

Abstract
Hippocampal sclerosis of aging (HS-Aging) is a neurodegenerative disease that mimics Alzheimer disease (AD) clinically and has a prevalence rivaling AD in advanced age. Whereas clinical biomarkers are not yet optimized, HS-Aging has distinctive pathological features that distinguish it from other diseases with “hippocampal sclerosis” pathology, such as epilepsy, cerebrovascular perturbations, and frontotemporal lobar degeneration. By definition, HS-Aging brains show neuronal cell loss and gliosis in the hippocampal formation out of proportion to AD-type pathology; it is strongly associated with aberrant TDP-43 pathology and arteriolosclerosis. Here, we describe 2 cases of “segmental” HS-Aging in which “sclerosis” in the hippocampus was evident only in a subset of brain sections by hematoxylin and eosin (H&E) stain. In these cases, TDP-43 pathology was more widespread on immunostained sections than the neuronal cell loss and gliosis seen in H&E stains. The 2 patients were cognitively intact at baseline and were tracked longitudinally over a decade using cognitive studies with at least 1 neuroimaging scan. We discuss the relevant HS-Aging literature, which indicates the need for a clearer consensus-based delineation of “hippocampal sclerosis” and TDP-43 pathologies in aged subjects.
Keywords: Biomarkers, Case study, Dementia, Frontotemporal lobar degeneration (FTLD), Hippocampal sclerosis, Neuropathology, Oldest-old
INTRODUCTION
Hippocampal sclerosis of aging (HS-Aging) is a neurodegenerative disease with high morbidity that usually affects individuals who survive past age 80 years (1–6). The diagnosis of HS-Aging rests primarily on neuropathological findings in hematoxylin and eosin (H&E) stains using consensus-based criteria (1, 7); these include cell loss, gliosis, and atrophy in the hippocampal formation that is out of proportion to Alzheimer disease (AD)–type pathology. HS-Aging is a common pathology among older individuals, but its clinical manifestations are often incorrectly attributed to AD (2, 4, 5, 8). Hence, there is a need for better understanding of the HS-Aging disease spectrum.
Although not part of current consensus-based diagnostic criteria, TAR-DNA binding protein 43 (TDP-43) pathology is strongly linked to HS-Aging (1, 5, 9–13). TDP-43 is a nucleotide-binding protein that is normally enriched in the nucleus of neurons (12, 14). In HS-Aging, affected neurons display loss of nuclear TDP-43, accumulation of cytoplasmic phosphorylated TDP-43 inclusion bodies, and aberrant TDP-43 in neurites (1). This aberrant TDP-43 immunostaining is often seen in hippocampal dentate granule cells, CA1 hippocampal sector, subiculum, and amygdala (1, 10). TDP-43 pathology is a key difference between HS-Aging and other diseases with hippocampal sclerosis (HS) pathology, such as epilepsy and vascular insufficiency, which lack aberrant TDP-43 staining (1, 15). We have found that if bilateral entorhinal cortex sections are assessed, approximately 90% of HS-Aging cases show aberrant TDP-43 pathology, whereas only 10% of non-HS-Aging cases show TDP-43 pathology (1). We have also shown that individuals with “unilateral” HS-Aging (by H&E stain) show aberrant TDP-43 inclusions in both the affected and contralateral side (1). This asymmetric HS (i.e., one-sided neuronal loss) observed in H&E-stained sections with bilateral TDP-43 immunopositivity provides insight into potentially early stages of HS-Aging pathology: hippocampal TDP-43 pathology without widespread “sclerosis” (5).
To raise awareness of the clinical-pathological features of TDP-43 immunoreactivity with only segmental “sclerotic” changes, we describe the findings from 2 research volunteer subjects from the University of Kentucky Alzheimer’s Disease Center (UK-ADC) longitudinal cohort. We define segmental HS-Aging as focal cell loss in the CA1 and/or subicular region(s), with astrocytosis seen on some but not all hippocampal sections on H&E and with aberrant TDP-43 immunostaining in the hippocampal formation. Both segmental HS-Aging cases were followed from initial cognitively intact status to death. Clinical, cognitive, imaging, and neuropathological findings are presented. We also review the relevant literature and emphasize that these cases are not unusual.
CLINICAL AND NEUROPATHOLOGICAL ASSESSMENTS
Details of UK-ADC recruitment, inclusion/exclusion criteria, clinical assessments, cognitive testing, and neuropathological protocols have been described (16, 17). Briefly, as part of a longitudinal study, the 2 individuals in this study consented to annual mental status testing, physical examinations, and postmortem brain donation (18). All protocols were performed with UK institutional review board approval. Clinical data from annual cognitive assessments were examined using normative values from baseline assessments of 648 healthy aging volunteers. Cognitive assessments reported here included measures of category verbal fluency, immediate memory and learning, and delayed recall scores incorporated from word list learning and paragraph tests (17, 19). Raw scores from each measure were transformed to T-scores based on the average baseline performance of the larger sample (19–21). The derived T-scores (mean = 50, SD = 10) incorporated adjustments for age, sex, and education. This method allowed for direct comparisons between each case. Mini-Mental State Exam (MMSE) scores (22) are presented as raw scores for comparative purposes. Neuroimaging studies were obtained at the request of the neurologist. Imaging modalities included magnetic resonance imaging (MRI)-T1 weighted imaging with or without contrast, MRI-T2-weighted imaging, and CT imaging without contrast.
Neuropathological assessments were performed at UK-ADC using previously described methodology (1, 18, 23). At least 28 sections were taken from various brain areas. At least 4 areas from the medial temporal lobes were sectioned: the amygdala, entorhinal cortex, and rostral and caudal hippocampi including the level of the lateral geniculate nucleus from both left and right sides. After formalin fixation and paraffin embedding, 8-μm-thick sections were stained with H&E. Immunostains for PHF-1 (gift from Dr. Peter Davies, Feinstein Institute for Medical Research, Manhasset, NY; 1:500 dilution) and amyloid-β (Vector Laboratories, Burlingame, CA; 1:100 dilution) were performed and used to assess neuritic plaques, amyloid β (Aβ), and neurofibrillary tangles following the National Institute on Aging-Alzheimer’s Association (NIA-AA) consensus recommendations (7). Anti-α-synuclein (Vector Laboratories; 1:40 dilution) immunohistochemistry was used for assessing Lewy body pathology. Anti-glial fibrillary acidic protein (Novocastra, Buffalo Grove, IL) immunostaining was performed to demonstrate reactive astrocytes. The anti-phospho TDP-43 antibody used was a gift from Dr. Manuela Neumann (University of Tubingen, Tubingen, Germany; 1:500 dilution).
RESEARCH SUBJECTS
Case “F”
An 82-year-old woman was initially recruited as a cognitively intact research volunteer. Her father had been clinically diagnosed with AD and died at age 73 years. Her medical history included hypertension, coronary artery disease, hyperlipidemia, and hypothyroidism, all of which were treated medically. She had smoked (25 pack/year history) and denied excessive alcohol drinking.
At the age of 88, she received a clinical consensus diagnosis of amnestic mild cognitive impairment (Fig. 1). Because of her cognitive decline, MRI T1- and T2-weighted images with and without contrast were obtained at age 88 (Fig. 2). Radiological impressions from images included moderate generalized cerebral atrophy with cerebellar atrophy, mild ex vacuo ventricular dilatation, and multiple confluent areas of supratentorial white matter hyperintensities. The latter finding was considered consistent with chronic microvascular infarcts.
FIGURE 1.

Mini-Mental State (MMSE) and T-scores from longitudinal neurocognitive assessments in animal fluency, immediate, and delayed memory tasks. For the T-scores, mean = 50, SD = 10. Standardization of t-scores is based on results from age-matched nondemented individuals. The age at which mild cognitive impairment (MCI) diagnosis, neuroimaging scans, and probable Alzheimer disease diagnosis were made are indicated by red, blue, and green asterisks, respectively. Note that memory scores declined more than verbal fluency.
FIGURE 2.

Cases F and M. (A–D) Magnetic resonance (MR) images for Case F at age 88. (A) Coronal T1-weighted image with contrast. Red arrows point to the hippocampi (A). Sagittal T1-weighted noncontrast image. Red arrow points to hippocampus (B). Transverse MR T1 weighted image with contrast. Red arrow points to the hippocampus (C). Transverse MR T1-weighted noncontrast image. Red asterisks indicate areas of white matter hyperintensities in the periventricular region (D). (E, F) Computerized tomography (CT) images for Case M at age 89 years. Transverse CT noncontrast image. Red arrows point to chronic subdural collection in the brain indicating a history of previous subdural hemorrhage (E). Transverse CT noncontrast image. Red asterisks indicate hippocampal regions (F).
Near her 90th birthday, she was having difficulties with finances and became paranoid, stating that people were stealing from her. Donepezil was initiated at that time. By age 91, she was no longer driving, although she was still performing simple chores in the home. Her cognition continued to decline, and she began to struggle increasingly with higher-order executive tasks. Her cognitive test results show a steady decline in delayed memory (1 SD below the mean), with preserved immediate memory and animal fluency tasks (Fig. 1). At age 94, she was diagnosed with probable AD and was wheelchair dependent. By this time, she required assistance with all activities of daily living including bathing. She died at age 95.
Neuropathological findings are shown in Figures 3 and 4. For comparison, the hippocampus of a nondemented individual is shown in Figure 3A. An unrelated HS-Aging case is shown in Figure 3D demonstrating diffuse neuronal loss, neuropil rarefaction (Fig. 3E), and astrocytosis (Fig. 3F). In contrast, Case F showed less generalized hippocampal shrinkage (Fig. 3G), with segmental neuronal cell loss in the CA1 region (Fig. 3H) and reactive astrocytosis (Fig. 3I). In Case F, HS-Aging pathology was predominantly localized in the CA1 region, whereas the comparison case in Figure 3D shows more widespread pathology extending into the subiculum. Case F showed abundant TDP-43-immunoreactive intracellular inclusions and neurites throughout the amygdala, entorhinal cortex, and hippocampus with bilateral TDP-43 immunoreactivity (Fig. 4D). TDP-43 inclusions were more widespread than neurites (blue outlined areas) in the brain sections (Fig. 4D).
FIGURE 3.
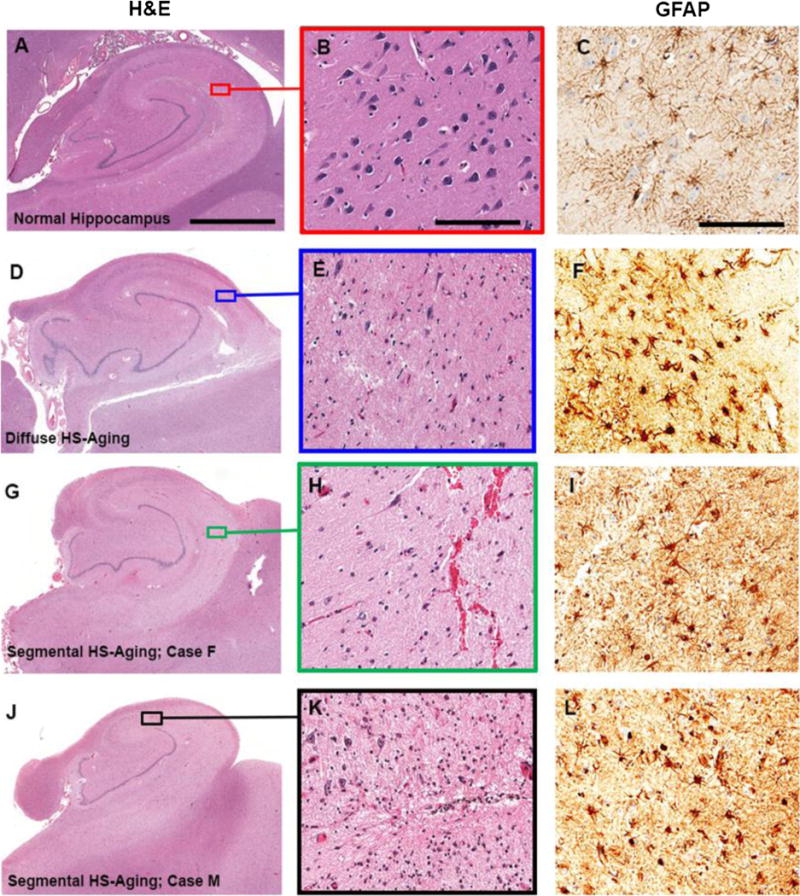
(A–L) Photomicrographs of hematoxylin and eosin-stained (A, B, D, E, G, H, J, K) and anti-glial fibrillary acidic protein (GFAP)-stained (C, F, I, L) hippocampal sections. Images illustrate either a normal hippocampus (A–C), diffuse hippocampal sclerosis of aging (HS-Aging) pathology (D–F), or segmental HS-Aging pathologies (G–L). (A) Low-power image demonstrates normal hippocampal structure. (B) Magnification of area in box in (A) shows a high-power image with abundant neuronal cells. (C) High-power image with few reactive astrocytes. (D) Low-power image demonstrates diffuse HS-Aging pathology: the hippocampus is shrunken with neuropil rarefaction extending into the subiculum. (E) Magnification of area in box in (D) shows a high-power image with marked neuronal cell loss and spongiosis. (F) Marked reactive astrocytosis in HS-Aging. (G, J) Low-power images show segmental HS-Aging pathology with a shrunken hippocampi and selective cell loss in CA1. (H, K) High-power images from boxed areas of (G) and (J), respectively, with less neuron loss and spongiosis compared with that seen in (E). (I, L) High-power images with reactive astrocytes. Scale bars: A, D, G, J, 3.5 mm; B, E, H, K, 200 μm; C, F, I, L, 150 μm.
FIGURE 4.

TAR-DNA binding protein 43 (TDP-43) immunoreactivity. (A) Schematic of the bilateral hippocampal formation levels for anti-TDP-43 immunostained sections of Cases F and M. The blue horizontal lines indicate approximate sectioning locations of the photomicrographs. I, V, amygdala; II, VI, entorhinal cortex; III, VII, rostral hippocampus; IV, VIII, caudal hippocampus. (B, C) Photomicrograph images of TDP-43 pathology. High-power photomicrograph of a neuron with TDP-43 positive multiple discrete granular cytosolic accumulations (possible “preinclusions;” green triangle), a potential marker of early HS-Aging pathology (B). High-power photomicrograph of TDP-43 positive intracellular inclusion bodies (red triangles) and neurites (blue arrows) (C). (D, E) Images of anti-TDP-43 immunostained serial sections from the hippocampal formation from Case F and Case M, respectively. Locations of intracellular inclusion bodies or granular cytosolic accumulations are represented by red or green triangles respectively. Locations of TDP-43 immunoreactive neurites are enclosed by the blue outline. Level VIII for Case M was not available. Scale bars: B, C, 50 μm; D, E, 5 mm.
Assessments using the NIA-AA consensus protocol for AD workup (7) revealed low levels of AD neuropathologic changes (Figure, Supplemental Digital Content 1, http://links.lww.com/NEN/A740): Braak I, Consortium to Establish a Registry for Alzheimer Disease (CERAD) “sparse,” and Thal stage 3. Her brain also showed evidence of multifocal cerebrovascular disease with acute infarcts in the right parietal and temporal lobes, insula, basal ganglia, and entorhinal cortices (the latter in focal areas). Remote microinfarcts were present in the left putamen, along with severe nonocclusive atherosclerosis in Circle of Willis, moderate cortical arteriolosclerosis, widening of Virchow Robin spaces, perivascular rarefaction in scattered areas, and other small blood vessel changes (not shown).
Case “M”
A 74-year-old man was initially recruited as a cognitively intact research volunteer. He had reported that his mother had dementia with an unknown age of diagnosis. His medical history was significant for hypertension and benign prostatic hypertrophy. He smoked (50 pack/years) and drank alcohol (3–4 drinks/day). His cognitive performance was relatively unremarkable until age 82, when he was diagnosed with mild cognitive impairment based on a consensus review of his symptoms (Fig. 1).
A workup at age 83 led to a diagnosis of mixed probable AD and vascular disease. He began treatment with donepezil at this time. At age 86, he began to struggle with higher order activities including driving. By age 87, he was no longer driving, but he was still intact with respect to most other activities of daily living; memantine was added to his medications at that time. By age 88, he needed help with most activities of daily living including bathing.
At his last UK-ADC clinic visit, his MMSE score was 19 with mild behavioral issues of impulsivity. Longitudinal cognitive test results showed a steady decline in immediate and delayed memory tasks (3 standard deviations below the mean by age 86) with preserved animal fluency performance (Fig. 1). After a fall at age 89, CT imaging without contrast was obtained. This image revealed a chronic left subdural hematoma, periventricular white matter hypodensity, and generalized brain volume loss (Fig. 2). The patient passed away later that year at age 89.
Neuropathological hippocampal autopsy findings are shown in Figures 3 and 4. There was focal hippocampal shrinkage (Fig. 3J), with segmental neuronal cell loss in the CA1 region (Fig. 3K) and astrocytic gliosis (Fig. 3L). Aberrant TDP-43 immunoreactivity was seen within the amygdala bilaterally and the left rostral and caudal entorhinal cortices (Fig. 4E). In addition, there were intracellular TDP-43-positive cytoplasmic “preinclusions” (green triangles) in the hippocampus bilaterally and in the right entorhinal cortex (Fig. 4E). TDP-43 intracellular inclusion bodies were more widespread than neurites (blue outlined areas) in the brain sections (Fig. 4E).
Assessments using the NIA-AA consensus protocol for AD workup (7) revealed an intermediate level of AD neuropathological changes Braak IV, CERAD “moderate,” and Thal phase 5 (Figure, Supplemental Digital Content 1, http://links.lww.com/NEN/A740). In addition to neurodegenerative disease pathologies, there was evidence of multifocal chronic cerebrovascular disease with moderate-to-severe multifocal atherosclerotic disease in the Circle of Willis, arteriolosclerosis, and other mild small vessel changes observable on histopathology. In some areas, there was widening of Virchow-Robin spaces and perivascular rarefaction. Incidental Lewy body pathology was found in the olfactory bulb (not shown).
DISCUSSION AND REVIEW OF THE LITERATURE
There is an evolving appreciation of the large impact of HS-Aging on public health, particularly among the oldest-old: approximately 10% to 25% of persons older than 85 demonstrate this pathology at autopsy (1–5, 8, 11, 24–29). Here, we describe the detailed findings in 2 subjects with segmental HS-Aging that may represent either early HS-Aging pathology or be those of a subset of cases that develop TDP-43 pathology without widespread hippocampal changes, thereby meeting current criteria for HS. These segmental HS-Aging patterns are frequently seen in our autopsy series—underscored by the observation that approximately one half of our HS-Aging cases are unilateral on H&E stains (1)—although pathogenesis of these patterns is not well understood.
There are inherent limitations of this study. The diagnosis of segmental HS-Aging was made primarily using H&E stain, but the “sclerotic” changes are difficult to discriminate with precision. Zinc transporter 3 immunostaining, choline acetyltransferase immunostaining, and Hirano silver staining have been used to delineate between CA3–CA2, CA2–CA1, and CA1–subiculum respectively, in HS cases (30). These stains can be used in future segmental HS-Aging studies to help determine which hippocampal regions are initially and/or most affected. The segmental HS-Aging pathology in these cases may not fully explain the cognitive declines seen years before death. Case F showed evidence of multifocal cerebrovascular disease, and Case M showed intermediate AD pathology with severe multifocal cerebrovascular disease, which may help explain the dementia. These cases indicate how frequent comorbidities, especially AD and cerebrovascular neuropathologic changes, could strongly alter the pattern of tested cognitive domains. Both cases in the present study showed TDP-43 pathology in the amygdala, entorhinal cortex, and hippocampus. According to reports from different research centers, hippocampal TDP-43 pathology with or without reported HS contributes to an additive component of cognitive impairment (14, 31–35). Therefore, it is reasonable to infer that the TDP-43 pathology seen in these individuals may partly explain their cognitive deterioration. Because of the lack of a clear consensus in the field on HS-Aging categorization and “boundary zones” between other neurodegenerative diseases, we highlight HS-Aging in relationship to prehippocampal sclerosis (pre-HpScl, a term that has been used to describe early HS-Aging pathology [36]), frontotemporal lobar degeneration (FTLD) with TDP-43 pathology (FTLD-TDP), AD and cerebrovascular pathologies.
Early HS-Aging or “Pre-HpScl”
Although information on HS-Aging is rapidly accumulating, nomenclature for this disease is not universal. One group refers to a disease category very similar to HS-Aging as “HpScl,” (2, 37, 38) and some may refer to this condition as “hippocampal sclerosis dementia” (39). In a very recent paper, a panel of experts discussed HS-Aging pathologic classification terminology (40); however, the average age of persons used in this consensus recommendation paper were younger (most less than 80 years old) than when HS-Aging prevalence is highest (1). Unfortunately, the term hippocampal sclerosis is itself potentially misleading. The pathologic features are not fully conveyed by the term sclerosis, which signifies “hardening.” Furthermore, the pathologic changes of HS-Aging generally extend beyond the hippocampus proper. We reiterate the important point that the term hippocampal sclerosis is widely used to describe multiple distinct diseases, including hippocampal pathology associated with epilepsy, hypoxia, hypoglycemia, FTLD, and others. Well more than 95% of PubMed citations linked to “hippocampal sclerosis” are unrelated to HS-Aging, HpScl, or HS dementia. Thus, to prevent confusion (whatever the eventual terminology ends up being), we recommend that it should include a component different from simply “hippocampal sclerosis.”
Recently, the term pre-HpScl was used to describe hippocampal pathology characterized by none to minimal neuronal loss or extracellular neurofibrillary tangles with abundant TDP-43 pathology (36). This condition may partially overlap with the “segmental” HS-Aging pathology seen in the 2 cases described here. Both articles present cases that show focal neuronal loss and gliosis in CA1 with more widespread TDP-43 pathology in the limbic system. These findings suggest that HS-Aging could be a progressive disease where the initial stages are characterized by the presence of TDP-43 pathology with or without some focal areas of cell loss (Fig. 5). More broadly, these findings may also be evidence for a presently unnamed “brain-wide” disease that is characterized by a spectrum of hippocampal TDP-43 pathologies (Fig. 5): TDP-43 pathology, TDP-43 pathology with segmental/focal sclerotic-type changes, and TDP-43 pathology with diffuse HS.
FIGURE 5.
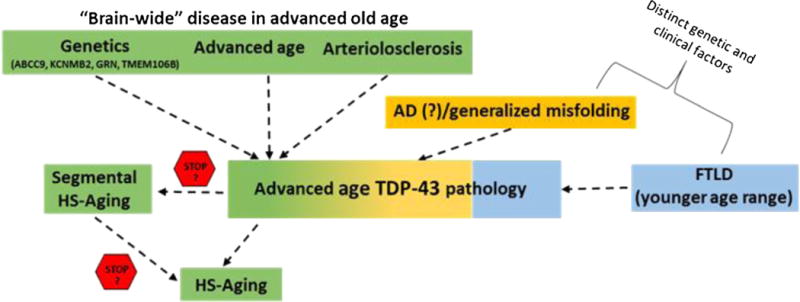
Schematic for neurodegenerative disease etiologies of TAR DNA binding protein-43 (TDP-43) pathology in advanced age. A currently unnamed “brain-wide” disease occurring in advanced age that is associated with certain gene polymorphisms (ABCC9, KCNMB2, GRN, TMEM106B) and characterized by a pathologic spectrum that includes arteriolosclerosis, TDP-43 pathology, segmental HS-Aging, and widespread HS-Aging. The disease(s) may comprise a continuous spectrum or 3 separate variants as indicated by the stop signs. A subset of cases may be due to or exacerbated by AD pathology leading to misfolding and aggregation of proteins including TDP-43. Frontotemporal lobar degeneration may lead to the accumulation of TDP-43 in some cases, even in advanced age. AD, Alzheimer disease; FLTD, frontotemporal lobar degeneration; HS-Aging, hippocampal sclerosis of aging.
Frontotemporal lobar degeneration–TDP
Aberrant TDP-43 is seen in 65% to 90% of HS-Aging cases (1, 10). Because of the high prevalence of TDP-43 pathology in HS-Aging cases there is some support for the hypothesis that HS-Aging is closely related to FTLD-TDP (41). In one study, the prevalence of hippocampal sclerosis in FTLD-TDP was 42% (42). Moreover, the slender nontapering TDP-43 neurites observed in FTLD-TDP hippocampi (“type A” pattern) resemble those seen in HS-Aging (42). In a different study, more than 70% of hippocampal sclerosis cases with TDP-43 pathology had neurites and inclusion bodies the morphology of which resembled that found in FTLD-TDP (10). Similarly, in Lewy body dementia (LBD) cases with HS-Aging, the TDP-43 immunohistochemistry pattern was similar to the FTLD-TDP type A pattern (36). We also note that some human genetic polymorphisms (in GRN and TMEM106B) are risk factors for both FTLD-TDP and HS-Aging pathologies (33, 37, 43–45). The TMEM106B polymorphism (rs1990622) was also recently shown to be a risk factor for non-HS TDP-43 pathology (46). These findings could be argued to support the possibility that HS-Aging and TDP-43 pathology in older people are either pathogenetically linked or represent a frank variant of FTLD.
Although there are areas of overlap, FTLD and HS-Aging also differ in clinical symptoms, genetic risk factors (discussed in greater detail below), and pathological characteristics. For example, patients with FTLD pathology show clinical manifestations and die at much younger ages than those with HS-Aging pathology (8, 11). HS-Aging cases tend to lack either the behavioral variant FTD or aphasia symptoms (1, 8), although late in their disease courses, both of the current 2 cases had features previously linked to frontal cortical dysfunction (i.e., delusions in Case F and impulsivity in Case M) (47–49). HS-Aging patients were previously shown to demonstrate a group-level neurocognitive profile characterized by higher verbal fluency scores 2 to 5 years before death, as seen in Case F and Case M, dissimilar to nontauopathic FTLD cases (8).
Pathologically, TDP-43 proteinopathy is specific neither to FTLD-TDP nor HS-Aging because TDP-43 pathology can be seen in Alexander disease, low-grade glial neoplasms, AD/Down syndrome, and brain trauma (15, 50–54). Thus (perhaps analogous to tau protein and neurofibrillary tangles), there appears to be a “reactive” aspect to TDP-43 pathology, although the pathology also seems deleterious when present. Frontotemporal lobar degeneration–TDP cases with HS showed more severe cortical and brainstem atrophy than HS-Aging cases, which localized to the hippocampus (11). HS-Aging cases had lower synaptophysin immunoreactivity, greater astrocytic reactivity and microglial reactivity compared with FTLD-TDP with HS cases (11). It has been suggested that HS-Aging in aged individuals may be a variant of FTLD-TDP (55); however, this does not seem to make sense because if this were true, there should be a very large cohort of individuals who ultimately express the full-fledged FTLD-TDP clinical picture among the aged, and this is not the case. Frontotemporal lobar degeneration is quite rare: there are only approximately 20,000–30,000 cases in America (75), around half with FTLD-TDP. This contrasts with HS-Aging, which is extremely prevalent, perhaps affecting more than a million Americans (5, 6, 29, 76). Furthermore, there is not evidence that the hippocampal pathology shows early clinical-temporal pattern in FTLD-TDP cases. These findings support the notion that HS-Aging is a distinct pathology from FTLD-TDP, although both diseases show TDP-43 immunoreactivity and HS (Fig. 5).
Alzheimer Disease
Because TDP-43 pathology is seen in many cases that also have abundant AD pathology, there may be a subset of HS-Aging cases that are best categorized as a variant of AD. TDP-43 positivity can be seen in 14% to 57% of AD cases (14, 34, 53, 54). More specifically in one study, TDP-43 pathology was seen 9% of familial AD cases, 10% in early onset AD cases, and 29% in late-onset AD cases (53). Another study showed that increasing TDP-43-immunoreactive pathology correlates with increasing Braak neurofibrillary tangle stages (10). It has also been shown that some TDP-43 inclusions seen in AD brains colocalize with phospho-tau in the entorhinal cortex and dentate fascia (10). Because in vivo and in vitro studies have shown that Aβ and/or tau can promote the misfolding/polymerization of polypeptides (e.g., α-synuclein) (59–61), it can be speculated that this could occur in late-stages of AD brain pathology (10). It remains to be seen whether the “TAD” staging system of TDP-43 pathology in AD brains or in brains with dementia with Lewy bodies (DLB) pathology (36) is directly relevant to the staging of TDP-43 pathology in HS-Aging. Because all of these pathologic changes (i.e., HS-Aging/TDP-43 pathology, AD, and DLB) are relatively prevalent in community-based samples evaluated to date (in strong contrast to FTLD), they could be coexistent rather than directly linked pathologies (39, 62–65).
A counterargument against a specific link between HS-Aging and AD pathologies is that HS-Aging has no association with APOE or with any other AD-related risk allele (2, 6, 23, 66). Thus far, there also does not seem to be a gender-based predisposition for HS-Aging (8). It would be expected that many individuals would manifest both AD and HS-Aging pathologies at autopsy because both pathologies are prevalent in older populations.
Cerebrovascular Disease
Another brain process that has been associated with HS-Aging pathology is cerebrovascular disease. Dickson and colleagues described cerebrovascular disease pathology in a cohort of 13 individuals with HS-Aging (67). Arteriosclerosis, atherosclerosis, subcortical arteriosclerotic leukoencephalopathy, and cerebral amyloid angiopathy were observed in these cases (3). We recently showed that arteriolosclerosis, characterized by thickened and/or dysmorphic arterioles in the brain, is associated with HS-Aging pathology; in the gray matter, the mean vessel wall thickness was significantly larger in HS-Aging cases compared with that of non–HS-Aging cases (68).
There is circumstantial support from genetic studies for a mechanism linking cerebrovascular pathology with HS-Aging. The only 2 genomics studies that used HS in aged persons as a genome-wide association analysis endophenotype reported risk alleles at genomic loci in potassium channel regulating genes ABCC9 and KCNMB2, previously unrelated to FTLD (43, 69). We recently replicated the observation that ABCC9 polymorphism is associated with HS-Aging pathology (44). The ABCC9 polypeptide is physiologically active in arteriolar smooth muscle (70, 71). Therefore, it is possible that ABCC9 dysregulation leads to arteriolosclerosis, which then may contribute to the pathogenesis of HS-Aging. ABCC9/SUR2 is an attractive candidate for therapeutic strategies because it is a well-established “druggable target.” Both agonists (nicorandil, diazoxide, and iptakalim) and antagonists (sulfonylurea drugs) have been applied in clinical trials (72, 73). Their potential for repurposing for HS-Aging is an active research area in our laboratory.
Coincidentally, both cases in the present study showed cerebrovascular disease including arteriolosclerosis, which may have partially accounted for their progressive cognitive decline. In sum, we infer that the association between arteriolosclerosis and HS-Aging provides added support for a “brain-wide” disease that affects both small blood vessels and hippocampal structures and leads to TDP-43 pathology in the aged brain (Fig. 5).
CONCLUSIONS
We present 2 cases that show what we consider to be either early stages of HS-Aging pathology or possibly a variant of a brain-wide disease characterized by arteriolosclerosis and TDP-43 pathology affecting the limbic system (Fig. 5). These 2 cases taken together with other articles describing hippocampal TDP-43 pathology in LBD and AD brains (36, 74) suggest the need for a consensus definition regarding the spectrum of pathologies with TDP-43 and HS seen in the aged human brain. HS-Aging is a prevalent neurodegenerative disease, the specific genetic, neuroimaging, symptomatology and pathologic characteristics of which are only beginning to be understood. We conclude that careful clinical-neuropathological correlations may assist in the overall goal of developing strategies to diagnose and treat individuals who have this debilitating illness.
Supplementary Material
Acknowledgments
This work was supported by the following National Institute of Health grants: P30 AG028383, R01 AG038651, R01 AG042419, and T32 AG 000242.
Footnotes
Supplemental digital content is available for this article. Direct URL citations appear in the printed text and are provided in the HTML and PDF versions of this article on the journal’s Web site (www.jneuropath.com).
References
- 1.Nelson PT, Schmitt FA, Lin Y, et al. Hippocampal sclerosis in advanced age: clinical and pathological features. Brain. 2011;134:1506–18. doi: 10.1093/brain/awr053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pao WC, Dickson DW, Crook JE, et al. Hippocampal sclerosis in the elderly: genetic and pathologic findings, some mimicking Alzheimer disease clinically. Alzheimer Dis Assoc Disord. 2011;25:364–8. doi: 10.1097/WAD.0b013e31820f8f50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dickson DW, Davies P, Bevona C, et al. Hippocampal sclerosis: a common pathological feature of dementia in very old (> or = 80 years of age) humans. Acta Neuropathol. 1994;88:212–21. doi: 10.1007/BF00293396. [DOI] [PubMed] [Google Scholar]
- 4.Zarow C, Sitzer TE, Chui HC. Understanding hippocampal sclerosis in the elderly: epidemiology, characterization, and diagnostic issues. Curr Neurol Neurosci Rep. 2008;8:363–70. doi: 10.1007/s11910-008-0057-3. [DOI] [PubMed] [Google Scholar]
- 5.Nelson PT, Smith CD, Abner EL, et al. Hippocampal sclerosis of aging, a prevalent and high-morbidity brain disease. Acta Neuropathol. 2013;126:161–77. doi: 10.1007/s00401-013-1154-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Leverenz JB, Agustin CM, Tsuang D, et al. Clinical and neuropathological characteristics of hippocampal sclerosis: a community-based study. Arch Neurol. 2002;59:1099–106. doi: 10.1001/archneur.59.7.1099. [DOI] [PubMed] [Google Scholar]
- 7.Montine TJ, Phelps CH, Beach TG, et al. National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease: a practical approach. Acta Neuropathol. 2012;123:1–11. doi: 10.1007/s00401-011-0910-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brenowitz WD, Monsell SE, Schmitt FA, et al. Hippocampal sclerosis of aging is a key Alzheimer’s disease mimic: clinical-pathologic correlations and comparisons with both alzheimer’s disease and non-tauopathic frontotemporal lobar degeneration. J Alzheimer’s Dis. 2014;39:691–702. doi: 10.3233/JAD-131880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Amador-Ortiz C, Dickson DW. Neuropathology of hippocampal sclerosis. Handb Clin Neurol. 2008;89:569–72. doi: 10.1016/S0072-9752(07)01253-5. [DOI] [PubMed] [Google Scholar]
- 10.Amador-Ortiz C, Lin WL, Ahmed Z, et al. TDP-43 immunoreactivity in hippocampal sclerosis and Alzheimer’s disease. Ann Neurol. 2007;61:435–45. doi: 10.1002/ana.21154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Amador-Ortiz C, Ahmed Z, Zehr C, et al. Hippocampal sclerosis dementia differs from hippocampal sclerosis in frontal lobe degeneration. Acta Neuropathol. 2007;113:245–52. doi: 10.1007/s00401-006-0183-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Neumann M, Sampathu DM, Kwong LK, et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314:130–3. doi: 10.1126/science.1134108. [DOI] [PubMed] [Google Scholar]
- 13.Arai T, Hasegawa M, Akiyama H, et al. TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem Biophys Res Commun. 2006;351:602–11. doi: 10.1016/j.bbrc.2006.10.093. [DOI] [PubMed] [Google Scholar]
- 14.Keage HA, Hunter S, Matthews FE, et al. TDP-43 in the population: prevalence and associations with dementia and age. J Alzheimer’s Disease. 2014;42:641–50. doi: 10.3233/JAD-132351. [DOI] [PubMed] [Google Scholar]
- 15.Lee EB, Lee VM, Trojanowski JQ, et al. TDP-43 immunoreactivity in anoxic, ischemic and neoplastic lesions of the central nervous system. Acta Neuropathol. 2008;115:305–11. doi: 10.1007/s00401-007-0331-5. [DOI] [PubMed] [Google Scholar]
- 16.Markesbery WR, Schmitt FA, Kryscio RJ, et al. Neuropathologic substrate of mild cognitive impairment. Arch Neurol. 2006;63:38–46. doi: 10.1001/archneur.63.1.38. [DOI] [PubMed] [Google Scholar]
- 17.Schmitt FA, Nelson PT, Abner E, et al. University of Kentucky Sanders-Brown healthy brain aging volunteers: donor characteristics, procedures and neuropathology. Curr Alzheimer Res. 2012;9:724–33. doi: 10.2174/156720512801322591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nelson PT, Jicha GA, Schmitt FA, et al. Clinicopathologic correlations in a large Alzheimer disease center autopsy cohort: neuritic plaques and neurofibrillary tangles “do count” when staging disease severity. J Neuropathol Exp Neurol. 2007;66:1136–46. doi: 10.1097/nen.0b013e31815c5efb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mathews M, Abner E, Caban-Holt A, et al. CERAD practice effects and attrition bias in a dementia prevention trial. Int Psychogeriatrics. 2013;25:1115–23. doi: 10.1017/S1041610213000367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Abner EL, Dennis BC, Mathews MJ, et al. Practice effects in a longitudinal, multi-center Alzheimer’s disease prevention clinical trial. Trials. 2012;13:217. doi: 10.1186/1745-6215-13-217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mathews M, Abner E, Kryscio R, et al. Diagnostic accuracy and practice effects in the National Alzheimer’s Coordinating Center Uniform Data Set neuropsychological battery. Alzheimer’s Dement. 2014;10:675–83. doi: 10.1016/j.jalz.2013.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Folstein MF, Folstein SE, McHugh PR. “Mini-mental state”. A practical method for grading the cognitive state of patients for the clinician. J Psych Res. 1975;12:189–98. doi: 10.1016/0022-3956(75)90026-6. [DOI] [PubMed] [Google Scholar]
- 23.Nelson PT, Smith CD, Abner EA, et al. Human cerebral neuropathology of type 2 diabetes mellitus. Biochim Biophys Acta. 1792;2009:454–69. doi: 10.1016/j.bbadis.2008.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zarow C, Wang L, Chui HC, et al. MRI shows more severe hippocampal atrophy and shape deformation in hippocampal sclerosis than in Alzheimer’s disease. Int J Alzheimer’s Dis. 2011;2011:483972. doi: 10.4061/2011/483972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Thom M. Hippocampal sclerosis: progress since Sommer. Brain Pathol. 2009;19:565–72. doi: 10.1111/j.1750-3639.2008.00201.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Attems J, Jellinger KA. Hippocampal sclerosis in Alzheimer disease and other dementias. Neurology. 2006;66:775. doi: 10.1212/01.wnl.0000200959.50898.26. [DOI] [PubMed] [Google Scholar]
- 27.Kuslansky G, Verghese J, Dickson D, et al. Hippocampal sclerosis: cognitive consequences and contribution to dementia. Neurology. 2004;62:A128–9. [Google Scholar]
- 28.Zabar Y, Carson KA, Troncoso JC, et al. Dementia due to hippocampal sclerosis: clinical features and comparison to Alzheimer’s disease. Neurology. 1998;50:A59–60. [Google Scholar]
- 29.Zarow C, Weiner MW, Ellis WG, et al. Prevalence, laterality, and comorbidity of hippocampal sclerosis in an autopsy sample. Brain Behav. 2012;2:435–42. doi: 10.1002/brb3.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hatanpaa KJ, Raisanen JM, Herndon E, et al. Hippocampal sclerosis in dementia, epilepsy, and ischemic injury: differential vulnerability of hippocampal subfields. J Neuropathol Exp Neurol. 2014;73:136–42. doi: 10.1097/OPX.0000000000000170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Robinson JL, Geser F, Corrada MM, et al. Neocortical and hippocampal amyloid-beta and tau measures associate with dementia in the oldest-old. Brain. 2011;134:3708–15. doi: 10.1093/brain/awr308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nelson PT, Abner EL, Schmitt FA, et al. Modeling the association between 43 different clinical and pathological variables and the severity of cognitive impairment in a large autopsy cohort of elderly persons. Brain Pathol. 2010;20:66–79. doi: 10.1111/j.1750-3639.2008.00244.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Murray ME, Cannon A, Graff-Radford NR, et al. Differential clinicopathologic and genetic features of late-onset amnestic dementias. Acta Neuropathol. 2014;128:411–21. doi: 10.1007/s00401-014-1302-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Josephs KA, Whitwell JL, Weigand SD, et al. TDP-43 is a key player in the clinical features associated with Alzheimer’s disease. Acta Neuropathol. 2014;127:811–24. doi: 10.1007/s00401-014-1269-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wilson RS, Yu L, Trojanowski JQ, et al. TDP-43 pathology, cognitive decline, and dementia in old age. JAMA Neurol. 2013;70:1418–24. doi: 10.1001/jamaneurol.2013.3961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Aoki N, Murray ME, Ogaki K, et al. Hippocampal sclerosis in Lewy body disease is a TDP-43 proteinopathy similar to FTLD-TDP Type A. Acta Neuropathol. 2015;129:53–64. doi: 10.1007/s00401-014-1358-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dickson DW, Baker M, Rademakers R. Common variant in GRN is a genetic risk factor for hippocampal sclerosis in the elderly. Neurodegener Dis. 2010;7:170–4. doi: 10.1159/000289231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Probst A, Taylor KI, Tolnay M. Hippocampal sclerosis dementia: a reappraisal. Acta Neuropathol. 2007;114:335–45. doi: 10.1007/s00401-007-0262-1. [DOI] [PubMed] [Google Scholar]
- 39.Blass DM, Hatanpaa KJ, Brandt J, et al. Dementia in hippocampal sclerosis resembles frontotemporal dementia more than Alzheimer disease. Neurology. 2004;63:492–7. doi: 10.1212/01.wnl.0000133008.89613.82. [DOI] [PubMed] [Google Scholar]
- 40.Rauramaa T, Pikkarainen M, Englund E, et al. Consensus recommendations on pathologic changes in the hippocampus: a postmortem multicenter inter-rater study. J Neuropathol Exp Neurol. 2013;72:452–61. doi: 10.1097/NEN.0b013e318292492a. [DOI] [PubMed] [Google Scholar]
- 41.Hatanpaa KJ, Blass DM, Pletnikova O, et al. Most cases of dementia with hippocampal sclerosis may represent frontotemporal dementia. Neurology. 2004;63:538–42. doi: 10.1212/01.wnl.0000129543.46734.c0. [DOI] [PubMed] [Google Scholar]
- 42.Hatanpaa KJ, Bigio EH, Cairns NJ, et al. TAR DNA-binding protein 43 immunohistochemistry reveals extensive neuritic pathology in FTLD-U: a midwest-southwest consortium for FTLD study. J Neuropathol Exp Neurol. 2008;67:271–9. doi: 10.1097/NEN.0b013e31816a12a6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nelson PT, Estus S, Abner EL, et al. ABCC9 gene polymorphism is associated with hippocampal sclerosis of aging pathology. Acta Neuropathol. 2014;127:825–43. doi: 10.1007/s00401-014-1282-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nelson PT, Wang WX, Partch AB, et al. Reassessment of risk genotypes (GRN, TMEM106B, and ABCC9 variants) associated with hippocampal sclerosis of aging pathology. J Neuropathol Exp Neurol. 2015;74:75–84. doi: 10.1097/NEN.0000000000000151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nicholson AM, Finch NA, Wojtas A, et al. TMEM106B p. T185S regulates TMEM106B protein levels: implications for frontotemporal dementia. J Neurochem. 2013;126:781–91. doi: 10.1111/jnc.12329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yu L, De Jager PL, Yang J, et al. The TMEM106B locus and TDP-43 pathology in older persons without FTLD. Neurology. 2015;84:927–34. doi: 10.1212/WNL.0000000000001313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Neary D, Snowden JS, Gustafson L, et al. Frontotemporal lobar degeneration: a consensus on clinical diagnostic criteria. Neurology. 1998;51:1546–54. doi: 10.1212/wnl.51.6.1546. [DOI] [PubMed] [Google Scholar]
- 48.Snowden JS, Rollinson S, Thompson JC, et al. Distinct clinical and pathological characteristics of frontotemporal dementia associated with C9ORF72 mutations. Brain. 2012;135:693–708. doi: 10.1093/brain/awr355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mendez MF, Joshi A, Tassniyom K, et al. Clinicopathologic differences among patients with behavioral variant frontotemporal dementia. Neurology. 2013;80:561–8. doi: 10.1212/WNL.0b013e3182815547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Walker AK, Daniels CM, Goldman JE, et al. Astrocytic TDP-43 pathology in Alexander disease. J Neurosci. 2014;34:6448–58. doi: 10.1523/JNEUROSCI.0248-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ling H, Holton JL, Lees AJ, et al. TDP-43 pathology is present in most post-encephalitic parkinsonism brains. Neuropathol Appl Neurobiol. 2014;40:654–7. doi: 10.1111/nan.12067. [DOI] [PubMed] [Google Scholar]
- 52.McKee AC, Gavett BE, Stern RA, et al. TDP-43 proteinopathy and motor neuron disease in chronic traumatic encephalopathy. J Neuropathol Exp Neurol. 2010;69:918–29. doi: 10.1097/NEN.0b013e3181ee7d85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Davidson YS, Raby S, Foulds PG, et al. TDP-43 pathological changes in early onset familial and sporadic Alzheimer’s disease, late onset Alzheimer’s disease and Down’s syndrome: association with age, hippocampal sclerosis and clinical phenotype. Acta Neuropathol. 2011;122:703–13. doi: 10.1007/s00401-011-0879-y. [DOI] [PubMed] [Google Scholar]
- 54.Lippa CF, Rosso AL, Stutzbach LD, et al. Transactive response DNA-binding protein 43 burden in familial Alzheimer disease and Down syndrome. Arch Neurol. 2009;66:1483–8. doi: 10.1001/archneurol.2009.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Onyike CU, Pletnikova O, Sloane KL, et al. Hippocampal sclerosis dementia: an amnesic variant of frontotemporal degeneration. Dementia Neuropsychol. 2013;7:83–7. doi: 10.1590/S1980-57642013DN70100013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Schneider JA, Aggarwal NT, Barnes L, et al. The neuropathology of older persons with and without dementia from community versus clinic cohorts. J Alzheimer’s Dis. 2009;18:691–701. doi: 10.3233/JAD-2009-1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sonnen JA, Larson EB, Crane PK, et al. Pathological correlates of dementia in a longitudinal, population-based sample of aging. Ann Neurol. 2007;62:406–13. doi: 10.1002/ana.21208. [DOI] [PubMed] [Google Scholar]
- 58.Tschanz JT, Treiber K, Norton MC, et al. A population study of Alzheimer’s disease: findings from the Cache County Study Group. Care Manag J. 2005;6:107–14. doi: 10.1891/cmaj.6.2.107. Summer. [DOI] [PubMed] [Google Scholar]
- 59.Giasson BI, Forman MS, Higuchi M, et al. Initiation and synergistic fibrillization of tau and alpha-synuclein. Science. 2003;300:636–40. doi: 10.1126/science.1082324. [DOI] [PubMed] [Google Scholar]
- 60.Lippa CF, Fujiwara H, Mann DM, et al. Lewy bodies contain altered alpha-synuclein in brains of many familial Alzheimer’s disease patients with mutations in presenilin and amyloid precursor protein genes. Am J Pathol. 1998;153:1365–70. doi: 10.1016/s0002-9440(10)65722-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rosenberg CK, Pericak-Vance MA, Saunders AM, et al. Lewy body and Alzheimer pathology in a family with the amyloid-beta precursor protein APP717 gene mutation. Acta Neuropathol. 2000;100:145–52. doi: 10.1007/s004019900155. [DOI] [PubMed] [Google Scholar]
- 62.Barker WW, Luis CA, Kashuba A, et al. Relative frequencies of Alzheimer disease, Lewy body, vascular and frontotemporal dementia, and hippocampal sclerosis in the State of Florida Brain Bank. Alzheimer Dis Assoc Disord. 2002;16:203–12. doi: 10.1097/00002093-200210000-00001. [DOI] [PubMed] [Google Scholar]
- 63.White L, Petrovitch H, Hardman J, et al. Cerebrovascular pathology and dementia in autopsied Honolulu-Asia Aging Study participants. Ann NY Acad Sci. 2002;977:9–23. doi: 10.1111/j.1749-6632.2002.tb04794.x. [DOI] [PubMed] [Google Scholar]
- 64.Schneider JA, Arvanitakis Z, Bang W, et al. Mixed brain pathologies account for most dementia cases in community-dwelling older persons. Neurology. 2007;69:2197–204. doi: 10.1212/01.wnl.0000271090.28148.24. [DOI] [PubMed] [Google Scholar]
- 65.Bennett DA, Schneider JA, Arvanitakis Z, et al. Neuropathology of older persons without cognitive impairment from two community-based studies. Neurology. 2006;66:1837–44. doi: 10.1212/01.wnl.0000219668.47116.e6. [DOI] [PubMed] [Google Scholar]
- 66.Troncoso JC, Kawas CH, Chang CK, et al. Lack of association of the apoE4 allele with hippocampal sclerosis dementia. Neurosci Lett. 1996;204:138–40. doi: 10.1016/0304-3940(96)12331-4. [DOI] [PubMed] [Google Scholar]
- 67.Uchikado H, Lin WL, DeLucia MW, et al. Alzheimer disease with amygdala Lewy bodies: a distinct form of alpha-synucleinopathy. J Neuropathol Exp Neurol. 2006;65:685–97. doi: 10.1097/01.jnen.0000225908.90052.07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Neltner JH, Abner EL, Baker S, et al. Arteriolosclerosis that affects multiple brain regions is linked to hippocampal sclerosis of ageing. Brain. 2014;137:255–67. doi: 10.1093/brain/awt318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Beecham GW, Hamilton K, Naj AC, et al. Genome-wide association meta-analysis of neuropathologic features of Alzheimer’s disease and related dementias. PLoS Genet. 2014;10:e1004606. doi: 10.1371/journal.pgen.1004606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Nichols CG, Singh GK, Grange DK. KATP channels and cardiovascular disease: suddenly a syndrome. Circulation Res. 2013;112:1059–72. doi: 10.1161/CIRCRESAHA.112.300514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Flagg TP, Enkvetchakul D, Koster JC, et al. Muscle KATP channels: recent insights to energy sensing and myoprotection. Physiol Rev. 2010;90:799–829. doi: 10.1152/physrev.00027.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Gribble FM, Reimann F. Differential selectivity of insulin secretagogues: mechanisms, clinical implications, and drug interactions. J Diabetes Complications. 2003;17(2 Suppl):11–5. doi: 10.1016/s1056-8727(02)00272-6. [DOI] [PubMed] [Google Scholar]
- 73.Lawson K. Potassium channel openers as potential therapeutic weapons in ion channel disease. Kidney Internat. 2000;57:838–45. doi: 10.1046/j.1523-1755.2000.00923.x. [DOI] [PubMed] [Google Scholar]
- 74.Josephs KA, Murray ME, Whitwell JL, et al. Staging TDP-43 pathology in Alzheimer’s disease. Acta Neuropathol. 2014;127:441–50. doi: 10.1007/s00401-013-1211-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Knopman DS, Roberts RO. Estimating the number of persons with frontotemporal lobar degeneration in the US population. J Mol Neurosci. 2011;45:330–5. doi: 10.1007/s12031-011-9538-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Nag S, Yu L, Capuano AW, et al. Hippocampal sclerosis and TDP-43 pathology in aging and Alzheimer disease. Ann Neurol. doi: 10.1002/ana.24388. [epub ahead of print February 23 2015] [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.