

Abstract
We have reported earlier that the non-viral Sleeping Beauty (SB) transposon system can mediate genomic integration and long-term reporter gene expression in human primary peripheral blood (PB) T cells. In order to test whether this system can be used for genetically modifying both PB T cells and umbilical cord blood (UCB) T cells as graft-versus-leukemia effector cells, an SB transposon was constructed to coexpress a single-chain chimeric antigen receptor (CAR) for human CD19 and CD20. PB and UCB were nucleofected with the transposon and a transposase plasmid, activated and then expanded in culture using anti-CD3/CD28 beads. Stable dual-gene expression was confirmed in both T-cell types, permitting enrichment by positive selection with Rituxan. The engineered CD4+ T cells and CD8+ T cells both exhibited specific cytotoxicity against CD19+ leukemia and lymphoma cell lines, as well as against CD19 transfectants, and produced high-levels of antigen-dependent Th1 (but not Th2) cytokines. The in vivo adoptive transfer of genetically engineered T cells significantly reduced tumor growth and prolonged the survival of the animal. Taken together, these data indicate that T cells from PB and UCB can be stably modified using a non-viral DNA transfer system, and that such modified T cells may be useful in the treatment of refractory leukemia and lymphoma.
INTRODUCTION
The Sleeping Beauty (SB) transposon system has emerged as an effective genetic tool to achieve high-level, persistent transgene expression from a non-viral plasmid vector.1,2 SB is a “cut-and-paste” DNA transposon of the Tc1/mariner superfamily, and was reconstructed from sequences of teleost fish.1 The SB transposase mediates transposition by recognition of short inverted/directed repeat sequences that make up the termini of a constructed SB transposon. SB transposons have been known to exhibit efficient transposition in cells from a wide range of vertebrates, including in cultured mammalian cells,1,2,3 mouse liver and lung tissue,5,6 mouse embryonic stem cells,7 and mouse embryos, thereby opening up potential for applications in germ-line transgenesis and insertional mutagenesis.8–12
For T-cell gene transfer and therapy applications, the SB transposon system offers several advantages over the widely used virus-based or conventional mammalian DNA vectors.2 First, the use of the SB transposon system is simple, and the transposons are also easy and inexpensive to manufacture. Second, the efficiency of SB-mediated stable gene transfer is considerably higher than that of conventional DNA-mediated random integration.3 Third, in contrast to retroviral mediated gene transfer, there is no need for prior T-cell activation when using SB. Thereby, the duration of ex vivo culture is reduced, and alterations in T-cell phenotypes and functions are minimal.
Although we have demonstrated that the SB transposon system can mediate stable expression of reporter genes in 5–20% of human primary CD4 and CD8 T cells without prior activation or drug selection,13 neither the expression of a therapeutic gene nor other potentially useful sources of therapeutic cells [e.g., umbilical cord blood (UCB) T cells] were examined after SB-mediated gene transfer. This study was conducted to determine whether the SB transposon system can be used for efficiently expressing therapeutic genes in human T cells from peripheral blood (PB) and UCB. Such cells could potentially be used in acute lymphoblastic leukemia (ALL), in lymphoma patients at high risk for relapse after hematopoietic stem cell transplantation, and as a strategy to enhance the graft-versus-leukemia/lymphoma effects.14
For this purpose, we coexpressed, in a SB transposon, a single-chain chimeric antigen receptor (CAR) for CD19, a universal antigen for B-lineage leukemia and lymphoma, and human CD20. CD20 was chosen as a coexpressing molecule for 2 reasons: (i) so as to enrich positively transfected T cells and (ii) to serve as a “suicide” strategy for eliminating infused T cells, using an clinically approved antibody that targets CD20 (Rituxan).
RESULTS
SB-mediated coexpression of CD19 CAR and CD20 in human primary T cells
A bidirectional promoter consisting of phosphoglycerate kinase (PGK) and minimal cytomegalovirus (mCMV) was used for coexpressing CD19 CAR (CD19scFv-4-lBB-CD3ζ) and CD20, respectively, in a SB transposon vector. CD19scFv-4-lBB-CD3ζ (19BBζ Figure 1a) is composed of an anti-CD19 scFv, human CD8 hinge and transmembrane domain, intracellular human 4-1BB domain, and the cytoplasmic domain of human CD3 ζ-chain.15 In order to verify the SB construct, pT2/P19BBζmCCD20 (19BBζ/CD20) (Figure 1b) was transfected into 293T cells. Flow cytometric analysis demonstrated that transfected 293T cells coordinately expressed both CD19 CAR and CD20 on the cell surface 24–48 hours after transfection (Figure 1b).
Figure 1. Cell surface expression of chimeric antigen receptor (CAR) and CD20 after transfection.
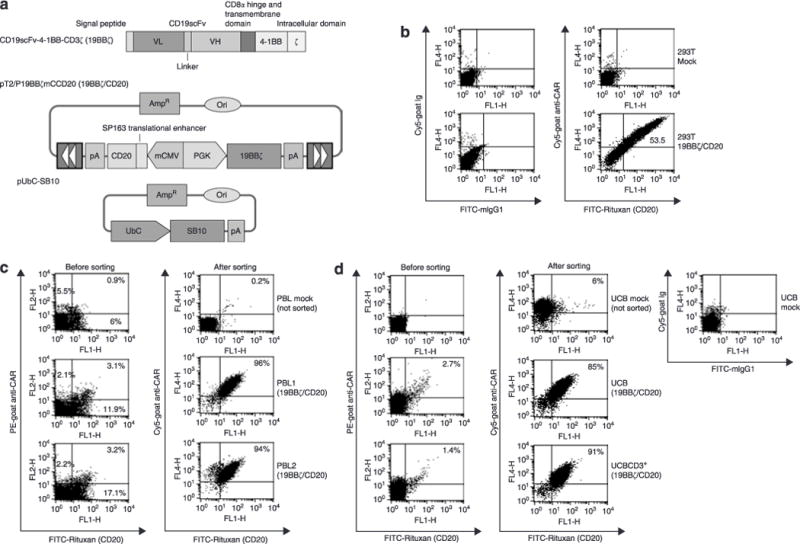
(a) Schematic representation of the CAR for CD19 and the Sleeping Beauty (SB) transposon system used in this study. PGK, phosphoglycerate kinase. mCMV, human cytomegalovirus (CMV) minimal core promoter element, (b) Expression of CAR and CD20 in 293T cells. 293T cells (8 × 105 per well in 12-well plates) were transfected with 2 μg of the SB transposon (19BBζ/CD20) without the SB10, using a standard calcium phosphate precipitation method. Twenty-four to forty-eight hours after transfection, the cells were stained with CAR [goat anti-mouse immunoglobulin G F(ab′)2] and CD20 (Rituxan) antibodies and analyzed by flow cytometry. Untransfected 293T cells were used as mock control. (c) Expression of CAR and CD20 in PBL from two donors (PBL1 and PBL2). (d) CAR and CD20 expression in umbilical cord blood (UCB) T cells. Note: neither mock treated PBL cells nor mock-treated UCB cells were sorted, and they were used for staining control only. A background CAR staining was observed in mock UCB cells. FITC, fluorescein isothiocyanate; PE, phycoerythrin.
Freshly isolated peripheral blood lymphocytes (PBLs) from two healthy donors (PBL1 and PBL2) were nucleofected with the SB transposon 19BBζ/CD20, either alone or along with an SB10 transposase expression plasmid (pUbC-SBl0; SB10) (Figure 1a). Three weeks after transfection, the PBL that had been transfected with the SB CD19 CAR transposon alone did not stain positive for either CAR or CD20 (data not shown). However, PBL1 and PBL2 transfected with both the transposon and SB10 showed that ~3% of cells expressed both CAR and CD20 (Figure 1c). After flow cytometric sorting for CAR and CD20, 96% of PBL1 and 94% of PBL2 showed coexpression of CAR and CD20 (Figure 1c). This high dual-gene expression was also confirmed in unfractionated UCB T cells and CD3+ UCB T cells after nucleofection with the SB CD19 CAR transposon plus SB10 (Figure 1d). Prior to cell sorting, 1–3% of UCB T cells expressed both CAR and CD20. After sorting, >85% of UCB cells were positive for CAR and CD20. Furthermore, engineered T cells from PBL and UCB stably expressed CAR and CD20, as evidenced by (i) stable expression of CAR/CD20 and specific cytotoxicity against CD19+ target cells over a 2–3 month culture, and (ii) sequencing of transposon:chromosome junctions using a splinkerette polymerase chain reaction (PCR) technique. Table 1 shows that 18 and 14 clones derived from the engineered T cells from PBL and UCB, respectively, contained novel chromosomal sequences at TA sites, the bench mark for SB transposition. These junction sequences (29 out of 32 clones) were mapped to their locations in human chromosomes. Overall, these results, in agreement with our earlier study involving reporter genes,13 demonstrate that the SB transposon system can stably coexpress CD19 CAR and CD20 in human T cells from PBL and UCB.
Table 1.
Mapping of transposition sites in SB-engineered T cells
| Clones | IR/DR (L) | Chromosome location | Clones | IR/DR (R) | Chromosome location |
|---|---|---|---|---|---|
| PBL2-19BBζ/CD20 | |||||
| 1 | GATGGACAGATAGATGAAC AGATGAACACATACAGTTG | 6 p21.32 | 10 | CAACTGTAACTTACCTTTC TGCTTCAGAAACCCCGCCC | 1 q42.12 |
| 2 | AGGTACAAAAACCTATCAA GAATTCCATATTACAGTTG | 5q31.1 | 11 | CAACTGTACATCCATGGT CCCTTAAGCGGAGCCCTA | ND |
| 3 | TATCTGTATATGAGTGTGC GTATAGGTACATACAGTTG | 9 q22.2 | 12 | CAACTGTATATGCTTAAAGC AAAAGGAACAAAGAGAGG | 2 q37.3 |
| 4 | CACAAAAGAAACATAAGCC AGAAGTACAATTACAGTTG | 14q24.3 | 13 | CAACTGTAAAAGCTATTTC TATAACTGCTCTCTGCCAA | 1 p21.1 |
| 5 | GAATGTAAGTGAGGTCCAG GTGAGCACATTTACAGTTG | llq23.3 | 14 | CAACTGTATCTATCTATGT ATCCATGGTCCCTTAAGCG | Ypll.32orXp22.33 |
| 6 | ATTTGTTACATATAGCACCC CTGGGAAATGTACAGTTG | 8 q24.13 | 15 | CAACTGTATTTCTGATGCC CTTCTCCTTGTCTGCCCCA | 2 q37.3 |
| 7 | TAAAAAGGAAGGAGGAAAA TCATACACCAATACAGTTG | 18qll.l | 16 | CAACTGTATATGTGAATAG TGTAGATTGTATGGGTTCT | 8 q24.22 |
| 8 | CAATCCGTAACTGTGGGTA GGTAGAGTTGTTACAGTTG | 14 q24.1 | 17 | CAACTGTAAAAGCTATTTC TATAACTGCTCTCTGCCAA | 1 p21.1 |
| 9 | CAGGCCACACCCTGGCAGT CACAAAGCACATACAGTTG | 9 q33.3 | 18 | CAACTGTACATGGTCC CTTAAGCGGAGCCCT | 14 q22.2 |
| UCBCD3+-19BBζ/CD20 | |||||
| 21 | ACCTCTGTTGCTGGAGAAA ATTAAACCACATACAGTTG | 2q31.3 | 28 | CAACTGTATATAGTCAGGG TCTTGCTCTGTTGCTCACA | Xq22.3 |
| 22 | TAGTTCAAAATAGGGAGAG CAAATTTCAAATACAGTTG | 1 q21.3 | 29 | CAACTGTAAATCTTCCCAT GGTCCCTTAAGCGGAGCCC | ND |
| 23 | ATGGTCCTACTGGGAGGGC CAGCAGTCTTATACAGTTG | 2 q22.3 | 30 | CAACTGTATATTTCCCTGA AAAGAAGTAATTCCCATTA | 6q26 |
| 24 | CTTTGGCCTGTGGAAAATC TCTATCACATATACAGTTG | 19 ql3.41 | 31 | CAACTGTATATACACATGG TCCCTTAAGCGGAGCCCT | ND |
| 25 | TGGGAGGGGCTCACAGTGG CTTCAAAGATATACAGTTG | 5 q35.3 | 32 | CAACTGTATTGCCACTTAC TTTTGCCAACAGGGTAATA | 2pll.2 |
| 26 | GTCACTTTCCAGGAGAAAG GGTGGCCCATATACAGTTG | 3 q25.1 | 33 | CAACTGTAAGTGTCCTTCA GTGGATGAACACATAAAGA | 18pll.21 |
| 27 | AGAGATAGGCTTTCTAACT GATTAAACACATACAGTTG | 2 q32.3 | 34 | CAACTGTACCAAATTCTAG TAAAACCGTTTGTTTTTCA | 7qll.23 |
Bfal or N/alll was used for digesting genomic DNA from engineered PBL and umbilical cord blood T cells to recover left and right inverted/directed repeat (IR/DR): chromosome junction sequences, respectively. Sequences in bold print represent the transposon junction region. ND, not determined because of short genomic sequences from cloned polymerase chain reaction products. The length of genomic sequences was between 25 and 263 base pairs (bp).
SB-engineered T cells specifically kill CD19+ leukemia and lymphoma cells
Figure 2a shows that both PBL1 and PBL2 engineered with the SB CD19 CAR efficiently lysed CD19+ B-cell lymphoma Daudi cells and two B-ALL cell lines (HPB-null and RS4:11) in an effector/target, dose-dependent manner. In contrast, CD19− erythroleukemia K562 cells were not killed. In order to further demonstrate that killing of target cells is CD19-specific, CD19+ K562 transfectants were generated, using either the full-length CD19 complementary DNA (cDNA) (K562-FCD19) or a cytoplasmic domain-deleted CD19 (K562-ACD19).16 Expression of CD19 was confirmed by flow cytometry. In addition, enhanced green fluorescent protein (enhanced GFP) cDNA was also transfected into K562 cells (K562-GFP) (Figure 2b). Both engineered PBL1 and PBL2 (19BBζ/CD20) were as efficient at specific killing of K562-FCD19 and K562-ΔCD19 as they were at killing Daudi cell lines. In contrast, K562-GFP or K562 were not killed (Figure 2c). Moreover, these cells did not kill CD19− erythroleukemia TF-1 and multiple myeloma U266 lines. In a manner similar to engineered PBL, engineered UCB T cells also killed CD19+ target cells including K562-FCD19, K562-ΔCD19, Daudi and HPB-null, but not CD19− K562 and K562-GFP cells (Figure 2d). In addition, killing of target cells by these engineered T cells from PBL and UCB was completely blocked by ethylene glycol tetraacetic acid and MgCl2. It is well known that perforin released from cytotoxic T cells polymerizes to form a pore on the target membrane in a Ca2+-dependent fashion and then granzymes and granulysin induce apoptosis. However, anti-CD19 antibodies (BU12 and HD37),17 either alone (data not shown) or together (Figure 2e), failed to block T-cell mediated killing of CD19+ target cells. This could be because these antibodies may bind to different epitopes on the CD19 molecule that recognized by CD19 CAR. Furthermore, engineered T cells from PBL and UCB expressed Fas but not FasL, whereas the target cells (K562, K562-FCD19, and Daudi) had no detectable expression of either Fas or FasL (data not shown). These data suggest that perforin may play an important role in target-cell destruction by the SB-engineered T cells. We conclude that SB-modified T cells from PBL and UCB expressing CD19 CAR/CD20 can kill CD19+ leukemia and lymphoma cells as well as CD19 K562 transfectants, and that this killing is CD19-specific and perforin-mediated.
Figure 2. Sleeping Beauty-engineered T cells specifically kill CD19+ leukemia and lymphoma cells.
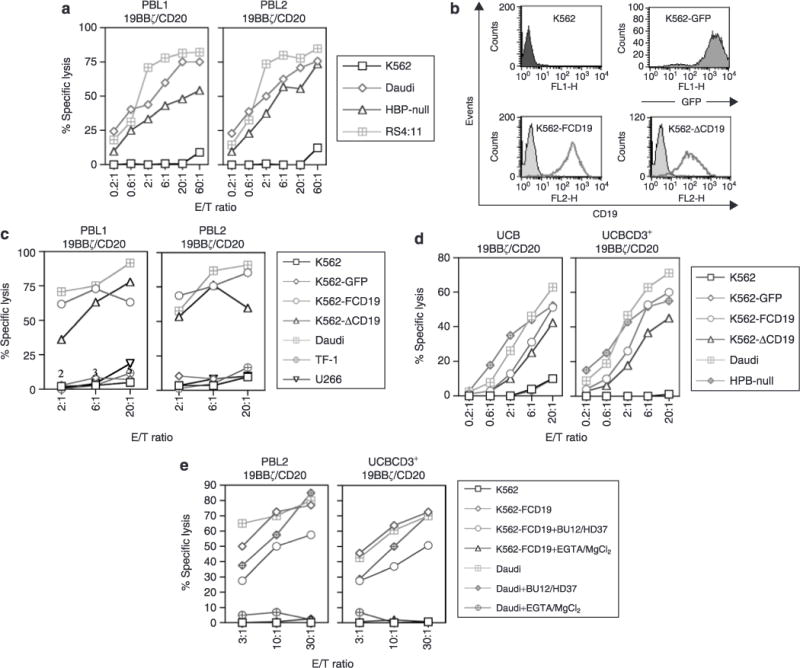
(a) Cytotoxicity against CD19+ target cells shown by engineered PBL from two donors (PBL1 and PBL2). (b) Expression of CD19 on K562 transfectants. (c) Cytotoxicity shown by the engineered PBL against K562 transfectants. (d) CD19 specificity of the engineered umbilical cord blood (UCB) T cells, (e) Engineered T cell killing of target cells inhibited by the perforin pathway inhibitors. Similar results were obtained in at least three independent assays. EGTA, ethylene glycol tetraacetic acid; E/T ratio, effector/target ratio; GFP, green fluorescent protein.
SB-transfected T cells can be enriched by Rituxan
In order to determine whether SB-transfected PBL can be enriched using Rituxan, freshly isolated PBL (PBL5 and PBL6) were nucleofected with SB transposon 19BBζ/CD20 plus SB10 and then expanded for 3 weeks prior to positive selection using a biotin-Rituxan/antibiotin bead procedure. As shown in Figure 3a, 1–3% of PBL5 and PBL6 were positive for both CAR and CD20. After one round of column purification, ~85% of cells were CAR+ (Figure 3a) and ~70–90% cell recovery of the engineered PBL and UCB cells can be achieved (data not shown). As expected, when these cells were expanded they specifically killed CD19+ but not CD19− targets.
Figure 3. Selection of the Sleeping Beauty-engineered T cells using Rituxan.
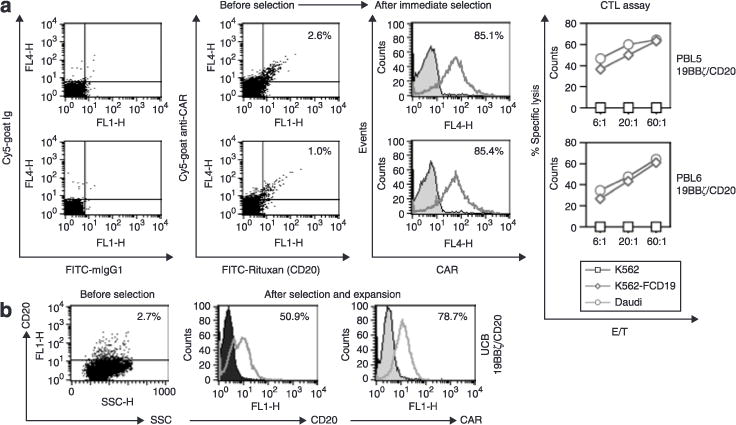
(a) PBL. The optimal amount of biotin-Rituxan used for selection of transfected PBL and umbilical cord blood (UCB) T cells was predetermined by staining T cells of PBL (PBL2-19BBζ/CD20) and UCB (UCBCD3+-19BBζ/CD20) expressing 19BBζ/CD20 with a series of biotin-Rituxan antibody dilutions. Two to four weeks after nucleofection, the T cells were harvested, washed once with magnetic cell sorting buffer (Miltenyi), resuspended, and incubated with human FcR blocking reagent (Miltenyi) and biotin-Rituxan (1–2 μg per 106 cells). The cells were washed and mixed with antibiotin microbeads (Miltenyi). After being washed, the cell pellet was resuspended and run through MS or LS columns (Miltenyi). (b) UCB. This experiment was repeated seven times and two times, respectively, with PBL and UCB, yielding similar results. CAR, chimeric antigen receptor; E/T ratio, effector/target ratio; FITC, fluorescein isothiocyanate; Ig, immunoglobulin; SSC, side scatter.
Using the same approach, we also performed selection of transfected UCB T cells transposed with SB 19BBζ/CD20. Approximately 50% of the UCB T cells were enriched for expression of transgene CD20 as compared to ~3% CD20+ in mock transfected UCB cells (Figure 3b). We observed that mock UCB cells, but not mock-transfected PBL cells, stained positive when an anti-CAR polyclonal antibody was used rather than when an isotype antibody was used (Figure 1d; data not shown). This shows that there is some background CAR staining of UCB T cells. Nevertheless, we conclude that CD20 can serve as a selection marker in SB-engineered T cells and, by a simple bead selection, transfected T cells from PBL and UCB can be enriched to at least 85 and 50% purity, respectively.
Both CD4 and CD8 SB-engineered T cells kill CD19+ target cells
In order to test whether engineered CD4 T cells are cytolytic for CD19+ target cells, PBL2-19BBζ/CD20 and UCBCD3+-19BBζ/CD20 engineered T cells were separated into CD4 and CD8 subsets using a fluorescence-activated cell sorting sorter. Surprisingly, CD4 subsets from engineered PBL and UCB killed CD19+ major histocompatibility complex class II− CD19 transfected K562 cells and CD19+ major histocompatibility complex class II+ Daudi cells as potently as their CD8 counterparts did, whereas neither CD4 nor CD8 UCB mock cells could kill (Figure 4a). Flow cytometric analysis demonstrated that the expressions of perforin, granzyme A, and granzyme B in engineered CD4 T cells of PBL and UCBs were 1.5 to 2-fold lower than for their CD8 counterparts (Figure 4b). Furthermore, the addition of ethylene glycol tetraacetic acid and MgCl2 in the cytotoxic T-lymphocyte assay blocked target-cell lysis by CD4+ and CD8+ engineered UCB T cells (data not shown). This shows that CD4 and CD8 T-cell mediated killing occurs through the perforin-granzyme pathway. We conclude that engineered CD4 and CD8 T cells generated from PBL and UCB are functionally cytolytic.
Figure 4. Sleeping Beauty-engineered CD4+ and CD8+ T cells kill CD19+ target cells.
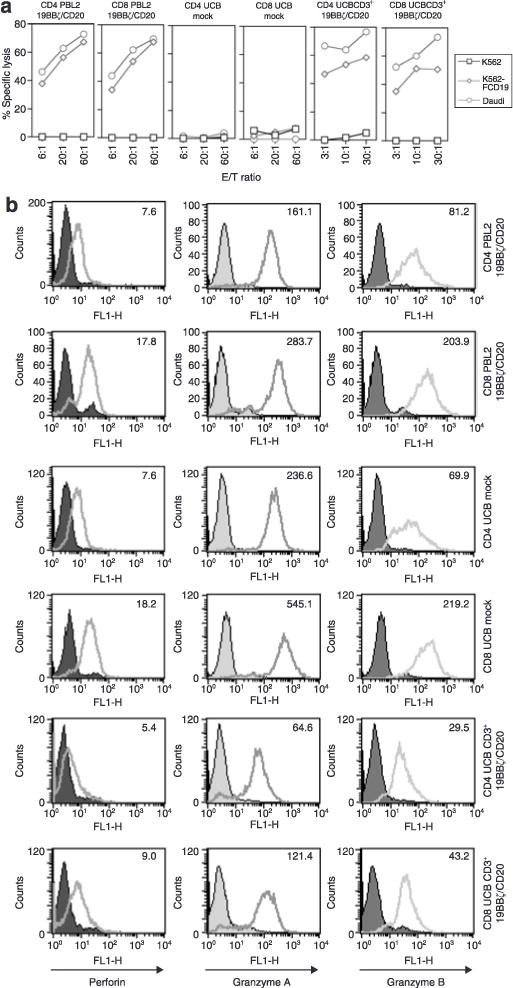
(a) Cytotoxicity assay of sorted subsets of CD4 and CD8 T cells after SB engineering of umbilical cord blood (UCB) and PBL with the SB 19BBζ/CD20 transposon. (b) Expression of cytolytic molecules in subsets of CD4 and CD8 T cell subsets. The mean fluorescence intensity was displayed inside the histograms. Similar results were obtained from at least two independent assays. E/T ratio, effector/target ratio.
SB-engineered T cells produce Th1 cytokines
Both UCB-19BBζ/CD20 and UCBCD3+-19BBζ/CD20 T cells produced high-levels of Thl cytokines, including interleukin-2 (IL-2), IFN-γ, TNF-α, and granulocyte-macrophage colonystimulating factor, but not Th2 cytokines such as IL-4 and IL-10 (Figure 5). These engineered UCB T cells also produced IL-5, IL-6, IL-8, monocyte chemotactic protein-1, macrophage inflammatory protein-lα, macrophage inflammatory protein-1β, and RANTES in response to CD19 antigen stimulation. However, there was no significant production of IL-17, IL-1α, IL-1β IL-1ra, epithelial cell-derived neutrophil-activating protein-78, fibroblast growth factor-basic, granulocyte colony-stimulating factor, Tpo, and vesicular endothelial growth factor (data not shown). This response is completely CAR-CD19-dependent, as is clear from the fact that UCB mock cells or engineered UCB T cells cocultured with CD19− K562 cells were incapable of releasing Thl cytokines. In agreement with the cytokine profiling in UCB T cells, two populations of PBL engineered with 19BBζ/CD20 also produced Thl but not Th2 cytokines (Figure 5). These data support the use of CD19 CAR containing the 4-1BB signaling domain as a potential therapeutic gene for redirected UCB T-cell therapy of B-lineage leukemia and lymphoma.
Figure 5. Cytokine profiling of the engineered T cells.
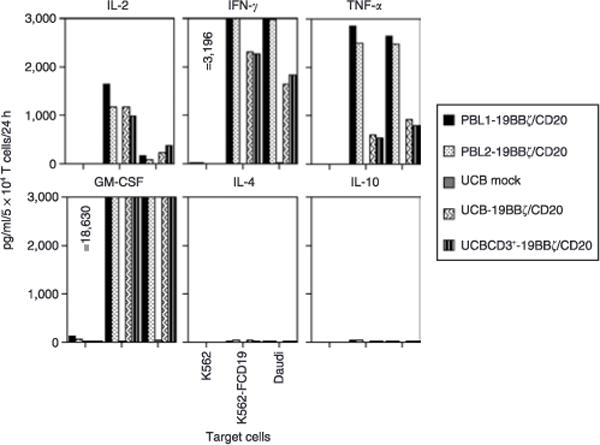
Cytokine release assays were performed by coculture of 5 × 104 T cells with 2 × 104 target cells per well in duplicate in 96-well flat-bottom plates at a final volume of 200 μl T-cell media. After 24 hours, the supernatant was assayed for cytokine production using Fluorokine MAP Immunoarray. Similar data were also obtained from another assay. CM-CSF, granulocyte-macrophage colony-stimulating factor; IFN-γ, interferon-γ; IL-2, interleukin-2; TNF-α, tumor necrosis factor-α; UCB, umbilical cord blood.
SB-engineered T cells can mount antitumor responses in vivo
In order to assess the antitumor effect of SB-engineered T cells in vivo, we employed a time-dynamic bioluminescence imaging technique in live mice. A Daudi cell line was transduced with a lentiviral vector carrying firefly luciferase (hffluc) and nerve growth factor receptor transgenes (Supplementary Materials and Methods), and then sorted for nerve growth factor receptor expression (Supplementary Figure Sla and b). The transduced Daudi cells (Daudi-LVhfflucN) were tested to confirm nerve growth factor receptor expression, and were found to express a high-level of ffluc activity (Supplementary Figure S1c). Daudi-LVhfflucN cells were lysed as efficiently as the Daudi parent line by PBL2-19BBζ/CD20 and UCBCD3+-19BBζ/CD20 (Supplementary Figure S1d). Phenotypic analysis showed that both PBL2-19BBζ/CD20 and UCBCD3+-19BBζ/CD20 were ~95% CD4+, CD25high, CD27low, CD28low, CD45RAlow, CD45ROhigh, CD62Llow, CCR7− effector memory cells18 (data not shown), because anti-CD3/28 bead preferentially expands CD4 T cells.19 Non-obese diabetic/severe combined immunodeficiency (NOD/SCID) mice were irradiated and then injected intraperitoneally with Daudi-LVhfflucN (Figure 6a). Figure 6b shows that one infusion of PBL2-19BBζ/CD20 cells was able to induce tumor regression. The administration of two infusions of the T cells was even more effective at inhibiting tumor growth (adjusted P = 0.011 on day 7). Additional T-cell infusions (three) or IL-2 administration did not improve efficacy beyond that achieved with two T-cell infusions. Furthermore, none of the treatments was able to eliminate the tumor completely, and tumor relapse occurred rapidly, around day 11. On day 7, there was a significant difference between animals that had been given tumor cells only, as compared with animals given tumor + PB T cells (adjusted P = 0.001), and with animals given tumor + PB T cells + IL-2 (adjusted P = 0.011) (Figure 6c). Also, on day 11, there was a significant difference between animals that had been given tumor cells only and animals that received tumor + PB T cells (adjusted P = 0.008). There was no significant difference among these groups on days 18 and 35. In addition, as Figure 6d shows, all of the tumor-bearing mice that had been treated with three infusions of SB-engineered PB T cells without IL-2 survived up to a time point between day 91 and day 110, whereas the untreated tumor-bearing mice all died between days 51 and 61. A log-rank test showed a significant difference of median time to death between these two groups (P = 0.0026).
Figure 6. In vivo antitumor responses by the engineered T cells.
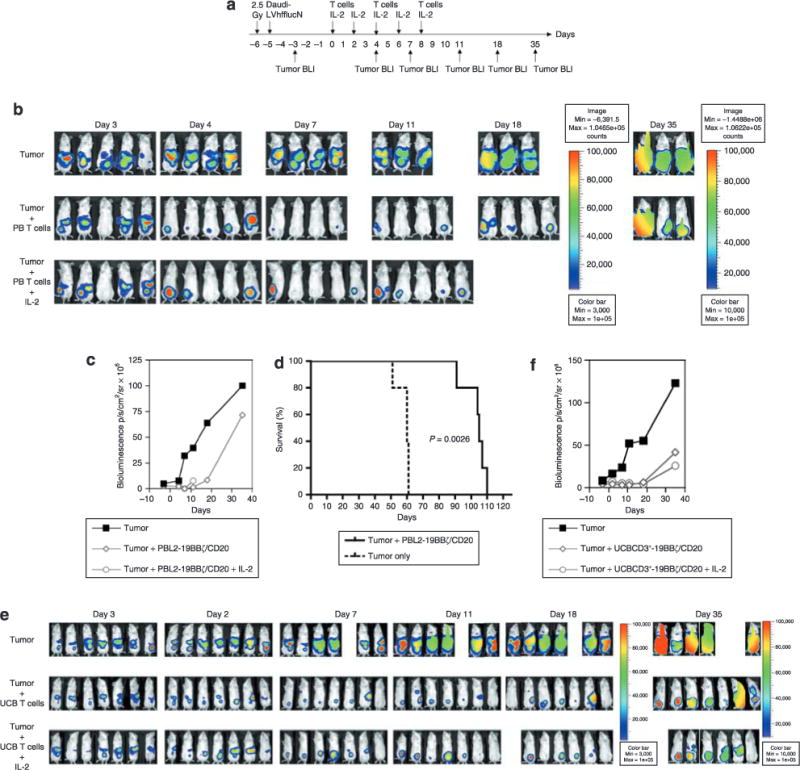
(a) The experimental schedule. (b) Bioluminescent imaging of tumor growth in non-obese diabetic/severe combined immunodeficiency (NOD/SCID) mice (three groups, n = 5 each) treated with engineered PBL expressing 19BBζ/CD20 with or without interleukin-2 (IL-2) injections. The mice in the first group (n = 5) received no treatment. The mice in the second group (n = 5) received three intraperitoneal infusions of PBL2-19BBζ/CD20 T-cells (5 × 106/mouse) on days 0, 4, and 8. A third group (n = 5) of mice received three intraperitoneal infusions of PBL2-19BBζ/CD20 T cells (5×106/mouse) on days 0, 4, and 8 along with human IL-2 (Chiron, 2.5 × 104 IU/mouse) intraperitoneally on days 0, 2, 4, 6, and 8. Note: the third group of mice treated with the engineered PBL and IL-2 died on day 17 after the first T cell infusion because of an accidental malfunction of the drinking water equipment. The level of tumor in one mouse from each of the T-cell groups was measured as 61,336 and 84,799 photons/s/cm2/steradian (p/s/cm2/sr). (c) Bioluminescence intensity of the mice treated with engineered PBL. (d) Animal survival after T-cell therapy. One group (n = 5) received no treatment. The other group (n = 5) received three intraperitoneal infusions of PBL2-19BBζ/CD20 T-cells (5×106/mouse) on days 0, 4, and 8. The mice were not imaged, but monitored for survival instead. The P value from log-rank test is 0.0026. The median time to death for untreated tumor-bearing mice, and for mice treated with engineered PBL, was 60 days [95% confidence interval (CI) is (51, 61)] and 105 days [95% CI is (91, 110)], respectively, (e) Bioluminescent imaging of tumor growth in mice treated with the engineered umbilical cord blood (UCB) T cells with or without IL-2. On day -6, the NOD/SCID mice were γ-irradiated (2.5 Gy, Cesium-137) and then injected intraperitoneally with 106 Daudi-LVhfflucN on the following day. On day -3, mice receiving Daudi-LVhfflucN were examined for tumor engraftment by bioluminescent imaging (BLI). On day 0, the first group of mice (n = 7) received no treatment. The second and third groups (n = 7 each) received three intraperitoneal infusions of UCBCD3+-19BBζ/CD20 (10×106/mouse) or UCB mock on days 0, 4, and 8, respectively. The fourth and fifth groups (n = 7 each) received three intraperitoneal infusions of UCBCD3+-19BBζ/CD20 or UCB mock (10×106/mouse) on days 0, 4, and 8, plus human IL-2 on those same days. Tumor BLI was performed every 4 days after the first T-cell infusion. Note: Tumor-bearing mice mock-treated with UCB (the third group), and those mock-treated with UCB and receiving IL-2 (the fifth group), showed early tumor regression after the first T-cell infusion. However, the tumors rebounded rapidly to the level of those in the untreated mice (data not shown), (f) Bioluminescence intensity of the mice treated with the engineered UCB T cells. PB, peripheral blood.
Figure 6e and f show that adoptive transfer of SB-engineered UCB T cells with or without IL-2 significantly reduced tumor growth (adjusted P < 0.01). IL-2 injections did not improve antitumor responses mediated by the UCB T cells (adjusted P > 0.05). It may therefore be concluded that the engineered T cells from PBL and UCB expressing CD19 CAR containing the 4-IBB and TCR ζ signaling domains can induce a short-duration (~10 days) antitumor response in vivo. Moreover, the engineered PBL and UCB (data not shown) also prolong survival of the animal. These experiments confirm that in vitro SB-engineered PBL T cells and UCB T cells can function as antitumor effector cells in vivo.
DISCUSSION
This report presents data that support four novel findings. First, the SB transposon system can mediate stable CD19 CAR and CD20 expression in T cells from both PBL and UCB. Second, the SB-engineered PBL T cells and UCB T cells can be enriched in vitro by Rituxan. Third, these engineered T cells from PBL and UCB displayed a specific cytotoxicity against CD19+ leukemia and lymphoma cells in vitro as well as in live mice, and produced Thl (but not Th2) cytokines. Fourth, the engineered CD4 T cells from both PBL and UCB killed CD19+ target cells as efficiently in vitro as did their CD8 T-cell counterparts.
We initially constructed an SB transposon and a lentiviral vector containing EFlα/mCMV promoter to regulate CD19 CAR and CD20 expression; this failed to express CD20 (but not CAR) in both 293T and PBL. Furthermore, the addition of a QBI translational enhancer element or 20 and 70 bp spacers in an SB bidirectional transposon just upstream of the CD20 ATG codon did not improve CD20 expression (X.H. and X.Z., unpublished data). With the use of a PGK/mCMV promoter, we were able to achieve coordinate expression of CD19 CAR and CD20 in 293T cells as well as in human primary T cells from T cells from PBL and UCB. The inclusion of a translational enhancer upstream of the CD20 coding sequence enhanced the expression 2.5-fold in comparison with the identical construct lacking this translational enhancer (data not shown). We also observed that PGK/mCMV-regulated expression of CAR/CD20 using the SB transposon system was highest in 293T cells, intermediate in PBL T cells, and modest in UCB T cells (Figure 1b–d; data not shown). This differential expression of the transgene in different cell types may be attributable to specific cellular transcriptional regulation or to different levels of gene transfer in these host cells as Supplementary Figure S2 shows that gene transfer efficiency in PBL transfected with SB transposons was slightly higher than that in UCB.
Genetic modification of human T cells with CD20 has been documented as a strategy to purify and lyse transduced cells with anti-CD20 antibodies.20–24 We applied this strategy to coexpress CD19 CAR in human T cells using the SB transposon system, and demonstrated that the transfected T cells from both PBL and UCB can be enriched to at least 85 and 50% purity, respectively, using Rituxan. However, we failed to demonstrate that the engineered T cells from PBL and UCB, expressing CD19 CAR/CD20, were lysed by Rituxan and complement in vitro (Supplementary Materials and Methods) even in the presence of a high concentration (500 μg/ml) of Rituxan, whereas a low concentration (7.8 μg/ml) of Rituxan with complement can readily kill Daudi cells (Supplementary Figure S3a). Clearly, CD20 levels on the engineered PBL were not of sufficient magnitude to observe in vitro complement-mediated lysis, because flow cytometric analysis revealed that surface CD20 expression was highest (mean fluorescence intensity = 726) on Daudi cells, intermediate (mean fluorescence intensity = 41) on the engineered PBL, and lowest (mean fluorescence intensity = 4) on RS4:11 when 12.5 μg/ml of fluorescein isothiocyanate (FITC)-Rituxan was used for cell staining (Supplementary Figure S3b). These data are in agreement with those from a recent study showing that complement-induced cell death by Rituxan depends on CD20 expression levels (approximately mean fluorescence intensity = 540 on CEM T-cell line is required to achieve 50% of cell lysis), and acts complementary to antibody-dependent cellular cytotoxicity,23 two major mechanisms for Rituxan-mediated cytotoxicity in vivo. While in vivo studies will have to await future approaches toward increasing CD20 levels by utilizing strong promoters such as Caggs13 or 2A-like sequences,25 or reversing a dual-gene expression order in the SB transposon with EFlα/mCMV promoter, the advantages of the currently achievable levels of CD20 expression nevertheless permit efficient in vitro selection of transfected T cells.
Our data, which are in agreement with those of other reports, demonstrate that SB-CD19 CAR-engineered T cells from PBL and UCB kill CD19+ leukemia and lymphoma cells.15,26–28 These T cells also mount antitumor responses and prolong survival in NOD/SCID mice, although tumor eventually re-emerges. The shortness of duration of the antitumor effect mediated by these T cells may be because (i) the majority of the infused T cells are CD4+ effector memory cells that may undergo apoptosis after adoptive transfer, and (ii) they achieve a low-level of IL-2 production to Daudi cells, but not to K562-FCD19 cells (Figure 5). Because IL-2 is essential for proliferation, survival, and differentiation of naïve T cells into effector and memory cells,29,30 our future experiments will attempt to increase IL-2 production by adding other costimulatory molecules with CD19 CAR and to expand both CD4 and CD8 T cells using artificial antigen-presenting cells,31 so as to further explore the possibility of enhancing the T-cell response in both our model and a recently described systemic model.32
Unexpectedly, we found that engineered CD4 T cells from both PBL and UCB kill CD19+ major histocompatibility complex class II+ and class II− target cells as potently as do their CD8 T counterparts. This is an important observation, given that most clinical trials with genetically modified T cells are focused on CD8 T cells or clones.27,28 Adoptive infusion of only CD8 T cells may be less efficacious because a large body of literature suggests that CD4 T cells are equally important in antitumor immunity.33 Therefore our approach will be to infuse both CD4 and CD8 T cells that can directly kill tumor cells in addition to producing antitumor Thl cytokines. This may be an effective approach in the development of cellular therapies for other tumor targets in addition to B-cell leukemia and lymphoma as described in this paper.
MATERIALS AND METHODS
Cell culture
K562 (erythroleukemia), TF-1 (erythroleukemia), and U266 (multiple myeloma) were provided by Ivan Borrello. HBP-null (pre B-ALL) and RS4:11 (infant B-ALL) were obtained from John Kersey and Tucker LeBien. Daudi (B-cell Burkitt lymphoma) was purchased from American Type Culture Collection (Manassas, VA). These cell lines were maintained in Epstein-Barr virus medium consisting of Rosewell Park Memorial Institute 1640, 10% fetal bovine serum, 1% nonessential amino acid, 1 mmol/1 sodium pyruvate, 2 mmol/1 L-glutamine, 50 U/ml penicillin, and 50 μg/ml streptomycin.
Cloning of human CD20 cDNA
Total RNA was isolated from Daudi cells using the RNeasy Mini Kit (Qiagen, Valencia, CA) and standard reverse transcription was performed using Superscript II (Invitrogen, Carlsbad, CA). PCR was performed using Platinum PfxDNA polymerase (Invitrogen) with CD20 specific primers: sense, 5′-taggatccatgacaacacccagaaattca gtaa-3′ and antisense, 5′-tgatctagattaaggagagctgtcattttctattgg-3′. The PCR product was digested, subcloned into pcDNA3 vector (Invitrogen), and sequenced. The sequence was completely matched with GenBank accession no. X07203.
SB CD19scFv transposons and transposase-encoding plasmid
The SB-CD19 CAR transposon (Figure 1a) was constructed using standard molecular cloning techniques and the inverted/directed repeat sequences previously described.34 pT2/P19BBζmCCD20 (19BBζ/CD20) was assembled into the SB transposon trans vectors in which the 19BBζ gene is coexpressed with human CD20, using a bidirectional promoter consisting of the human PGK promoter and a mCMV core promoter element, respectively.35 In addition, the QBI SP163 translational enhancer element was placed directly upstream of the ATG initiation codon of the human CD20 coding sequence. pUbC-SBl0 (Figure 1c) contains the human ubiquitin C promoter regulating SB transposase expression.
Human T-cell gene transfer
Human T-cell gene transfer was carried out using a Nucleofector device (Program U14) with the human T-cell nucleofector kit (Amaxa, Gaithersburg, MD) previously described.13 PBL were obtained from buffy coat purchased from Memorial Blood Centers (Minneapolis, MN) or from blood taken from healthy donors after informed consent. The UCB cells obtained were those discarded by the Duke University Cord Blood Center, St. Louis Blood Center, New York Blood Center, and the Red Cross in the Twin Cities. Mononuclear cells from PB or UCB, or CD3 bead-purified UCB T cells (Miltenyi) (5 × 106 cells) were mixed with 5 μg of SB-CD19 CAR transposon and 15 μg of the SB10 transposase plasmid and nucleofected. This combination was predetermined in two PBL batchesh transfected with 5 g of CAR/CD20 transposon and different amounts (0, 2.5, 5, 10, 15, and 20 g) of SB10 (data not shown). Twenty-four hours after nucleofection, the cells were activated using anti-CD3 and anti-CD28 beads (anti-CD3/28 beads) for 3–5 days.19 After removal of the beads, the activated T-cells were maintained in human T-cell medium consisting of Rosewell Park Memorial Institute-1640 (Invitrogen), heat-inactivated (56 °C, 30 minutes) 10% fetal bovine serum (HyClone, Carlsbad, CA) or human serum (SeraCare Life Sciences, Oceanside, CA), 10mmol/l HEPES, 2 mmol/1 L-glutamine, 50μmol/l β-mercaptoethanoL 50 U/ml penicillin, and 50 μg/ml streptomycin, supplemented with IL-2 (50IU/ml; Chiron, Emeryville, CA) and IL-7 (l0ng/ml; National Cancer Institute Biological Resources Branch, Rockville, MD), and re-stimulated once every 10–14 days with anti-CD3/28 beads or OKT3 (Ortho Biotech, Raritan, NJ) as previously described.19,36 All donors provided informed consent as approved by the University of Minnesota Institutional Review Board.
Flow cytometric analysis
F(ab′)2 fragment of goat anti-mouse immunoglobulin G (anti-CAR) conjugated with FITC, or phycoerythrin, or Cyanine (Cy5), and the control F(ab′)2 fragment of ChromPure goat immunoglobulin G isotype antibodies were purchased from Jackson ImmunoResearch (West Grove, PA). Mouse immunoglobulin Gl conjugated with FITC or phycoerythrin or Alexa Fluor 647, mouse immunoglobulin G2b isotypes, anti-human perforin, granzyme A and B, CD4, CD8, CD19, and CD20 were purchased from BD Biosciences (San Jose, CA). Flow cytometric analysis was carried out on the BD FACSCaliber system using CellQuest software (BD Biosciences).
Conjugation of Rituxan with FITC or Biotin and selection of transfected T cells with Rituxan
Rituxan (Rituximab; Genentech, South San Francisco, CA) was purchased from the University Pharmacy. EZ-label FITC protein labeling kit and EZ-link sulfo-NHS-LC-biotinylation kit (Pierce, Rockford, IL) were used for conjugating Rituxan with FITC and biotin, respectively. Protein concentrations of FITC-Rituxan or biotin-Rituxan were determined using the protein assay kit (Bio-Rad, Hercules, CA) and bovine serum albumin standard (Pierce). Positive selection of transfected T cells with biotin-Rituxan and anti-biotin microbeads was carried out in accordance with the manufacturer’s instructions (Miltenyi).
Cytotoxicity and cytokine release assays
A T-cell cytotoxicity assay was carried out using a chromium (51Cr) release assay.37 For inhibition of target-cell recognition by anti-CD19 monoclonal antibodies (BU12 and HD37),17 target cells were preincubated for 45 minutes with monoclonal antibodiess (50 μg/ml) before subsequent addition of effector cells. For blockade of perforin-mediated cytolysis, ethylene glycol tetraacetic acid (4 mmol/1) and MgCl2 (2 mmol/1) were added in the 51Cr-release assay. Cytokine release assays were performed using Fluorokine MAP Immunoarray (R&D Systems, Minneapolis, MN) and the Luminex 100 analyzer (Luminex, Austin, TX).
PCR cloning of insertion sites
The splinkerette PCR techniques used for recovering flanking transposon inserts on the 5′ and 3′ sites have been described earlier.6,8,13 Supplementary Table S1 summarizes the linker and primer sequences. Nested PCR products were cloned into TOPO XL PCR Cloning Kit (Invitrogen) and sequenced. The resulting sequences were subjected to human BLAT search at http://genome.ucsc.edu.
Generation of CD19 KS62 transfectants
Human full-length CD19 cDNA (GenBank accession no. BC006338) in a pOTB7-hCD19 vector was purchased from Open Biosystems (Huntsville, AL), cut with EcoRI and Xhol, and subcloned into the pcDNA3 vector. In order to generate a truncated form of CD19 (ΔCD19), which lacks a cytoplasmic domain, pOTB7-hCD19 was cut, blunted, and subcloned into pcDNA3. K562 transfectants were generated by transfection of K562 cells in 24-well plates with pcDNA3-FCD19, pcDNA3-ΔCD19, and pEGFP-N3 (Clontech, Mountain View, CA) using LipofectAMINE 2000 (Invitrogen).
In vivo antitumor assays
Eight- to ten-week-old female NOD/SCID (NOD.CB17-Pkrdcscid/J, stock number: 001303) mice were purchased from the Jackson Laboratory (Bar Harbor, ME), and all experiments involving animals was conducted at the University of Minnesota Animal Facility in accordance with institutional guidelines. On day 0, the mice were divided into three groups: (i) a group to receive no treatment, (ii) a group to receive T-cell infusions, and (iii) a group to receive T-cell infusions plus IL-2. On day -6, the NOD/SCID mice were γ-irradiated (2.5 Gy, Cesium-137), and then injected intraperitoneally with 106 Daudi-LVhfflucN on the following day. On day -3, the mice that had received Daudi-LVhfflucN were examined for tumor engraftment using bioluminescent imaging. Tumor bioluminescent imaging was performed every 4 days after the first T-cell infusion, after injecting sodium pentobarbital followed by D-luciferin (Xenogen, Hopkinton, MA). Images were collected and analyzed using the Xenogen-IVIS Imaging System. A constant region-of-interest was drawn over the tumor region, the intensity of the signal was measured as total photon flux normalized for exposure time and surface area, and expressed in units of photons/s/cm2/steradian.
Statistical analysis
A linear mixed model was applied to for analyzing the dataset in Figure 6c and f. In order to make the outcome (intensity) closer to normal distribution, a natural log transformation was used. All analyses were based on a natural log–transformed outcome variable. The number of days elapsed after treatment was treated as a discrete variable. The interaction term of treatment administered and number of days after treatment was included, because it was significant. A baseline value of intensity in the model was also adjusted, and a random subject effect was introduced. A covariance matrix was set as a spatial power structure after selection of the model. P values from pair-wise comparisons are adjusted using the Tukey method. Statistical analyses were conducted using SAS 9.1 software (Cary, NC). P < 0.05 were considered statistically significant.
Supplementary Material
Figure S2. Gene transfer efficiency in PBL and UCB.
Figure S3. Complement mediated cell death by Rituxan.
Figure S1. Lentiviral transduced Daudi cells expressing humanized firefly luciferase remain susceptible to cytolysis mediated by CAR/CD20 engineered T cells.
Table S1. Linker and primer sequences used in a splinkerette PCR.
Acknowledgments
We thank Luigi Naldini and Mario Amendola (San Raffaele Telethon Institute for Gene Therapy, Milan, Italy) for providing us with the bidirectional PGK/mCMV promoter, Daniel Vallera (University of Minnesota, Minneapolis, MN) for the anti-CD19 antibodies, Pablo Rubinstein (New York Blood Center, New York, NY) for cord blood units, Arlys Clements (University of Minnesota, Minneapolis, MN) for secretarial assistance, and Cindy Eide (University of Minnesota, Minneapolis, MN) or editing the paper. This work was supported by grants from the Alliance for Cancer Gene Therapy Young Investigator Award, the G & P Foundation for Cancer Research, the National Blood Foundation, the Sidney Kimmel Foundation for Cancer Research Kimmel Scholar Program and, in part, by the Children’s Cancer Research Fund in Minneapolis and the University of Minnesota Medical School Dean’s Commitment (X.Z.). X.Z. is a recipient of an American Society of Hematology Junior Faculty Scholar Award. This investigation was conducted in a facility constructed with support from a Research Facilities Improvement Program Grant (C06 CA062526-01) from the National Center for Research Resources, National Institutes of Health (NIH) (X.Z.) and NIH R01 CA72669 (B.R.B.). C.H.J. is an inventor of a reagent (CD3/CD28 beads) used in the manufacturing of SB-engineered T cells, and therefore may potentially receive royalties from their commercial use. R.S.M. has a financial interest in Discovery Genomics. The other authors declare no competitive financial interests. This work was presented in abstract no. 722 at the 48th annual meeting of the American Society of Hematology, Orlando, Florida, 9 December 2006, and abstract no. 380 at the Keystone Symposium on the Potent New Anti-Tumor Immunotherapies, Banff, Canada, 28 March 2007.
Footnotes
SUPPLEMENTARY MATERIAL
Materials and Methods.
References
- 1.Ivies Z, Hackett PB, Plasterk RH, Izsvak Z. Molecular reconstruction of Sleeping Beauty, a Tc1-like transposon from fish, and its transposition in human cells. Cell. 1997;91:501–510. doi: 10.1016/s0092-8674(00)80436-5. [DOI] [PubMed] [Google Scholar]
- 2.Izsvak Z, Ivies Z. Sleeping Beauty transposon: biology and applications for molecular therapy. Mol Ther. 2004;9:147–156. doi: 10.1016/j.ymthe.2003.11.009. [DOI] [PubMed] [Google Scholar]
- 3.Geurts AM, Yang Y, Clark KJ, Liu G, Cui Z, Dupuy AJ, et al. Gene transfer into genomes of human cells by the Sleeping Beauty transposon system. Mol Ther. 2003;8:108–117. doi: 10.1016/s1525-0016(03)00099-6. [DOI] [PubMed] [Google Scholar]
- 4.Yant SR, Wu X, Huang Y, Garrison B, Burgess SM, Kay MA. High-resolution genome-wide mapping of transposon integration in mammals. Mol Cell Biol. 2005;25:2085–2094. doi: 10.1128/MCB.25.6.2085-2094.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yant SR, Meuse L, Chiu W, Ivies Z, Izsvak Z, Kay MA. Somatic integration and long-term transgene expression in normal and heamophilic mice using a DNA transposon system. Nat Cenet. 2000;25:35–41. doi: 10.1038/75568. [DOI] [PubMed] [Google Scholar]
- 6.Belur LB, Frandsen JL, Dupuy AJ, Ingbar DH, Largaespada DA, Hackett PB, et al. Gene insertion and long-term expression in lung mediated by the Sleeping Beauty transposon system. Mol Ther. 2003;8:501–507. doi: 10.1016/s1525-0016(03)00211-9. [DOI] [PubMed] [Google Scholar]
- 7.Luo G, Ivies Z, Izsvak Z, Bradley A. Chromosomal transposition of a Tc1/mariner-like element in mouse embryonic stem cells. Proc Natl Acad Sci USA. 1998;95:10769–10773. doi: 10.1073/pnas.95.18.10769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dupuy AJ, Clark K, Carlson CM, Fritz S, Davidson AE, Markley KM, et al. Mammalian germ-line transgenesis by transposition. Proc Natl Acad Sci USA. 2002;99:4495–4499. doi: 10.1073/pnas.062630599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Collier LS, Carlson CM, Ravimohan S, Dupuy AJ, Largaespada DA. Cancer gene discovery in solid tumours using transposon-based somatic mutagenesis in the mouse. Nature. 2005;436:272–276. doi: 10.1038/nature03681. [DOI] [PubMed] [Google Scholar]
- 10.Dupuy AJ, Akagi K, Largaespada DA, Copeland NG, Jenkins NA. Mammalian mutagenesis using a highly mobile somatic Sleeping Beauty transposon system. Nature. 2005;436:221–226. doi: 10.1038/nature03691. [DOI] [PubMed] [Google Scholar]
- 11.Keng VW, Yae K, Hayakawa T, Mizuno S, Uno Y, Yusa K, et al. Region-specific saturation germline mutagenesis in mice using the Sleeping Beauty transposon system. Nat Methods. 2005;2:763–769. doi: 10.1038/nmeth795. [DOI] [PubMed] [Google Scholar]
- 12.Kitada K, Ishishita S, Tosaka K, Takahashi R, Ueda M, Keng VW, et al. Transposon-tagged mutagenesis in the rat. Nat Methods. 2007;4:131–133. doi: 10.1038/nmeth1002. [DOI] [PubMed] [Google Scholar]
- 13.Huang X, Wilber AC, Bao L, Tuong D, Tolar J, Orchard P, et al. Stable gene transfer and expression in human primary T cells by the Sleeping Beauty transposon system. Blood. 2006;107:483–491. doi: 10.1182/blood-2005-05-2133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Barker JN, Wagner JE. Umbilical-cord blood transplantation for the treatment of cancer. Nat Rev Cancer. 2003;3:526–532. doi: 10.1038/nrc1125. [DOI] [PubMed] [Google Scholar]
- 15.Imai C, Mihara K, Andreansky M, Nicholson IC, Pui CH, Greiger TL, et al. Chimeric receptor with 4-1BB signaling capacity provoke potent cytotoxicity against acute lymphoblastic leukemia. Leukemia. 2004;18:676–684. doi: 10.1038/sj.leu.2403302. [DOI] [PubMed] [Google Scholar]
- 16.Mahmoud MS, Fujii R, Ishikawa H, Kawano MM. Enforced CD19 expression leads to growth inhibition and reduced tumorigenicity. Blood. 1999;94:3551–3558. [PubMed] [Google Scholar]
- 17.Vallera DA, Elson M, Brechbiel MW, Dusenbery KE, Burns LJ, Jaszcz WB, et al. Radiotherapy of CD19 expressing Daudi tumors in nude mice with Yttrium-90-labeled anti-CDI 9 antibody. Cancer Biother Radiopharm. 2004;19:11–23. doi: 10.1089/108497804773391630. [DOI] [PubMed] [Google Scholar]
- 18.Sallusto F, Geiginat J, Lanzavecchia A. Central memory and effector memory T cell subsets: function, generation, and maintenance. Ann Rev Immunol. 2004;22:745–763. doi: 10.1146/annurev.immunol.22.012703.104702. [DOI] [PubMed] [Google Scholar]
- 19.Levine BL, Bernstein WB, Aronson NE, Schlienger K, Cotte J, Perfetto S, et al. Adoptive transfer of costimulated CD4+ T cells induces expansion of peripheral T cells and decreased CCR5 expression in HIV infection. Nat Med. 2002;8:47–53. doi: 10.1038/nm0102-47. [DOI] [PubMed] [Google Scholar]
- 20.Introna M, Barbul AM, Bambacioni F, Casati C, Gaipa G, Bernasconi S, et al. Genetic modification of human T cells with CD20: a strategy to purify and lyse transduced cells with anti-CD20 antibodies. Hum Gene Ther. 2000;11:611–620. doi: 10.1089/10430340050015798. [DOI] [PubMed] [Google Scholar]
- 21.Serafini M, Manganini M, Borleri G, Bonamino M, Imberti L, Biondi A, et al. Characterization of CD20-transduced T lymphocytes as an alternative suicide gene therapy approach for the treatment of graft-versus-host disease. Hum Gene Ther. 2004;15:63–76. doi: 10.1089/10430340460732463. [DOI] [PubMed] [Google Scholar]
- 22.Serafini M, Ronamino M, Golay J, Introna M. Elongation factor 1 (EFIa) promoter in a lentiviral backbone improves expression of the CD20 suicide gene in primary T lymphocytes allowing efficient rituximab-mediated lysis. Haematologica. 2004;89:86–95. [PubMed] [Google Scholar]
- 23.Van Meerten T, van Rijn RS, HoI S, Hagenbeek A, Ebeling SB. Complement-induced cell death by Rituximab depends on CD20 expression level and acts complementary to antibody-dependent cellular cytotoxicity. Clin Cancer Res. 2006;12:4027–4035. doi: 10.1158/1078-0432.CCR-06-0066. [DOI] [PubMed] [Google Scholar]
- 24.Van Meeten T, Claessen MJ, Hagenbeek A, Ebeling SB. The CD20/alphaCD20 “suicide” system: novel vectors with improved safety and expression profiles and efficient elimination of CD20-transgenic T cells. Gene Ther. 2006;13:789–797. doi: 10.1038/sj.gt.3302705. [DOI] [PubMed] [Google Scholar]
- 25.Donnelly MLL, Hughes LE, Luke G, Mendoza H, ten Dam E, Gani D, et al. The “cleavage” activities of foot-and-mouse disease virus 2A site-directed mutants and naturally occurring “2A-like” sequences. J Gen Virol. 2001;82:1027–1041. doi: 10.1099/0022-1317-82-5-1027. [DOI] [PubMed] [Google Scholar]
- 26.Brentjens RJ, Latouche JB, Stantos E, Marti F, Gong MC, Lyddane C, et al. Eradication of systemic B-cell tumors by genetically targeted human T lymphocytes co-stimulated by CD80 and interleukein-15. Nat Med. 2003;9:279–286. doi: 10.1038/nm827. [DOI] [PubMed] [Google Scholar]
- 27.Cooper LJN, Topp MS, Serrano LM, Gonzalez S, Chang WC, Araceli N, et al. T-cell clones can be rendered specific for CD19: toward the selective augmentation of the graft-versus-B-lineage leukemia effect. Stood. 2003;101:1637–1644. doi: 10.1182/blood-2002-07-1989. [DOI] [PubMed] [Google Scholar]
- 28.Serrano LM, Pfeiffer T, Olivares S, Numbenjapon T, Bennitt J, Kim D, et al. Differentiation of naive cord blood T cells into CD19-specific cytolytic effectors for posttransplantation adoptive immunotherapy. Stood. 2006;107:2643–2651. doi: 10.1182/blood-2005-09-3904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Williams MA, Tyznik AJ, Bevan MJ. lnterleukin-2 signals during priming are required for secondary expansion of CD8+ memory T cells. Nature. 2006;441:890–893. doi: 10.1038/nature04790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kamimura D, Bevan MJ. Naive CD8+ T cells differentiate into protective memory-like cells after IL-2-anti-IL-2 complex treatment in vivo. J Exp Med. 2007;204:1803–1812. doi: 10.1084/jem.20070543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Suhoski MM, Golovina TN, Aqui NA, Tai VC, Varela-Rohena A, Milone MC, et al. Engineering artificial antigen-presenting cells to express a diverse array of co-stimulatory molecules. Mol Ther. 2007;15:981–988. doi: 10.1038/mt.sj.6300134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Brentjens RJ, Santos E, Nikhamin Y, Yeh R, Matsushita M, Perle KL, et al. Genetically targeted T cells eradicate systmetic acute lymphoblastic leukemia xenografts. Clin Cancer Res. 2007;13:5426–5435. doi: 10.1158/1078-0432.CCR-07-0674. [DOI] [PubMed] [Google Scholar]
- 33.Gerloni M, Zanetti M. CD4 T cells in tumor immunity. Springer Semin Immunopathol. 2005;27:37–48. doi: 10.1007/s00281-004-0193-z. [DOI] [PubMed] [Google Scholar]
- 34.Cui Z, Geurts AM, Liu G, Kaufman CD, Hackett PB. Structure-function analysis of inverted terminal repeats for the Sleeping Beauty transposon. J Mol Biol. 2002;318:1221–1235. doi: 10.1016/s0022-2836(02)00237-1. [DOI] [PubMed] [Google Scholar]
- 35.Amendola M, Venneri MA, Biffi A, Vigna E, Naldini L. Coordinate dual-gene transgenesis by lentiviral vectors carrying synthetic bidirectional promoters. Nat Biotechnol. 2004;23:108–116. doi: 10.1038/nbt1049. [DOI] [PubMed] [Google Scholar]
- 36.Riddell SR, Greenberg PD. The use of anti-CD3 and anti-CD28 monoclonal antibodies to clone and expand human antigen-specific T cells. J Immunol Methods. 1990;128:189–201. doi: 10.1016/0022-1759(90)90210-m. [DOI] [PubMed] [Google Scholar]
- 37.Zhou X, Jun DY, Thomas AM, Huang X, Huang LQ, Mautner J, et al. Diverse CD8+ T-cell responses to renal cell carcinoma antigens in patients treated with an autologous granulocyte-macrophage colony-stimulating factor gene-transduced renal tumor cell vaccine. Cancer Res. 2005;65:1079–1088. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S2. Gene transfer efficiency in PBL and UCB.
Figure S3. Complement mediated cell death by Rituxan.
Figure S1. Lentiviral transduced Daudi cells expressing humanized firefly luciferase remain susceptible to cytolysis mediated by CAR/CD20 engineered T cells.
Table S1. Linker and primer sequences used in a splinkerette PCR.