

Summary
The studies of stem cell behavior and differentiation in a developmental context is complex, time-consuming and expensive, and for this reason, cell culture remains a method of choice for developmental and regenerative biology and mechanistic studies. Similar to ES cells, iPS cells have the ability to differentiate into endothelial cells (ECs), and the route for differentiation appears to mimic the developmental process that occurs during the formation of an embryo. Traditional EC induction methods from embryonic stem (ES) cells rely mostly on the formation the embryoid body (EB), which employs feeder or feeder-free conditions in the presence or absence of supporting cells. Similar to ES cells, iPS cells can be cultured in feeder-layer or feeder-free conditions. Here, we describe the iPS cell culture methods and induction differentiation of these cells into ECs. We use anti-mouse Flk1 and anti-mouse VE-cadherin to isolate and characterize mouse ECs, because these antibodies are commercially available and their use has been described in the literature, including by our group. The ECs produced by this method have been used by our laboratory, and we have demonstrated their in vivo potential. We also discuss how iPS cells differ in their ability to differentiate into endothelial cells in culture.
Keywords: Angiogenesis, Endothelial cells, iPS cells, Nanog, Oct4, Sox2, Klf4, Flk1, CD31, VE-cadherin
1. Introduction
Totipotent embryonic stem cells (ESCs) can give rise to differentiated and specialized cells with restricted developmental potential (1-7). At some point, the specialized cells no longer differentiate or dedifferentiate, and this state has been referred to as terminal differentiation. The process of terminal differentiation has been thought to be an irreversible process. In contrast to this long-held view, retroviral-mediated, forced expression of key transcription factors such as Klf4, c-Myc, Nanog, Oct4, and Sox2 into somatic cells, such as fibroblasts, can convert (reprogram) these cells into induced pluripotent stem (iPS) cells (8-12). The additive activities of these transcription factors were thought to be necessary and sufficient to reprogram human or mouse somatic cells to iPS cells. In addition to these classical transcription factors described by the Yamanaka and Thomson groups (8,9,11), additional transcription factors and miRNAs and small molecules have been added to the list (12-15). Accordingly, a combination of two or three transcription factors (often called Yamanaka factors) may be sufficient to reprogram fibroblast cells into human or mouse iPS cells. For example, in some cell types, Oct4 and Sox2 might be sufficient to establish an iPS cell line (16), while in others, Sox2 is dispensable (17,18). It is apparently clear that Oct4 occupies the most upstream position in terms of its ability to reprogram somatic cells, while other Yamanaka factors are required for developmental differentiation events downstream of Oct4 (19,20). More recently, forced expression of the transcription factors Sall4, Nanog, Esrrb, and Lin28 in mouse fibroblast cells have been shown to generate high-quality iPS cells (21). The mechanisms of differentiation in iPS and ES cells could differ from those of various iPS cells derived from different somatic cells, but their similarities and differences have not been precisely delineated. Currently, the underlying mechanisms of iPS generation remain an area of great interest.
Upon orthotopic implantation into nude mice, similar to embryonic stem cells (ESCs), iPS cells form teratomas (8-11). Immunohistochemical analyses of the teratoma sections using markers for the three germ layers, e.g., ectoderm, mesoderm and endoderm, provide a good indication of iPS cell stemness. In addition, functional tests, including tetraploid complementation assays and the production of chimeric and germline mice, establish that iPS cells can acquire an ESC-like state (8-11,21,22). Therefore, it is not surprising that genuine interest for the application of iPS technology has emerged in many areas of regenerative, reparative and transplantation medicine. Nevertheless, inefficiency remains the main bottleneck for converting somatic cells into iPS cells, e.g., of 1000-10,000 somatic cells, only a single iPS cell can be fully reprogrammed using the most efficient method. For this reason, the production of patient-derived stem cells, is not only an expensive task but also remains an uphill battle. Although a retroviral method is considered the most efficient way to produce iPS cells, chimeric mice and mice derived through the use of these iPS cells often produce tumors (8-11). One of the caveats of this approach is that the retroviruses, for instance, long terminal repeats are known to integrate randomly into the genome, which could activate oncogenes or inactivate tumor suppressor genes to initiate neoplastic transformation. Thus, these observations have provided the impetus to the development of non-integrating vectors such as piggyback, episomal non-integrating and non-integrating Sendai Virus as well as mini-genes and small-molecule compounds (23-27). Thus, the development of a highly efficient iPS reprogramming technique that also evades these undesirable genetic alterations should be a rewarding research endeavor.
The observations that iPS cells have the capacity to self-renew and undergo differentiation in response to specific growth factors in culture dishes make these cells an ideal source of progenitor cells for cell-based therapy, drug screening and disease modeling, thus they have vast therapeutic potential. Therefore, in our laboratory, we have used iPS cells as a source for VE-cadherin+ and Flk1+ endothelial cells (ECs) and showed their ability to incorporate into CD31+ neovessels in Matrigel plugs (28) and into newly formed blood vessels in a mouse model of hind limb ischemia (28) and in Matrigel plug assays. Thus, based purely upon our recent publication (28), here, we outline methods for iPS culture, including the conditions used to differentiate iPS cells into ECs as well as for the isolation, purification, and characterization of VE-cadherin+ and Flk1+ ECs.
2. Materials
A clean cell culture laboratory with laminar flow and vacuum connected to a liquid waste container (a glass flask) through a HEPA filter, an electrical outlet for a pipette aid, and a gas connection for a Bunsen burner. Some media and growth factors are filter-sterilized using a 0.2-μm filter. Please ensure that the iPS cell lines you are using do not secrete live virus or are contaminated with mycoplasma. Please follow aseptic techniques throughout the procedure.
2.1 Equipment
For all cell culture work, we recommend a Zeiss Primo Vert (Carl Zeiss MicroImaging Inc., Thornwood, NY), Nikon TS100 or Olympus CKX41 inverted phase contrast cell culture microscope, with long working distance (LWD) lenses and 5X, 10X and 20X magnifications. Preferably with a digital camera for recording images.
Refrigerated cell culture centrifuge
Water bath (20 Liters), 37°C
Humidified CO2 (5%) cell culture incubator set at 37°C
Liquid N2 storage tank
Frosty freezing container (Nalgene)
Nalgene System cryovials (1.8 ml)
Flow cytometer. We use Beckman Coulter MoFlo (Becton Dickinson, Franklin Lakes, NJ).
70% ethanol in a spray bottle
2.2 iPS Cell-freezing Medium
Prepare cell-freezing medium, which contains 30% fetal bovine serum (FBS), 10-20% Dimethyl Sulfoxide (DMSO), 60% DMEM (alternatively, use ES-DMEM).
Store at +4°C for 3 months.
2.3 Medium and Growth Factors
Cell culture dishes: 6-, 12- and 24-well cell culture-certified dishes.
Sterile serological pipettes (1-, 5-, and 10-ml; all disposable plastics must be sterile.
iPS cells can be generated in the laboratory using commercially available Yamanaka factors. The mouse induced Pluripotent Stem (iPS) cells (iMZ-9 and iMZ-21 lines) used in this example were obtained through Material Transfer Agreement (MTA) from Dr. Kristin Baldwin (The Scripps Research Institute, La Jolla, CA).
Embryonic Stem (ES) cell-qualified Fetal Calf Serum (FCS) (Invitrogen)
Attachment factor (AF) from Life Technologies
Human placenta-derived type IV collagen (Millipore/Chemicon)
Dulbecco’s Phosphate-buffered saline (DPBS, pH 7.4, with no Ca++ or Mg++)
TrypLE™ Select Cell Dissociation Reagent (Life Technologies). For passaging with a feeder layer, use type IV collagenase (Invitrogen) diluted with DMEM-F12.
Falcon cell strainer (70-μm nylon mesh)
2 mM Ethylenediaminetetraacetic Acid (EDTA) in PBS, pH 7.4.
Recombinant Leukemia Inhibitory Factor (LIF; ESGRO®; Millipore, Billerica, MA).
High-glucose Dulbecco’s Modified Eagle Medium (DMEM, Invitrogen, Carlsbad, CA). Alternatively, use KnockOut DMEM (Life Technologies).
iPS cell Complete Medium: prepare high glucose Dulbecco’s modified Eagle’s Medium (DMEM) (alternatively, use Knockout-DMEM (Invitrogen, Carlsbad, CA) containing 15% ES-qualified FCS, 1.0 mM Sodium Pyruvate, 2.5 mM Glutamax (Invitrogen), 0.1 mM non-essential amino acids, penicillin (10.0 μg/ml), streptomycin (5.0 μg/ml), 0.1 mM β-mercaptoethanol (Invitrogen), and 1000 U/ml recombinant leukemia inhibitory factor (LIF). Filter sterilize the solution using a 0.22-μm filter, aliquot the solution into 100 ml volumes and store them at +4°C for 1 week.
Mitomycin-treated mouse embryonic fibroblast cells (MEFs) can be purchased or prepared in house. Alternatively, use irradiated MEFs. These MEFs can be used for up to 3-4 passages. Store aliquots of Mitomycin-treated or irradiated MEFs in liquid nitrogen.
A refrigerated cell culture centrifuge should be within close proximity of the cell culture room.
All cell culture and stem cell media should be stored inside a +4°C refrigerator. Do not store the cell culture media with E. coli bacterial plates or leftover E. coli cultures.
Some universities may have preferred vendors for cell culture dishes, stem cell culture media and growth factors.
3. Methods
3.1 Aseptic Techniques and Biohazard Procedures
We emphasize here that ES and iPS cell culture is an expensive undertaking. Therefore, it is important to minimize waste in stem cell and biological research. With regard to compliance and safety issues, please follow your institutional guidelines and rules. The following general points may be applied to all stem cell and iPS culture laboratories.
Make sure there is no mycoplasma contamination in iPS cells.
We highly recommend a dedicated stem cell culture area with low traffic that is secured behind a closed door. Strict cleanliness is required at all times, including a regular schedule for the removal of used pipettes, dishes, and any liquid or solid wastes from the cell culture room. All cell culture waste should be removed on a daily basis. This cleanliness will minimize possible contamination from mycoplasma, yeast, bacteria or fungi. Cell culture areas often experience too much human traffic; therefore, effort should be made to minimize unnecessary movement. Before and after work, spray 70% ethanol and clean the inside of the cell culture hood. Leave the UV lamp on overnight. The best time to work is in the morning, as the hood had been exposed to UV-lamp overnight.
Cell culture hoods are usually equipped with HEPA filters with a 4-5-year shelf life. The cell culture HEPA filter should be cleaned and re-certified on a regular basis. All personnel must wear a clean laboratory coat and latex or nitrile gloves before using the cell culture hood. The 37°C water bath must be cleaned on a regular basis, and the shelves of the CO2 incubator should be autoclaved every month.
Every institute has an oversight committee for blood-borne pathogens, laboratory safety, recombinant DNA, animal and stem cell research. All personnel working in a wet-bench laboratory must have completed the required training. Some universities require yearly re-certification, and the laboratory safety and recombinant DNA oversight committees may also conduct online training and or classroom training and instructions.
If you are new to the cell culture laboratory or if you are training a new member of the laboratory, please provide clear instructions about cleanliness and aseptic techniques. There should be no mouth pipetting or discarding of waste in the regular trash or dumping into a laboratory sink.
All liquid wastes must be neutralized and disinfected with Clorox (bleach, final concentration 15%). All solid wastes, including serological pipettes, dishes, and paper towels, should be autoclaved in a biohazard bag and disposed according to the institutional rules.
Finally, absence of warning does not mean that everything is safe and sterile.
3.2 Culture Conditions for iPS cells
We grow iPS cells in feeder-free conditions (28). High-quality iPS cells grow well if under optimal conditions. Like ES cells, the cell doubling time of iPS cells is short, 18-20 hours. However, if the density of the iPS cells is too high, they tend to grow as 3D cellular aggregates. Cellular aggregates of iPS cells more often give rise to cells with heterogeneous morphologies and may be indistinguishable from parental iPS cells. It is important that iPS cells be sub-cultured every 4-5 days to maintain their growth in exponential phase. If iPS cells are not growing in exponential phase, it is be desirable to periodically clone them. We recommend freezing iPS cells in several aliquots on a regular basis.
3.2.1 iPS cell culture
This protocol is optimized for >105 iPS cells per 6-well cell culture dish. We recommend recording phase-contrast images of ES and iPS cell growth every other day during the course of culture.
Prepare iPS cell complete culture media as described above. If using a commercially available kit, add all components to the 500-ml basal media container. If the kit has no Penicillin, Streptomycin and GlutaMax solutions, add these reagents to the media prior to use.
Warm the media in a 37°C water bath for 30-60 minutes. Do not keep the media in the water bath for more than 1 hour at 37°C, as prolonged exposure to 37°C will reduce the effectiveness of the growth factors and GlutaMax.
iPS cells can be maintained in an undifferentiated state on mitomycin-C-arrested or irradiated MEFs. Mitotically arrested MEFs are plated 3–4 days prior to passaging of iPS cells.
Add 100 μl of attachment factor (AF) solution to the well, after 5 minutes, remove by aspirating. If plating multiple cell lines, AF can be kept on the dish for as long as 1 hour. Add 5 ml of sterile 1X PBS to the dish, swirl side-to-side, and thereafter remove the PBS by aspirating. Excess AF should be removed by washing with PBS; however, some laboratories do not recommend washing with PBS. AF-coated wells are good for use for up to 24 hours.
Label all dishes or wells with the cell line name and number, date and passage #.
Add 2 ml of warm, complete media to the well (6-well plate). If using MEFs as a feeder layer, the MEF media must be removed before the iPS cells can be plated. The iPS cells can be cultured in MEF dishes conditioned with iPS medium. We recommend conditioning new feeder dishes prior to passaging iPS cells into them.
Thaw a frozen cryovial containing iPS cells (if stored in liquid N2) by immersing the vial in a 37°C water bath for 5 minutes or to the point when the ice crystals disappear. Spray the cryovial with 70% ethanol, remove the excess ethanol using a clean Kim-Wipe or paper towel, and bring the cryovial to the sterile cell culture hood.
Empty the contents of the cryovial into 15-ml sterile Falcon centrifuge tube containing warm complete media.
Centrifuge the cell suspension at ~1000 rpm (400 × g) in a refrigerated centrifuge for 5 minutes. After centrifugation, discard the supernatant and save the cell pellet.
Add 6 ml of iPS cell media to the tube, and completely resuspend the cell pellet.
Remove the iPS cell suspension using a 5-ml pipette; thereafter, dispense 2 ml into each of 3 wells of a 6-well plate.
Return the dish to the humidified 37°C CO2 incubator.
The next day, aspirate the media to remove the dead cells and gently add pre-warmed media. Examine the cells under the microscope.
The iPS cell cycle is approximately 18-20 hours, and iPS cell colonies (three-dimensional structures) should be visible within 3-4 days. While growing, do not let two or more colonies fuse or mix because this event triggers differentiation.
Cells should be passaged after 3-4 days. If iPS cells dominate the culture as 3D structures, the iPS cells must be passaged by enzymatic dissociation. However, if the cells are too dense or growing quickly, some of the cells will differentiate spontaneously at the periphery of the 3D colony. In that case, the 3D iPS colonies must be separated manually from the differentiated cells (these often have an adherent phenotype) using the “car-wash method” with a cell scraper. If 3D iPS structures (cellular aggregates) dominate the culture, we recommend expanding the iPS cells into 35-mm dishes as described below.
3.2.2 Passaging iPS cells
To maintain high-quality iPS cells, we recommend passaging the iPS cells on a regular basis. Maintenance of high-quality iPS cells in culture will be crucial for endothelial differentiation experiments. Under carefully monitored iPS cell culture conditions, iPS cells retain pluripotency and self-renewing capacity.
Prepare dishes as described in 3.3.4 and 3.3.4.
Pre-warm the complete media as needed.
Label the wells with the cell line name, number, date, passage #, and split ratio.
Using a microscope, examine the morphology of the iPS cell colonies. Remove any differentiated cells from the iPS cell colonies to be passaged. Both ES and iPS cells can be dissociated enzymatically or manually by scraping the cell colonies. If your wells have more than 25 colonies/well in a 6-well plate, we recommend manually scraping the iPS cell colonies.
Aspirate the media from the wells using a Pasteur pipette and rinse the well once with DPBS without Ca++ and Mg++. If working with multiple cell lines or wells, change the pipette for each dish or each well.
Add 200 μL of pre-warmed TrypLE ™ (without Phenol red, Life Technologies) cell dissociation solution to the dish containing the iPS cell colonies. The volume of the cell detachment solution can be adjusted depending on the size of the well or dish.
Incubate the cells for 4-5 minutes in a CO2 incubator at 37°C. If the cells are detaching from the dish, the cell dissociation solution is likely inactive. If that is the case, please use a new batch of TrypLE cell dissociation solution.
After detachment, collect all cells into a 15-ml sterile centrifuge tube. Resuspend the cells in complete media and centrifuge at 1000 rpm (400 × g) for 5 minutes at +4°C in a refrigerated centrifuge.
Discard the supernatant by aspirating, and resuspend the cell pellet by gently adding the appropriate volume of media. If the cells form clumps, pass the cell suspension through a Falcon Cell strainer (70-μm nylon mesh).
Replate the cells at the appropriate density (1:2 ratio) and return the dishes to a CO2 incubator at 37°C.
Change the media every 24 hours.
If large-scale amounts of iPS cells are needed, repeat steps 1-11.
3.2.3 Freezing iPS cells
Freezing aliquots of high-quality iPS cells in liquid nitrogen should be performed on a regular basis to maintain reliable stocks of iPS cells. We recommend changing the media 3-4 hours before freezing iPS cells.
Detach the iPS cells using TrypLE cell dissociation solution, as described in step 3.4.6.
Resuspend the cell pellet (2 × 106 cells/ml) in cell-freezing reagent (30% FBS, 10% DMSO, 60% DMEM). If preparing fresh cell-freezing solution, cool the solution on ice for 20 minutes prior to use. Remember DMSO is a toxic solvent. Open the DMSO container inside the laminar flow hood.
Use a permanent marker to label the cryovials with the cell line name, passage number, date, and your initials.
Transfer the vials to a Frosty freezing cryocontainer filled with isopropanol.
Leave the Frosty freezing container in a -86°C freezer overnight.
On the next day, transfer the cryovials to a liquid nitrogen tank.
3.3 Induction of Differentiation of iPS cells into Endothelial Cells (ECs)
There are several methods in the literature that describe the induction of differentiation of ES and iPS cells into ECs (29-37). Depending upon the main objectives of the downstream experiments, the methods for inducing ES and iPS cells could be different (29-37). To minimize contamination of unknown cells, we use monoclonal antibodies that recognize the extracellular domains of Flk-1 and VE-cadherin. It is important to note that, in vitro, iPS-derived ECs express the Flk-1 and VE-cadherin endothelial markers, which are commonly used to distinguish adult ECs from both mature and immature cell sources. It would be fair to state here that, currently, there is no definitive marker for endothelial progenitor cells (EPCs) that can be used for re-vascularization therapy or therapeutic angiogenesis. In the following section, we describe a method for isolating Flk-1+ and VE-cadherin+ cells from mouse iPS cells. In addition to VE-cadherin and Flk1, we use functional assays, including tube formation and Matrigel plug assays. We have also tested the efficacy of these cells using a mouse model of hind limb ischemia.
3.3.1 Materials
Cell dissociation solution: 1.0 mM EDTA in PBS, pH 7.4 or TrypLE
Type IV collagen-coated dishes (BD Bioscience, San Jose, CA)
Serum-free basal medium: 75% Iscove’s Modified Dulbecco’s Media (IMDM) and 25% Ham’s F12 medium (Life Technologies)
B-27 Supplement (no Vitamin A)
Bovine Serum Albumin (BSA) (Life Technologies)
α-Monothioglycerol (MTG) (Sigma, St Louis, MO)
Ascorbic Acid (Sigma, St Louis, MO) (42)
Human BMP-4 (R&D Systems)
Human VEGF165 (Miltenyi Biotec)
Human Basic FGF (Millipore)
Rat anti-mouse Flk1 antibody (clone avas12a, BD Bioscience) and goat-anti-mouse VE-cadherin (R&D Systems, Minneapolis, MN) antibody
Secondary antibodies conjugated to Alexa-Flour, APC, FITC or PE
3.3.2 Differentiation and Sorting of ECs
Given that ECs are anchorage-dependent cells, the induction of ES and iPS cell differentiation entails the use of a defined supporting matrix or purified extracellular matrix (ECM) protein (See notes 1 and 2). Thus, to initiate differentiation in ES and iPS cells, we used type IV collagen as a supporting ECM protein, which is known to induce differentiation in ES cells to mesodermal lineages. We recommend recording the differentiating cells in culture every 24 hours using an inverted cell culture phase contrast microscope. Prepare in advance to do FACS sorting, e.g., sign-up and coordinate with the core facility, because the cell sorter must be aseptically cleaned. Alternative to FACS, antibody-coated e.g., anti-CD31, magnetic beads may be used. To isolate ECs, all antibodies must be FACS validated and free of sodium azide or any preservatives. If the antibody contains sodium azide, it must be dialyzed and filter sterilized. As commercial antibodies are expensive, we recommend using antibodies produced by a bioreactor or cell culture supernatant of hybridoma cells that secrete monoclonal antibodies for routine isolation of large scale ECs. The Flk1 hybridoma can be purchased from ATCC or may be obtained through the Iowa Developmental Hybridoma Bank.
Serum-free medium: prepare with 75% IMDM, 25% Ham’s F12 medium, B-27 Supplement, 0.05% BSA, 4.5×104 M MTG and 0.5 mM ascorbic acid, human BMP-4 (2 ng/ml), human VEGF165 (50 ng/ml) and human basic FGF (10 ng/ml).
Coat a 35-mm dish with type IV collagen (5.0 μm/ml) for 1 hour, wash with sterile 1X DPBS, pH 7.4. Alternatively, use pre-made type IV collagen-coated dishes from BD Biosciences.
Plate actively growing, high-quality iPS cells (see step 3.2.2.12) at a density of 3.5 × 104/35-mm well onto the type IV collagen-coated dishes.
Adherent cells will emerge as soon as 2-3 days. Change the differentiation media every 24 hours and take pictures every 24 hours.
The expression of Flk-1 begins to appear as early as 2.0-3.0 days, while VE-cadherin expression begins at 2.5-3.5 days (Figure 1). For optimal expression of Flk-1 and VE-cadherin proteins, we recommend culturing these cells up to day 4-5.
Physically remove 3D cellular aggregates, as these are a mixture of cells.
Add cell dissociation solution (1 mM EDTA, pH 7.4) to the dish and incubate for 20 minutes in a CO2 incubator at 37°C.
To stop the action of EDTA or cell dissociation enzyme, add complete serum-free media (differentiation media); thereafter, centrifuge at 400 × g for 5 minutes in a refrigerated centrifuge.
After centrifugation, wash the cells in cold phosphate-buffered saline (PBS). If clumps appear, pass the cells through a Falcon cell strainer (70-μm pore size).
To isolate high-quality ECs, the detached cells are made into a single-cell suspension as described in step 9 and are thereafter subjected to FACS or magnetic separation.
The cell suspension is adjusted to 2 × 106 cells/ml. Use an appropriate concentration of primary antibody (1-2 μg/ml). Incubate on ice for 1 hour (in the dark if the antibody is directly conjugated to Alexa fluor); thereafter, wash once with cold PBS, and save the cell pellet.
Next, resuspend the cell pellet in secondary antibody solution, incubate on ice for 30-60 minutes (in dark), wash once with PBS, and resuspend the cells in FACS buffer.
For FACS, please have negative and positive control samples and determine the gating parameters for cell sorting. Gating should be fixed at the level of fluorescence above which almost all cells are negative. These parameters can be adjusted depending on the FACS machine and objective of the downstream experiments as well as on the experience of the investigator. We use the Moflo FACS high-speed sorter.
As ECs are larger than leukocytes, adjust the autofluorescence to the first quadrant of the fluorescence to be monitored while gating the ECs and ensure that the cells of interest are included in the gate. If required, check the fluorescence of the positive control sample and compensate. Initiate the acquisition of Flk1+ and VE-cadherin+ ECs; 105/sample is optimal. Save the data and analyze the dot blot of the gated ECs.
If ECs are not 100% pure, subject these cells through one more cycle of FACS with the anti-Flk1 and anti-VE-cadherin antibodies (Figure 2).
Figure 1. Induction of Flk1+VE-cadherin+ vascular EC progenies from iPS and ES cells.
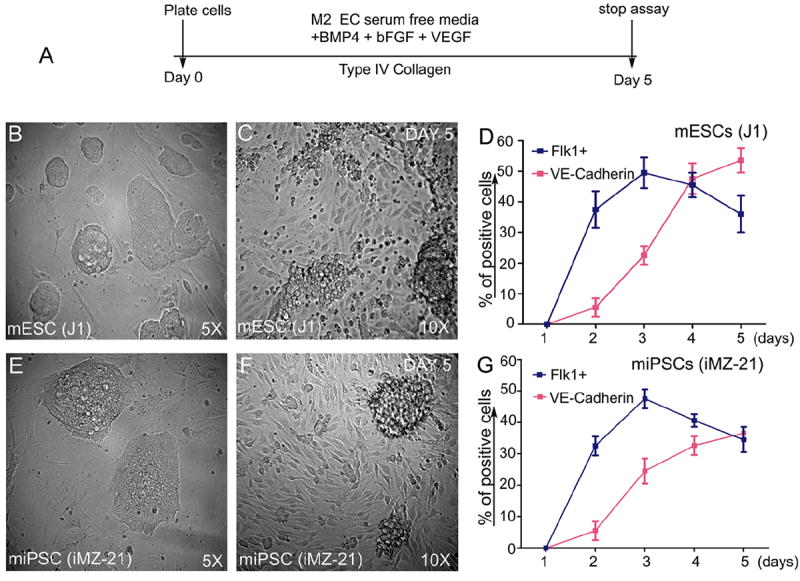
Timeline of emergence of Flk1+VE-cadherin+ vascular ECs. (A). Undifferentiated mES (J1 line) or miPS (iMZ-21) cells were cultured for 5 d in IV Col-coated dishes in media containing BMP4, bFGF, and VEGF165 to induce generation of vascular EC progenies. (B&C). Representative phase contrast microscopy of mES cell-derived adherent vascular progenies after d 5 in culture at indicated magnifications (B&C). Representative phase contrast microscopy of miPS cell-derived vascular progenies after day 5 in culture at the indicated magnifications (E&F). FACS profile of the emergence of Flk1+VE-cadherin+ vascular progenies (D&G). All experiments were repeated >5 times. Data indicate the mean ± S.E.M. n=5. (Reprinted from Kohler EE et al., PloS One 2013 Dec 30;8(12):e85549).
Figure 2. FACS analysis of emerging vascular EC progenies from mES and iPS cells.
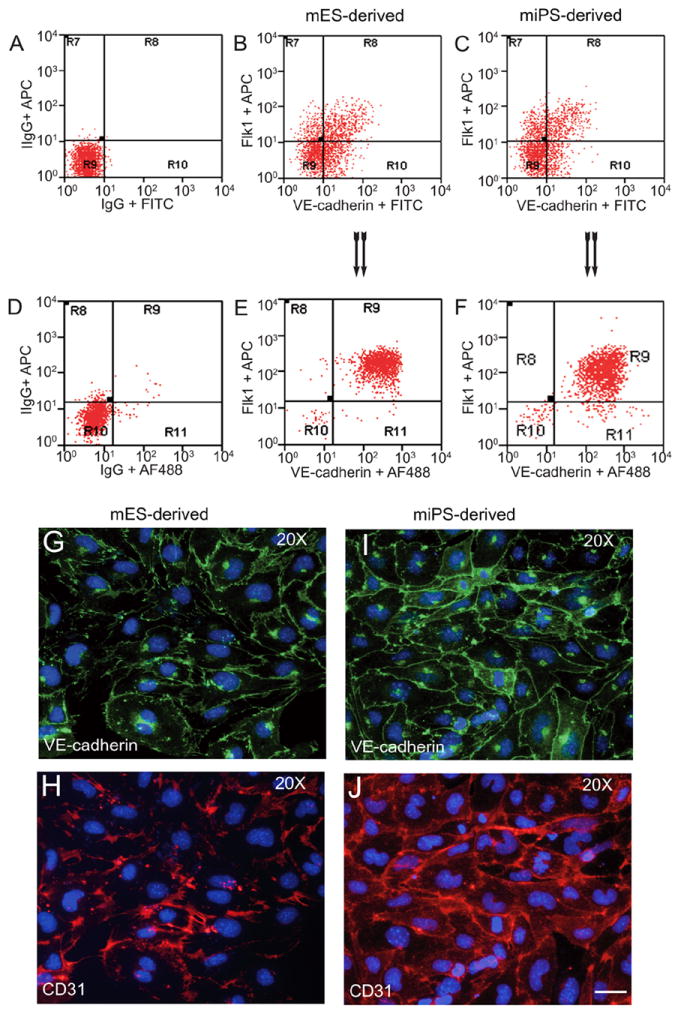
Adherent cells (2×105) were detached and subjected to two-step FACS-aided purification. Control FACS profile on day 5 of cells derived from mES (J1) cells (A). Representative FACS profiles of day 5, with vascular progenies assessed using anti-Flk1 and anti-VE-cadherin antibodies obtained from mES (J1) cells (B) and derived from miPS (iMZ-21) cells (C); Control FACS profile on day 5 of cells derived from miPS (iMZ-21) cells (D). Representative FACS after the second step of purification derived from mES (E) and iPS cells (F). The yield of Flk1+VE-cadherin+ after the second round of FACS was 100% for both mES and miPS-derived vascular progenies. Morphology of mES- and miPS-derived vascular ECs (G-J). Flk1+VE-cadherin+ vascular progenies derived from mES and miPS cells were cultured overnight in IV Col-coated dishes, immunostained with anti-VE-cadherin (green) and anti-CD31 (red) of cells derived from mES cells (G&H) and miPS cells (I&J). DAPI, nucleus (blue). Magnifications are as indicated; the scale bar is 200 μm. Experiments were repeated 3 times. (Reprinted from Kohler EE et al., PloS One 2013 Dec 30;8(12):e85549).
To determine the phenotypic characteristics and colony formation of the sorted cells, plate the cells onto type IV collagen-coated coverslips in the presence of serum-free differentiation media. Use rat anti-mouse CD41 to assess hematopoietic cells and anti-VE-cadherin and anti-CD31 antibodies to determine the identity of the ECs. FACS-sorted cells can be passaged 1-2 times. We recommend further characterization of these cells using anti-VE-cadherin, anti-CD31, anti-Flk1, and anti-vWF antibodies. In addition, gene expression profiling, proteomics, and epigenetic studies can be carried out. In all of these characterization experiments, positive and negative controls must be included.
Acknowledgments
The research in the authors’ laboratory is supported by grants from the National Institutes of Health (NIH) and the American Heart Association (AHA) to KKW.
Footnotes
Based purely upon our recent publication (28), here, we have described iPS cell culture and the induction of EC differentiation in vitro as well as the isolation of Flk1- and VE-cadherin-positive ECs. Unlike tumor cell culture, one has to be patient with iPS cell culture.
It is necessary to have live iPS cells and optimal cell culture conditions. The quality of the media and growth factors could alter the behavior of the iPS cells and their differentiation potential. However, if the conditions are optimal, the iPS cells will grow so that important experiments can be carried-out in a timely manner.
There are a multiplicity of protocols for the differentiation and characterization of ECs. However, the quality of iPS cells is critical for the emergence of ECs. It is important to realize that not all iPS cell lines will give rise to the same number of ECs, and we have found that some iPS cell lines perform better than others. For this reason, we recommend starting with a high-quality iPS cell culture, optimal growth and maintenance conditions with no contamination. If the FACS-sorted cells are not clean, these cells will quickly die in culture or become contaminated. If contamination occurs, we recommend thawing a new aliquot of cells and begin everything anew. We do not recommend sorted ECs be passaged more than 1-2 times. Currently, we have not established iPS-derived ECs as cell lines.
To assess the functionality of ECs, several different assays can be carried-out, including in vitro Matrigel tube formation assays and in vivo Matrigel plug angiogenesis assays (28). For in vivo studies, these cells can be transfected by a retrovirus encoding red fluorescent protein (RFP) (28). For all functional studies, we recommend the use of freshly isolated cells.
According to the PloS One Open Access Journal policy no permission is required to reproduce text and the Figures (Kohler EE et al., PLoS One 8(12):e85549.)
References
- 1.Evans MJ, Kaufman MH. Establishment in culture of pluripotential cells from mouse embryos. Nature. 1981;292:154–156. doi: 10.1038/292154a0. [DOI] [PubMed] [Google Scholar]
- 2.Bradley A, Robertson E. Embryo-derived stem cells: a tool for elucidating the developmental genetics of the mouse. Curr Top Dev Biol. 1986;20:357–371. doi: 10.1016/s0070-2153(08)60675-4. [DOI] [PubMed] [Google Scholar]
- 3.Nagy A, Gócza E, Diaz EM, Prideaux VR, Iványi E, Markkula M, Rossant J. Embryonic stem cells alone are able to support fetal development in the mouse. Development. 1990;110(3):815–821. doi: 10.1242/dev.110.3.815. [DOI] [PubMed] [Google Scholar]
- 4.Nagy A, Rossant J, Nagy R, Abramow-Newerly W, Roder JC. Derivation of completely cell culture-derived mice from early-passage embryonic stem cells. Proc Natl Acad Sci U S A. 1993;90(18):8424–8428. doi: 10.1073/pnas.90.18.8424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wilmut I, Schnieke AE, McWhir J, Kind AJ, Campbell KH. Viable offspring derived from fetal and adult mammalian cells. Nature. 1997;385:810–813. doi: 10.1038/385810a0. [DOI] [PubMed] [Google Scholar]
- 6.Thomson JA, Itskovitz-Eldor J, Shapiro SS, Waknitz MA, Swiergiel JJ, Marshall VS, Jones JM. Embryonic stem cell lines derived from human blastocysts. Science. 1998;282(5391):1145–1147. doi: 10.1126/science.282.5391.1145. [DOI] [PubMed] [Google Scholar]
- 7.Amit M, Carpenter MK, Inokuma MS, Chiu CP, Harris CP, Waknitz MA, Itskovitz-Eldor J, Thomson JA. Clonally derived human embryonic stem cell lines maintain pluripotency and proliferative potential for prolonged periods of culture. Dev Biol. 2000;227(2):271–278. doi: 10.1006/dbio.2000.9912. [DOI] [PubMed] [Google Scholar]
- 8.Waddington CH. The Strategy of the Genes. London: Geo Allen and Unwin; 1957. [Google Scholar]
- 9.Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126(4):663–676. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- 10.Okita K, Ichisaka T, Yamanaka S. Generation of germline-competent induced pluripotent stem cells. Nature. 2007;448(7151):313–317. doi: 10.1038/nature05934. [DOI] [PubMed] [Google Scholar]
- 11.Yu J, Vodyanik MA, Smuga-Otto K, Antosiewicz-Bourget J, Frane JL, Tian S, Nie J, Jonsdottir GA, Ruotti V, Stewart R, Slukvin II, Thomson JA. Induced pluripotent stem cell lines derived from human somatic cells. Science. 2007;318(5858):1917–1920. doi: 10.1126/science.1151526. [DOI] [PubMed] [Google Scholar]
- 12.Wernig M, Meissner A, Foreman R, Brambrink T, Ku M, Hochedlinger K, Bernstein BE, Jaenisch R. In vitro reprogramming of fibroblasts into a pluripotent ES-cell-like state. Nature. 2007;448(7151):318–324. doi: 10.1038/nature05944. [DOI] [PubMed] [Google Scholar]
- 13.Wang Y, Baskerville S, Shenoy A, Babiarz JE, Baehner L, Blelloch R. Embryonic stem cell-specific microRNAs regulate the G1-S transition and promote rapid proliferation. Nat Genet. 2008;40:1478–1483. doi: 10.1038/ng.250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Judson RL, Babiarz JE, Venere M, Blelloch R. Embryonic stem cell-specific microRNAs promote induced pluripotency. Nat Biotechnol. 2009;27:459–461. doi: 10.1038/nbt.1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Subramanyam D, Lamouille S, Judson RL, Liu JY, Bucay N, Derynck R, Blelloch R. Multiple targets of miR-302 and miR-372 promote reprogramming of human fibroblasts to induced pluripotent stem cells. Nat Biotechnol. 2011;29(5):443–448. doi: 10.1038/nbt.1862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gonzalez F, Boue S, Belmonte JC. Methods for making induced pluripotent stem cells: reprogramming a la carte. Nat Rev Genet. 2011;12:231–242. doi: 10.1038/nrg2937. [DOI] [PubMed] [Google Scholar]
- 17.Giorgetti A, et al. Generation of induced pluripotent stem cells from human cord blood using OCT4 and SOX2. Cell Stem Cell. 2009;5:353–357. doi: 10.1016/j.stem.2009.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Utikal J, Maherali N, Kulalert W, Hochedlinger K. Sox2 is dispensable for the reprogramming of melanocytes and melanoma cells into induced pluripotent stem cells. J Cell Sci. 2009;122:3502–3510. doi: 10.1242/jcs.054783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Eminli S, Utikal J, Arnold K, Jaenisch R, Hochedlinger K. Reprogramming of neural progenitor cells into induced pluripotent stem cells in the absence of exogenous Sox2 expression. Stem Cells. 2008;26(10):2467–2474. doi: 10.1634/stemcells.2008-0317. [DOI] [PubMed] [Google Scholar]
- 20.Kim JB, Greber B, Araúzo-Bravo MJ, Meyer J, Park KI, Zaehres H, Schöler HR. Direct reprogramming of human neural stem cells by OCT4. Nature. 2009;461:649–643. doi: 10.1038/nature08436. [DOI] [PubMed] [Google Scholar]
- 21.Li Y, Zhang Q, Yin X, Yang W, Du Y, Hou P, Ge J, Liu C, Zhang W, Zhang X, Wu Y, Li H, Liu K, Wu C, Song Z, Zhao Y, Shi Y, Deng H. Generation of iPSCs from mouse fibroblasts with a single gene, Oct4, and small molecules. Cell Res. 2011;21(1):196–204. doi: 10.1038/cr.2010.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Theunissen TW, Jaenisch R. Molecular control of induced pluripotency. Cell Stem Cell. 2014;14(6):720–734. doi: 10.1016/j.stem.2014.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Boland MJ, Hazen JL, Nazor KL, Rodriguez AR, Gifford W, Martin G, Kupriyanov S, Baldwin KK. Adult mice generated from induced pluripotent stem cells. Nature. 2009;461:91–94. doi: 10.1038/nature08310. [DOI] [PubMed] [Google Scholar]
- 24.Woltjen K, Michael IP, Mohseni P, Desai R, Mileikovsky M, Hämäläinen R, Cowling R, Wang W, Liu P, Gertsenstein M, Kaji K, Sung HK, Nagy A. piggyback transposition reprograms fibroblasts to induced pluripotent stem cells. Nature. 2009;458(7239):766–770. doi: 10.1038/nature07863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kaji K, Norrby K, Paca A, Mileikovsky M, Mohseni P, Woltjen K. Virus-free induction of pluripotency and subsequent excision of reprogramming factors. Nature. 2009;458(7239):771–775. doi: 10.1038/nature07864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Woltjen K, Hämäläinen R, Kibschull M, Mileikovsky M, Nagy A. Transgene-free production of pluripotent stem cells using piggyBac transposons. Methods Mol Biol. 2011;767:87–103. doi: 10.1007/978-1-61779-201-4_7. [DOI] [PubMed] [Google Scholar]
- 27.Papapetrou EP, Sadelain M. Generation of transgene-free human induced pluripotent stem cells with an excisable single polycistronic vector. Nat Protoc. 2011;6:1251–1273. doi: 10.1038/nprot.2011.374. [DOI] [PubMed] [Google Scholar]
- 28.Kohler EE, Wary KK, Li F, Chatterjee I, Urao N, Toth PT, Ushio-Fukai M, Rehman J, Park C, Malik AB. Flk1+ and VE-cadherin+ endothelial cells derived from iPSCs recapitulates vascular development during differentiation and display similar angiogenic potential as ESC-derived cells. PLoS One. 2013;8(12):e85549. doi: 10.1371/journal.pone.0085549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yamashita J, Itoh H, Hirashima M, Ogawa M, Nishikawa S, Yurugi T, Naito M, Nakao K, Nishikawa S. Flk1-positive cells derived from embryonic stem cells serve as vascular progenitors. Nature. 2000;408:92–96. doi: 10.1038/35040568. [DOI] [PubMed] [Google Scholar]
- 30.Levenberg S, Golub JS, Amit M, Itskovitz-Eldor J, Langer R. Endothelial cells derived from human embryonic stem cells. Proc Natl Acad Sci U S A. 2002;99:4391–4396. doi: 10.1073/pnas.032074999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Choi KD, Yu J, Smuga-Otto K, Salvagiotto G, Rehrauer W, Vodyanik M, Thomson J, Slukvin I. Hematopoietic and endothelial differentiation of human induced pluripotent stem cells. Stem Cells. 2009;27(3):559–67. doi: 10.1634/stemcells.2008-0922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yin L, Ohanyan V, Pung YF, Delucia A, Bailey E, Enrick M, Stevanov K, Kolz CL, Guarini G, Chilian WM. Induction of vascular progenitor cells from endothelial cells stimulates coronary collateral growth. Circ Res. 2012;110(2):241–52. doi: 10.1161/CIRCRESAHA.111.250126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yang HM, Moon SH, Choi YS, Park SJ, Lee YS, Lee HJ, Kim SJ, Chung HM. Therapeutic efficacy of human embryonic stem cell-derived endothelial cells in humanized mouse models harboring a human immune system. Arterioscler Thromb Vasc Biol. 2013;33:2839–2849. doi: 10.1161/ATVBAHA.113.302462. [DOI] [PubMed] [Google Scholar]
- 34.Adams WJ, Zhang Y, Cloutier J, Kuchimanchi P, Newton G, Sehrawat S, Aird WC, Mayadas TN, Luscinskas FW, García-Cardeña G. Functional vascular endothelium derived from human induced pluripotent stem cells. Stem Cell Reports. 2013;1(2):105–13. doi: 10.1016/j.stemcr.2013.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Prasain N, Lee MR, Vemula S, Meador JL, Yoshimoto M, Ferkowicz MJ, Fett A, Gupta M, Rapp BM, Saadatzadeh MR, Ginsberg M, Elemento O, Lee Y, Voytik-Harbin SL, Chung HM, Hong KS, Reid E, O’Neill CL, Medina RJ, Stitt AW, Murphy MP, Rafii S, Broxmeyer HE, Yoder MC. Differentiation of human pluripotent stem cells to cells similar to cord-blood endothelial colony-forming cells. Nat Biotechnol. 2014;32(11):1151–7. doi: 10.1038/nbt.3048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Belair DG, Whisler JA, Valdez J, Velazquez J, Molenda JA, Vickerman V, Lewis R, Daigh C, Hansen TD, Mann DA, Thomson JA, Griffith LG, Kamm RD, Schwartz MP, Murphy WL. Human Vascular Tissue Models Formed from Human Induced Pluripotent Stem Cell Derived Endothelial Cells. Stem Cell Rev. 2014 doi: 10.1007/s12015-014-9549-5. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kusuma S, Facklam A, Gerecht S. Characterizing Human Pluripotent-Stem-Cell-Derived Vascular Cells for Tissue Engineering Applications. Stem Cells Dev. 2014 doi: 10.1089/scd.2014.0377. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]