

Abstract
Background
Upregulation of voltage-gated-calcium-channel α2δ1 subunit post spinal nerve ligation injury (SNL) or in α2δ1-overexpressing transgenic (Tg) mice correlates with tactile allodynia, a pain state mediated mainly by Aβ sensory fibers forming synaptic connections with deep dorsal horn neurons. It is not clear however whether dysregulated α2δ1 alters deep dorsal horn synaptic neurotransmission that underlies tactile allodynia development post nerve injury.
Methods
Tactile allodynia was tested in the SNL and α2δ1 Tg models. Miniature excitatory/inhibitory postsynaptic currents were recorded in deep dorsal horn (DDH) neurons from these animal models using whole cell patch clamp slice recording techniques..
Results
There was a significant increase in the frequency, but not amplitude, of miniature excitatory postsynaptic currents (mEPSC) in DDH neurons that correlated with tactile allodynia in SNL and α2δ1 Tg mice. Gabapentin, an α2δ1 ligand that is known to block tactile allodynia in these models, also normalized mEPSC frequency dose-dependently in DDH neurons from SNL and α2δ1 Tg mice. In contrast, neither frequency nor amplitude of miniature inhibitory postsynaptic currents (mIPSC) was altered in DDH neurons from SNL and α2δ1 Tg mice.
Conclusion
Our data suggest that α2δ1 dysregulation is highly likely contributing to tactile allodynia through a pre-synaptic mechanism involving facilitation of excitatory synaptic neurotransmission in deep dorsal horn of spinal cord.
Introduction
Neuropathic pain, or pain states derived from injuries of the peripheral or central nervous system, usually manifests hypersensitivity to innocuous (allodynia) or noxious (hyperalgesia) stimulation (Woolf & Mannion, 1999; Zimmermann, 2001; Costigan et al., 2009; Baron et al., 2010). It remains as an unmet clinical problem because current treatments are not effective for many patients, and often associated with severe side effects. Development of the next generation of medications for neuropathic pain management relies on our better understanding of neuropathic pain mechanisms.
Dysregulation of the voltage-gated-calcium-channel (VGCC) α2δ1 subunit (α2δ1) in sensory pathways is believed to play a role in mediating neuropathic pain states. α2δ1 is one of auxiliary subunits of VGCC that modulate calcium channel properties in vivo and in vitro (Tanabe et al., 1987; Klugbauer et al., 2003; Yaksh, 2006; Park & Luo, 2010). Spinal nerve ligation (SNL) injury induces α2δ1 upregulation in dorsal root ganglia and associated segments of dorsal spinal cord that contributes to pain modulation (Luo, 2000; Luo et al., 2001; Li et al., 2004; Li et al., 2006; Bauer et al., 2009; Nguyen et al., 2009). Blocking this injury-induced neuroplasticity by intrathecal treatments with α2δ1 antisense oligodeoxynucleotide or gabapentin (GBP), a ligand for α2δ1, has been shown to block spinal nerve injury induced pain states (Luo et al., 2002; Li et al., 2004; Bauer et al., 2009). Moreover, α2δ1 overexpression in transgenic (Tg) mice mimics behavioral hypersensitivities observed in the SNL model (Luo et al., 2002; Li et al., 2006; Nguyen et al., 2009). Together, it is highly likely that increased α2δ1 expression contributes to behavioral hypersensitivities post nerve injury.
Importantly, central sensitization is a common mechanism of different chronic pain states (Woolf & Salter, 2000). Factors contributing to central sensitization may include enhanced primary afferent excitability (Gracely et al., 1992; Matzner & Devor, 1994), glutamatergic input to dorsal spinal cord (Kohno et al., 2003; Wang et al., 2007), or impaired spinal inhibitory transmission (Moore et al., 2002b; Baba et al., 2003; Coull et al., 2003). We recently found that α2δ1 upregulation post SNL enhances excitatory pre-synaptic input into superficial dorsal horn (SDH, laminae I–II) that contributes to pain states transmitted through nociceptive sensory afferents such as myelinated Aδ- and unmyelinated C-type sensory afferents (Zhou & Luo, 2014). However, it is not known if similar changes also occur in deep dorsal horn (DDH, laminae III–V), where neurons receive mainly innocuous tactile input from large Aβ-sensory afferents. Maladaptive changes of synaptic neurotransmission in DDH post injury may also contribute to specific pain states such as tactile allodynia (Brown, 1982; Basbaum et al., 2009; Todd, 2010).
In this study, we examined the miniature excitatory (mEPSC) and inhibitory (mIPSC) post-synaptic currents in DDH neurons from spinal cord slices of SNL mice with tactile allodynia (Kim & Chung, 1992). In order to determine if injury-induced changes, if any, are modulated by elevated α2δ1, but not other injury factors, we compared the data with that from injury-free α2δ1 Tg mice with similar tactile allodynia (Li et al., 2006).
Methods and materials
Animals
Adult male mice (8–12 wks) with 129sv background for sham and SNL procedures were purchased from Charles River laboratories, Inc. (Wilmington, MA). The α2δ1 Tg mice were generated and characterized as described in our previous studies (Li et al., 2006). Briefly, a neuronal specific thy-1 promoter, derived from deletion of exon 3 and its flanking introns from the murine thy-1.2 gene (Vidal et al., 1990), was used to drive over expression of the mouse brain α2δ1 cDNA (Genbank accession number U73484) in neuronal tissues of the α2δ1 Tg mice (Feng et al., 2000). Even though the α2δ1 level in neuronal tissues of the Tg mice was compatible to that induced by nerve injury in the SNL model (Li et al., 2006), the α2δ1 Tg mice appeared fertile, physically normal with respect to grooming, social interactions, and feeding, and showed no detectable signs of abnormality such as motor defects, ataxia, tremor, or seizure. The Tg mice were backcrossed to the 129sv background for over 10 generations. Only age-matched (8–12 wks) male α2δ1 Tg and littermate wild-type (WT) mice were used for the experiments. All animal care and experiments were performed according to protocols approved by the Institutional Animal Care Committees of the University of California Irvine.
Spinal nerve ligation surgery
Unilateral SNL was performed as described by Kim and Chung (Kim & Chung, 1992). Briefly, mice were deeply anesthetized with air-mixed isoflurane (5% for induction, and 2% for maintenance). The left L4 spinal nerve, which is equivalent anatomically to L5 spinal nerve in rat (Rigaud et al., 2008), was exposed and ligated tightly with a 6.0 silk suture. We performed sham ligation by exposing the left L4 spinal nerve and loosely winding a suture around the nerve without ligation.
Von Frey filament stimulation
Paw withdrawal thresholds were determined by the up–down method (Dixon, 1980) using a set of von Frey monofilaments (Stoelting Co., Wood Dale, IL). Briefly, each mouse was habituated in a test compartment with a mesh floor for at least 30 min until exploratory behavior has stopped or decreased to a minimal level. The first von Frey filament (0.41g) was applied to the plantar surface of the hindpaw until it buckled slightly. If a withdrawal response was observed within 5 second, the next lower weight filament was used. Conversely, if the filament failed to elicit a withdrawal response, the next filament with a higher weight was applied. After the first change in response occurred, this paradigm continued until a total of six responses, starting from the one before the change in response, were recorded. In the case of four consecutive positive responses to filaments with decreasing weight, or three consecutive negative responses to filaments with increasing weight, a score of 0.01 g or 3.0 g, respectively, was assigned. These responses were used to calculate the 50% withdrawal threshold as described previously (Luo et al., 2001).
Spinal cord slice preparation
Isoflurane anesthetized mice were decapitated, and the L4 lumber region of the spinal cords was removed, transversely cut with a vibratome (VT-1200, Leica Inc.) into 300 μm slices in ice-cold modified, sucrose-based artificial cerebral spinal fluid (SACSF) saturated with 95% O2/5% CO2 (carbogen). The SACSF contained (mM): 250 sucrose, 2.5 KCl, 1.2 NaH2PO4, 1.2 MgCl2, 2.4 CaCl2, 26 NaHCO3 and 11 glucose. Slices were first incubated in SACSF at 31 °C for at least one hr before being kept at room temperature in carbogenated ACSF containing (mM) 126 NaCl, 2.5 KCl, 1.2 NaH2PO4, 1.2 MgCl2, 2.4 CaCl2, 26 NaHCO3 and 11 glucose for at least 30 min, then placed in a recording chamber and continually perfused with carbogen-saturated ACSF at a flow rate of 2.0 ml/min. 1μM tetrodotoxin (TTX) was applied continually when mEPSC/mIPSC were recorded.
Patch clamp recording
DDH neurons in slices were visualized with an upright microscope (Eclipse FN1, Nikon) with near-infrared illumination. All DDH recordings were made in the grey matter dorsal lateral to the central canal and at least 100 μm away from bottom of lamina II. The neuron locations were confirmed under low magnification after recording. The intracellular solution for recording mEPSC contains (mM): 135 potassium gluconate, 5 KCl, 5 EGTA, 0.5 CaCl2, 10 HEPES, 2 Mg-ATP, and 0.1 GTP; that for recordeding mIPSC contains (mM): 70 potassium gluconate, 65 KCl, 5 EGTA, 0.5 CaCl2, 10 HEPES, 2 Mg-ATP, 0.1 GTP. These solutions were adjusted to pH 7.2 with Tris-base, osmolarity 300 mOsm. The junction potential was nulled between the patch pipette and bath solution before gigaseal formation. Series resistance was monitored throughout the experiment without compensation (Multiclamp 700B) and changes in series resistance (15–30 MΩ) more than 20% during whole-cell recording would lead to data elimination. Electrophysiological signals were recorded with MultiClamp 700B amplifiers (Axon Instruments, Molecular Devices, Union City, CA), Digidata 1440 analog-to-digital converters (Axon Instruments) and pClamp 10.2 software (Axon Instruments). Data were sampled at 10 kHz and filtered at 2 kHz. All recordings were done at a temperature of 32 ± 0.5°C.
The slices were stabilized for 5 min, then mEPSC/mIPSC were recorded, counted and analyzed using clampfit 10.3 (Molecular Devices). For drug treatment, mEPSC frequency baseline mean values were obtained for 5 min during the control period, and the mean values for drug application were obtained over the peak drug response for 2 min. The mEPSC frequencies during and after drug applications were compared to their baseline mean values. Drug effects were expressed as percentage changes (mean ± SEM) over the baseline values.
Statistics analysis
All data were expressed as mean ± SEM. Differences between two groups were compared with student’s t-test. One-way or Two-way ANOVA analysis with Bonferroni post hoc test was used for multi-group comparisons where necessary. p < 0.05 was considered statistically significant.
Results
Upregulation of α2δ1 induces tactile allodynia
We first examined if tactile allodynia, shown as reduced hindpaw thresholds to light touch was manifested similarly in both SNL and α2δ1 Tg mice by measuring hindpaw sensitivity to von Frey filament stimuli in both models. In the SNL model, paw withdrawal thresholds were significantly decreased in the injury side of SNL mice within one week post injury while there was no significant change in the injury side of sham mice during the same period (Fig. 1A). The tactile allodynia state correlates with upregulation of α2δ1 proteins in dorsal root ganglia and associated spinal dorsal horn reported previously (Zhou & Luo, 2014). The time course of SNL-induced tactile allodynia was similar to that previously reported (Luo et al., 2001; Luo et al., 2002; Li et al., 2004). Similarly, α2δ1 overexpression in the Tg mice also caused tactile allodynia compared with their WT littermates (Fig. 1B) (Li et al., 2006). In combination with previous findings that blocking α2δ1 expression or activity with intrathecal α2δ1 antisense oligodeoxynucleotides, or the α2δ1 ligand gabapentin (GBP), respectively, reverses behavioral hypersensitivity in both SNL and α2δ1 Tg animals (Li et al., 2004; Li et al., 2006), these findings suggest that elevated spinal α2δ1 mediates tactile allodynia.
Figure 1. Similar tactile allodynia were observed in unilateral L4 SNL and α2δ1 Tg mice.
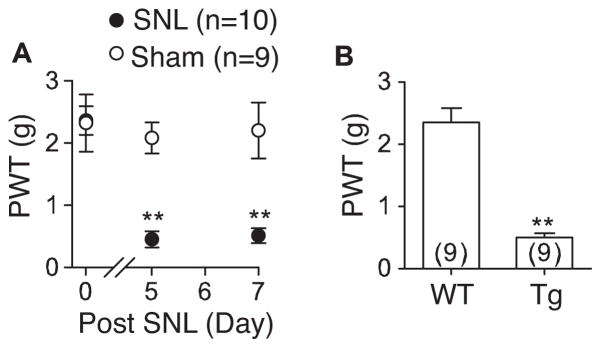
Tactile allodynia was measured by applying von Frey filaments to the plantar surface of the hindpaw as described. A. Hindpaw withdrawal thresholds (PWT) to light touch in the ipsilateral hindpaws of sham and SNL mice were tested in a blinded fashion at designated time points post surgery. B. Similarly tested adult α2δ1 Tg mice also showed decreased PWT compared with their WT littermates. Data presented are the means ± SEM from the number of animals indicated. **p < 0.01 compared with control values by one-way ANOVA test.
SNL or elevated α2δ1 enhanced excitatory synaptic transmission in DDH neurons through a pre-synapitc mechanism
To the best of our knowledge, the effects of SNL injury in DDH neuron synaptic transmission are not yet investigated or reported, which however could play a significant role in the processing of tactile allodynia. Accordingly, we examined mEPSC in deep dorsal horn neurons from L4 spinal cord slices of the surgery side (SNL or sham) 5–7 days post operation when SNL mice displayed allodynia. AP-5 (50 μM), biccuculine (10 μM), strychnine (1 μM), and tetrodotoxin (TTX 1μM) were added to block NMDA receptors, GABAA receptors, glycine receptors, and TTX-sensitive sodium channels, respectively. Bath application of 20 μM CNQX, a non-NMDA receptor antagonist, at the end of the recording abolished mEPSCs (data not shown), confirming that mEPSC from DDH neurons was mediated through AMPA/Kainate receptors.
Our data indicated that mEPSC frequency was increased in deep dorsal horn neurons from the injury side of SNL mice compared with that from sham control mice (Fig. 2A), resulting in a leftward shift in the cumulative probability curve of interevent intervals (Fig. 2B), but not amplitude (Fig. 2C) of mEPSC. These corresponded to a ~100% increase in mEPSC average frequency, likely reflecting increased glutamate release frequency from pre-synaptic sites (Fig. 2D), but no significant change in mEPSC amplitude (Fig. 2E), a measurement of post-synaptic neuron excitability. Importantly, GBP (10–100 μM) could reversibly normalize SNL-induced mEPSC frequency in DDH neurons dose-dependently without affecting that in sham mice (Fig. 2F and G). These data support that SNL enhanced excitatory synaptic transmission in DDH through a pre-synaptic mechanism.
Figure 2. L4 spinal nerve ligation in mice led to enhancement of mEPSC frequency in DDH neurons that could be blocked by gabapentin.

mEPSC from injury side of L4 deep dorsal horn neurons of sham and SNL mice was recorded 5 – 7 days post surgery. A. Representative traces of mEPSC from DDH neurons of sham and SNL mice, respectively. B and C. Cumulative probability curves of mEPSC interevent intervals (B) and amplitude (C) from Sham and SNL mice, respectively. p = 0.001 (k-s test) for a comparison of mEPSC interevent intervals between sham and SNL mice. p = 0.21 for a comparison of mEPSC amplitude between sham and SNL mice. D and E. Summary of mEPSC frequency (D) and amplitude (E) in L4 DDH neurons from injury side of SNL and sham mice. **p < 0.01 compared with sham L4 neurons by Student’s t-test. F. Time courses for the effects of 100μM GBP on mEPSC frequency from sham and SNL neurons. G. Dose-dependent normalization by GBP on mEPSC frequency on SNL neurons. *p < 0.05, **p < 0.01 compared with pre-GBP treatment; #p < 0.05 compared with 10μM treatment by one-way ANOVA test. Dotted line represents percentage of mEPSC frequency in sham mice shown in D. Summarized data are shown as the means ± SEM from the number of neurons indicated in the parentheses.
Since SNL caused dysregulation of a lot of genes in DRG and spinal cord (Wang et al., 2002; Valder et al., 2003), it was not clear if SNL-induced α2δ1 was responsible for the increased pre-synaptic excitatory input that correlated with tactile allodynia. We addressed this question by examining whether elevated α2δ1 alone without the influence from other injury factors is sufficient to enhance mEPSC frequency in DDH of the binjury-free α2δ1 Tg mice. Overexpression of α2δ1 in neuronal tissues, including spinal cord and DRG, in the α2δ1 Tg mice leads to similar behavioral hypersensitivity as that observed in the SNL model (Li et al., 2006), thus, the injury-free α2δ1 Tg mice could be a complementary model to the SNL model in investigating the role of α2δ1 upregulation in pain state processing. Similar to that in SNL mice, α2δ1 upregulation in the Tg mice resulted in a significant increase in frequency (Fig. 3A) of mEPSC with a leftward shift in cumulative probability curve of interevent interals (Fig. 3B), but no change in mEPSC amplitude (Fig. 3C) compared with that from the WT mice. These changes corresponded to a ~128% higher average mEPSC frequency (Fig. 3D), but no significant change in average mEPSC amplitude (Fig. 3E) in the Tg mice compared with that in WT mice. GBP (10–100μM) dose-dependently attenuated mEPSC frequency in DDH neurons from the Tg mice without affecting that from WT mice with the highest concentration tested (100μM) (Fig. 3F and G). In combination with similar findings from the SNL mice, these data support a role of elevated α2δ1 in enhancing excitatory synaptic transmission in DDH through a pre-synaptic mechanism.
Figure 3. Enhanced mEPSC in DDH neurons from α2δ1 Tg mice that could be blocked by gabapentin.

mEPSC from L4 DDH neurons of adult WT and α2δ1 Tg mice was recorded as described. A. Representative traces of mEPSC from WT and α2δ1 Tg DDH neurons, respectively. B and C. Cumulative probability curves of mEPSC interevent intervals (B) and amplitude (C) from WT and Tg DDH neurons, respectively. p = 0.001 (k-s test) for a comparison of mEPSC interevent intervals between WT and Tg mice. p = 0.53 for a comparison of mEPSC amplitude between WT and Tg mice. D and E. Summary data of average mEPSC frequency (D) and amplitude (E), respectively, from WT and Tg DDH neurons. **p < 0.01 compared with WT group by Student’s t-test. F. Time courses for the effects of 100μM GBP on mEPSC frequency from WT and Tg L4 DDH neurons. G. Dose-dependent normalization of mEPSC frequency in L4 Tg neurons by GBP. **p < 0.01 compared with pre-GBP treatment; #p < 0.01 compared with 10μM GBP treatment by one-way ANOVA test. Dotted line represents percentage of mEPSC frequency in sham mice shown in D. Summarized data are shown as the means ± SEM from the number of neurons indicated in the parentheses.
Unaltered inhibitory synaptic transmission in DDH neurons from SNL and α2δ1 Tg mice
Besides glutamatergic excitatory innervations, DDH neurons also receive GABAergic and Glycinergic innervations from inhibitory interneurons in spinal dorsal horn (Braz et al., 2014). To test if α2δ1 upregulation in SNL (5–7 days post injury) and α2δ1 Tg mice also affected inhibitory synaptic transmission in DDH neurons, we examined mIPSC in the presence of TTX (1μM) from L4 spinal slices of these models. During the recording, mEPSC was blocked with DNQX (20 μM) and APV (50 μM). The remaining mIPSC could be eliminated by bicuculline (10 μM) and strychnine (1 μM) (data not shown) confirming a suitable recording condition for examining mIPSC (Fig. 4A).
Figure 4. mIPSC was not altered in DDH neurons from SNL and α2δ1 Tg mice.

mIPSC from L4 dorsal horn neurons of WT and α2δ1 Tg mice or injury side of sham and SNL mice 5–7 days post surgery was recorded as described. A. Representative traces of mIPSC from a sham and a SNL DDH neuron, respectively. B, C. Cumulative probability curves of mIPSC interevent intervals and amplitude, respectively, from sham and SNL mice. D, E. Summary of average mIPSC frequency (D) and amplitude (E), respectively, from DDH neurons of sham and SNL mice. F, G. Cumulative probability curves of mIPSC interevent intervals (F) and amplitude (G), respectively, from WT and α2δ1 Tg DDH neurons. H, I. Summary of average mIPSC frequency (H) and amplitude (I), respectively, from WT and α2δ1 Tg DDH neurons. p > 0.1 (k-s test) for all the comparisons between sham and SNL as well as between WT and α2δ1 Tg mice.
Interestingly, there is no significant difference in mIPSC either between the SNL and Sham mice, or between the α2δ1 Tg and WT mice. The distribution of interevent intervals and amplitude of mIPSC did not shift in SNL mice compared with that from the sham mice (Fig. 4B, C), nor in α2δ1 Tg mice compared with that from the WT mice (Fig. 4F, G). The average mIPSC frequencies and amplitudes in DDH neurons were similar between SNL/sham (Fig. 4D, E) or Tg/WT mice (Fig. 4H, I). These data suggest that increased α2δ1 in neither the SNL nor the α2δ1 Tg mouse model affects inhibitory synaptic transmission in deep dorsal spinal cord.
Discussion
Peripheral nerve injury induces neuroplasticity changes in deep dorsal horn of spinal cord that may contribute to neuropathic allodynia, a painful state of hypersensitivity to innocuous light touch sensation, which is normally transmitted by Aβ-sensory afferents projecting to and forming synapse connections with DDH neurons (Braz et al., 2014). These changes may include, but are not limited to, dysregulation of genes (Dobremez et al., 2005), suppressing inhibitory and/or enhancing facilitatory descending modulations (Suzuki et al., 2004; Rahman et al., 2008), attenuated glutamate uptake (Binns et al., 2005), and increased pain-inducing peptides in DDH neurons (Nitzan-Luques et al., 2011). However, to the best of our knowledge, it is not yet known if peripheral nerve injury alters excitatory and/or inhibitory miniature synaptic transmission in deep dorsal horn, which can play a critical role in mediating neuropathic allodynia. We provide data here to indicate that spinal nerve ligation injury leads to increased frequency of mEPSC in deep dorsal horn neurons, which is gabapentin-reversible, and neuropathic tactile allodynia. Similar maladaptive changes in DDH mEPSC and tactile allodynia also occur in injury-free transgenic mice with α2δ1 overexpression in neuronal tissues. In contrast, neither spinal nerve ligation injury nor α2δ1 overexpression causes any significant change in DDH mIPSC. Together, these data support that peripheral nerve injury enhances excitatory pre-synaptic input into DDH neurons under non-stimulating conditions, most likely through α2δ1 upregulation, that may play a role in neuropathic allodynia.
Several lines of evidence support our conclusion. First, α2δ1 proteins are highly upregulated in DRG after SNL injury, then translocated to primary afferent pre-synaptic terminals in dorsal spinal cord during the development of neuropathic allodynia (Li et al., 2004; Bauer et al., 2009). Second, blocking this injury-induced neuroplasticity with intrathecal treatments of α2δ1 antisense oligodeoxynucleotides, or GBP prevents (Boroujerdi et al., 2008) and reverses (Luo et al., 2002; Li et al., 2004; Bauer et al., 2009) neuropathic allodynia. Third, α2δ1 overexpression in injury-free Tg mice is sufficient to induce behavioral hypersensitivity and dorsal horn neuron hyperexcitability with similar pharmacological profiles as that observed in the SNL model (Li et al., 2006; Chang et al., 2013; Chang et al., 2014; Zhou & Luo, 2014).
To our knowledge, this is the first study revealing maladaptive changes in DDH miniature synaptic transmission underlying nerve injury-induced mechanical allodynia. Even though we have recently reported that peripheral nerve injury induces similar changes in superficial dorsal horn (SDH) and behavioral nociceptions in SNL and α2δ1 Tg mice (Zhou & Luo, 2014), SDH and DDH have functional differences in mediating sensory signal processing since neurons in these locations receive distinct sensory input from different primary afferents (Light & Perl, 1979; Brown, 1982; Millan, 1999; Graham et al., 2007; Braz et al., 2014). The SDH neurons receive primary afferent inputs mainly from small myelinated Aδ and unmyelinated C-fibers carrying nociceptive, including thermal and cold, information. In contrast, DDH neurons receive inputs mainly from large Aβ myelinated fibers carrying tactile information and synaptic inputs from SDH neurons. Neuron populations and synaptic connections may also differ in superficial and deep laminae. Compared with a high percentage of nociceptive specific neurons in the superficial dorsal horn (lamina I–II), wide dynamic range (WDR) neurons in deep dorsal horn are responsive to a large range of sensory modalities (thermal, chemical and mechanical) and represent an important component in spinal sensory/pain transmission (Brown, 1982; Basbaum et al., 2009; Todd, 2010; Braz et al., 2014). WDR neurons exhibit a frequency-dependent, progressive increase in neuronal excitability in response to repeated electrical stimulation of afferent C-fibers (windup phenomenon) (Herrero et al., 2000). In addition, excitatory and inhibitory interneurons also form specific circuitries in SDH and DDH with projection, WDR neurons or between each other to modulate sensory information processing at the spinal cord level, which in turn, is subjected to descending modulation from the higher central nervous system (Braz et al., 2014).
Our previous data from the injury-free α2δ1 Tg mice indicate that elevated neuronal α2δ1 expression leads to exaggerated and prolonged WDR neuron firing in response to mechanical and thermal stimuli, but normal windup response to electrical stimuli, suggesting that elevated α2δ1 mediates DDH neuron aberrant activities and pain states through a mechanism independent of abnormal C-fiber stimulation (Li et al., 2006). Findings from this study indicate that increased mEPSC frequency is found in both SNL and α2δ1 Tg mice that have common correlates of elevated α2δ1 proteins and behavioral hypersensitivities (Luo et al., 2002; Li et al., 2004; Li et al., 2006). GBP can dose-dependently normalize aberrant mEPSC frequency in both models (Figs. 2F, 2G, 3F, 3G) in a dose-range achievable in plasma when GBP is given orally to patients (Ben-Menachem et al., 1992; Bryans & Wustrow, 1999). While GBP binds to both α2δ1 and α2δ2 proteins (Klugbauer et al., 2003), it is less likely that α2δ2 is involved in this process since SNL only induces DRG α2δ1, but not α2δ2, upregulation (Bauer et al., 2009) and GBP is not affecting mEPSC frequency in sham and WT control mice (Figs. 2F, 3F). This is consistent with previous studies suggesting that GBP only exhibits therapeutic effect under pathological conditions (Ben-Menachem et al., 1992; Field et al., 1997; Stanfa et al., 1997; Bryans & Wustrow, 1999; Moore et al., 2002a). The fast action of GBP on mEPSC frequency supports its effects on synaptic transmission, rather than trafficking of VGCC (Hendrich et al., 2008). In addition, GBP at this dose-range is able to inhibit evoked Ca2+ influx in cortical synaptosomal suspensions (Fink et al., 2000; Meder & Dooley, 2000), and excitatory synaptic transmission in hyperalgesic spinal cord (Patel et al., 2000). Together, these findings support that peripheral nerve injury-induced α2δ1 mediates neuropathic allodynia at least at the spinal cord level by enhancing excitatory pre-synaptic input into deep dorsal horn.
The mechanism underlying α2δ1 modulation on excitatory synaptic transmission in deep dorsal spinal cord reminds elusive. A large body of emerging evidence indicates that α2δ1 subunit is a multifunctional protein. It not only regulates VGCC functions, but is also critical for VGCC-independent functions. Since miniature glutamate release does not require Ca2+ influx into pre-synaptic terminals, α2δ1 may have other regulatory functions or interactions with pre-synaptic proteins involved in synaptic neurotransmission. Data from recent studies show that α2δ1 is the receptor for thrombospondin, an extracellular matrix protein secreted by astrocytes, in promoting excitatory synaptogenesis in the CNS, which is GBP sensitive, but VGCC-independent (Eroglu et al., 2009). In addition, α2δ1 interacts with a serotonergic descending facilitation pathway at the spinal level that is critical in mediating central sensitization and neuropathic allodynia/thermal hyperalgesia (Chang et al., 2013), as well as GBP efficacy in reversing pain states (Suzuki et al., 2005). Therefore, it is possible that α2δ1 modulation on mEPSC frequency requires integration of multiple pathways that could be GBP-sensitive, but VGCC-independent. This is supported by recent findings that α2δ1 modulates EPSC frequency in ventromedial hypothalamus neurons through a VGCC-independent mechanism that underlies GBP-induced weight gain (Cordeira et al., 2014).
The contribution of spinal dorsal horn inhibitory circuitry to pain processing is equally important as that from excitatory circuitry. However, findings related to contribution of spinal inhibitory circuitry to neuropathic pain state processing remain inconsistent. While suppression of inhibitory synaptic transmission in the dorsal spinal cord has been reported in certain chronic pain states such as tactile allodynia and hyperalgesia (Yaksh, 1989; Kontinen et al., 2001), other studies have shown that neither GABAergic nor glycinergic IPSC is changed in neuropathic rodents (Polgar et al., 2003; Polgar et al., 2005; Wang et al., 2007), which is similar to our findings that behavioral hypersensitivity in neither the SNL nor α2δ1 Tg mice correlates with any change in DDH mIPSC (Fig. 4). This discrepancy could be due to the differences in experimental conditions, neuron sampling locations, animal stains and pain models. In addition, our data cannot exclude the possibility that evoked IPSC, but not mIPSC, is altered in these allodynic models. Nevertheless, our findings provide new evidences indicating that α2δ1 upregulation in spinal cord differentially modulate excitatory and inhibitory synaptic transmission.
In summary, our data indicate that peripheral nerve injury can enhance excitatory synaptic neurotransmission in DDH by enhancing mEPSC through a pre-synaptic mechanism, most likely mediated by elevated α2δ1 expression, which in turn contributes to neuropathic allodynia. Blocking this maladaptive change post nerve injury may provide an alternative strategy in managing modality specific nociception.
Acknowledgments
Funding Sources: This study was supported in part by grants NS40135, NS064341 and DE021847 from the National Institutes of Health (Z.D. Luo).
Footnotes
Author Contributions:
In addition to the following contributions from each author, both authors discussed the results, commented on the manuscript and approved for the submission.
C.Z. contributed to conception, design of the study, data acquisition, analysis, and interpretation, drafting, editing the manuscript.
Z.D.L. contributed to conception, design and overall supervision of the study. He also performed data analysis, interpretation, drafting and revising the manuscript.
- It is known that elevated α2δ1 proteins in spinal cord contribute to nerve injury induced neuropathic pain states.
- Findings from this study support that elevated spinal α2δ1 proteins increase excitatory pre-synaptic input to deep dorsal horn neurons, underlying the development of neuropathic allodynia post nerve injury.
References
- Baba H, Ji RR, Kohno T, Moore KA, Ataka T, Wakai A, Okamoto M, Woolf CJ. Removal of GABAergic inhibition facilitates polysynaptic A fiber-mediated excitatory transmission to the superficial spinal dorsal horn. Mol Cell Neurosci. 2003;24:818–830. doi: 10.1016/s1044-7431(03)00236-7. [DOI] [PubMed] [Google Scholar]
- Baron R, Binder A, Wasner G. Neuropathic pain: diagnosis, pathophysiological mechanisms, and treatment. Lancet Neurol. 2010;9:807–819. doi: 10.1016/S1474-4422(10)70143-5. [DOI] [PubMed] [Google Scholar]
- Basbaum AI, Bautista DM, Scherrer G, Julius D. Cellular and molecular mechanisms of pain. Cell. 2009;139:267–284. doi: 10.1016/j.cell.2009.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer CS, Nieto-Rostro M, Rahman W, Tran-Van-Minh A, Ferron L, Douglas L, Kadurin I, Sri Ranjan Y, Fernandez-Alacid L, Millar NS, Dickenson AH, Lujan R, Dolphin AC. The increased trafficking of the calcium channel subunit alpha2delta-1 to presynaptic terminals in neuropathic pain is inhibited by the alpha2delta ligand pregabalin. J Neurosci. 2009;29:4076–4088. doi: 10.1523/JNEUROSCI.0356-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Menachem E, Persson LI, Hedner T. Selected CSF biochemistry and gabapentin concentrations in the CSF and plasma in patients with partial seizures after a single oral dose of gabapentin. Epilepsy Res. 1992;11:45–49. doi: 10.1016/0920-1211(92)90020-t. [DOI] [PubMed] [Google Scholar]
- Binns BC, Huang Y, Goettl VM, Hackshaw KV, Stephens RL., Jr Glutamate uptake is attenuated in spinal deep dorsal and ventral horn in the rat spinal nerve ligation model. Brain Res. 2005;1041:38–47. doi: 10.1016/j.brainres.2005.01.088. [DOI] [PubMed] [Google Scholar]
- Boroujerdi A, Kim HK, Lyu YS, Kim DS, Figueroa KW, Chung JM, Luo ZD. Injury discharges regulate calcium channel alpha-2-delta-1 subunit upregulation in the dorsal horn that contributes to initiation of neuropathic pain. Pain. 2008;139:358–366. doi: 10.1016/j.pain.2008.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braz J, Solorzano C, Wang X, Basbaum AI. Transmitting pain and itch messages: a contemporary view of the spinal cord circuits that generate gate control. Neuron. 2014;82:522–536. doi: 10.1016/j.neuron.2014.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown AG. The dorsal horn of the spinal cord. Q J Exp Physiol. 1982;67:193–212. doi: 10.1113/expphysiol.1982.sp002630. [DOI] [PubMed] [Google Scholar]
- Bryans JS, Wustrow DJ. 3-substituted GABA analogs with central nervous system activity: a review. Med Res Rev. 1999;19:149–177. doi: 10.1002/(sici)1098-1128(199903)19:2<149::aid-med3>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- Chang E, Chen X, Kim M, Gong N, Bhatia S, Luo ZD. Differential Effects of Voltage-Gated Calcium Channel Blockers on Calcium Channel Alpha-2-Delta-1 Subunit Protein Mediated Nociception. Eur J Pain. 2014 doi: 10.1002/ejp.585. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang EY, Chen X, Sandhu A, Li CY, Luo ZD. Spinal 5-HT3 receptors facilitate behavioural hypersensitivity induced by elevated calcium channel alpha-2-delta-1 protein. Eur J Pain. 2013;17:505–513. doi: 10.1002/j.1532-2149.2012.00221.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordeira JW, Felsted JA, Teillon S, Daftary S, Panessiti M, Wirth J, Sena-Esteves M, Rios M. Hypothalamic dysfunction of the thrombospondin receptor alpha2delta-1 underlies the overeating and obesity triggered by brain-derived neurotrophic factor deficiency. J Neurosci. 2014;34:554–565. doi: 10.1523/JNEUROSCI.1572-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costigan M, Scholz J, Woolf CJ. Neuropathic pain: a maladaptive response of the nervous system to damage. Annu Rev Neurosci. 2009;32:1–32. doi: 10.1146/annurev.neuro.051508.135531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coull JA, Boudreau D, Bachand K, Prescott SA, Nault F, Sik A, De Koninck P, De Koninck Y. Trans-synaptic shift in anion gradient in spinal lamina I neurons as a mechanism of neuropathic pain. Nature. 2003;424:938–942. doi: 10.1038/nature01868. [DOI] [PubMed] [Google Scholar]
- Dixon WJ. Efficient analysis of experimental observations. Annu Rev Pharmacol Toxicol. 1980;20:441–462. doi: 10.1146/annurev.pa.20.040180.002301. [DOI] [PubMed] [Google Scholar]
- Dobremez E, Bouali-Benazzouz R, Fossat P, Monteils L, Dulluc J, Nagy F, Landry M. Distribution and regulation of L-type calcium channels in deep dorsal horn neurons after sciatic nerve injury in rats. Eur J Neurosci. 2005;21:3321–3333. doi: 10.1111/j.1460-9568.2005.04177.x. [DOI] [PubMed] [Google Scholar]
- Eroglu C, Allen NJ, Susman MW, O’Rourke NA, Park CY, Ozkan E, Chakraborty C, Mulinyawe SB, Annis DS, Huberman AD, Green EM, Lawler J, Dolmetsch R, Garcia KC, Smith SJ, Luo ZD, Rosenthal A, Mosher DF, Barres BA. Gabapentin receptor alpha2delta-1 is a neuronal thrombospondin receptor responsible for excitatory CNS synaptogenesis. Cell. 2009;139:380–392. doi: 10.1016/j.cell.2009.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng G, Mellor RH, Bernstein M, Keller-Peck C, Nguyen QT, Wallace M, Nerbonne JM, Lichtman JW, Sanes JR. Imaging Neuronal Subsets in Transgenic Mice Expressing Multiple Spectral Variants of GFP. Neuron. 2000;28:41–51. doi: 10.1016/s0896-6273(00)00084-2. [DOI] [PubMed] [Google Scholar]
- Field MJ, Oles RJ, Lewis AS, McCleary S, Hughes J, Singh L. Gabapentin (neurontin) and S-(+)-3-isobutylgaba represent a novel class of selective antihyperalgesic agents. Brit J Pharmacol. 1997;121:1513–1522. doi: 10.1038/sj.bjp.0701320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fink K, Meder W, Dooley DJ, Gothert M. Inhibition of neuronal Ca(2+) influx by gabapentin and subsequent reduction of neurotransmitter release from rat neocortical slices. Br J Pharmacol. 2000;130:900–906. doi: 10.1038/sj.bjp.0703380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gracely RH, Lynch SA, Bennett GJ. Painful neuropathy: altered central processing maintained dynamically by peripheral input. Pain. 1992;51:175–194. doi: 10.1016/0304-3959(92)90259-E. [DOI] [PubMed] [Google Scholar]
- Graham BA, Brichta AM, Callister RJ. Moving from an averaged to specific view of spinal cord pain processing circuits. J Neurophysiol. 2007;98:1057–1063. doi: 10.1152/jn.00581.2007. [DOI] [PubMed] [Google Scholar]
- Hendrich J, Van Minh AT, Heblich F, Nieto-Rostro M, Watschinger K, Striessnig J, Wratten J, Davies A, Dolphin AC. Pharmacological disruption of calcium channel trafficking by the alpha2delta ligand gabapentin. Proc Natl Acad Sci U S A. 2008;105:3628–3633. doi: 10.1073/pnas.0708930105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrero JF, Laird JM, Lopez-Garcia JA. Wind-up of spinal cord neurones and pain sensation: much ado about something? Prog Neurobiol. 2000;61:169–203. doi: 10.1016/s0301-0082(99)00051-9. [DOI] [PubMed] [Google Scholar]
- Kim SH, Chung JM. An experimental model for peripheral neuropathy produced by segmental spinal nerve ligation in the rat. Pain. 1992;50:355–363. doi: 10.1016/0304-3959(92)90041-9. [DOI] [PubMed] [Google Scholar]
- Klugbauer N, Marais E, Hofmann F. Calcium channel alpha2delta subunits: differential expression, function, and drug binding. J Bioenerg Biomembr. 2003;35:639–647. doi: 10.1023/b:jobb.0000008028.41056.58. [DOI] [PubMed] [Google Scholar]
- Kohno T, Moore KA, Baba H, Woolf CJ. Peripheral nerve injury alters excitatory synaptic transmission in lamina II of the rat dorsal horn. J Physiol. 2003;548:131–138. doi: 10.1113/jphysiol.2002.036186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kontinen VK, Stanfa LC, Basu A, Dickenson AH. Electrophysiologic evidence for increased endogenous gabaergic but not glycinergic inhibitory tone in the rat spinal nerve ligation model of neuropathy. Anesthesiology. 2001;94:333–339. doi: 10.1097/00000542-200102000-00024. [DOI] [PubMed] [Google Scholar]
- Li CY, Song YH, Higuera ES, Luo ZD. Spinal dorsal horn calcium channel alpha2delta-1 subunit upregulation contributes to peripheral nerve injury-induced tactile allodynia. J Neurosci. 2004;24:8494–8499. doi: 10.1523/JNEUROSCI.2982-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li CY, Zhang XL, Matthews EA, Li KW, Kurwa A, Boroujerdi A, Gross J, Gold MS, Dickenson AH, Feng G, Luo ZD. Calcium channel alpha2delta1 subunit mediates spinal hyperexcitability in pain modulation. Pain. 2006;125:20–34. doi: 10.1016/j.pain.2006.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Light AR, Perl ER. Reexamination of the dorsal root projection to the spinal dorsal horn including observations on the differential termination of coarse and fine fibers. J Comp Neurol. 1979;186:117–131. doi: 10.1002/cne.901860202. [DOI] [PubMed] [Google Scholar]
- Luo ZD. Rat dorsal root ganglia express distinctive forms of the alpha2 calcium channel subunit. Neuroreport. 2000;11:3449–3452. doi: 10.1097/00001756-200011090-00010. [DOI] [PubMed] [Google Scholar]
- Luo ZD, Calcutt NA, Higuera ES, Valder CR, Song YH, Svensson CI, Myers RR. Injury type-specific calcium channel alpha 2 delta-1 subunit up-regulation in rat neuropathic pain models correlates with antiallodynic effects of gabapentin. J Pharmacol Exp Ther. 2002;303:1199–1205. doi: 10.1124/jpet.102.041574. [DOI] [PubMed] [Google Scholar]
- Luo ZD, Chaplan SR, Higuera ES, Sorkin LS, Stauderman KA, Williams ME, Yaksh TL. Upregulation of dorsal root ganglion (alpha)2(delta) calcium channel subunit and its correlation with allodynia in spinal nerve-injured rats. J Neurosci. 2001;21:1868–1875. doi: 10.1523/JNEUROSCI.21-06-01868.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matzner O, Devor M. Hyperexcitability at sites of nerve injury depends on voltage-sensitive Na+ channels. J Neurophysiol. 1994;72:349–359. doi: 10.1152/jn.1994.72.1.349. [DOI] [PubMed] [Google Scholar]
- Meder WP, Dooley DJ. Modulation of K(+)-induced synaptosomal calcium influx by gabapentin. Brain Res. 2000;875:157–159. doi: 10.1016/s0006-8993(00)02610-x. [DOI] [PubMed] [Google Scholar]
- Millan MJ. The induction of pain: an integrative review. Prog Neurobiol. 1999;57:1–164. doi: 10.1016/s0301-0082(98)00048-3. [DOI] [PubMed] [Google Scholar]
- Moore KA, Baba H, Woolf CJ. Gabapentin-- actions on adult superficial dorsal horn neurons. Neuropharmacology. 2002a;43:1077–1081. doi: 10.1016/s0028-3908(02)00226-5. [DOI] [PubMed] [Google Scholar]
- Moore KA, Kohno T, Karchewski LA, Scholz J, Baba H, Woolf CJ. Partial peripheral nerve injury promotes a selective loss of GABAergic inhibition in the superficial dorsal horn of the spinal cord. J Neurosci. 2002b;22:6724–6731. doi: 10.1523/JNEUROSCI.22-15-06724.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen D, Deng P, Matthews EA, Kim DS, Feng G, Dickenson AH, Xu ZC, Luo ZD. Enhanced pre-synaptic glutamate release in deep-dorsal horn contributes to calcium channel alpha-2-delta-1 protein-mediated spinal sensitization and behavioral hypersensitivity. Mol Pain. 2009;5:6. doi: 10.1186/1744-8069-5-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nitzan-Luques A, Devor M, Tal M. Genotype-selective phenotypic switch in primary afferent neurons contributes to neuropathic pain. Pain. 2011;152:2413–2426. doi: 10.1016/j.pain.2011.07.012. [DOI] [PubMed] [Google Scholar]
- Park J, Luo ZD. Calcium channel functions in pain processing. Channels (Austin) 2010;4:510–517. doi: 10.4161/chan.4.6.12869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel MK, Gonzalez MI, Bramwell S, Pinnock RD, Lee K. Gabapentin inhibits excitatory synaptic transmission in the hyperalgesic spinal cord. Br J Pharmacol. 2000;130:1731–1734. doi: 10.1038/sj.bjp.0703530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polgar E, Hughes DI, Arham AZ, Todd AJ. Loss of neurons from laminas I–III of the spinal dorsal horn is not required for development of tactile allodynia in the spared nerve injury model of neuropathic pain. J Neurosci. 2005;25:6658–6666. doi: 10.1523/JNEUROSCI.1490-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polgar E, Hughes DI, Riddell JS, Maxwell DJ, Puskar Z, Todd AJ. Selective loss of spinal GABAergic or glycinergic neurons is not necessary for development of thermal hyperalgesia in the chronic constriction injury model of neuropathic pain. Pain. 2003;104:229–239. doi: 10.1016/s0304-3959(03)00011-3. [DOI] [PubMed] [Google Scholar]
- Rahman W, D’Mello R, Dickenson AH. Peripheral nerve injury-induced changes in spinal alpha(2)-adrenoceptor-mediated modulation of mechanically evoked dorsal horn neuronal responses. J Pain. 2008;9:350–359. doi: 10.1016/j.jpain.2007.11.010. [DOI] [PubMed] [Google Scholar]
- Rigaud M, Gemes G, Barabas ME, Chernoff DI, Abram SE, Stucky CL, Hogan QH. Species and strain differences in rodent sciatic nerve anatomy: implications for studies of neuropathic pain. Pain. 2008;136:188–201. doi: 10.1016/j.pain.2008.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanfa LC, Singh L, Williams RG, Dickenson AH. Gabapentin, ineffective in normal rats, markedly reduces C-fibre evoked responses after inflammation. Neuroreport. 1997;8:587–590. doi: 10.1097/00001756-199702100-00002. [DOI] [PubMed] [Google Scholar]
- Suzuki R, Rahman W, Hunt SP, Dickenson AH. Descending facilitatory control of mechanically evoked responses is enhanced in deep dorsal horn neurones following peripheral nerve injury. Brain Res. 2004;1019:68–76. doi: 10.1016/j.brainres.2004.05.108. [DOI] [PubMed] [Google Scholar]
- Suzuki R, Rahman W, Rygh LJ, Webber M, Hunt SP, Dickenson AH. Spinal-supraspinal serotonergic circuits regulating neuropathic pain and its treatment with gabapentin. Pain. 2005;117:292–303. doi: 10.1016/j.pain.2005.06.015. [DOI] [PubMed] [Google Scholar]
- Tanabe T, Takeshima H, Mikami A, Flockerzi V, Takahashi H, Kangawa K, Kojima M, Matsuo H, Hirose T, Numa S. Primary structure of the receptor for calcium channel blockers from skeletal muscle. Nature. 1987;328:313–318. doi: 10.1038/328313a0. [DOI] [PubMed] [Google Scholar]
- Todd AJ. Neuronal circuitry for pain processing in the dorsal horn. Nat Rev Neurosci. 2010;11:823–836. doi: 10.1038/nrn2947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valder CR, Liu JJ, Song YH, Luo ZD. Coupling gene chip analyses and rat genetic variances in identifying potential target genes that may contribute to neuropathic allodynia development. J Neurochem. 2003;87:560–573. doi: 10.1046/j.1471-4159.2003.02016.x. [DOI] [PubMed] [Google Scholar]
- Vidal M, Morris R, Grosveld F, Spanopoulou E. Tissue-specific control elements of the Thy-1 gene. Embo Journal. 1990;9:833–840. doi: 10.1002/j.1460-2075.1990.tb08180.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Sun H, Della Penna K, Benz RJ, Xu J, Gerhold DL, Holder DJ, Koblan KS. Chronic neuropathic pain is accompanied by global changes in gene expression and shares pathobiology with neurodegenerative diseases. Neuroscience. 2002;114:529–546. doi: 10.1016/s0306-4522(02)00341-x. [DOI] [PubMed] [Google Scholar]
- Wang XL, Zhang HM, Chen SR, Pan HL. Altered synaptic input and GABAB receptor function in spinal superficial dorsal horn neurons in rats with diabetic neuropathy. J Physiol. 2007;579:849–861. doi: 10.1113/jphysiol.2006.126102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolf CJ, Mannion RJ. Neuropathic pain: aetiology, symptoms, mechanisms, and management. Lancet. 1999;353:1959–1964. doi: 10.1016/S0140-6736(99)01307-0. [DOI] [PubMed] [Google Scholar]
- Woolf CJ, Salter MW. Neuronal plasticity: increasing the gain in pain. Science. 2000;288:1765–1769. doi: 10.1126/science.288.5472.1765. [DOI] [PubMed] [Google Scholar]
- Yaksh TL. Behavioral and autonomic correlates of the tactile evoked allodynia produced by spinal glycine inhibition: effects of modulatory receptor systems and excitatory amino acid antagonists. Pain. 1989;37:111–123. doi: 10.1016/0304-3959(89)90160-7. [DOI] [PubMed] [Google Scholar]
- Yaksh TL. Calcium channels as therapeutic targets in neuropathic pain. J Pain. 2006;7:S13–30. doi: 10.1016/j.jpain.2005.09.007. [DOI] [PubMed] [Google Scholar]
- Zhou C, Luo ZD. Electrophysiological characterization of spinal neuron sensitization by elevated calcium channel alpha-2-delta-1 subunit protein. Eur J Pain. 2014;18:649–658. doi: 10.1002/j.1532-2149.2013.00416.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann M. Pathobiology of neuropathic pain. Eur J Pharmacol. 2001;429:23–37. doi: 10.1016/s0014-2999(01)01303-6. [DOI] [PubMed] [Google Scholar]