 3 | RA | Stable | 47.3 | 38.1 | 52.7 | 44.8 | |
| AoDILD | 45.7 | 42.5 | 64.8 | 47.8 | |||
| HC | 22.8 (2.5) | 19.3 (1.8) | 24.8 (4.5) | 22.7 (0.5) | |||
| C-1 | C-2 | C-5 | C-6 | ||||
| 4 | PM/DM | Stable | 28.9 | 23.0 | 21.1 | 54.7 | |
| AoDILD | 28.6 | 25.4 | 25.9 | 59.8 | |||
| 5 | RA | Stable | 403.0 | 27.3 | 30.5 | 47.8 | |
| AoDILD | 28.3 | 24.3 | 28.9 | 58.7 | |||
| HC | 22.8 (2.5) | 19.3 (1.8) | 19.2 (0.8) | 45.4 (5.6) | |||
Notes: The mode fluorescence intensity (MFI) of cells gated for granulocytes in the scattergram is shown. Average values of healthy control group (n = 3) are also shown. Standard deviations are shown in parenthesis.
Abbreviations: GIFT, granulocyte immunofluorescence test; AoDILD, acute-onset diffuse interstitial lung disease; RA, rheumatoid arthritis; PM/DM, polymyositis/dermatomyositis; HC, healthy control.
Auto-Ab profiles of sera pooled from patients with collagen disease in the stable and AoDILD states
Auto-Ab levels in pooled sera were compared between the AoDILD and stable states with ProtoArray (Fig. 4A, Supplementary Table S2). According to the Z-factor cutoff value of 0.4, auto-Abs for 767 probes (689 proteins) were present in both the AoDILD and stable states. Auto-Abs for a further 865 probes (827 proteins) were found in the AoDILD but not in the stable state. Reciprocally, auto-Ab for 82 probes (80 proteins) were found in the stable but not in the AoDILD state. For the remaining 7,769 probes (6,566 proteins), there were no auto-Abs detected in either state. The number of auto-Abs present only in the sera from patients with AoDILD was greater than the number of auto-Abs only present in the stable state. Anti-HLA-B or C Abs were detected neither in the AoDILD nor stable states with ProtoArray. Thus, auto-Ab profiles were different in AoDILD and stable states.
Figure 4.
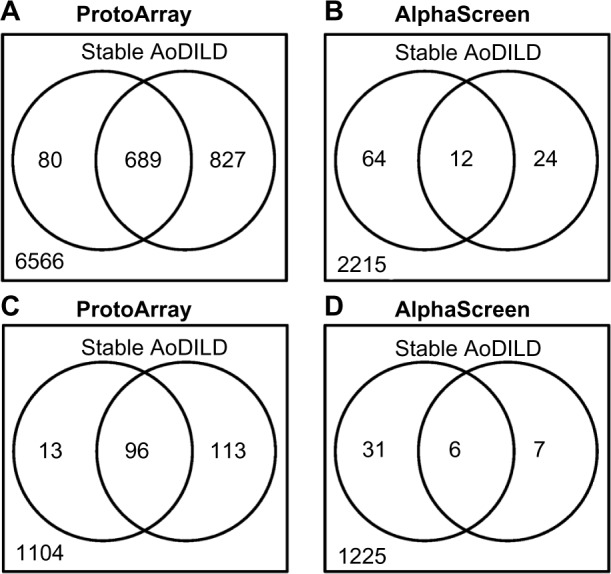
Auto-Abs present in the pooled sera from collagen disease patients with acute-onset diffuse ILD (AoDILD). The numbers of auto-Abs detected with (A) ProtoArray and (b) AlphaScreen in pooled sera from patients with collagen disease in the stable (left circle) and AoDILD states (right circle). The numbers of auto-Abs for proteins overlapped between (C) ProtoArray and (D) AlphaScreen in pooled sera from patients with collagen disease in the stable (left circle) and AoDILD states (right circle).
Auto-Ab levels of the pooled sera were also compared between the AoDILD and stable states using AlphaScreen (Fig. 4B, Supplementary Table S3). The average ± SD values of negative control signals in this assay were 199.6 ± 10.9; the cutoff level was 232.2. According to this cutoff value, auto-Abs were detected for 12 probes (12 proteins) in both the AoDILD and stable states and for 24 probes (24 proteins) in the AoDILD but not stable state. Auto-Abs for 64 probes (64 proteins) were seen in sera of the stable state but not of the AoDILD state, whereas no Ab was found in either state for 2,326 probes (2,215 proteins). Thus, the number of auto-Abs present only in the stable state was greater than those present only in the AoDILD state. This reflects the differences in the antigens selected in the two different assay systems; ProtoArray is an array-based binding assay in solid phase, whereas AlphaScreen is a bead-based proximity assay in liquid phase.
Auto-Ab levels for proteins overlapped between ProtoArray and AlphaScreen were analyzed between the AoDILD and stable states (Fig. 4C and D). In ProtoArray, auto-Abs were detected for 120 probes (96 proteins) in both the AoDILD and stable states, and for 119 probes (113 proteins) in the AoDILD but not stable state. Auto-Abs for 13 probes (13 proteins) were seen in the sera of the stable state but not of the AoDILD state, whereas no Ab was found in either state for 1,337 probes (1,104 proteins). In AlphaScreen, auto-Abs were detected for six probes (six proteins) in both the AoDILD and stable states, and for seven probes (seven proteins) in the AoDILD but not stable state. Auto-Abs for 31 probes (31 proteins) were seen in the sera of the stable state but not of the AoDILD state, whereas no Ab was found in either state for 1,297 probes (1,225 proteins).
GST capture ELISA on individual sera from patients with collagen disease in the stable and AoDILD states
Eight potential auto-antigens [ATPBD1C, AYTL1, C12orf32 (RHNO1), CD44, CD55, IL13RA2, MUC20, SYT1] were selected for further analysis of auto-Ab target proteins in individual patient serum, based on the AlphaScreen data. The eight antigens are thought to be related to immunological response, apoptosis, inflammation, DNA repair, fibrosis, or lung. Auto-Ab levels in individual sera were also compared between the AoDILD and stable states using this GST capture ELISA for the eight selected antigens (Table 5). The results show that the auto-Ab levels against AYTL1 marginally decreased in the AoDILD state, whereas they increased in the AoDILD states in ProtoArray and AlphaScreen assays. Thus, no apparent change of auto-Ab levels was observed in the AoDILD state.
Table 5.
Auto-Abs detected by GST capture ELISA in the individual sera of the collagen disease patients in the stable and AoDILD state.
| STABLE | AoDILD | P | |
|---|---|---|---|
| ATPBD1C | 0.410 (0.342) | 0.380 (0.352) | 0.2641 |
| AYTL1 | 0.600 (0.374) | 0.533 (0.386) | 0.0480 |
| C12orf32 [RHNO1] | 0.748 (0.466) | 0.691 (0.502) | 0.0926 |
| CD44 | 0.228 (0.219) | 0.217 (0.221) | 0.7064 |
| CD55 | 0.464 (0.433) | 0.465 (0.483) | 0.3395 |
| IL13RA2 | 0.308 (0.325) | 0.276 (0.294) | 0.3395 |
| MUC20 | 0.164 (0.219) | 0.156 (0.208) | 0.3130 |
| SYT1 | 0.555 (0.368) | 0.490 (0.326) | 0.2312 |
Notes: Average values of absorbance in each group are shown. Standard deviations are shown in parenthesis. Differences were tested by Wilcoxon signed-rank test.
Abbreviations: AoDILD, acute-onset diffuse interstitial lung disease; Ab, antibody; GST, glutathione S-transferase; ELISA, enzyme-linked immunosorbent assay.
Discussion
Several studies have shown that auto-Ab profiles are altered in various diseases and have explored the possibility of using them for diagnosis or to evaluate treatments.27–32 Few studies have focused on comprehensive auto-Ab profiling in CVD-ILD, although some auto-Abs are known to be associated with this disease condition.7–9 We attempted to diagnose the presence of CVD-ILD, one of the potentially life-threatening manifestations of collagen diseases, using auto-Ab profiles. To the best of our knowledge, this is the first report of auto-Ab profiles in CVD-ILD. We found that anti-MICA Ab levels were significantly higher in the ILD(+)RA group (Table 2). The MICA/Class I ratio could be a better marker for ILD in RA than KL-6 (Fig. 2). Anti-Class I Ab levels were lower in the AoDILD state (Tables 3 and 5). Thus, we found that auto-Ab profiles are altered in CVD-ILD.
It was already well known that solid organ transplantation causes the production of anti-HLA donor-specific Abs in recipients, accelerating graft rejection.33 The pathogenesis of chronic rejection caused by anti-HLA Abs induced after organ transplantation is thought to be similar to that of autoimmune diseases.34 Although anti-HLA Abs observed in our study (Tables 2 and 3) are not allele-specific, they may explain some aspects of the pathogenesis of collagen diseases.
MICA is a non-classical HLA class I antigen encoded in the HLA region of chromosome 6 and is highly polymorphic. MICA is a ligand of NKG2D, one of the natural killer (NK) receptors expressed on T cells and NK cells.35,36 Pulmonary epithelial cells infected with Pseudomonas aeruginosa upregulate cell-surface expression of ligands of NKG2D, which play important roles in the clearance of the bacteria.37,38 Escherichia coli and Mycobacterium tuberculosis infection also increase cell-surface expression of NKG2D ligands.39,40 Anti-MICA Abs in patients with collagen disease may block the NKG2D–MICA interaction and prevent the stimulation of T cells or NK cells for clearance of bacteria, causing sustained inflammation followed by the fibrotic changes of the lung (Fig. 5).
Figure 5.
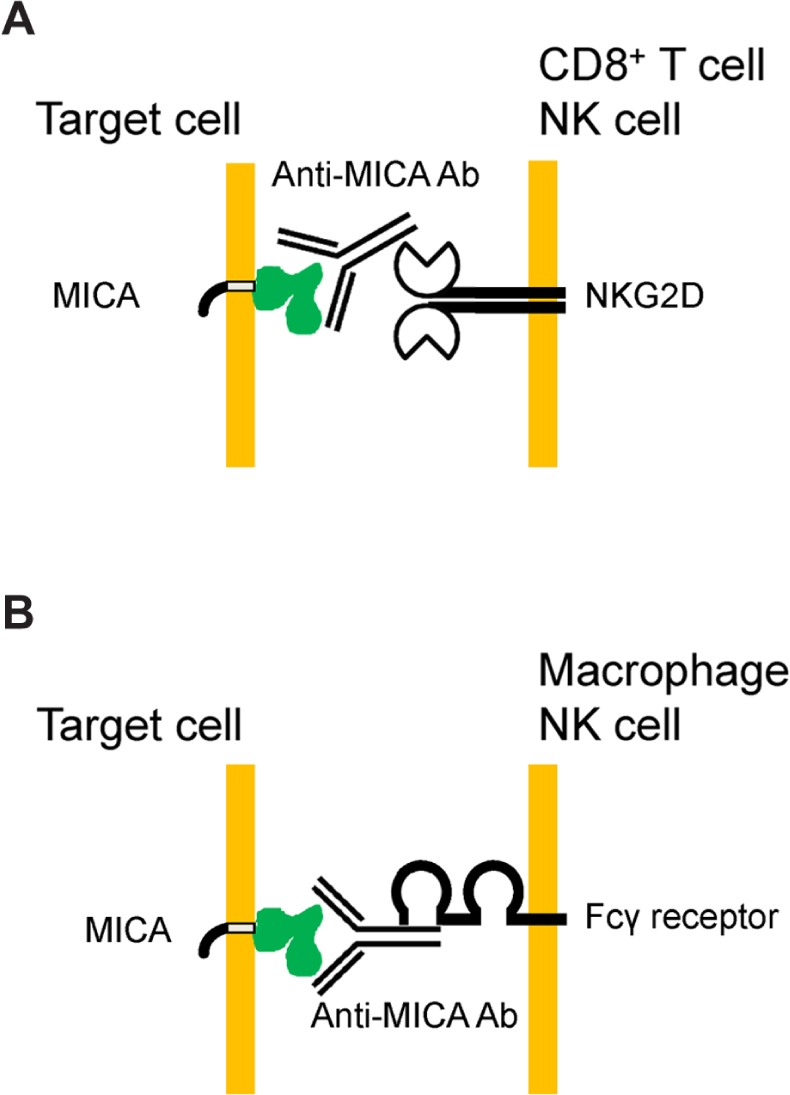
Hypotheses on the roles of anti-MICA Abs. (A) Anti-MICA Abs block the NKG2D–MICA interaction and prevent the stimulation of CD8+ T cells or NK cells for clearance of microorganisms, causing sustained inflammation followed by the fibrotic changes of the lung. (b) Chronically produced auto-Abs for MICA in RA patients may cause injury of the pulmonary epithelial cells expressing MICA by antibody-dependent cellular cytotoxicity.
TRALI is a life-threatening adverse effect of transfusion,12 which is thought to be caused by anti-HLA or anti-granulocyte Abs. Slowly but steadily produced auto-Abs for nonclassical HLA class I antigens may cause CVD-ILD in RA, and rapid infusion of allo-Abs recognizing HLA antigens of recipients may cause acute lung injury. It is also known that anti-HLA class I Abs stimulate pulmonary endothelial cells and cause lung injury.41,42 In the present study, anti-HLA or anti-granulocyte Abs were detected in sera of the patients with collagen diseases, though only limited data for anti-granulocyte Abs were obtained because of the difficulty of conducting GIFT on many patients. However, we did not find any evidence implicating these Abs in causing ILD in patients with collagen disease. It was also reported that kidney transplantation can result in the generation of anti-MICA Abs in recipients; these Abs caused graft rejection.43 Since the mechanism of chronic graft rejection is similar to that of autoimmune diseases,34 chronically produced auto-Abs for nonclassical HLA class I in RA patients may also cause injury of the pulmonary epithelial cells expressing MICA. The unknown roles of anti-MICA Abs in RA patients should be further elucidated.
The conformation of antigens screened in ProtoArray, AlphaScreen, or GST capture ELISA would not be maintained, though antigens screened in LABScreen Mixed Beads or GIFT were human cell surface antigens with physiological conformation. Thus, conformational epitopes of anti-MICA Abs in the sera of the patients were detected in this study. Other auto-Abs than rheumatoid factors or anti-citrullinated peptide Abs were frequently detected in sera of RA patients,27 as anti-MICA Abs were found in this study. The role of these auto-Abs still remains unknown and needs to be revealed.
The AUC value of the ROC curve for the MICA/Class I ratio was 0.912 (Fig. 2) but only 0.853 for KL-6 in this study, suggesting that the former is a better ILD marker in RA. Further replication studies on the MICA/Class I ratio are needed to confirm these results. Because of the limited sample size in the present study, the evaluation of auto-Ab profiles should be confirmed in future independent work. Further large-scale auto-Ab profiling would be expected to provide better markers for CVD-ILD. In conclusion, this is the first report of auto-Ab profiles of CDV-ILD. The MICA/Class I ratio could be a better marker than KL-6 for diagnosing CVD-ILD in RA.
Supplementary Material
Supplementary Table S1. Antigens coated on fluorescence beads of LABScreen Mixed Lot#016.
Supplementary Table S2. Auto-antibodies detected with ProtoArray in the pooled sera from the collagen disease patients in the stable and acute-onset diffuse interstitial lung disease (AoDILD) states.
Supplementary Table S3. Auto-antibodies detected with AlphaScreen in the pooled sera from the collagen disease patients in the stable and acute-onset diffuse interstitial lung disease (AoDILD) states.
Acknowledgments
We thank Dr. Kazuhiro Matsuoka (Cell-Free Science and Technology Research Center, Ehime University), Dr. Teruyuki Kadonishi (Proteo-Medicine Research Center, Ehime University), and Ms. Hiromi Hayakawa (Clinical Research Center for Allergy and Rheumatology, Sagamihara Hospital) for providing technical assistance. We also thank Ms. Mayumi Yokoyama and Ms. Tomomi Hanawa (Clinical Research Center for Allergy and Rheumatology, Sagamihara Hospital) for secretarial assistance.
Footnotes
PEER REVIEW: Six peer reviewers contributed to the peer review report. Reviewers’ reports totaled 1,103 words, excluding any confidential comments to the academic editor.
ACADEMIC EDITOR: Karen Pulford, Editor in Chief
FUNDING: The work was supported by a Grant-in-Aid for Scientific Research (C) (22591090) from the Japan Society for the Promotion of Science, Health and Labour Science Research Grants from the Ministry of Health, Labour, and Welfare of Japan, Grants-in-Aid for Clinical Research from the National Hospital Organization, as well as research grants from the Daiwa Securities Health Foundation, the Japan Research Foundation for Clinical Pharmacology, the Nakatomi Foundation, the Takeda Science Foundation, the Mitsui Sumitomo Insurance Welfare Foundation, and the following pharmaceutical companies: Abbott Japan Co., Ltd., Astellas Pharma Inc., Chugai Pharmaceutical Co., Ltd., Eisai Co., Ltd., Mitsuibishi Tanabe Pharma Corporation, Merck Sharp and Dohme Inc., Pfizer Japan Inc., Takeda Pharmaceutical Company Limited, and Teijin Pharma Limited. The funders had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
COMPETING INTERESTS: HF has the following conflicts of interest, and the following funders are supported wholly or in part by the indicated pharmaceutical companies. The Japan Research Foundation for Clinical Pharmacology is run by Daiichi Sankyo; the Takeda Science Foundation is supported by an endowment from Takeda Pharmaceutical Company; and the Nakatomi Foundation was established by Hisamitsu Pharmaceutical Co., Inc. The Daiwa Securities Health Foundation was established by Daiwa Securities Group Inc.; and Mitsui Sumitomo Insurance Welfare Foundation was established by Mitsui Sumitomo Insurance Co., Ltd. HF received honoraria from Ajinomoto Co., Inc., Daiichi Sankyo Co., Ltd., Dainippon Sumitomo Pharma Co., Ltd., Pfizer Japan Inc., and Takeda Pharmaceutical Company. HF holds a patent for a predicting method for drug-induced interstitial lung disease and HLA. NT is supported by SENSHIN Medical Research Foundation, which is supported by an endowment from Mitsubishi Tanabe Pharma Corporation, and received honoraria from Eisai Co., Ltd., Daiichi Sankyo Co., Ltd., and Asahi Kasei Corporation. ST was supported by research grants from nine pharmaceutical companies: Abbott Japan Co., Ltd., Astellas Pharma Inc., Chugai Pharmaceutical Co., Ltd., Eisai Co., Ltd., Mitsubishi Tanabe Pharma Corporation, Merck Sharp and Dohme Inc., Pfizer Japan Inc., Takeda Pharmaceutical Company Limited, and Teijin Pharma Limited. ST received honoraria from Asahi Kasei Pharma Corporation, Astellas Pharma Inc., AbbVie GK., Chugai Pharmaceutical Co., Ltd., Ono Pharmaceutical Co., Ltd., Mitsubishi Tanabe Pharma Corporation, and Pfizer Japan Inc. KM is an employee of VERITAS Corporation. The other authors declare no financial or commercial conflict of interest.
Paper subject to independent expert blind peer review. All editorial decisions made by independent academic editor. Upon submission manuscript was subject to anti-plagiarism scanning. Prior to publication all authors have given signed confirmation of agreement to article publication and compliance with all applicable ethical and legal requirements, including the accuracy of author and contributor information, disclosure of competing interests and funding sources, compliance with ethical requirements relating to human and animal study participants, and compliance with any copyright requirements of third parties. This journal is a member of the Committee on Publication Ethics (COPE).
Author Contributions
Conceived and designed the experiments: HF, MN, NT, ST. Performed the experiments: HF, SO, KM, FN, SF, YT. Analyzed the data: HF. Contributed reagents/materials/analysis tools: HF, KS, AK, NF, TS, KT, MN, ST. Wrote the manuscript: HF, NT, ST. All authors read the final manuscript and approved the same.
REFERENCES
- 1.Turesson C, Jacobsson L, Bergstrom U, Truedsson L, Sturfelt G. Predictors of extra-articular manifestations in rheumatoid arthritis. Scand J Rheumatol. 2000;29(6):358–64. doi: 10.1080/030097400447552. [DOI] [PubMed] [Google Scholar]
- 2.Koduri G, Norton S, Young A, et al. Interstitial lung disease has a poor prognosis in rheumatoid arthritis: results from an inception cohort. Rheumatology. 2010;49(8):1483–9. doi: 10.1093/rheumatology/keq035. [DOI] [PubMed] [Google Scholar]
- 3.Kameda H, Tokuda H, Sakai F, et al. Clinical and radiological features of acute-onset diffuse interstitial lung diseases in patients with rheumatoid arthritis receiving treatment with biological agents: importance of Pneumocystis pneumonia in Japan revealed by a multicenter study. Intern Med. 2011;50(4):305–13. doi: 10.2169/internalmedicine.50.4508. [DOI] [PubMed] [Google Scholar]
- 4.Oka S, Furukawa H, Shimada K, et al. Serum biomarker analysis of collagen disease patients with acute-onset diffuse interstitial lung disease. BMC Immunol. 2013;14(9):9. doi: 10.1186/1471-2172-14-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ohnishi H, Yokoyama A, Kondo K, et al. Comparative study of KL-6, surfactant protein-A, surfactant protein-D, and monocyte chemoattractant protein-1 as serum markers for interstitial lung diseases. Am J Respir Crit Care Med. 2002;165(3):378–81. doi: 10.1164/ajrccm.165.3.2107134. [DOI] [PubMed] [Google Scholar]
- 6.Furukawa H, Oka S, Takehana K, et al. Plasma amino acid profiles in collagen disease patients with interstitial lung disease. Immunome Res. 2013;9(1):1000064. [Google Scholar]
- 7.Mori S, Koga Y, Sugimoto M. Different risk factors between interstitial lung disease and airway disease in rheumatoid arthritis. Respir Med. 2012;106(11):1591–9. doi: 10.1016/j.rmed.2012.07.006. [DOI] [PubMed] [Google Scholar]
- 8.Koga T, Fujikawa K, Horai Y, et al. The diagnostic utility of anti-melanoma differentiation-associated gene 5 antibody testing for predicting the prognosis of Japanese patients with DM. Rheumatology (Oxford) 2012;51(7):1278–84. doi: 10.1093/rheumatology/ker518. [DOI] [PubMed] [Google Scholar]
- 9.Hamaguchi Y, Fujimoto M, Matsushita T, et al. Common and distinct clinical features in adult patients with anti-aminoacyl-tRNA synthetase antibodies: heterogeneity within the syndrome. PLoS One. 2013;8(4):e60442. doi: 10.1371/journal.pone.0060442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sato S, Hoshino K, Satoh T, Fujita T, Kawakami Y, Kuwana M. RNA helicase encoded by melanoma differentiation-associated gene 5 is a major autoantigen in patients with clinically amyopathic dermatomyositis: association with rapidly progressive interstitial lung disease. Arthritis Rheum. 2009;60(7):2193–200. doi: 10.1002/art.24621. [DOI] [PubMed] [Google Scholar]
- 11.Matsushita T, Hasegawa M, Fujimoto M, et al. Clinical evaluation of anti-aminoacyl tRNA synthetase antibodies in Japanese patients with dermatomyositis. J Rheumatol. 2007;34(5):1012–8. [PubMed] [Google Scholar]
- 12.Silliman CC, Ambruso DR, Boshkov LK. Transfusion-related acute lung injury. Blood. 2005;105(6):2266–73. doi: 10.1182/blood-2004-07-2929. [DOI] [PubMed] [Google Scholar]
- 13.Furukawa H, Oka S, Shimada K, et al. Association of human leukocyte antigen with interstitial lung disease in rheumatoid arthritis: a protective role for shared epitope. PLoS One. 2012;7(5):e33133. doi: 10.1371/journal.pone.0033133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Arnett FC, Edworthy SM, Bloch DA, et al. The American Rheumatism Association 1987 revised criteria for the classification of rheumatoid arthritis. Arthritis Rheum. 1988;31(3):315–24. doi: 10.1002/art.1780310302. [DOI] [PubMed] [Google Scholar]
- 15.Subcommittee for Scleroderma Criteria of the American Rheumatism Association Diagnostic and Therapeutic Criteria Committee Preliminary criteria for the classification of systemic sclerosis (scleroderma) Arthritis Rheum. 1980;23(5):581–90. doi: 10.1002/art.1780230510. [DOI] [PubMed] [Google Scholar]
- 16.Bohan A, Peter JB. Polymyositis and dermatomyositis (first of two parts) N Engl J Med. 1975;292(7):344–7. doi: 10.1056/NEJM197502132920706. [DOI] [PubMed] [Google Scholar]
- 17.Hashimoto S, Nakajima F, Kamada H, et al. Relationship of donor HLA antibody strength to the development of transfusion-related acute lung injury. Transfusion. 2010;50(12):2582–91. doi: 10.1111/j.1537-2995.2010.02779.x. [DOI] [PubMed] [Google Scholar]
- 18.Verheugt FW, von dem Borne AE, Decary F, Engelfriet CP. The detection of granulocyte alloantibodies with an indirect immunofluorescence test. Br J Haematol. 1977;36(4):533–44. doi: 10.1111/j.1365-2141.1977.tb00994.x. [DOI] [PubMed] [Google Scholar]
- 19.Babel I, Barderas R, Diaz-Uriarte R, Martinez-Torrecuadrada JL, Sanchez-Carbayo M, Casal JI. Identification of tumor-associated autoantigens for the diagnosis of colorectal cancer in serum using high density protein microarrays. Mol Cell Proteomics. 2009;8(10):2382–95. doi: 10.1074/mcp.M800596-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang JH, Chung TD, Oldenburg KR. Confirmation of primary active substances from high throughput screening of chemical and biological populations: a statistical approach and practical considerations. J Comb Chem. 2000;2(3):258–65. doi: 10.1021/cc9900706. [DOI] [PubMed] [Google Scholar]
- 21.Matsuoka K, Komori H, Nose M, Endo Y, Sawasaki T. Simple screening method for autoantigen proteins using the N-terminal biotinylated protein library produced by wheat cell-free synthesis. J Proteome Res. 2010;9(8):4264–73. doi: 10.1021/pr9010553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Madin K, Sawasaki T, Ogasawara T, Endo Y. A highly efficient and robust cell-free protein synthesis system prepared from wheat embryos: plants apparently contain a suicide system directed at ribosomes. Proc Natl Acad Sci U S A. 2000;97(2):559–64. doi: 10.1073/pnas.97.2.559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sawasaki T, Ogasawara T, Morishita R, Endo Y. A cell-free protein synthesis system for high-throughput proteomics. Proc Natl Acad Sci U S A. 2002;99(23):14652–7. doi: 10.1073/pnas.232580399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tanaka Y, Komori H, Mori S, et al. Evaluating the role of rheumatoid factors for the development of rheumatoid arthritis in a mouse model with a newly established ELISA system. Tohoku J Exp Med. 2010;220(3):199–206. doi: 10.1620/tjem.220.199. [DOI] [PubMed] [Google Scholar]
- 25.Lenth RV. Statistical power calculations. J Anim Sci. 2007;85(13 suppl):E24–9. doi: 10.2527/jas.2006-449. [DOI] [PubMed] [Google Scholar]
- 26.Steinbrocker O, Traeger CH, Batterman RC. Therapeutic criteria in rheumatoid arthritis. J Am Med Assoc. 1949;140(8):659–62. doi: 10.1001/jama.1949.02900430001001. [DOI] [PubMed] [Google Scholar]
- 27.Charpin C, Arnoux F, Martin M, et al. New autoantibodies in early rheumatoid arthritis. Arthritis Res Ther. 2013;15(4):R78. doi: 10.1186/ar4255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Packard TA, Li QZ, Cosgrove GP, Bowler RP, Cambier JC. COPD is associated with production of autoantibodies to a broad spectrum of self-antigens, correlative with disease phenotype. Immunol Res. 2013;55(1–3):48–57. doi: 10.1007/s12026-012-8347-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Motts JA, Shirley DL, Silbergeld EK, Nyland JF. Novel biomarkers of mercury-induced autoimmune dysfunction: a cross-sectional study in Amazonian Brazil. Environ Res. 2014;132:12–8. doi: 10.1016/j.envres.2014.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Le Roux S, Devys A, Girard C, Harb J, Hourmant M. Biomarkers for the diagnosis of the stable kidney transplant and chronic transplant injury using the ProtoArray(R) technology. Transplant Proc. 2010;42(9):3475–81. doi: 10.1016/j.transproceed.2010.09.006. [DOI] [PubMed] [Google Scholar]
- 31.Schweitzer B, Meng L, Mattoon D, Rai AJ. Immune response biomarker profiling application on ProtoArray protein microarrays. Methods Mol Biol. 2010;641:243–52. doi: 10.1007/978-1-60761-711-2_14. [DOI] [PubMed] [Google Scholar]
- 32.Auger I, Balandraud N, Rak J, Lambert N, Martin M, Roudier J. New autoantigens in rheumatoid arthritis (RA): screening 8268 protein arrays with sera from patients with RA. Ann Rheum Dis. 2009;68(4):591–4. doi: 10.1136/ard.2008.096917. [DOI] [PubMed] [Google Scholar]
- 33.Suciu-Foca N, Reed E, D’Agati VD, et al. Soluble HLA antigens, anti-HLA antibodies, and antiidiotypic antibodies in the circulation of renal transplant recipients. Transplantation. 1991;51(3):593–601. doi: 10.1097/00007890-199103000-00011. [DOI] [PubMed] [Google Scholar]
- 34.Nath DS, Basha HI, Mohanakumar T. Antihuman leukocyte antigen antibody-induced autoimmunity: role in chronic rejection. Curr Opin Organ Transplant. 2010;15(1):16–20. doi: 10.1097/MOT.0b013e3283342780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yabe T, McSherry C, Bach FH, et al. A multigene family on human chromosome 12 encodes natural killer-cell lectins. Immunogenetics. 1993;37(6):455–60. doi: 10.1007/BF00222470. [DOI] [PubMed] [Google Scholar]
- 36.Spear P, Wu MR, Sentman ML, Sentman CL. NKG2D ligands as therapeutic targets. Cancer Immun. 2013;13(8):8. [PMC free article] [PubMed] [Google Scholar]
- 37.Borchers MT, Harris NL, Wesselkamper SC, et al. The NKG2D-activating receptor mediates pulmonary clearance of Pseudomonas aeruginosa. Infect Immun. 2006;74(5):2578–86. doi: 10.1128/IAI.74.5.2578-2586.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wesselkamper SC, Eppert BL, Motz GT, Lau GW, Hassett DJ, Borchers MT. NKG2D is critical for NK cell activation in host defense against Pseudomonas aeruginosa respiratory infection. J Immunol. 2008;181(8):5481–9. doi: 10.4049/jimmunol.181.8.5481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tieng V, Le Bouguenec C, du Merle L, et al. Binding of Escherichia coli adhesin AfaE to CD55 triggers cell-surface expression of the MHC class I-related molecule MICA. Proc Natl Acad Sci U S A. 2002;99(5):2977–82. doi: 10.1073/pnas.032668099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rausch A, Hessmann M, Holscher A, et al. Interleukin-15 mediates protection against experimental tuberculosis: a role for NKG2D-dependent effector mechanisms of CD8+ T cells. Eur J Immunol. 2006;36(5):1156–67. doi: 10.1002/eji.200535290. [DOI] [PubMed] [Google Scholar]
- 41.Jaramillo A, Smith CR, Maruyama T, Zhang L, Patterson GA, Mohanakumar T. Anti-HLA class I antibody binding to airway epithelial cells induces production of fibrogenic growth factors and apoptotic cell death: a possible mechanism for bronchiolitis obliterans syndrome. Hum Immunol. 2003;64(5):521–9. doi: 10.1016/s0198-8859(03)00038-7. [DOI] [PubMed] [Google Scholar]
- 42.Matsui T, Inui N, Suda T, Chida K. Anti-endothelial cell antibodies in patients with interstitial lung diseases. Respir Med. 2008;102(1):128–33. doi: 10.1016/j.rmed.2007.07.024. [DOI] [PubMed] [Google Scholar]
- 43.Zou Y, Stastny P, Susal C, Dohler B, Opelz G. Antibodies against MICA antigens and kidney-transplant rejection. N Engl J Med. 2007;357(13):1293–300. doi: 10.1056/NEJMoa067160. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table S1. Antigens coated on fluorescence beads of LABScreen Mixed Lot#016.
Supplementary Table S2. Auto-antibodies detected with ProtoArray in the pooled sera from the collagen disease patients in the stable and acute-onset diffuse interstitial lung disease (AoDILD) states.
Supplementary Table S3. Auto-antibodies detected with AlphaScreen in the pooled sera from the collagen disease patients in the stable and acute-onset diffuse interstitial lung disease (AoDILD) states.