

Abstract
Autosomal Dominant Polycystic Kidney Disease (ADPKD) is the most common monogenic kidney disease and the fourth leading cause of end-stage renal disease, responsible for 5–10% of cases. The disease is characterized by relentless development and growth of cysts causing progressive kidney enlargement associated with hypertension, pain, reduced quality of life, and eventually kidney failure. It is caused by mutations to PKD1 or PKD2, encoding polycystin-1 and polycystin-2, respectively. Their function and the molecular mechanisms responsible for the development of polycystic kidney disease are not well understood. The objective of this review is to synthesize a large body of literature that examines how reduction of functional PC1 or PC2 at the primary cilia and/or the endoplasmic reticulum directly disrupts intracellular calcium signaling and indirectly disrupts calcium regulated cAMP and purinergic signaling. We propose a hypothetical model where dysregulated metabolism of cAMP and purinergic signaling increase the sensitivity of principal cells in collecting ducts and of tubular epithelial cells in the distal nephron to the constant tonic action of vasopressin. The resulting magnified response to vasopressin further enhances the disruption of calcium signaling initiated by mutations to PC1 or PC2 and activates downstream signaling pathways responsible for impaired tubulogenesis, cell proliferation, increased fluid secretion and interstitial inflammation.
Introduction
Autosomal Dominant Polycystic Kidney Disease (ADPKD) is the most common monogenic kidney disease and the fourth leading cause of end-stage renal disease, responsible for 5–10% of cases.1,2 It affects all ethnic groups and is a major burden for public health. It is genetically heterogeneous with two loci identified, PKD1 (16p13.3), encoding polycystin-1 (PC1) and PKD2 (4q22), encoding PC2. The disease is characterized by relentless development and growth of cysts causing progressive kidney enlargement associated with hypertension, abdominal fullness and pain, episodes of cyst hemorrhage, gross hematuria, nephrolithiasis, cyst infections, and reduced quality of life. Despite continuous destruction of the parenchyma, compensatory hyperfiltration of the surviving glomeruli maintains renal function within the normal range for decades. Only when the majority of nephrons have been destroyed, renal function declines, typically after the fourth decade of life, and end-stage renal disease eventually ensues in the majority of patients. Medical advances have improved the treatment of patients with ADPKD, but today this remains restricted to the detection and treatment of complications and renal replacement therapy. Research on Polycystic Kidney Disease (PKD) increased exponentially after the discovery of the PKD1 and PKD2 genes in 1994 and 1996. Targets for intervention have been identified and tested in animal models and some clinical trials have provided modest but encouraging results.3–5 Nevertheless, the function of the polycystins and the molecular mechanisms responsible for the development of PKD remain poorly understood and better therapies are needed.
The objective of this review is to synthesize a large body of literature that examines how reduction of functional PC1 or PC2 (1) directly disrupts calcium signaling at specific cellular compartments, i.e primary cilia (2) and/or endoplasmic reticulum (3), and indirectly disrupts calcium regulated cyclic adenosine monophosphate (cAMP) (4) and purinergic signaling (5). We propose a hypothetical model where dysregulated cAMP metabolism and purinergic signaling increase the sensitivity of principal cells in collecting ducts and of tubular epithelial cells in the distal nephron to the continuous tonic action of vasopressin (6), further enhancing the disruption of calcium signaling initiated by mutations to PC1 or PC2 (7) and activating downstream signaling pathways responsible for impaired tubulogenesis, cell proliferation, increased fluid secretion and interstitial inflammation (8). In vivo studies addressing the role of calcium and cAMP in PKD are consistent with this hypothetical model (9). The following nine sections examine the different components of this hypothesis in detail. A cartoon illustrating this hypothesis is presented in Figure 1.
Figure 1. Hypothetical roles of calcium, vasopressin and purinergic signaling in ADPKD.

Polycystin-1 and polycystin-2 the primary cilia and endoplasmic reticulum regulate intracellular calcium signaling directly and through their interaction with other calcium channel proteins. Reduced intracellular calcium caused by mutations to polycystin-1 or polycystin-2 enhances the generation of cAMP by calcium inhibitable adenylyl cyclase 6 and inhibits the destruction of cAMP by calcium dependent phosphodiesterase 1 and cyclic guanosine monophosphate (cGMP) inhibited phosphodiesterase 3 which control the cAMP pool responsive to vasopressin stimulation. The reduction of physiological calcium oscillations also blunts the release of ATP and its action on P2Y2, thus abolishing a negative feedback loop that normally limits the vasopressin V2 receptor-dependent activation of adenylyl cyclase 6. Undefined cellular mechanisms (possibly disruption of the calcium dependent insertion of aquaporin-2 into the apical membrane) and/or disruption of medullary architecture by cysts interfere with urine concentration and increase circulating levels of vasopressin. Thus, altered cAMP metabolism and purinergic signaling, along with increased levels of circulating vasopressin, markedly enhanced the constant tonic effect of vasopressin on the V2 receptors in collecting duct principal cells and distal nephron epithelial cells. Vasopressin-driven activation of protein kinase A enhances the phosphorylation of polycystin-2, ryanodine receptors and inositol 1,4,5-trisphosphate receptors, increases the leakage of calcium across the endoplasmic reticulum membrane, and further disrupts intracellular calcium signaling. The reduction in intracellular calcium determines a striking switch in the cellular response to cAMP from suppression to marked stimulation of proliferation. Thus, in the setting of reduced intracellular calcium sustained activation of cAMP and protein kinase A activate downstream signaling pathways responsible for impaired tubulogenesis, cell proliferation, increased fluid secretion and interstitial inflammation, characteristic of the cystic phenotype.
AC6: adenylyl cyclase 6 ; PDE: Phosphodiesterase; PKA: Protein kinase A; RYR: Ryanodine receptor; AQP2: Aquaporin 2; PLC: Phospholipase C; PIP2: phosphatidylinositol (4,5)-bisphosphate ; IP3: Inositol 1,4,5-trisphosphate; DAG: Diacylglycerol; PKC: Protein Kinase C; Gs and Gi refer to guanosine nucleotide-binding proteins s and i, respectively. Yellow indicates molecules that are reduced in PKD; blue indicates molecules that are increased in PKD.
1. Polycystins and calcium signaling
PC1 (4303aa; ∼600kDa, uncleaved and glycosylated) is a receptor-like protein with a large extracellular region (3074aa) that comprises a number of domains involved in protein-protein and protein-carbohydrate interactions (Figure 2). PC1 also has 11 transmembrane domains and a cytoplasmic tail. The last six transmembrane spans of PC1 share sequence homology with PC2 although PC1 has not been shown to function directly as a channel protein. Auto-proteolytic cleavage of PC1 at the G protein coupled receptor proteolytic site (GPS) domain is an important step to form a functional protein.6–8 After embryonic development, the full-length protein is rarely seen, with a ∼130kDa C-terminal (CT) and two large N-terminal (NT) products; NT and CT are thought to stay associated after cleavage. The two NT products are glycoforms, one mature form that traffics through the Golgi and is thought to be localized at the plasma membrane/cilia or secreted and one that may be retained in the endoplasmic reticulum.9–11 PC1 has been proposed to undergo further proteolytic cleavages, one that generates a P100 intramembranous fragment with six membrane spanning domains that interacts with Stromal Interaction Molecule 1 (STIM1) and inhibits store operated calcium entry. The other two generate COOH-terminal tail (CTT) fragments capable of entering the nucleus and either binding to transcription factors T-cell factor or TCF and CCAAT-enhancer-binding protein homologous protein or CHOP (35 kDa CTT fragment) preventing coactivation by P300 or binding to coactivator P100 (17 kDa fragment) and activating the transcription factor signal transducer and activator of transcription 6 (STAT6).12–16 The importance of these latter cleaved products to the pathogenesis of ADPKD has not been elucidated. Several studies have used exogenous expression of chimeric proteins containing the CTT of PC1 fused to a membrane-targeted protein. The results of these studies are difficult to interpret and in some cases may recapitulate the function of the native protein while in other cases have dominant negative effects (Table 1).17–24
Figure 2. Predicted structures of polycystin-1 (PC1), left, and polycystin-2 (PC2), right.
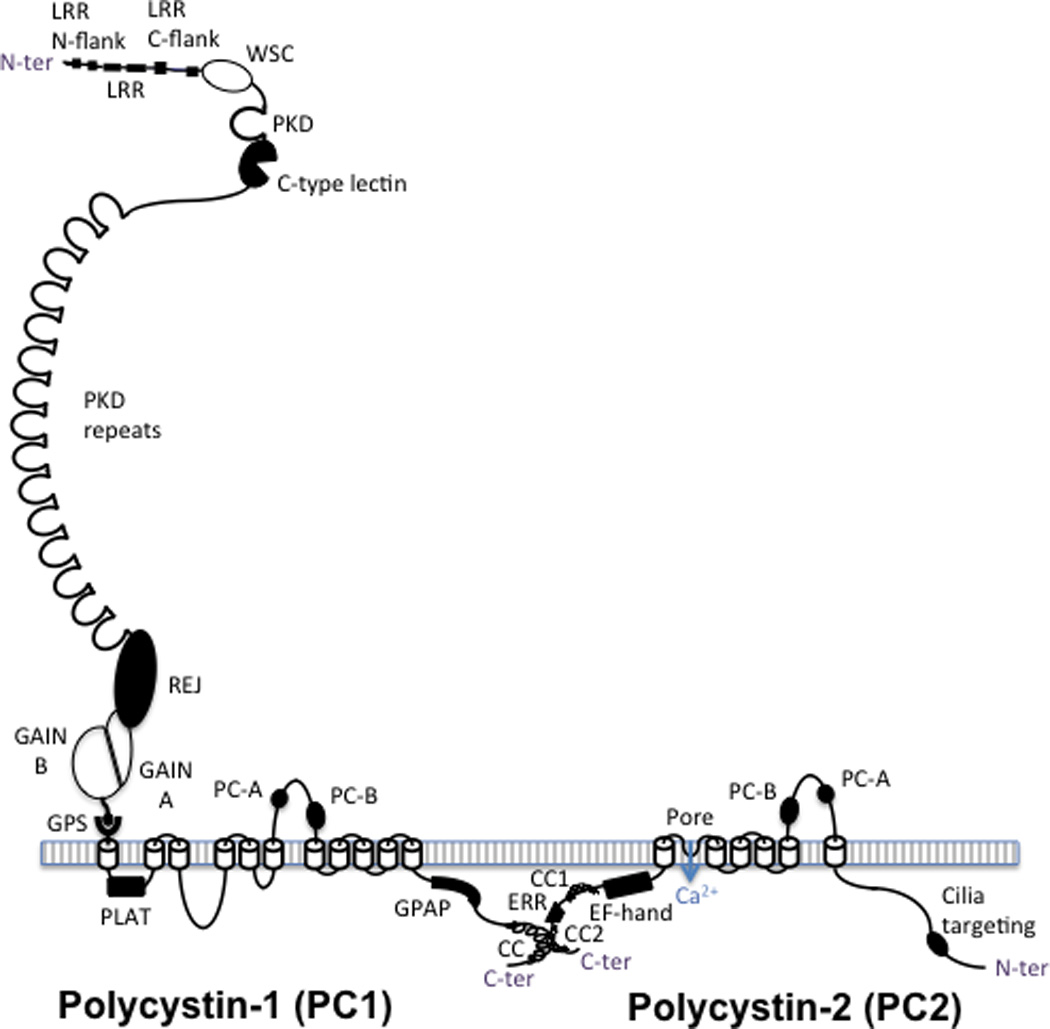
PC1 has a large ectodomain with a number of recognized domains sequentially located from the N terminus of the protein (after the signal peptide is cleaved): leucine rich repeats (LRR), flanked by N and C terminal domains, the cell wall integrity and stress-response component (WSC) domain, polycystic kidney disease (PKD) protein repeats, a C-type lectin, the receptor for egg jelly (REJ) domain, the G-protein coupled receptor (GPCR) proteolysis site (GPS), where PC1 is cleaved, and the associated GPCR autoproteolysis inducing (GAIN) domain, with A and B subdomains. PC1 is associated with the membrane with 11 transmembrane domains with the PC1, lipoxygenase, alpha-toxin (PLAT) domain found in the first cytoplasmic loop. The last 6 transmembrane region of PC1 is homologous to the transmembrane region of PC2, with the polycystin homologous motifs A and B found in the large extracellular loops of both proteins. The ∼200aa cytoplasmic tail contains a G-protein activating peptide (GPAP) and a coiled coil (CC) domain that interacts with the second coiled coil domain (CC2) found in the C-ter tail of PC2. PC2 is a TRP-like calcium channel that has an EF-hand and an endoplasmic reticulum retention (ERR) signal also in the C-tail and a proposed cilia targeting sequence in the N-terminal tail.
Table 1.
Articles using plasma membrane anchored chimeric proteins to study the function of the COOH-terminal tail of Polycystin 1
| Reference | Model system | Findings |
|---|---|---|
| Sutters 200117 | M1 cells; dexamethasone inducible sIg7-PC1-193 | sIg7-PC1-193 transforms the response to cAMP from growth inhibition to growth stimulation. |
| Vandorpe 200218 | Xenopus oocytes and HEK cells; CD16.7-PC1(CTT constructs) | CD16.7-PKD1(1–226), (115–226), (115–203) or (115–189), but not CD16.7-PKD1(1–92), (1–115), (1–155) or CD16.7 (control) up-regulate a nonselective Ca2+-permeable cation conductance. |
| Aguiari 200319 | HEK-293 and HeLa cells; Trk-PC1(CTT constructs) | Trk-PC1-1-226 or 26-226 enhance ATP-evoked (HEK) and histamine-evoked (HeLA) cytosolic Ca2+ transients (upregulation of PC2 or other intracellular cation channel activity?). |
| Hooper 200321 | M1 cells; dexamethasone inducible sIg7-PC1-193 | sIg7-PC1-193 enhances ATP induced, but not forskolin induced, chloride secretory currents. |
| Wildman 200320 | M1 cells; dexamethasone inducible sIg7-PC1-193 | sIg7-PC1-193 prolongs the decay phase of ATP induced Ca2+ transients and the duration of ATP stimulated Cl-conductance. |
| Puri 200422 | HEK cells; sIg7-PC1(CTT constructs) | PC1 constructs containing the heterotrimeric G protein binding and activation domain activate Gαq and PLC, releasing of Ca2+ from intracellular stores, activating store-operated Ca2+ entry, and activating calcineurin and NFAT. |
| Manzati 200523 | HEK-293 cells; Trk-PC1(26–226) | Trk-PC1(26–226) expressing cells have higher basal Ca2+ concentration and 1% FBS or ATP induced Ca2+ transients and proliferative responses. |
| Xu 200724 | NK cells; CD16.7-PC1(115–226). | CD16.7-PKD1(115–226) accelerates the decay of flow-induced Ca2+ transients. |
PC2 (968aa; ∼110kDa) is a six transmembrane, large conductance, calcium permeable, non-selective cation channel of the transient receptor potential (TRP) family.25,26 PC1 and PC2 interact via their C-terminal tails. The resulting PC complex is thought to localize to primary cilia and play a role intracellular calcium regulation.6,27–29 PC2 is mainly localized in the endoplasmic reticulum with its interaction with PC1 important for its translocation to the plasma membrane and primary cilia. Recent evidence indicates that this interaction is essential for PC1 maturation.30,31 The interaction of PC1 and PC2 with other calcium channel and sensor proteins provides support to the hypothesis that the polycystins function in the regulation of intracellular calcium homeostasis. For example, PC1 interacts with the inositol 1,4,5-trisphosphate receptor (IP3R) and the stromal interaction molecule 1 (STIM1), while PC2 interacts with IP3R, the ryanodine receptor (RyR), TRPV4 and TRPC1, 3, 4 and 7.32–39
2. Role of the polycystins in primary cilia
The function of the polycystins in primary cilia has been proposed to be critical in the pathogenesis of PKD. Data supporting this view are that LOV1 or location of vulva 1 (homolog of PC1) and PC2 in C. elegans are found in primary cilia of male-specific sensory neurons and mediate male sensory behaviors, that mice homozygous for a hypomorphic Ift88Tg737Rpw mutation resulting in absent or stunted primary cilia have polycystic kidneys, and that bending of the primary cilium by fluid flow or directly with a micropipette elicits an intracellular calcium signaling response. .40–42 Additional experiments showed that PC1 and PC2, influx of extracellular calcium, and RyR-mediated release of intracellular calcium mediate the transduction of fluid flow shear stress into calcium transients in kidney epithelial cells 43 These observations led to the ciliocentric hypothesis of PKD, according to which PC1 senses the bending of the primary cilium and activates tightly associated PC2 calcium channels. The local calcium influx in the cilium subsequently triggers calcium induced calcium release from intracellular stores and store operated calcium entry. The absence of flow sensing in renal epithelia causes PKD.
Recent observations have challenged the validity of this hypothesis. Knockdown of TRPV4, which forms mechanosensitive heteromeric complexes with PC2, abolishes the intracellular calcium response to fluid flow shear stress but does not result in a cystic phenotype in mice or zebrafish.38 Studies using genetically engineered calcium indicators have shown that primary cilia are specialized calcium signaling organelles with calcium concentrations much higher than those in the cytoplasm and that the amount of calcium that can diffuse from the cilia to the cytoplasm in response to bending of the cilia is too miniscule to trigger calcium induced calcium release from intracellular stores.44 Changes of calcium concentration in the cilia can be dissociated from those in the cytoplasm and a lag time between the calcium increase in the cilia and in the cytoplasm raises the possibility of an intermediate step.45 The shear stress of flow on epithelial cell monolayers differs from the mechanical effect of flow in isolated perfused tubules which includes stretch in addition to shear stress.46 Extracellular adenosine triphosphate (ATP) acting on purinergic (P2) receptors has recently emerged as a potential candidate to mediate both shear- and stretch-induced calcium responses.46–48 A recent study using Madin-Darby canine kidney (MDCK) cells showed that the intracellular calcium response to shear stress was already present in unciliated cells on culture day 1 and was magnified in ciliated cells on day 7, but that this enhanced response was dependent on higher expression of purinergic P2Y2 and P2×7 receptors rather than the presence of cilia per se.49
3. Role of the polycystins in the endoplasmic/sarcoplasmic reticulum
PC1 and particularly PC2 are found in the endoplasmic/sarcoplasmic reticulum membrane and contribute to the regulation of calcium release from intracellular stores. Studies aimed at elucidating the role of the polycystins in this process and how it is altered in ADPKD are summarized in Table 2.12,32–34,39,50–65 The results of these studies are not always consistent, likely due to different experimental conditions. For example, reduced IP3-induced calcium release has been observed in experiments where PC1 was either knocked down or overexpressed.32,33,60 While some studies have shown that PC1 or PC2 haploinsufficiency disrupts intracellular calcium homeostasis,66–68 particularly in myocytes,51,54,65 others have found no evidence for this.69
Table 2.
Articles using endogenous, exogenously expressed or knockdown of polycystins to study intracellular calcium stores and release.
| Reference | Model system | Findings |
|---|---|---|
| Koulen 200250 | LLC-PK1 cells. Exogenous expression of full-length PC2, C-terminal truncated PC2 or missense PC2 mutant. Microsomes reconstituted onto lipid bilayers. | WT PC2 is functions as calcium-activated high conductance, divalent cation permeable channel. Truncated forms of PC2 traffic to the plasma membrane and retain channel activity. Missense PC2 mutant retains the subcellular localization in the ER but has no channel activity. Vasopressin (70 nM) induces IP3R–mediated [Ca2+]I transients in LLC-PK1 cells overexpressing PC2 of greater amplitude and duration compared to cells transfected with empty vector. |
| Qian 200351 | Endogenous PC2. Mouse Pkd2+/− and +/+ VSMCs. | Mouse Pkd2+/− VSMCs have lower basal [Ca2+]i, sarcoplasmic reticulum Ca2+ store, and store operated Ca2+ channel activity compared with wild-type VSMCs. |
| Cai 200452 | LLC-PK1 cells. Exogenous expression of PC2 or PC-2 S812A. Microsomes reconstituted onto lipid bilayers. | Constitutive phosphorylation at Ser812 makes the PC2 channel more sensitive to activation by calcium and the duration and amplitude of vasopressin (100 nM) induced intracellular Ca2 transients. |
| Aguiari 200453 | Endogenous PC1 and PC2. B-lymphoblastoid cells. | Lower platelet-activating factor induced Ca2+ transients and cell proliferation in PC1 or PC2 deficient cells. |
| Gao 200454 | WT, Pkd2 null and RyR mutant Drossophila larvae. | PC2 cooperates with the RyR to mediate optimal rate of larval body wall contraction. Pkd2 haploinsufficiency lowers smooth muscle contractility by 53%. |
| Koulen 200555 | WT and pkd2 null C. elegans ER vesicles incorporated onto lipid bilayers. | C. elegans pc2 is activated by 100 nM-1 mM cytosolic Ca2+ and inactivated by higher levels. Lower IP3R- or RyR-mediated Ca2+ release from pc2 deficient compared to WT microsomes. Longer half-rise and half-decay of Ca2+ transients in pc2 null cells. |
| Li 200556 | Xenopus oocytes. Exogenous expression of PC2 (WT, mutant, C-terminus) and IP3R. UV photolysis of caged IP3 in the absence of extracellular Ca2+. | PC2 interacts with IP3R through its C terminus. Exogenous PC2 (but not PC2 mutants) prolongs IP3-dependent Ca2+ release from intracellular stores. A channel dead mutant lowers the amplitude of IP3-induced Ca2 transients. Expression of PC2 C-terminus prolongs the half-decay time (due to dysfunctional closure of IP3R?) but reduces transient amplitude (due to ER Ca2+ depletion?). |
| Hooper 200564 | MDCK cells. Exogenous expression of full length PC1 | PC1 overexpression accelerates the decay of Ca2+ transients in response to ATP. |
| Anyatonwu 200734 | Embryonic mouse Pkd2−/− and +/+ cardiomyocytes | PC2 interacts with RyR2, stabilizes closed state and inhibits Ca2+ release. Loss of inhibition of RyR2 by PC2 in PC2-deficient cardiac myocytes reduces amplitude and increases frequency of Ca2+ oscillations and reduces SR calcium stores. |
| Geng 200857 | LLC-PK1 cells. Exogenous expression of WT PC2, Δ(5–72)PC2 or empty vector. ER vesicles incorporated onto lipid bilayers. | Syntaxin-5 interacts with PC2 and blocks its channel activity. Expression of PC2 increases resting Ca2+ above control. Expression of a PC2 mutant that does not bind Stx5 further increases resting cytosolic Ca2+ levels, decreases ER Ca2+ stores, and reduces vasopressin (200 nM)-induced Ca2+ release from ER stores. |
| Weber 200858 | MDCK cells. Exogenous expression of PC1. Fura and fura 2-AM microfluorimetry. | PC1 attenuates the Ca2+ peak and rate of decay of luminal ER calcium following inhibition of SERCA (with no effect on mitochondrial Ca2+uptake or extrusion of Ca2+ across the plasma membrane). PC1 inhibits Ca2+ leak across the ER membrane. |
| Wegierski 200959 | Xenopus oocytes; HEK, HeLA cells (exogenous PC2 expression). MDCK cells (shRNA Pkd2 knockdown). | PC2 transfection or depletion of PC2 has opposite effects (decrease or increase) ER Ca+ stores. PC2 regulates the ER Ca2+ gateway to apoptosis by decreasing the Ca2+ concentration in the ER. |
| Morel 200965 | MousePkd1+/+ andPkd1−/− VSMCs | Denuded aortic muscle from Pkd1+/−mice has lower basal intracellular Ca2+ compared to that of Pkd1+/+mice. Ca2+ entry after caffeine induced SR Ca2+ depletion was reduced in Pkd1+/− aortic muscle, but basal SR Ca2+ store was not reduced. |
| Li 200932 | Xenopus oocytes, MDCK cells. Exogenous PC1 expression (fragments or full-length). | A PC1 fragment consisting (last six TM domains and C-terminal tail) interacts with the IP3-binding domain of IP3R and reduces the amplitude of IP3-induced Ca2+ transients. WT PC1 decreases ATP-induced Ca2+ transients. |
| Miyagi 200939 | LLC-PK1 cells. Exogenous expression of WT PC2 and PC2 with a C-terminus truncating mutation. | WT PC2 localizes to ER; PC2 with truncated C-terminus localizes to plasma membrane. PC2 (but not truncated PC2) enhances acetylcholine-induced, ER Ca2+ release in the absence of external Ca2+. Enhanced Ca2+ entry after reintroduction of external Ca2+ in cells expressing truncated PC2 and co-expressing TRPC3 or TRPC7. |
| Plotnikova 201063 | HEK cells. Exogenous expression of PC2 and Aurora A kinase. | Cotransfection of Aurora kinase A in PC2 transfected cells reduces the amplitude of vasopressin induced calcium transients |
| Woodward 201012 | Xenopus oocytes, MDCK and CHO cells. Expression of WT PC1, P100 (fragment generated by cleavage within the third intracellular loop). | PC1 and P100 (but not a mutant P100 lacking the C-terminal 76 amino acids) interfere with STIM1 puncta formation after ER Ca2+ depletion and inhibit SOC. |
| Santoso 201133 | MCDK, M1 cells. Exogenous expression of WT PC1 or PC1R4227X. | PC2 and STIM1 interact with IP3R, enhancing inhibiting ER Ca2+ release. PC1 reduces the interaction of IP3R with PC2 and enhances the interaction of STIM1 with IP3R thus 1) inhibiting ER Ca2+ release, 2) enhancing traffic of PC2 to the plasma membrane and entry of extracellular Ca2+, and 3) inhibiting store operated calcium entry. Mutations in PC1 may increase the interaction between PC2 and IP3R, increase ER release of Ca2+, reduce Ca2+ influx through the plasma membrane and lead to depletion of ER Ca2+. Mutations in PC2 may reduce Ca2+ influx across the plasma membrane and store operated calcium entry (increased interaction of PC1 and STIM1) leading to low intracellular Ca2+. |
| Mekahli 201260 | Permeabilized, non-mitochondrial calcium store loaded, conditionally immortalized human PTE cells. PKD1 and/or PKD2 knockdown. | Simultaneous presence of PC1 and PC2 to amplify IP3-induced ER Ca2+ release. PC1 and possibly PC2 inhibit the passive Ca2+ leak from the ER. |
| Paavola 201361 | Pkd2−/− and pkd2+/+ zebrafish. Microdissected hearts. | Pkd2 null cardiac myocytes display prolonged Ca2+ transient rise and decay times, slow Ca2+ cycling, and reduced SR Ca2+ stores, leading to reduced heart rate and stroke volume, pericardial and abdominal edema, pulse alternans, and AV block. |
| Streets 201362 | HEK and MDCK cells; Xenopus. Exogenous WT PC2, PC2(S829A) or PC2(S829D). | PKA phosphorylates PC2 at Ser829. PC1 through its interaction with PC2 and PP1a facilitates the dephosphorylation of pSer829. PC2 Ser829 phosphorylation is increased in cystic human and mouse tissue. Phosphorylation of PC2 at Ser829 is necessary to rescue a pkd2 knockdown phenotype in Xenopus. The phosphomimic S829D mutant shows increased ATP-stimulated Ca2+ transients and loss of growth suppression. PC2 while hyperphosphorylation may contribute to cystogenesis. |
a) Polycystin-2
PC2 functions as an endoplasmic reticulum calcium release channel activated by increases in calcium concentration.50 Constitutive phosphorylation by casein kinase II at Ser812 increases the sensitivity of the channel to calcium activation on the cytosolic side.52 Aurora A kinase and protein kinase A (PKA) phosphorylate PC2 at Ser829, enhancing the amplitude of ATP-induced calcium release.62,63 In contrast, co-transfection of Aurora kinase A in PC2 transfected human embryonic kidney (HEK) cells reduced the amplitude of vasopressin induced calcium transients, but the effect of Ser829 phosphorylation was not directly tested.63 Syntaxin-5 binds to and prevents leaking of calcium from endoplasmic reticulum stores.57 PC2 interacts physically and functionally with IP3R and RyR.34,39 In most studies PC2 has been found to enhance the amplitude of IP3R and RyR mediated calcium transients and to inhibit the leakage of calcium from endoplasmic reticulum stores,34 although in one study exogenous expression of PC2 was associated with increased leakage of calcium from the endoplasmic reticulum.59 Another study suggests that the simultaneous presence of PC1 and PC2 is required to amplify IP3-induced calcium release.60 Interestingly, knockdown of IP3R1 or IP3R3, like the knockdown of PC2, induces cell proliferation, apoptosis, and cyst formation in a collagen-matrigel three-dimensional culture system using pig kidney epithelial (LLC-PK) cells.70 Several studies have shown that store operated calcium entry is reduced in PC2 defective cells34,51,55,57,71 and it has been suggested that the interaction between STIM1 (a transmembrane sensor for calcium concentration in the endoplasmic reticulum lumen) and ORAi (the pore-forming unit of a plasma membrane channel that permits calcium entry in response to a reduction in the endoplasmic reticulum calcium concentration) is uncoupled in these cells.71 Although PC2 is mostly located in the endoplasmic/sarcoplasmic reticulum membrane, truncated forms lacking the endoplasmic/sarcoplasmic reticulum retention domain translocate to the plasma membrane.50
b) Polycystin-1
The function of PC1 in the endoplasmic/sarcoplasmic reticulum is complex and not well understood. PC1 has been shown to interact with the IP3R and STIM1 in the endoplasmic/sarcoplasmic reticulum.12,32,33 Exogenous expression of full-length PC1 decreased the ATP-induced, IP3R–mediated increase in cytosolic calcium in MDCK cells.32 In contrast, the simultaneous presence of PC1 and PC2 was required to elicit an IP3-induced calcium release in conditionally immortalized, plasma membrane-permeabilized human proximal-tubule epithelial cells.60 In another study PC1 was shown to inhibit calcium leak across the endoplasmic reticulum membrane.58 More recently PC1 has been proposed to play a more complex role in the regulation of calcium release from the endoplasmic reticulum through its interactions with PC2 and STIM1.33 Exogenous expression of PC1 reduces the interaction of IP3R with PC2 and enhances the interaction of STIM1 with IP3R leading to inhibition of endoplasmic reticulum calcium release, enhancement of PC2 traffic to the plasma membrane and entry of extracellular calcium, and inhibition of store operated calcium entry. According to this model, mutations in PC1 and PC2 lead to depletion of endoplasmic reticulum calcium by different mechanisms. PC1 mutations increase the interaction between PC2 and IP3R, increase calcium release from the endoplasmic reticulum, and reduce calcium influx through the plasma membrane, while PC2 mutations reduce calcium influx across the plasma membrane and enhance the interaction of PC1 and STIM1, thus inhibiting store operated calcium entry.
Despite the diversity of conclusions reached in these studies, most studies are consistent with the hypothesis that the polycystins by themselves and through their interaction with other calcium channels in the endoplasmic/sarcoplasmic reticulum function to prevent the leaking of calcium across the endoplasmic/sarcoplasmic reticulum membrane and depletion of intracellular stores, thus maintaining the amplitude of physiological calcium oscillations.34,58 Either loss or gain of function may disrupt the physiological role of the polycystins and lead to a cystic phenotype.11,72–75
4. Calcium and dysregulation of cAMP signaling in PKD
Numerous animal models of PKD show elevated levels of cAMP in the kidney 11,76–79 as well as in cholangiocytes,80 vascular smooth muscle cells,81 and choroid plexus.82 Tissue levels of cAMP are determined by the activities of membrane-bound and soluble adenylyl cyclases (ACs) and phosphodiesterases (PDEs). Both are subject to complex regulatory mechanisms, e.g, membrane bound ACs are under the positive or negative control of G protein–coupled receptors [GPCRs] and extracellular ligands.
Several hypotheses have been advanced to account for the increased levels of cAMP in PKD tissues, most directly linked to the dysregulation of intracellular calcium signaling. A prevailing hypothesis contends that reduced calcium in PKD affects both cAMP synthesis and hydrolysis. cAMP synthesis is increased in PKD due to activation of calcium-inhibitable AC6. cAMP hydrolysis is decreased in PKD due to inhibition of calcium/calmodulin-dependent PDE1.77,81,83 Levels of cyclic guanosine monophosphate also increase with limited PDE1 activity, and contribute to elevated cAMP levels by inhibiting cyclic guanosine monophosphate-inhibitable PDE3.
The importance of AC6 to PKD is demonstrated by the occurrence of a milder cystic disease in mice with a collecting duct-specific knockout of both AC6 and Pkd1 compared to knockout of Pkd1 alone.84 The induction of pronephric cysts, hydrocephalus, and body curvature in pde1a zebrafish morphants, its rescue by human PDE1A RNA and PKA inhibitors, the aggravation of the cystic phenotype in pkd2 morphants by morpholino induced depletion of PDE1, and the partial rescue of pkd2 morphants by PDE1A RNA support the role of PDE1 in PKD.85 Notably, the pool of cAMP generated in response to vasopressin is mainly catabolized by PDE1 and the accumulation of cAMP in response to vasopressin is markedly increased when intracellular calcium is reduced, mainly due to lower PDE1 activity.86,87 PDE3, which localizes to ER membranes,88,89 and catabolizes cAMP pools that control cell proliferation90–92 and cystic fibrosis transmembrane conductance regulator (CFTR) driven chloride secretion,93,94 may also be involved. Other hypothetical mechanisms linked to disrupted calcium signaling include: 1) Dysfunction of a ciliary protein complex comprising A-kinase anchoring protein 150, AC5/6, PC2, PDE4C, and PKA, such that decreased PC-2 mediated calcium entry activates AC5/6 and inhibits PDE4C.95 2) Depletion of the ER calcium stores, such that STIM1 oligomerizes, translocates to the plasma membrane, and recruits and activates AC6.71
5. Calcium and dysregulation of purinergic signaling in PKD
Renal tubular epithelial cells, like neuronal and neuroendocrine cells, exhibit constitutive ATP release and spontaneous calcium oscillations.46–48 These two processes are reciprocally regulated since ATP release occurs by calcium dependent vesicular exocytosis while the calcium oscillations are abolished by ATP scavengers or P2 receptor antagonists. ATP acts on ligand-gated P2X receptors and on G protein–coupled P2Y receptors. P2Y2 and P2×7 are expressed in collecting ducts and in MDCK cells. P2X receptors are calcium-permeable, nonselective cation channels that, when activated, increase intracellular calcium. P2Y receptors are Gq/phospholipase C (PLC)-coupled receptors; PLC cleaves phosphatidylinositol (4,5)-bisphosphate (PIP2) into IP3 and diacylglycerol. IP3 acts on IP3R to release calcium and diacylglycerol activates protein kinase C (PKC). P2 receptors require approximately 1 to 10 µM of extracellular nucleotides for full activation. High concentrations of released ATP sufficient to activate purinergic receptors likely exist close to the cell membrane. Primary and immortalized ADPKD cyst cells exhibit markedly reduced flow-sensitive ATP release.96 Furthermore, the intracellular calcium response to exogenously added ATP is moderately reduced in immortalized ADPKD cyst cells, possibly due to reduced mRNA and protein levels of P2×7 as also observed in immature, unciliated MDCK cells. ATP release in response to shear stress has also been shown to be reduced in collecting duct principal cells derived from an Oak Ridge polycystic kidney (orpkTg737) mouse model that lack normal apical cilia as compared to rescued cells exhibiting intact primary cilia.97 Knockdown of PC2 markedly blunts the ATP-dependent increase in intracellular calcium elicited by shear stress or by mechanical stretch in endothelial cells.98 It is plausible but not proven that the defective release of ATP in ADPKD derived cyst cells is a consequence of the altered intracellular calcium homeostasis in PKD.
6. Continuous, tonic effect of vasopressin in polycystic kidney disease
As a result of terrestrial adaptation, tetrapods are constantly subjected to the tonic action of vasopressin on V2 receptors. This effect is enhanced in patients with ADPKD because the circulating levels of vasopressin are often increased in patients with this disease.99–101 Plasma levels of copeptin, a surrogate for vasopressin, have also been found to be elevated in ADPKD patients compared to healthy individuals after donation of a kidney for transplantation despite higher glomerular filtration rate (GFR) in the ADPKD patients.102 Furthermore, plasma copetin levels have been shown to correlate with the severity of PKD in cross-sectional studies and with the rate of disease progression in prospective studies.103–105 The cause of the elevated levels of vasopressin and copeptin is thought to be impaired urinary-concentrating capacity, which is one of the earliest if not the earliest manifestation of ADPKD and can be detected in patients with normal glomerular filtration rate and even in children with ADPKD.99,106 The cause of this concentration defect has not been fully elucidated. It cannot be explained by reduced generation of cAMP or expression of aquaporin-2, as both cAMP level and aquaporin-2 expression are increased in animal models of PKD.77,107,108 It is interesting to note that urine-concentrating capacity is not reduced, or is even slightly increased, in Pkd1+/− mice which have very few renal cysts.68 In contrast, in a rapidly progressive PKD model, the cpk mouse, the development of polyuria occurs alongside overexpression of genes involved in urinary concentration in collecting ducts, and this overexpression precedes the development of cysts.107 Similarly, in diphenylamine- or diphenylthiazole-induced PKD models, polyuria occurs before collecting duct dilation.109,110 Additionally, renal ciliopathies in which macroscopic cysts feature less prominently, such as autosomal recessive PKD, nephronophthisis, and Bardet-Biedl syndrome, are also characterized by the early development of impaired renal concentrating capacity. Some studies have shown an abnormal intracellular distribution of aquaporin-2, suggesting that its trafficking or recycling may be altered in PKD.107,111 In the final analysis, alteration of molecular pathways within collecting duct cells paired with cystic distortion of the countercurrent mechanism, probably underlie the urine-concentrating defect.
a) Vasopressin and cAMP signaling
Studies in the last decade have shown that circulating vasopressin acting on vasopressin V2 receptors in the basolateral cell membranes and/or, less likely, urine vasopressin acting on V2 receptors in the primary cilia exert a strong modulatory effect on the development of PKD. Vasopressin is the main agonist of AC in freshly dissociated collecting ducts.112 Administration of the V2 receptor agonist DDAVP increases the renal levels of cAMP and aggravates the development of PKD rodent models orthologous to autosomal recessive PKD (ARPKD, PCK rat) and ADPKD caused by PKD1 (Pkd1RC/RC mouse) or PKD2 (Pkd2WS25/− mouse) mutations.113 Genetic elimination of circulating vasopressin markedly inhibits the development of the renal cystic disease in a rat model (PCK rat) while exogenous administration of DDAVP fully recovers the cystic phenotype.114 Vasopressin V2 receptor antagonists have been shown to inhibit the development of the disease in five rodent models orthologous to human ARPKD, ADPKD and nephronophthisis.77,108,115–117 Finally, a randomized double blind clinical trial showed that the V2 receptor antagonist tolvaptan, given over 3 years, slowed the increase in kidney volume and the decline in kidney function and lowered the frequency of disease related events in 18–50 year-old patients with ADPKD, baseline estimated creatinine clearance ≥60 ml/min, and total kidney volume ≥750 ml.3
b) Vasopressin and purinergic signaling
Physiological concentrations of vasopressin induce RyR-dependent calcium oscillations in inner medullary collecting duct (IMCD) cells, trafficking and exocytotic insertion of aquaporin-2 into the apical membrane, and release of ATP in sufficient concentrations to stimulate the luminal P2 receptors.118,119 Vasopressin V2 receptor antagonists reduce the urinary ATP concentration in mice.120 Release of ATP may initiate a negative feedback loop that limits the effect of vasopressin since ATP inhibits vasopressin induced water permeability in the collecting duct. This is due to activation of the phosphoinositide signaling pathway and a PKC and Gi-dependent mechanism.121,122 Possibly calcium-dependent activation of phospholipase A2 resulting in increased generation of arachidonic acid and prostaglandin E2 (PGE2) acting on Prostaglandin E Receptor 3 (EP3) may also play a role.123 Genetic deletion of the P2Y2 receptor results in increased expression of aquaporin-2 and urea transporters (UT)-A1 and UT-A2, and in increased urine concentration under basal conditions and following the administration of vasopressin.124 This feedback mechanism may be defective in the cystic epithelium, further up-regulating cAMP and PKA signaling which in the setting of reduced intracellular calcium leads to the proliferative and secretory phenotype of the cystic epithelium.
7. Self-perpetuating disruption of calcium signaling in ADPKD
While dysregulation of intracellular calcium homeostasis directly linked to mutated PC1 or PC2 may be responsible for the upregulation of cAMP and PKA signaling, sustained activation of PKA may result in a maladaptive response that further disrupts calcium signaling. PKA is a component of the IP3R macromolecular signaling complex, and PKA phosphorylation of IP3R increases its channel activity in planar lipid bilayers.125,126 PKA is also a component of RyR macromolecular complexes, and PKA phosphorylation of RyR promotes the dissociation of the inhibitory protein calstabin enhancing the channel activity.127,128 The physical interaction of PC2 with the RyR2 receptor has been shown to stabilize the RyR2 receptor in the closed position, and the knockdown of PC2 enhances the leak of calcium from the sarcoplasmic reticulum leading to cardiomyopathy.61 PKA also phosphorylates and enhances the channel activity of PC2.62 Dephosphorylation of PC2 is mediated by PC1 binding through the recruitment of protein phosphatase-1 alpha and unopposed cAMP stimulated hyperphosphorylation of PC2 in the absence of functional PC1 may contribute to cyst initiation in PKD1 patients62. Sustained maladaptive upregulation of cAMP and PKA signaling may lead to hyperphosphorylation of IP3R, RyR and PC2, depletion of endoplasmic reticulum calcium stores, reduced calcium transients, and development of PKD. Thus, PKD may bear some similarity to heart failure and muscle fatigue129 where persistent catecholaminergic stimulation and PKA induced hyperphosphorylation of RyR2 or RyR1 makes these channels leaky and depletes the SR calcium stores,130–132 causing heart failure or muscle fatigue, respectively.
8. Downstream effects from the dysregulation of intracellular calcium and cAMP signaling
The dysregulation of intracellular calcium and cAMP signaling may be responsible for many of the features characteristic of the cystic phenotype, reviewed in detail elsewhere.133 The cross-talk of calcium and cAMP signaling seems to be particularly important for the control of cell proliferation. cAMP exerts opposite effects on cell proliferation in different cell types. Whereas cAMP and PKA signaling inhibit cellular proliferation in normal human kidney cortical cells, they activate pro-proliferative pathways (extracellular signal-regulated kinase [ERK]) in cells derived from polycystic kidneys.134,135 Treatment of normal human kidney or murine collecting duct cells with calcium channel blockers replicates the proliferative response of the ADPKD cells to cAMP.136 Conversely, calcium-elevating treatments (calcium channel activators or ionophores) restore the antimitogenic response to cAMP of ADPKD or ARPKD cyst-derived cells. Thus, there is overwhelming evidence that intracellular calcium controls the cellular proliferative response to cAMP. Understanding the mechanisms involved is critical to understanding the pathogenesis of PKD. Yamaguchi et al showed that inhibition of calcium-dependent phosphatidylinositol 3-kinase activity and downstream inhibition of Akt in mouse collecting duct epithelial cells allows B rapidly accelerated fibrosarcoma (B-Raf) and ERK to be activated in a PKA-, Src-, and Ras-dependent manner and underlie the proliferative response to cAMP in the setting of reduced calcium.119 Consistent with these observations other studies have shown that knockdown of PC1 or PC2, reduction in intracellular calcium and downregulation of calcium calmodulin kinase II (CaMKII) inhibit Akt and activate ERK,137,138 and that knockdown of CaMKII induces pronephric cysts in zebrafish.139 Similarly, inhibition of PKA-associated proliferation by elevation of intracellular calcium in cholangiocytes derived from PCK rats has been shown to be dependent on the activation of PI3K and Akt.140 On the other hand, enhanced phosphorylation of PI3K and Akt have been found in cystic tissues from patients and rodent models of PKD.141–145 Activation of mammalian target of rapamycin (mTOR) signaling downstream of PKA through ERK-mediated phosphorylation of tuberin146,147 has been linked to transcriptional activation of aerobic glycolysis, increased levels of ATP, and together with ERK-dependent inhibition of liver kinase B1, inhibition of AMP kinase, which may further enhance mTOR signaling.148–151 Phosphorylation and inhibition of glycogen synthase kinase 3β and direct phosphorylation and stabilization of β-catenin by PKA enhance Wnt/β-catenin signaling may contribute to the development of PKD.152–155 PKA-dependent upregulation of cAMP response element binding transcription factor, paired box gene 2, and signal transducer and activator of transcription 3 also contribute to the proliferative phenotype of the cystic epithelium.156–161
Upregulation of PKA-stimulated, CFTR-mediated, chloride-driven fluid secretion plays an important role in the progression of the cystic disease.162,163 Recent studies suggest that the calcium-dependent chloride channel anoctamin-1 (ANO1) activated through stimulation of Gq-coupled purinergic receptors (P2YR) may also play an important role.164,165 ANO1 is strongly expressed in human ADPKD kidneys. Inhibitors of anoctamin ion channels and knockdown of ANO1 inhibit forskolin-driven MDCK or metanephric organ culture cystogenesis. Possibly, upregulation of CFTR- and ANO1-driven chloride secretion act synergistically downstream from PKA activation since forskolin stimulates the translocation of ANO1 to the apical membrane and PKA phosphorylation enhances the sensitivity of the IP3R to IP3. It is also possible that the relative importance of CFTR- and ANO1-driven chloride secretion in PKD depends on the animal species as it does in cystic fibrosis.166
9. In vivo studies addressing the role of calcium in Polycystic Kidney Disease
The literature reviewed here provides strong support to the prevailing belief that disruption of intracellular calcium signaling plays a central role in the pathogenesis of ADPKD. This view is supported by an increasing number of in vivo studies that have focused on the ability of calcium aimed interventions to modify the course of PKD as summarized in Table 2.38,167–171,138,172–176
Treatments that increase intracellular calcium have in general shown a protective effect. Cyclic AMP-induced cystogenesis in metanephric organ culture is inhibited by a calcium ionophore.175 Although the knockout of TRPV4 does not induce a cystic phenotype, TRPV4 activators significantly alleviate cystic disease development in PCK rats.173,176 The administration of triptolide, a drug that activates the PC2 channel by a poorly understood mechanism, has an inhibitory effect on the development of PKD in Pkd1−/− embryos and in conditional and kidney specific Pkd1 knockout mice.167,168,172 Calcimimetic agents have a beneficial effect in Cy/+ Han:SPRD rats and pcy mice,170,174 but no significant effect (except for reduced fibrosis) in PCK rats and Pkd2WS25/− mice, possibly because the potential beneficial effect of the drug was offset by marked hypocalcemia.171,177 On the other hand, interventions that lower intracellular calcium aggravate the development of PKD. The administration of verapamil aggravated cyst development in Cy/+ Han:SPRD rats._ENREF_134169 CaV1.2 zebrafish morphants have dilated pronephric ducts.138 Lentiviral transfection of CaV1.2 shRNA aggravates the cystic phenotype in Pkd1+/− mice.138
Conclusions
ADPKD, a leading cause of end-stage kidney disease, is caused by mutations to PKD1 or PKD2 encoding PC1 and PC2, respectively. The function of the polycystins and the molecular mechanisms responsible for the development of the disease are not well understood. Substantial evidence indicates that a reduction of functional PC1 or PC2 at the primary cilia and/or the endoplasmic reticulum directly disrupts intracellular calcium signaling and indirectly disrupts calcium regulated cAMP and purinergic signaling. This review presents a hypothetical model where dysregulated metabolism of cAMP and purinergic signaling increase the sensitivity of principal cells in collecting ducts and of tubular epithelial cells in the distal nephron to the constant tonic action of vasopressin, further enhancing the disruption of calcium signaling initiated by mutations to PC1 or PC2 and activating downstream signaling pathways responsible for impaired tubulogenesis, cell proliferation, increased fluid secretion and interstitial inflammation.
Table 3.
Articles examining the role of calcium in Polycystic Kidney Disease in vivo.
| Reference | Model system | Findings |
|---|---|---|
| Leuenroth 2007167 | Pkd1 KO mice. Triptolide. | Pkd2−/− epithelial cells are less susceptible to triptolide induced cell death. Triptolide binds to PC2, induces IP3R dependent Ca2+ release in Pkd1+/− and Pkd1−/− but not in Pkd2−/− cells, increases p21 expression and arrests growth of Pkd1−/− kidney cells. Treatment of pregnant mice from E10.5 lowers the cyst burden of Pkd1/− embryonic kidneys. |
| Kottgen 200838 | Xenopus oocytes, MDCK and HEK cells.Trpv4 and Pkd2 mutant mice. | Knockdown of Trpv4 (lentiviral siRNA) abolishes flow induced Ca2+ transients. Trpv4 morpholinos by themselves or in combination with low concentrations of pkd2 morpholinos do not induce cyst formation in zebrafish pronephros. Trpv4-deficient mice do not develop cysts. |
| Leuenroth 2008168 | Pkd1flox/−;Ksp-Cre mice. Triptolide. | Treatment with triptolide from P1 through P8 lowers kidney weight, cyst burden, epithelial cell proliferation, and BUN levels at postnatal day 8. Treatment from P8 through P12 achieves a slight but significant reduction in the number of cysts. |
| Nagao 2008169 | Cy/+ Han:SPRD rats. Verapamil. | Verapamil increases B-Raf and ERK phosphorylation, tubular epithelial cell proliferation and apoptosis, kidney weight and cyst burden. |
| Gattone 2009170 | Cy/+ Han:SPRD rats. Calcimimetic R-568, high calcium or both. | R-568 and to a lesser extent high calcium lower kidney volume, cyst burden, fibrosis and serum BUN at 38, but not at 34 weeks of age. |
| Wang 2009171 | PCK rats and Pkd2WS25/− mice. Calcimimetic R -568. | No effect on kidney weight, cyst volume or plasma BUN. Significant reduction in renal interstitial fibrosis in PCK rats. Potential benefit from R-568 might have been offset by marked hypocalcemia. |
| Leuenroth 2010172 | Pkd1flox/flox;Mx1Cre mice. Triptolide. | Treatment with triptolide from P16 to P35 lowers tubular epithelial cell proliferation, cyst burden and BUN levels at P35. |
| Gradilone 2010173 | WT and PCK cholangiocyte cell lines and primary cultures; bile ducts; PCK rats. TRPV4 activators. | Trpv4 stimulation increases cytosolic Ca2+, activates PI3K and Akt, inhibits ERK, and decreases cell proliferation and cyst growth in vitro. Trpv4 activation decreases cyst growth and fibrosis in vivo. |
| Chen 2011174 | Immortalized human ARPKD renal epithelial cells; pcy mice. Calcimimetic R-568. | R-568 increased intracellular Ca2+ and decreases cAMP accumulation and cell proliferation. R-568 treatment from 4 to 15 weeks decreases kidney cAMP, weight, cyst volume, and fibrosis when given. Treatment at later stages is less effective. |
| Mahajan 2012175 | Metanephric organ culture. Ca2+ ionophores and channel agonists. | A23187 and phorbol-12-myristate-13-acetate inhibit 8-Br-cAMP induced cystogenesis in metanephric organ culture. |
| Zaica 2013176 | PCK and WT SD rats. Freshly microdissected cyst walls and split-open collecting ducts. TRPV4 activator. | Lower resting cytosolic Ca2+ in cyst cells. TRPV4 activator diminishes cyst development and growth in PCK rats. |
| Jin 2014138 | Zebrafish; WT and Pkd1+/−mice. Transfection with CaV1.2 shRNA-lentivirus. | Cav1.2 zebrafish morphants have dilated pronephric ducts. Lentiviral CaV1.2 shRNA transfection aggravates the cystic phenotype in Pkd1+/−mice. |
Key points.
Polycystin 1 and polycystin 2 in the primary cilia and endoplasmic reticulum regulate intracellular calcium signaling.
In Polycystic Kidney Disease, reduced intracellular calcium enhances the generation and inhibits the destruction of cAMP, and blunts the release of ATP and purinergic signaling.
Altered cAMP metabolism and purinergic signaling in collecting duct principal cells and distal nephron epithelial cells markedly increases the sensitivity of these cells to the constant tonic effects of vasopressin.
Undefined cellular mechanisms and/or disruption of medullary architecture by cysts interfere with urine concentration and increase circulating levels of vasopressin.
Vasopressin and cAMP driven activation of protein kinase A enhances the phosphorylation of polycystin-2, ryanodine receptors and inositol 1,4,5-trisphosphate receptors, increases the leakage of calcium across the endoplasmic reticulum membrane, and further disrupts intracellular calcium signaling.
The reduction in intracellular calcium that results from PKD1 or PKD2 mutations causes a striking switch in the cellular response to cAMP from suppression to marked stimulation of proliferation.
A better appreciation of the mechanisms responsible for this phenotypic switch is critical for the understanding of Polycystic Kidney Disease.
Preclinical trials targeting calcium or cAMP signaling and clinical trials targeting cAMP signaling have provided encouraging results, providing support for their pathophysiological relevance in Polycystic Kidney Disease.
ACKNOWLEDGMENTS
Supported in part by grants from the National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health (DK044863 and DK090728).
Biographies
Fouad T. Chebib, MD, is a nephrology fellow at Mayo Clinic, Rochester, Minnesota. He received his medical degree from the University of Balamand, Lebanon. He spent two years at Beth Israel Deaconess Medical center in Boston, MA in the molecular renal division under the mentorship of Dr. Seth Alper. He trained for internal medicine residency at Saint Elizabeth’s medical center, Boston, MA. He has special interest in polycystic kidney disease (PKD) and is currently a research fellow at the Mayo Translational PKD Center under the mentorship of Dr. Vicente Torres. He is studying calcium signaling in ADPKD and its potential therapeutic implications.
Caroline R. Sussman, PhD, is Assistant Professor of Physiology in the Division of Nephrology and Hypertension, Mayo Clinic, Rochester, Minnesota. She received her PhD in Physiology from the University of Connecticut in 1997, which was followed by postdoctoral training at Yale University in New Haven, CT and Case Western Reserve University in Cleveland, OH. Her research has included renal transport, kidney and central nervous system development, and Epidermal Growth Factor Receptors. Her current research focuses on cAMP and calcium signaling pathways, and phosphodiesterases in PKD using zebrafish and mammalian models.
Xiaofang Wang, MD, is a Research Associate in the Division of Nephrology and Hypertension, Mayo Clinic, Rochester, Minnesota. She received her medical degree from Shanxi Medical University in 1986 and a medical science degree from Peking Union Medical College in 1991. She was an Assistant Professor of Pathophysiology, at the Peking Union Medical College and the Chinese Academy of Medical Sciences during 1991–1994 and a research fellow at the Mayo clinic during 1994–2000. Her main research interest is PKD and has worked in close association with Dr. Vicente Torres for the last fourteen years.
Peter C. Harris, PhD, is Professor of Biochemistry/Molecular Biology and Medicine in the Division of Nephrology and Hypertension, Mayo Clinic, Rochester, Minnesota. The main focus of Dr. Harris’ career has been the study of PKD. He was head of the European Consortium that identified the PKD1 gene in 1994, and in 2002 he identified the gene for ARPKD. Genes for Meckel syndrome were identified in 2006 and 2011. He has published over 200 peer-reviewed articles and reviews mainly on PKD, and hosts the ADPKD Mutation Database at Mayo. In 2003, he received the inaugural Lillian Jean Kaplan Prize for PKD and in 2008 the Homer Smith Award from the American Society of Nephrology.
Vicente E. Torres, MD, PhD, is Professor of Medicine in the Division of Nephrology and Hypertension, Mayo Clinic, Rochester, Minnesota. He has served as chair of the Division of Nephrology and Hypertension and Director of the NIH funded Mayo Kidney Disease Research Training Grant, and currently directs the Mayo Translational PKD Center. His research has focused on the study of PKD. He has published over 250 peer-reviewed articles mainly on PKD and is a principal investigator for the NIH funded CRISP imaging and HALT-PKD clinical trial consortia, and for Industry funded clinical trials of vasopressin V2 receptor antagonists. In 2007, he was awarded the Lillian Jean Kaplan International Prize for Advancement in the Understanding of Polycystic Kidney Disease.
References
- 1.Torres VE, Harris PC, Pirson Y. Autosomal dominant polycystic kidney disease. Lancet. 2007;369:1287–1301. doi: 10.1016/S0140-6736(07)60601-1. [DOI] [PubMed] [Google Scholar]
- 2.Harris PC, Torres VE. Polycystic Kidney Disease. Annu Rev Med. 2009;60:321–327. doi: 10.1146/annurev.med.60.101707.125712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Torres VE, et al. Tolvaptan in Patients with Autosomal Dominant Polycystic Kidney Disease. N Engl J Med. 2012;367:2407–2418. doi: 10.1056/NEJMoa1205511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Caroli A, et al. Effect of long acting somatostatin analogue on kidney and cyst growth in autosomal dominant polycystic kidney disease (ALADIN): a randomised, placebo-controlled, multicentre trial. Lancet. 2013;382:1485–1495. doi: 10.1016/S0140-6736(13)61407-5. [DOI] [PubMed] [Google Scholar]
- 5.Schrier RS, et al. Angiotensin Blockade, Blood Pressure and Autosomal Dominant Polycystic Kidney Disease. N Engl J Med. 2014 Nov; doi: 10.1056/NEJMoa1402686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Qian F, et al. PKD1 interacts with PKD2 through a probable coiled-coil domain. Nature Genet. 1997;16:179–183. doi: 10.1038/ng0697-179. [DOI] [PubMed] [Google Scholar]
- 7.Arac D, et al. A novel evolutionarily conserved domain of cell-adhesion GPCRs mediates autoproteolysis. Embo J. 2012;31:1364–1378. doi: 10.1038/emboj.2012.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yu S, et al. Essential role of cleavage of Polycystin-1 at G protein-coupled receptor proteolytic site for kidney tubular structure. Proc Natl Acad Sci U S A. 2007;104:18688–18693. doi: 10.1073/pnas.0708217104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Qian F, et al. Cleavage of polycystin-1 requires the receptor for egg jelly domain and is disrupted by human autosomal-dominant polycystic kidney disease 1-associated mutations. Proc Natl Acad Sci U S A. 2002;99:16981–16986. doi: 10.1073/pnas.252484899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hogan M, et al. Characterization of PKD protein-positive exosome-like vesicles. J Am Soc Nephrol. 2009;20:278–288. doi: 10.1681/ASN.2008060564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hopp K, et al. Functional polycystin-1 dosage governs autosomal dominant polycystic kidney disease severity. J Clin Invest. 2012;122:4257–4273. doi: 10.1172/JCI64313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Woodward OM, et al. Identification of a polycystin-1 cleavage product, P100, that regulates store operated Ca entry through interactions with STIM1. PLoS ONE. 2010;5:e12305. doi: 10.1371/journal.pone.0012305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chauvet V, et al. Mechanical stimuli induce cleavage and nuclear translocation of the polycystin-1 C terminus. J Clin Invest. 2004;114:1433–1443. doi: 10.1172/JCI21753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bertuccio CA, et al. Polycystin-1 C-terminal cleavage is modulated by polycystin-2 expression. J Biol Chem. 2009;284:21011–21026. doi: 10.1074/jbc.M109.017756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Merrick D, et al. The gamma-secretase cleavage product of polycystin-1 regulates TCF and CHOP-mediated transcriptional activation through a p300-dependent mechanism. Dev Cell. 2012;22:197–210. doi: 10.1016/j.devcel.2011.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Low SH, et al. Polycystin-1, STAT6, and P100 function in a pathway that transduces ciliary mechanosensation and is activated in polycystic kidney disease. Dev Cell. 2006;10:57–69. doi: 10.1016/j.devcel.2005.12.005. [DOI] [PubMed] [Google Scholar]
- 17.Sutters M, et al. Polycystin-1 transforms the cAMP growth-responsive phenotype of M-1 cells. Kidney Int. 2001;60:484–494. doi: 10.1046/j.1523-1755.2001.060002484.x. [DOI] [PubMed] [Google Scholar]
- 18.Vandorpe DH, et al. Cation channel regulation by COOH-terminal cytoplasmic tail of polycystin-1: mutational and functional analysis. Physiol Genomics. 2002;8:87–98. doi: 10.1152/physiolgenomics.00092.2001. [DOI] [PubMed] [Google Scholar]
- 19.Aguiari G, et al. Expression of polycystin-1 C-terminal fragment enhances the ATP-induced Ca2+ release in human kidney cells. Biochem Biophys Res Commun. 2003;301:657–664. doi: 10.1016/s0006-291x(02)03011-5. [DOI] [PubMed] [Google Scholar]
- 20.Wildman SS, et al. The isolated polycystin-1 cytoplasmic COOH terminus prolongs ATP-stimulated Cl conductance through increased Ca2+ entry. Am J Physiol Renal Physiol. 2003;285:F1168–F1178. doi: 10.1152/ajprenal.00171.2003. [DOI] [PubMed] [Google Scholar]
- 21.Hooper KM, Unwin RJ, Sutters M. The isolated C-terminus of polycystin-1 promotes increased ATP-stimulated chloride secretion in a collecting duct cell line. Clin Sci (Lond) 2003;104:217–221. doi: 10.1042/CS20020239. [DOI] [PubMed] [Google Scholar]
- 22.Puri S, et al. Polycystin-1 activates the calcineurin/NFAT (nuclear factor of activated T-cells) signaling pathway. J Biol Chem. 2004;279:55455–55464. doi: 10.1074/jbc.M402905200. [DOI] [PubMed] [Google Scholar]
- 23.Manzati E, et al. The cytoplasmic C-terminus of polycystin-1 increases cell proliferation in kidney epithelial cells through serum-activated and Ca(2+)-dependent pathway(s) Experimental Cell Research. 2005;304:391–406. doi: 10.1016/j.yexcr.2004.10.023. [DOI] [PubMed] [Google Scholar]
- 24.Xu C, et al. Human ADPKD primary cyst epithelial cells with a novel, single codon deletion in the PKD1 gene exhibit defective ciliary polycystin localization and loss of flow-induced Ca2+ signaling. Am J Physiol Renal Physiol. 2007;292:F930–F945. doi: 10.1152/ajprenal.00285.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gonzalez-Perrett S, et al. Polycystin-2, the protein mutated in autosomal dominant polycystic kidney disease (ADPKD), is a Ca2+-permeable non-selective cation channel. Proc Natl Acad Sci USA. 2001;98:1182–1187. doi: 10.1073/pnas.98.3.1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vassilev PM, et al. Polycystin-2 is a novel cation channel implicated in defective intracellular Ca2+ homeostasis in polycystic kidney disease. Biochem Biophys Res Commun. 2001;282:341–350. doi: 10.1006/bbrc.2001.4554. [DOI] [PubMed] [Google Scholar]
- 27.Yu Y, et al. Structural and molecular basis of the assembly of the TRPP2/PKD1 complex. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:11558–11563. doi: 10.1073/pnas.0903684106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Feng S, Rodat-Despoix L, Delmas P, Ong AC. A single amino acid residue constitutes the third dimerization domain essential for the assembly and function of the tetrameric polycystin-2 (TRPP2) channel. J Biol Chem. 2011;286:18994–19000. doi: 10.1074/jbc.M110.192286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hanaoka K, et al. Co-assembly of polycystin-1 and-2 produces unique cation-permeable currents. Nature. 2000;408:990–994. doi: 10.1038/35050128. [DOI] [PubMed] [Google Scholar]
- 30.Kim H, et al. Ciliary membrane proteins traffic through the Golgi via a Rabep1/GGA1/Arl3-dependent mechanism. Nature communications. 2014;5:5482. doi: 10.1038/ncomms6482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gainullin VG, Hopp K, CJ W, Hommerding CJ, PC H. Polycystin-1 maturation requires polcystiv-2 in a dose-dependent manner. Journal of Clinical Investigation. 2014 doi: 10.1172/JCI76972. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li Y, et al. Polycystin-1 interacts with inositol 1,4,5-trisphosphate receptor to modulate intracellular Ca2+ signaling with implications for polycystic kidney disease. J Biol Chem. 2009;284:36431–36441. doi: 10.1074/jbc.M109.068916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Santoso NG, Cebotaru L, Guggino WB. Polycystin-1, 2, and STIM1 interact with IP(3)R to modulate ER Ca release through the PI3K/Akt pathway. Cell Physiol Biochem. 2011;27:715–726. doi: 10.1159/000330080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Anyatonwu GI, Estrada M, Tian X, Somlo S, Ehrlich BE. Regulation of ryanodine receptor-dependent calcium signaling by polycystin-2. Proc Natl Acad Sci USA. 2007;104:6454–6459. doi: 10.1073/pnas.0610324104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tsiokas L, et al. Specific association of the gene product of PKD2 with the TRPC1 channel. Proc Natl Acad Sci U S A. 1999;96:3934–3939. doi: 10.1073/pnas.96.7.3934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bai CX, et al. Formation of a new receptor-operated channel by heteromeric assembly of TRPP2 and TRPC1 subunits. EMBO Rep. 2008;9:472–479. doi: 10.1038/embor.2008.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Du J, Ding M, Sours-Brothers S, Graham S, Ma R. Mediation of angiotensin II-induced Ca2+ signaling by polycystin 2 in glomerular mesangial cells. Am J Physiol Renal Physiol. 2008;294:F909–F918. doi: 10.1152/ajprenal.00606.2007. [DOI] [PubMed] [Google Scholar]
- 38.Kottgen M, et al. TRPP2 and TRPV4 form a polymodal sensory channel complex. J Cell Biol. 2008;182:437–447. doi: 10.1083/jcb.200805124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Miyagi K, et al. A pathogenic C terminus-truncated polycystin-2 mutant enhances receptor-activated Ca2+ entry via association with TRPC3 and TRPC7. J Biol Chem. 2009;284:34400–34412. doi: 10.1074/jbc.M109.015149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Barr MM, Sternberg PW. A polycystic kidney-disease gene homologue required for male mating behaviour. Nature. 1999;401:386–389. doi: 10.1038/43913. [DOI] [PubMed] [Google Scholar]
- 41.Haycraft CJ, Swoboda P, Taulman PD, Thomas JH, Yoder BK. TheC. elegans homolog of the murine cystic kidney disease gene Tg737 functions in a ciliogenic pathway and is disrupted in osm-5 mutant worms. Development. 2001;128:1493–1505. doi: 10.1242/dev.128.9.1493. [DOI] [PubMed] [Google Scholar]
- 42.Praetorius HA, Spring KR. Bending the MDCK cell primary cilium increases intracellular calcium. J Membr Biol. 2001;184:71–79. doi: 10.1007/s00232-001-0075-4. [DOI] [PubMed] [Google Scholar]
- 43.Nauli SM, et al. Polycystins 1 and 2 mediate mechanosensation in the primary cilium of kidney cells. Nat Genet. 2003;33:129–137. doi: 10.1038/ng1076. [DOI] [PubMed] [Google Scholar]
- 44.Delling M, DeCaen PG, Doerner JF, Febvay S, Clapham DE. Primary cilia are specialized calcium signalling organelles. Nature. 2013;504:311–314. doi: 10.1038/nature12833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jin X, et al. Cilioplasm is a cellular compartment for calcium signaling in response to mechanical and chemical stimuli. Cell Mol Life Sci. 2014;71:2165–2178. doi: 10.1007/s00018-013-1483-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Praetorius HA, Leipziger J. Intrarenal purinergic signaling in the control of renal tubular transport. Annual Review of Physiology. 2010;72:377–393. doi: 10.1146/annurev-physiol-021909-135825. [DOI] [PubMed] [Google Scholar]
- 47.Praetorius HA, Leipziger J. Released nucleotides amplify the cilium-dependent, flow-induced [Ca2+]i response in MDCK cells. Acta physiologica. 2009;197:241–251. doi: 10.1111/j.1748-1716.2009.02002.x. [DOI] [PubMed] [Google Scholar]
- 48.Praetorius HA, Leipziger J. Primary cilium-dependent sensing of urinary flow and paracrine purinergic signaling. Seminars in Cell and Developmental Biology. 2013;24:3–10. doi: 10.1016/j.semcdb.2012.10.003. [DOI] [PubMed] [Google Scholar]
- 49.Rodat-Despoix L, Hao J, Dandonneau M, Delmas P. Shear stress-induced Ca(2)(+) mobilization in MDCK cells is ATP dependent, no matter the primary cilium. Cell Calcium. 2013;53:327–337. doi: 10.1016/j.ceca.2013.02.002. [DOI] [PubMed] [Google Scholar]
- 50.Koulen P, et al. Polycystin-2 is an intracellular calcium release channel. Nat Cell Biol. 2002;4:191–197. doi: 10.1038/ncb754. [DOI] [PubMed] [Google Scholar]
- 51.Qian Q, et al. Pkd2 haploinsufficiency alters intracellular calcium in vascular smooth muscle cells. Hum Mol Genet. 2003;12:1875–1880. doi: 10.1093/hmg/ddg190. [DOI] [PubMed] [Google Scholar]
- 52.Cai Y, et al. Calcium dependence of polycystin-2 channel activity is modulated by phosphorylation at Ser812. J Biol Chem. 2004;279:19987–19995. doi: 10.1074/jbc.M312031200. [DOI] [PubMed] [Google Scholar]
- 53.Aguiari G, et al. Deficiency of polycystin-2 reduces Ca2+ channel activity and cell proliferation in ADPKD lymphoblastoid cells. Faseb J. 2004;18:884–886. doi: 10.1096/fj.03-0687fje. [DOI] [PubMed] [Google Scholar]
- 54.Gao Z, Joseph E, Ruden DM, Lu X. Drosophila Pkd2 is haploid-insufficient for mediating optimal smooth muscle contractility. J Biol Chem. 2004;279:14225–14231. doi: 10.1074/jbc.M312223200. [DOI] [PubMed] [Google Scholar]
- 55.Koulen P, et al. Polycystin-2 accelerates Ca2+ release from intracellular stores in Caenorhabditis elegans. Cell Calcium. 2005;37:593–601. doi: 10.1016/j.ceca.2005.03.003. [DOI] [PubMed] [Google Scholar]
- 56.Li Y, Wright JM, Qian F, Germino GG, Guggino WB. Polycystin 2 interacts with type I inositol 1,4,5-trisphosphate receptor to modulate intracellular Ca2+ signaling. J Biol Chem. 2005;280:41298–41306. doi: 10.1074/jbc.M510082200. [DOI] [PubMed] [Google Scholar]
- 57.Geng L, et al. Syntaxin 5 regulates the endoplasmic reticulum channel-release properties of polycystin-2. Proc Natl Acad Sci U S A. 2008;105:15920–15925. doi: 10.1073/pnas.0805062105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Weber KH, et al. Heterologous expression of polycystin-1 inhibits endoplasmic reticulum calcium leak in stably transfected MDCK cells. Am J Physiol Renal Physiol. 2008;294:F1279–F1286. doi: 10.1152/ajprenal.00348.2007. [DOI] [PubMed] [Google Scholar]
- 59.Wegierski T, et al. TRPP2 channels regulate apoptosis through the Ca(2+) concentration in the endoplasmic reticulum. EMBO J. 2009;28:490–499. doi: 10.1038/emboj.2008.307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mekahli D, et al. Polycystin-1 and polycystin-2 are both required to amplify inositol-trisphosphate-induced Ca2+ release. Cell calcium. 2012;51:452–458. doi: 10.1016/j.ceca.2012.03.002. [DOI] [PubMed] [Google Scholar]
- 61.Paavola J, et al. Polycystin-2 mutations lead to impaired calcium cycling in the heart and predispose to dilated cardiomyopathy. Journal of molecular and cellular cardiology. 2013;58:199–208. doi: 10.1016/j.yjmcc.2013.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Streets AJ, Wessely O, Peters DJ, Ong AC. Hyperphosphorylation of polycystin-2 at a critical residue in disease reveals an essential role for polycystin-1-regulated dephosphorylation. Human Molecular Genetics. 2013;22:1924–1939. doi: 10.1093/hmg/ddt031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Plotnikova OV, Pugacheva EN, Golemis EA. Aurora A kinase activity influences calcium signaling in kidney cells. J Cell Biol. 2011;193:1021–1032. doi: 10.1083/jcb.201012061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hooper K, et al. Expression of polycystin-1 enhances endoplasmic reticulum calcium uptake and decreases capacitative calcium entry in ATP-stimulated MDCK cells. Am J Physiol Renal Physiol. 2005;289:F521–F530. doi: 10.1152/ajprenal.00355.2004. [DOI] [PubMed] [Google Scholar]
- 65.Morel N, et al. PKD1 haploinsufficiency is associated with altered vascular reactivity and abnormal calcium signaling in the mouse aorta. Pflugers Arch. 2009;457:845–856. doi: 10.1007/s00424-008-0561-y. [DOI] [PubMed] [Google Scholar]
- 66.Chang MY, et al. Haploinsufficiency of Pkd2 is associated with increased tubular cell proliferation and interstitial fibrosis in two murine Pkd2 models. Nephrol Dial Transplant. 2006;21:2078–2084. doi: 10.1093/ndt/gfl150. [DOI] [PubMed] [Google Scholar]
- 67.Parker E, et al. Hyperproliferation of PKD1 cystic cells is induced by insulin-like growth factor-1 activation of the Ras/Raf signalling system. Kidney Int. 2007;72:157–165. doi: 10.1038/sj.ki.5002229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ahrabi AK, et al. PKD1 haploinsufficiency causes a syndrome of inappropriate antidiuresis in mice. J Am Soc Nephrol. 2007;18:1740–1753. doi: 10.1681/ASN.2006010052. [DOI] [PubMed] [Google Scholar]
- 69.Nauli SM, et al. Loss of polycystin-1 in human cyst-lining epithelia leads to ciliary dysfunction. J Am Soc Nephrol. 2006;17:1015–1025. doi: 10.1681/ASN.2005080830. [DOI] [PubMed] [Google Scholar]
- 70.Kuo IY, et al. Cyst formation following disruption of intracellular calcium signaling. Proc Natl Acad Sci USA. 2014;111:14283–14288. doi: 10.1073/pnas.1412323111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Spirli C, et al. Altered store operated calcium entry increases cyclic 3’,5’-adenosine monophosphate production and extracellular signal-regulated kinases 1 and 2 phosphorylation in polycystin-2-defective cholangiocytes. Hepatology. 2012;55:856–868. doi: 10.1002/hep.24723. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 72.Lantinga-van Leeuwen IS, et al. Lowering of Pkd1 expression is sufficient to cause polycystic kidney disease. Hum Mol Genet. 2004;13:3069–3077. doi: 10.1093/hmg/ddh336. [DOI] [PubMed] [Google Scholar]
- 73.Pritchard L, et al. A human PKD1 transgene generates functional polycystin-1 in mice and is associated with a cystic phenotype. Hum Mol Genet. 2000;9:2617–2627. doi: 10.1093/hmg/9.18.2617. [DOI] [PubMed] [Google Scholar]
- 74.Thivierge C, et al. Overexpression of PKD1 causes polycystic kidney disease. Mol Cell Biol. 2006;26:1538–1548. doi: 10.1128/MCB.26.4.1538-1548.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Park EY, et al. Cyst formation kidney via B-RAF signaling in the PKD2 transgenic mice. J Biol Chem. 2009;284:7214–7222. doi: 10.1074/jbc.M805890200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Yamaguchi T, Nagao S, Kasahara M, Takahashi H, Grantham J. Renal accumulation and excretion of cyclic adenosine monophosphate in a murine model of slowly progressive polycystic kidney disease. Am J Kidney Dis. 1997;30:703–709. doi: 10.1016/s0272-6386(97)90496-0. [DOI] [PubMed] [Google Scholar]
- 77.Gattone VH, Wang X, Harris PC, Torres VE. Inhibition of renal cystic disease development and progression by a vasopressin V2 receptor antagonist. Nature Med. 2003;9:1323–1326. doi: 10.1038/nm935. [DOI] [PubMed] [Google Scholar]
- 78.Starremans PG, et al. A mouse model for polycystic kidney disease through a somatic in-frame deletion in the 5’ end of Pkd1. Kidney Int. 2008;73:1394–1405. doi: 10.1038/ki.2008.111. [DOI] [PubMed] [Google Scholar]
- 79.Smith LA, et al. Development of polycystic kidney disease in juvenile cystic kidney mice: insights into pathogenesis, ciliary abnormalities, and common features with human disease. J Am Soc Nephrol. 2006;17:2821–2831. doi: 10.1681/ASN.2006020136. [DOI] [PubMed] [Google Scholar]
- 80.Masyuk TV, Masyuk AI, Torres VE, Harris PC, Larusso NF. Octreotide inhibits hepatic cystogenesis in a rodent model of polycystic liver disease by reducing cholangiocyte adenosine 3’,5’-cyclic monophosphate. Gastroenterology. 2007;132:1104–1116. doi: 10.1053/j.gastro.2006.12.039. [DOI] [PubMed] [Google Scholar]
- 81.Kip SN, et al. [Ca2+]i reduction increases cellular proliferation and apoptosis in vascular smooth muscle cells: Relevance to the ADPKD phenotype. Circ Res. 2005;96:873–880. doi: 10.1161/01.RES.0000163278.68142.8a. [DOI] [PubMed] [Google Scholar]
- 82.Banizs B, et al. Altered pHi regulation and Na+/HCO3 transporter activity in choroid plexus of cilia-defective Tg737orpk mutant mouse. Am J Physiol Cell Physiol. 2007;292:C1409–C1416. doi: 10.1152/ajpcell.00408.2006. [DOI] [PubMed] [Google Scholar]
- 83.Wang X, Ward CJ, Harris PC, Torres VE. Cyclic nucleotide signaling in polycystic kidney disease. Kidney Int. 2010;77:129–140. doi: 10.1038/ki.2009.438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Rees S, et al. Adenylyl cyclase 6 deficiency ameliorates polycystic kidney disease. J Am Soc Nephrol. 2014;25:232–237. doi: 10.1681/ASN.2013010077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Sussman CR, et al. Phosphodiesterase 1A Modulates Cystogenesis in Zebrafish. J Am Soc Nephrol. 2014;25:2222–2230. doi: 10.1681/ASN.2013040421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Yamaki M, McIntyre S, Rassier ME, Schwartz JH, Dousa TP. Cyclic 3’,5’-nucleotide diesterases in dynamics of cAMP and cGMP in rat collecting duct cells. Am J Physiol. 1992;262:F957–F964. doi: 10.1152/ajprenal.1992.262.6.F957. [DOI] [PubMed] [Google Scholar]
- 87.Kusano E, et al. Effects of calcium on the vasopressin-sensitive cAMP metabolism in medullary tubules. Am J Physiol. 1985;249:F956–F966. doi: 10.1152/ajprenal.1985.249.6.F956. [DOI] [PubMed] [Google Scholar]
- 88.Kenan Y, Murata T, Shakur Y, Degerman E, Manganiello VC. Functions of the N-terminal region of cyclic nucleotide phosphodiesterase 3 (PDE 3) isoforms. Journal of Biological Chemistry. 2000;275:12331–12338. doi: 10.1074/jbc.275.16.12331. [DOI] [PubMed] [Google Scholar]
- 89.Shakur Y, et al. Membrane localization of cyclic nucleotide phosphodiesterase 3 (PDE3). Two N-terminal domains are required for the efficient targeting to, and association of, PDE3 with endoplasmic reticulum. Journal of Biological Chemistry. 2000;275:38749–38761. doi: 10.1074/jbc.M001734200. [DOI] [PubMed] [Google Scholar]
- 90.Grantham JJ. Renal cell proliferation and the two faces of cyclic adenosine monophosphate. J Lab Clin Med. 1997;130:459–460. doi: 10.1016/s0022-2143(97)90121-9. [DOI] [PubMed] [Google Scholar]
- 91.Matousovic K, Tsuboi Y, Walker H, Grande JP, Dousa TP. Inhibitors of cyclic nucleotide phosphodiesterase isozymes block renal tubular cell proliferation induced by folic acid. J Lab Clin Med. 1997;130:487–495. doi: 10.1016/s0022-2143(97)90125-6. [DOI] [PubMed] [Google Scholar]
- 92.Cheng J, et al. Lixazinone stimulates mitogenesis of Madin-Darby canine kidney cells. Exp Biol Med (Maywood) 2006;231:288–295. doi: 10.1177/153537020623100308. [DOI] [PubMed] [Google Scholar]
- 93.De Jonge HR, et al. cGMP inhibition of type 3 phosphodiesterase is the major mechanism by which C-type natriuretic peptide activates CFTR in the shark rectal gland. Am J Physiol Cell Physiol. 2014;306:C343–C353. doi: 10.1152/ajpcell.00326.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Penmatsa H, et al. Compartmentalized cyclic adenosine 3’,5’-monophosphate at the plasma membrane clusters PDE3A and cystic fibrosis transmembrane conductance regulator into microdomains. Molecular Biology of the Cell. 2010;21:1097–1110. doi: 10.1091/mbc.E09-08-0655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Choi YH, et al. Polycystin-2 and phosphodiesterase 4C are components of a ciliary A-kinase anchoring protein complex that is disrupted in cystic kidney diseases. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:10679–10684. doi: 10.1073/pnas.1016214108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Xu C, et al. Attenuated flow-induced ATP release contributes to absence of flow-sensitive, purinergic Cai2+ signaling in human ADPKD cyst epithelial cells. American journal of physiology. Renal physiology. 2009;296:F1464–F1476. doi: 10.1152/ajprenal.90542.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Hovater MB, et al. Loss of apical monocilia on collecting duct principal cells impairs ATP secretion across the apical cell surface and ATP-dependent and flow-induced calcium signals. Purinergic Signal. 2008;4:155–170. doi: 10.1007/s11302-007-9072-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.AbouAlaiwi WA, et al. Ciliary polycystin-2 is a mechanosensitive calcium channel involved in nitric oxide signaling cascades. Circ Res. 2009;104:860–869. doi: 10.1161/CIRCRESAHA.108.192765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Seeman T, et al. Renal concentrating capacity is linked to blood pressure in children with autosomal dominant polycystic kidney disease. Physiol Res. 2004;53:629–634. [PubMed] [Google Scholar]
- 100.Danielsen H, et al. Expansion of extracellular volume in early polycystic kidney disease. Acta Med Scand. 1986;219:399–405. doi: 10.1111/j.0954-6820.1986.tb03330.x. [DOI] [PubMed] [Google Scholar]
- 101.Michalski A, Grzeszczak W. The effect of hypervolemia on electrolyte level and and level of volume regulating hormones in patients with autosomal dominant polycystic kidney disease. Pol Arch Med Wewn. 1996;96:329–343. [PubMed] [Google Scholar]
- 102.Zittema D, et al. Kidney function and plasma copeptin levels in healthy kidney donors and autosomal dominant polycystic kidney disease patients. Clin J Am Soc Nephrol. 2014;9:1553–1562. doi: 10.2215/CJN.08690813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Boertien WE, et al. Relationship of Copeptin, a Surrogate Marker for Arginine Vasopressin, With Change in Total Kidney Volume and GFR Decline in Autosomal Dominant Polycystic Kidney Disease: Results From the CRISP Cohort. Am J Kidney Dis. 2013;61:420–429. doi: 10.1053/j.ajkd.2012.08.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Meijer E, et al. Copeptin, a surrogate marker of vasopressin, is associated with disease severity in autosomal dominant polycystic kidney disease. Clin J Am Soc Nephrol. 2011;6:361–368. doi: 10.2215/CJN.04560510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Boertien WE, et al. Copeptin, a surrogate marker for vasopressin, is associated with kidney function decline in subjects with autosomal dominant polycystic kidney disease. Nephrol Dial Transpl. 2012;27:4131–4137. doi: 10.1093/ndt/gfs070. [DOI] [PubMed] [Google Scholar]
- 106.Gabow P, et al. The clinical utility of renal concentrating capacity in polycystic kidney disease. Kidney Int. 1989;35:675–680. doi: 10.1038/ki.1989.38. [DOI] [PubMed] [Google Scholar]
- 107.Gattone VH, Maser RL, Tian C, Rosenberg JM, Branden MG. Developmental expression of urine concentration-associated genes and their altered expression in murine infantile-type polycystic kidney disease. Develop Gen. 1999;24:309–318. doi: 10.1002/(SICI)1520-6408(1999)24:3/4<309::AID-DVG14>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 108.Torres VE, et al. Effective treatment of an orthologous model of autosomal dominant polycystic kidney disease. Nature Med. 2004;10:363–364. doi: 10.1038/nm1004. [DOI] [PubMed] [Google Scholar]
- 109.Carone FA, Ozono S, Samma S, Kanwar YS, Oyasu R. Renal functional changes in experimental cystic disease are tubular in origin. Kidney Int. 1988;33:8–13. doi: 10.1038/ki.1988.2. [DOI] [PubMed] [Google Scholar]
- 110.Safouh M, Crocker JF, Vernier RL. Experimental cystic disease of the kidney. Sequential, functional, and morphologic studies. Lab Invest. 1970;23:392–400. [PubMed] [Google Scholar]
- 111.Hayashi M, et al. Expression and localization of the water channels in human autosomal dominant polycystic kidney disease. Nephron. 1997;75:321–326. doi: 10.1159/000189556. [DOI] [PubMed] [Google Scholar]
- 112.Yasuda G, Jeffries WB. Regulation of cAMP production in initial and terminal inner medullary collecting ducts. Kidney Int. 1998;54:80–86. doi: 10.1046/j.1523-1755.1998.00990.x. [DOI] [PubMed] [Google Scholar]
- 113.Hopp K, et al. Effects of hydration in rats and mice with polycystic kidney disease. Am J Physiol. 2015 doi: 10.1152/ajprenal.00345.2014. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Wang X, Wu Y, Ward CJ, Harris PC, Torres VE. Vasopressin directly regulates cyst growth in polycystic kidney disease. J Am Soc Nephrol. 2008;19:102–108. doi: 10.1681/ASN.2007060688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Wang X, Gattone V, 2nd, Harris PC, Torres VE. Effectiveness of vasopressin V2 receptor antagonists OPC-31260 and OPC-41061 on polycystic kidney disease development in the PCK rat. J Am Soc Nephrol. 2005;16:846–851. doi: 10.1681/ASN.2004121090. [DOI] [PubMed] [Google Scholar]
- 116.Meijer E, et al. Therapeutic potential of vasopressin V2 receptor antagonist in a mouse model for autosomal dominant polycystic kidney disease: optimal timing and dosing of the drug. Nephrol Dial Transplant. 2011;26:2445–2453. doi: 10.1093/ndt/gfr069. [DOI] [PubMed] [Google Scholar]
- 117.Hopp K, et al. Tolvaptan plus pasireotide shows enhanced efficacy in an ADPKD1 model. J Am Soc Nephrol. 2014 Jul 3; doi: 10.1681/ASN.2013121312. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Yip KP, Sham JS. Mechanisms of vasopressin-induced intracellular Ca2+ oscillations in rat inner medullary collecting duct. Am J Physiol Renal Physiol. 2011;300:F540–F548. doi: 10.1152/ajprenal.00544.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Odgaard E, Praetorius HA, Leipziger J. AVP-stimulated nucleotide secretion in perfused mouse medullary thick ascending limb and cortical collecting duct. Am J of Physiol Renal Physiol. 2009;297:F341–F349. doi: 10.1152/ajprenal.00190.2009. [DOI] [PubMed] [Google Scholar]
- 120.Rieg T, et al. Mice lacking P2Y2 receptors have salt-resistant hypertension and facilitated renal Na+ and water reabsorption. FASEB J. 2007;21:3717–3726. doi: 10.1096/fj.07-8807com. [DOI] [PubMed] [Google Scholar]
- 121.Kishore BK, Chou CL, Knepper MA. Extracellular nucleotide receptor inhibits AVP-stimulated water permeability in inner medullary collecting duct. Am J Physiol. 1995;269:F863–F869. doi: 10.1152/ajprenal.1995.269.6.F863. [DOI] [PubMed] [Google Scholar]
- 122.Teitelbaum I. Protein kinase C inhibits arginine vasopressin-stimulated cAMP accumulation via a Gi-dependent mechanism. Am J Physiol. 1993;264:F216–F220. doi: 10.1152/ajprenal.1993.264.2.F216. [DOI] [PubMed] [Google Scholar]
- 123.Welch BD, Carlson NG, Shi H, Myatt L, Kishore BK. P2Y2 receptor-stimulated release of prostaglandin E2 by rat inner medullary collecting duct preparations. Am J Physiol Renal Physiol. 2003;285:F711–F721. doi: 10.1152/ajprenal.00096.2003. [DOI] [PubMed] [Google Scholar]
- 124.Zhang Y, et al. Potential role of purinergic signaling in urinary concentration in inner medulla: insights from P2Y2 receptor gene knockout mice. Am J Physiol Renal Physiol. 2008;295:F1715–F1724. doi: 10.1152/ajprenal.90311.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.DeSouza N, et al. Protein kinase A and two phosphatases are components of the inositol 1,4,5-trisphosphate receptor macromolecular signaling complex. J Biol Chem. 2002;277:39397–39400. doi: 10.1074/jbc.M207059200. [DOI] [PubMed] [Google Scholar]
- 126.Wagner LE, 2nd, Joseph SK, Yule DI. Regulation of single inositol 1,4,5-trisphosphate receptor channel activity by protein kinase A phosphorylation. J Physiol. 2008;586:3577–3596. doi: 10.1113/jphysiol.2008.152314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Andersson DC, et al. Stress-induced increase in skeletal muscle force requires protein kinase A phosphorylation of the ryanodine receptor. J Physiol. 2012;590:6381–6387. doi: 10.1113/jphysiol.2012.237925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Marx SO, et al. PKA phosphorylation dissociates FKBP12.6 from the calcium release channel (ryanodine receptor): defective regulation in failing hearts. Cell. 2000;101:365–376. doi: 10.1016/s0092-8674(00)80847-8. [DOI] [PubMed] [Google Scholar]
- 129.Torres V. Vasopressin receptor antagonists, heart failure, and polycystic kidney disease. Ann Rev Med. 2015;66 doi: 10.1146/annurev-med-050913-022838. 32.10-32.16. [DOI] [PubMed] [Google Scholar]
- 130.Wehrens XH, et al. Ryanodine receptor/calcium release channel PKA phosphorylation: a critical mediator of heart failure progression. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:511–518. doi: 10.1073/pnas.0510113103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Morimoto S, et al. Protein kinase A-dependent phosphorylation of ryanodine receptors increases Ca2+ leak in mouse heart. Biochem Biophys Res Commun. 2009;390:87–92. doi: 10.1016/j.bbrc.2009.09.071. [DOI] [PubMed] [Google Scholar]
- 132.Marks AR. Calcium cycling proteins and heart failure: mechanisms and therapeutics. J Clin Invest. 2013;123:46–52. doi: 10.1172/JCI62834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Torres VE, Harris PC. Strategies targeting cAMP signaling in the treatment of polycystic kidney disease. J Am Soc Nephrol. 2014;25:18–32. doi: 10.1681/ASN.2013040398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Yamaguchi T, et al. cAMP stimulates the in vitro proliferation of renal cyst epithelial cells by activating the extracellular signal-regulated kinase pathway. Kidney Int. 2000;57:1460–1471. doi: 10.1046/j.1523-1755.2000.00991.x. [DOI] [PubMed] [Google Scholar]
- 135.Hanaoka K, Guggino W. cAMP regulates cell proliferation and cyst formation in autosomal polycystic kidney disease cells. J Am Soc Nephrol. 2000;11:1179–1187. doi: 10.1681/ASN.V1171179. [DOI] [PubMed] [Google Scholar]
- 136.Yamaguchi T, et al. Calcium restriction allows cAMP activation of the B-Raf/ERK pathway, switching cells to a cAMP-dependent growth-stimulated phenotype. J Biol Chem. 2004:40419–40430. doi: 10.1074/jbc.M405079200. [DOI] [PubMed] [Google Scholar]
- 137.Mekahli D, et al. Polycystin-1 but not polycystin-2 deficiency causes upregulation of the mTOR pathway and can be synergistically targeted with rapamycin and metformin. Pflugers Arch. 2014;466:1591–1604. doi: 10.1007/s00424-013-1394-x. [DOI] [PubMed] [Google Scholar]
- 138.Jin X, et al. L-type calcium channel modulates cystic kidney phenotype. Biochim Biophys Acta. 2014;1842:1518–1526. doi: 10.1016/j.bbadis.2014.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Rothschild SC, Francescatto L, Drummond IA, Tombes RM. CaMK-II is a PKD2 target that promotes pronephric kidney development and stabilizes cilia. Development. 2011;138:3387–3397. doi: 10.1242/dev.066340. [DOI] [PubMed] [Google Scholar]
- 140.Banales JM, et al. The cAMP effectors Epac and protein kinase a (PKA) are involved in the hepatic cystogenesis of an animal model of autosomal recessive polycystic kidney disease (ARPKD) Hepatology. 2009;49:160–174. doi: 10.1002/hep.22636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Fischer DC, et al. Activation of the AKT/mTOR pathway in autosomal recessive polycystic kidney disease (ARPKD) Nephrol Dial Transplant. 2009;24:1819–1827. doi: 10.1093/ndt/gfn744. [DOI] [PubMed] [Google Scholar]
- 142.Nishio S, et al. Pkd1 regulates immortalized proliferation of renal tubular epithelial cells through p53 induction and JNK activation. J Clin Invest. 2005;115:910–918. doi: 10.1172/JCI22850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Wahl PR, et al. Mitotic activation of Akt signalling pathway in Han:SPRD rats with polycystic kidney disease. Nephrology (Carlton) 2007;12:357–363. doi: 10.1111/j.1440-1797.2007.00811.x. [DOI] [PubMed] [Google Scholar]
- 144.Natoli TA, et al. Inhibition of glucosylceramide accumulation results in effective blockade of polycystic kidney disease in mouse models. Nature medicine. 2010;16:788–792. doi: 10.1038/nm.2171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Ren XS, et al. Activation of the PI3K/mTOR pathway is involved in cystic proliferation of cholangiocytes of the PCK rat. PloS one. 2014;9:e87660. doi: 10.1371/journal.pone.0087660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Distefano G, et al. Polycystin-1 regulates extracellular signal-regulated kinase-dependent phosphorylation of tuberin to control cell size through mTOR and its downstream effectors S6K and 4EBP1. Mol Cell Biol. 2009;29:2359–2371. doi: 10.1128/MCB.01259-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Spirli C, et al. Mammalian target of rapamycin regulates vascular endothelial growth factor-dependent liver cyst growth in polycystin-2-defective mice. Hepatology. 2010;51:1778–1788. doi: 10.1002/hep.23511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Rowe I, et al. Defective glucose metabolism in polycystic kidney disease identifies a new therapeutic strategy. Nature medicine. 2013;19:488–493. doi: 10.1038/nm.3092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Duvel K, et al. Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Molecular cell. 2010;39:171–183. doi: 10.1016/j.molcel.2010.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Yecies JL, Manning BD. Transcriptional control of cellular metabolism by mTOR signaling. Cancer research. 2011;71:2815–2820. doi: 10.1158/0008-5472.CAN-10-4158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Takiar V, et al. Activating AMP-activated protein kinase (AMPK) slows renal cystogenesis. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:2462–2467. doi: 10.1073/pnas.1011498108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Li M, et al. Cyclic AMP promotes neuronal survival by phosphorylation of glycogen synthase kinase 3beta. Molecular and cellular biology. 2000;20:9356–9363. doi: 10.1128/mcb.20.24.9356-9363.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Taurin S, Sandbo N, Qin Y, Browning D, Dulin NO. Phosphorylation of beta-catenin by cyclic AMP-dependent protein kinase. J Biol Chem. 2006;281:9971–9976. doi: 10.1074/jbc.M508778200. [DOI] [PubMed] [Google Scholar]
- 154.Saadi-Kheddouci S, et al. Early development of polycystic kidney disease in transgenic mice expressing an activated mutant of the β-catenin gene. Oncogene. 2001;20:5972–5981. doi: 10.1038/sj.onc.1204825. [DOI] [PubMed] [Google Scholar]
- 155.Qian CN, et al. Cystic renal neoplasia following conditional inactivation of apc in mouse renal tubular epithelium. J Biol Chem. 2005;280:3938–3945. doi: 10.1074/jbc.M410697200. [DOI] [PubMed] [Google Scholar]
- 156.Qin S, Taglienti M, Cai L, Zhou J, Kreidberg JA. c-Met and NF-kappaB-dependent overexpression of Wnt7a and −7b and Pax2 promotes cystogenesis in polycystic kidney disease. J Am Soc Nephrol. 2012;23:1309–1318. doi: 10.1681/ASN.2011030277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Talbot JJ, et al. Polycystin-1 regulates STAT activity by a dual mechanism. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:7985–7990. doi: 10.1073/pnas.1103816108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Leonhard WN, et al. Curcumin inhibits cystogenesis by simultaneous interference of multiple signaling pathways:in vivo evidence from a Pkd1-deletion model. Am J Physiol.Renal Physiol. 2011;300:F1193–F1202. doi: 10.1152/ajprenal.00419.2010. [DOI] [PubMed] [Google Scholar]
- 159.Takakura A, et al. Pyrimethamine inhibits adult polycystic kidney disease by modulating STAT signaling pathways. Human molecular genetics. 2011;20:4143–4154. doi: 10.1093/hmg/ddr338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Stayner C, et al. Pax2 gene dosage influences cystogenesis in autosomal dominant polycystic kidney disease. Human molecular genetics. 2006;15:3520–3528. doi: 10.1093/hmg/ddl428. [DOI] [PubMed] [Google Scholar]
- 161.Ostom L, Tang M, Grus P, Dressler G. Reduced Pax2 gene dosage increases apoptosis and slows the progression of renal cystic disease. Develop Biol. 2000;219:250–258. doi: 10.1006/dbio.2000.9618. [DOI] [PubMed] [Google Scholar]
- 162.Sullivan LP, Wallace DP, Grantham JJ. Epithelial transport in polycystic kidney disease. Physiol Rev. 1998;78:1165–1191. doi: 10.1152/physrev.1998.78.4.1165. [DOI] [PubMed] [Google Scholar]
- 163.Sullivan LP, Wallace DP, Grantham JJ. Chloride and fluid secretion in polycystic kidney disease. J Am Soc Nephrol. 1998;9:903–16. doi: 10.1681/ASN.V95903. [DOI] [PubMed] [Google Scholar]
- 164.Buchholz B, Teschemacher B, Schley G, Schillers H, Eckardt KU. Formation of cysts by principal-like MDCK cells depends on the synergy of cAMP- and ATP-mediated fluid secretion. Journal of Molecular Medicine. 2011;89:251–261. doi: 10.1007/s00109-010-0715-1. [DOI] [PubMed] [Google Scholar]
- 165.Buchholz B, et al. Anoctamin 1 induces calcium-activated chloride secretion and proliferation of renal cyst-forming epithelial cells. Kidney International. 2014;85:1058–1067. doi: 10.1038/ki.2013.418. [DOI] [PubMed] [Google Scholar]
- 166.Janga Y, Oha U. Anoctamin 1 in secretory epithelia. Cell Calcium. 2014;55:355–361. doi: 10.1016/j.ceca.2014.02.006. [DOI] [PubMed] [Google Scholar]
- 167.Leuenroth SJ, et al. Triptolide is a traditional Chinese medicine-derived inhibitor of polycystic kidney disease. Proc Natl Acad Sci U S A. 2007;104:4389–4394. doi: 10.1073/pnas.0700499104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Leuenroth SJ, Bencivenga N, Igarashi P, Somlo S, Crews CM. Triptolide reduces cystogenesis in a model of ADPKD. J Am Soc Nephrol. 2008;19:1659–1662. doi: 10.1681/ASN.2008030259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Nagao S, et al. Calcium channel inhibition accelerates polycystic kidney disease progression in the Cy/+ rat. Kidney Int. 2008;73:269–277. doi: 10.1038/sj.ki.5002629. [DOI] [PubMed] [Google Scholar]
- 170.Gattone VH, 2nd, et al. Calcimimetic inhibits late-stage cyst growth in ADPKD. J Am Soc Nephrol. 2009;20:1527–1532. doi: 10.1681/ASN.2008090927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Wang X, Harris PC, Somlo S, Batlle D, Torres VE. Effect of calcium-sensing receptor activation in models of autosomal recessive or dominant polycystic kidney disease. Nephrol Dial Transplant. 2009;24:526–534. doi: 10.1093/ndt/gfn527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Leuenroth SJ, Bencivenga N, Chahboune H, Hyder F, Crews CM. Triptolide reduces cyst formation in a neonatal to adult transition Pkd1 model of ADPKD. Nephrol Dial Transpl. 2010;25:2187–2194. doi: 10.1093/ndt/gfp777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Gradilone SA, et al. Activation of Trpv4 reduces the hyperproliferative phenotype of cystic cholangiocytes from an animal model of ARPKD. Gastroenterology. 2010;139:304–314. doi: 10.1053/j.gastro.2010.04.010. e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Chen NX, et al. Calcimimetics inhibit renal pathology in rodent nephronophthisis. Kidney International. 2011;80:612–619. doi: 10.1038/ki.2011.139. [DOI] [PubMed] [Google Scholar]
- 175.Mahajan N, et al. Calcium Ameliorates Renal Cyst Growth in Metanephric Organ Culture: A Morphological Study. J Environ Pathol Toxicol Oncol. 2012;31:285–293. doi: 10.1615/jenvironpatholtoxicoloncol.v31.i3.90. [DOI] [PubMed] [Google Scholar]
- 176.Zaica O, et al. TRPV4 Dysfunction Promotes Renal Cystogenesis in Autosomal Recessive Polycystic Kidney Disease. J Am Soc Nephrol. 2013;24:604–616. doi: 10.1681/ASN.2012050442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Torres VE. Type II calcimimetics and polycystic kidney disease: unanswered questions. J Am Soc Nephrol. 2009;20:1421–1425. doi: 10.1681/ASN.2009050501. [DOI] [PubMed] [Google Scholar]