

Abstract
XPG is a causative gene underlying the photosensitive disorder xeroderma pigmentosum group G (XP-G) and is involved in nucleotide excision repair. Here, we show that XPG knockdown represses epidermal growth factor (EGF)-induced FOS transcription at the level of transcription elongation with little effect on EGF signal transduction. XPG interacted with transcription elongation factors in concert with TFIIH, suggesting that the XPG-TFIIH complex serves as a transcription elongation factor. The XPG-TFIIH complex was recruited to promoter and coding regions of both EGF-induced (FOS) and housekeeping (EEF1A1) genes. Further, EGF-induced recruitment of RNA polymerase II and TFIIH to FOS was reduced by XPG knockdown. Importantly, EGF-induced FOS transcription was markedly lower in XP-G/Cockayne syndrome (CS) cells expressing truncated XPG than in control cells expressing wild-type (WT) XPG, with less significant decreases in XP-G cells with XPG nuclease domain mutations. In corroboration of this finding, both WT XPG and a missense XPG mutant from an XP-G patient were recruited to FOS upon EGF stimulation, but an XPG mutant mimicking a C-terminal truncation from an XP-G/CS patient was not. These results suggest that the XPG-TFIIH complex is involved in transcription elongation and that defects in this association may partly account for Cockayne syndrome in XP-G/CS patients.
INTRODUCTION
Nucleotide excision repair (NER) is an evolutionally conserved DNA repair pathway that removes bulky helix-distorting DNA damage, such as that induced by UV light (1). NER comprises two subpathways: global genome repair (GGR) and transcription-coupled repair (TCR). GGR removes DNA lesions throughout the genome, and TCR specifically removes them from the transcribed strand of active genes. GGR and TCR differ only in the way that they recognize DNA damage. GGR is initiated by UV-DDB- and XPC/RAD23B-mediated recognition of helix distortions inflicted by DNA damage, whereas TCR-specific factors are recruited when RNA polymerase IIo (RNAPIIo) stalls at a site of DNA damage. Subsequent core reactions, including damage excision, gap filling, and ligation, are common to both subpathways (1).
Defects in NER lead to autosomal recessive genetic disorders, such as xeroderma pigmentosum (XP) and Cockayne syndrome (CS) (2). XP is characterized by increased sensitivity to sunlight and development of skin cancer at an early age. Outside of patients harboring mutations in XPV, who are defective in translesion synthesis, seven complementation groups (XP-A to XP-G) that harbor mutations in XPA to XPG have been identified. CS is characterized by cutaneous photosensitivity, growth failure, impaired development of the nervous system, and premature aging but not by a significant increase in skin cancer. Two complementation groups have been identified in CS, termed CS-A and CS-B, which harbor mutations in CSA and CSB, respectively. XP-C and XP-E cells are defective only in GGR, while all other XP cells are defective in both GGR and TCR. In contrast, CS cells are selectively affected in TCR. Despite patients with these diseases being partially or completely defective in NER, the wide spectrum of pathological features in CS patients is not observed in NER-deficient XP patients, such as those with XP-A, and is not explained by defects in NER alone. Although the clinical features of CS may be caused by aberrant transcription (3), not much is known about the functions of NER factors in transcription.
One clue to understanding the functions of NER factors in transcription comes from rare cases in which XP-B, XP-D, and XP-G patients present with the features of CS in addition to those of XP (XP-B/CS, XP-D/CS, and XP-G/CS). XPB and XPD are subunits of TFIIH, a multifunctional complex involved in basal transcription, transactivation, cell cycle, and NER. TFIIH can be divided into two subcomplexes: the core TFIIH comprising XPB, XPD, p62, p52, p44, p34, and p8, and a cdk-activating kinase (CAK) subcomplex that contains cdk7, cyclin H, and MAT1 (4, 5). Therefore, the severe CS features observed in XP-B/CS and XP-D/CS patients may be due to defects in basal transcription, transactivation, and/or the cell cycle. XPG comprises 1,186 amino acids and serves as a structure-specific endonuclease in NER (6). XP-G/CS patients express a mutant XPG bearing a truncated C terminus, whereas XP-G patients express full-length XPG harboring missense mutations (7). We previously reported that XPG forms a stable complex with TFIIH, to facilitate transactivation of nuclear receptors, and that XPG C-terminal truncation (deletion of amino acids 926 to 1186 [Δ926–1186]) destabilizes the XPG-TFIIH complex, thereby reducing CAK-mediated phosphorylation and transactivation of nuclear receptors (8). In addition, NER factors are involved in the epigenetic regulation of gene expression (9–12), indicating an important role in gene transcription.
RNAPII-mediated transcription can be divided into three steps: initiation, elongation, and termination. Previously, gene expression was thought to be regulated mainly at the initiation step, with no further control over the elongation of nascent RNA. Recently, however, a dozen transcription elongation factors have been identified, indicating that the elongation step can also be regulated (13). In contrast to XPB, XPD, and XPG, which are known to regulate initiation, CSB is the sole NER protein that functions during elongation (14). Thus, the pathogenesis of CS is due not only to aberrant regulation of the initiation step but also to aberrant regulation of elongation.
Here, we examined the functions of XPG in epidermal growth factor (EGF)-induced FOS transcription, which is regulated at the level of transcription elongation. Our results show that XPG is required for EGF-induced FOS transcription and that XPG interacts with transcription elongation factors along with TFIIH. XPG knockdown markedly reduced EGF-induced TFIIH recruitment to the promoter and coding regions of FOS, suggesting that the XPG-TFIIH complex is involved in transcription elongation. The XPG-TFIIH complex also bound to promoter and coding regions of the housekeeping gene EEF1A1, indicating involvement in regulating a wide range of gene expression. Importantly, EGF-induced FOS transcription was significantly decreased in XP-G/CS cells and less significantly in XP-G cells, underscoring the importance of the XPG C terminus in transcription elongation. In addition, both wild-type (WT) XPG and full-length XPG harboring a missense mutation (derived from an XP-G patient) were recruited to FOS following EGF stimulation, whereas mutant XPG harboring a C-terminal deletion (derived from an XP-G/CS patient) was not. Taken together, these results suggest that the XPG-TFIIH complex is involved not only in initiation but also in elongation of transcription and that defects in both contribute to CS in XP-G/CS patients.
MATERIALS AND METHODS
Cell lines.
The cell lines used in this study were as follows: HeLa, HeLa stably expressing short hairpin RNA (shRNA) against luciferase or XPG (8), HEK293, and human primary fibroblasts (FS3, XP125LO, XP65BE, XP82DC, and XP20BE). Primary cells from patients were purchased from Coriell Cell Repositories. The cells were cultured in Dulbecco's modified Eagle's medium supplemented with antibiotics and 10% (HeLa and HEK293 cells) or 15% (primary fibroblasts) fetal bovine serum.
Establishment of HEK293 cells stably expressing recombinant XPG.
Green fluorescent protein (GFP) cDNA without a stop codon was amplified by PCR from pEGFP-N1 (Clontech) with the following primers: 5′-ATGGGTACCATGGTGAGCAAGGGCGAGGAG-3′ and 5′-ATGGGTACCCTTGTACAGCTCGTCCATGCC-3′. The PCR product was digested with KpnI and cloned into the KpnI site of pcDNA5/FRT-XPG (WT, A792V, or Δ926-1186)-FLAG-V5-His (8). The plasmids were sequenced to rule out misincorporations during PCR. HEK293 cells stably expressing recombinant XPGs were established using GFP-tagged XPG expression constructs and the Flp-In system (Life Technologies) according to the manufacturer's instructions.
Immunoprecipitation and immunoblotting.
For dithiobis(succinimidyl propionate) (DSP) (Thermo Scientific) cross-linking immunoprecipitation, human embryonic kidney 293 (HEK293) cells were washed twice with phosphate-buffered saline (PBS) containing 1 mM MgCl2 (PBS-Mg) and incubated at room temperature for 30 min with PBS-Mg containing 2 mM DSP. The cross-linking reaction was quenched by adding glycine. Cells were washed twice with PBS and lysed in RIPA buffer (10 mM Tris-HCl [pH 8.0], 140 mM NaCl, 0.1% SDS, 1% Triton X-100, 0.1% sodium deoxycholate, 1 mM EDTA [pH 8.0], and 0.5 mM EGTA) supplemented with protease inhibitor cocktail (Roche) and phosphatase inhibitor cocktail (Roche), followed by brief sonication. The cell lysates were cleared by centrifugation at 16,000 × g at 4°C for 15 min and then precleared by adding ∼1 mg protein to 25 μl of protein G UltraLink resin (Thermo Scientific) and incubated with 10 μl of anti-GFP antibody (full-length Aequorea victoria polyclonal; Clontech), 3 μl of anti-Spt5 serum (15), or 3 μl of preimmune serum at 4°C overnight. A 20-μl aliquot of 50% protein G UltraLink resin slurry containing 40 μg of herring sperm DNA and 40 μg of bovine serum albumin was added to each sample and incubated at 4°C for 3 h. Precipitates were washed sequentially as follows: three washes in RIPA buffer, three washes in 0.5 M RIPA buffer (RIPA buffer with 500 mM NaCl), two washes in LiCl wash buffer (10 mM Tris-HCl [pH 8.0], 0.25 M LiCl, 1% NP-40, 1% sodium deoxycholate, 1 mM EDTA [pH 8.0], 1 mM EGTA), and two washes in RIPA buffer. Bound materials were eluted with Laemmli buffer. With the anti-V5 antibody, Sepharose CL-4B (Sigma) and anti-V5 agarose (Sigma) were used for the preclear and immunoprecipitation steps, respectively.
The immunoblotting primary antibodies were as follows: anti-V5 (Invitrogen), anti-TFIIH p62 (H-10 or Q-19; Santa Cruz), anti-Spt5, anti-Tat-SF1 (BD Pharmingen), anti-MMS19 (Euromedex), anti-GFP (full-length A. victoria polyclonal; Clontech), anti-CTR9 (GeneTex), anti-Paf1 (GeneTex), anti-Cdk9 (EPR3119Y; GeneTex), anti-Rpb1 (H-224 or A-10; Santa Cruz), anti-Rpb1 (H5; Covance), anti-Rb (C-15; Santa Cruz), anti-Cdk7 (MO-1.1; GeneTex), anti-XPG (8H7 [Millipore] or A301-484A [Bethyl]), anti-XPD (H-150; Santa Cruz), anti-MAT1 (FL-309; Santa Cruz), anti-Elk-1 (Calbiochem), (anti-Elk-1 (pS383; Calbiochem), anti-extracellular signal-regulated kinase 1/2 (anti-ERK1/2; Calbiochem), anti-ERK1/2 (pT202/pY204, 12D4; Calbiochem), and anti-XPB (S-19; Santa Cruz). The secondary antibodies were horseradish peroxidase (HRP)-linked anti-mouse and anti-rabbit IgG (GE Healthcare) and HRP-linked anti-mouse IgM (Zymed). ECL and ECL plus kits (GE Healthcare) were used for detection.
RNA preparation and real-time PCR.
Cells were seeded to reach ∼50% confluence at the time of harvest. Subsequently, the cells were cultured for 18 to 24 h in the presence of 0.2% serum and either stimulated for 15 to 60 min with 0.1 μg/ml EGF (Peprotech) or left untreated. Total RNA was prepared using an RNeasy Plus Mini kit (Qiagen) and reverse transcribed to cDNA using a QuantiTect reverse transcription kit (Qiagen). Real-time PCR was performed with SYBR GreenER qPCR supermix (Invitrogen) or TaqMan gene expression master mix (Applied Biosystems). The primers for FOS were 5′-CATGGAGCTGAAGACCGAGC-3′ (forward) and 5′-AGCAGCGTGGGTGAGCTGAG-3′ (reverse), and the primers for GAPDH were 5′-CTGGCGTCTTCACCACCATGG-3′ (forward) and 5′-CATCACGCCACAGTTTCCCGG-3′ (reverse). The TaqMan gene expression assays (Life Technologies) used in this study were as follows: Hs99999140_m1 (FOS), Hs02758991_g1 (GAPDH), Hs00152928_m1 (EGR1), Hs00166165_m1 (EGR2), Hs00357891_s1 (JUNB), Hs00610256_g1 (DUSP1), Hs00153133_m1 (PTGS2), Hs01060665_g1 (ACTB), and Hs00265885_g1 (EEF1A1).
ChIP.
Chromatin immunoprecipitation (ChIP) was performed essentially as described previously (16). For two-step cross-linking ChIP, cells were either stimulated with 0.1 μg/ml EGF for 10 min or left untreated and then cross-linked with 2 mM disuccinimidyl glutarate (DSG) (Thermo Scientific) in PBS-Mg for 45 min at room temperature. After two washes with PBS-Mg, the cells were cross-linked with 1% formaldehyde in PBS-Mg at room temperature for 15 min. The cross-linking reaction was quenched by adding glycine. Nuclei were isolated from cross-linked cells, and chromatin was sheared to an average size of 500 bp using a Bioruptor UCD-200TM (Cosmo Bio). Aliquots of soluble chromatin were diluted 10-fold and immunoprecipitated with anti-Rpb1 (H224; Santa Cruz), anti-Spt5, anti-TBP (SI-1; Santa Cruz), anti-TFIIB (SI-1; Santa Cruz), anti-XPG (A301-484A; Bethyl), anti-XPD (H-150; Santa Cruz), anti-MAT1 (FL-309; Santa Cruz), anti-TFIIH p62 (Q-19; Santa Cruz), or anti-GFP (full-length A. victoria polyclonal; Clontech) antibodies or with rabbit IgG (Santa Cruz). Genomic DNA fragments in cross-linked samples and immunoprecipitates were analyzed by real-time PCR using the following primer pairs: for region u, 5′-GTCACCTCCTCTGGGACCTGTTT-3′ and 5′-CTGTCCCGACCCTCAGAGAGATT-3′; region a, 5′-GCCCCGTGACGTTTACACTCATTC-3′ and 5′-GAGAACATCATCGTGGCGGTTAG-3′; region b, 5′-CTGGCGTTGTGAAGACCATGAC-3′ and 5′-TCATCCTCTGTACTGGGCTCCTG-3′; region c, 5′-CACATCTTCCCTAGAGGGTTCCTG-3′ and 5′-CACACTCCATGCGTTTTGCTACA-3′; region d, 5′-CAATTGAACCGGTGCCTAGAGAA-3′ and 5′-CAAACCCGTTGCGAAAAAGAAC-3′; region e, 5′-AATTAAGGGCTGGGGACAAGGAA-3′ and 5′-ATACCACGTTCACGCTCAGCTTT-3′; region f, 5′-TGTGCCTGACCTCCCATATGTAAA-3′ and 5′-CCTGGGCAACAAGAACTCCATCT-3′; region g, 5′-GCCCTGGTTGGAGTGGAAGTTA-3′ and 5′-ACTTGAGCCCAGGAGTTGACCAG-3′; and region i, 5′-GCCTTAAGGTTTATACCAAAATCA-3′ and 5′-GGAAGGCACTGTTAAAGTTGAG-3′, in which the control primers (i) were directed against an intergenic region on chromosome 2. For absolute quantification, three serial dilutions of cross-linked sample (prior to immunoprecipitation) were assayed concurrently with the immunoprecipitated samples and the data were analyzed using SDS software (Applied Biosystems). For conventional ChIP, cells were cross-linked with 1% formaldehyde alone.
RESULTS
XPG is required for EGF-induced FOS transcription.
To evaluate the physiological function of XPG during transcription, we examined the effect of XPG knockdown using an EGF-induced FOS transcription assay. To avoid potential off-target effects, we used two types of XPG knockdown HeLa cells expressing different XPG shRNAs (8). Immunoblotting showed that XPG protein expression in these cell lines was dramatically reduced compared to that of WT HeLa cells or of control HeLa cells that stably express luciferase-targeted shRNA (Fig. 1A). In contrast, protein levels of the other TFIIH subunits, as well as Rpb1 and Cdk9, were comparable across all four cell lines. Next, we measured FOS and GAPDH transcription using quantitative PCR (Fig. 1B). GAPDH expression levels were unchanged in any of the conditions. Following EGF stimulation, FOS expression increased more than 150-fold in WT and shRNA control cells, whereas a markedly attenuated response was seen in both XPG knockdown cell types. We then examined whether this attenuated response was specific to FOS or common to all EGF-inducible genes (Fig. 1C). The results showed that the expression of other EGF-inducible genes after EGF stimulation was also affected in XPG knockdown cells, whereas the expression of housekeeping genes was not. Taken together, these results indicate that XPG plays an important role in EGF-induced FOS transcription.
FIG 1.

XPG is required for EGF-induced FOS transcription. (A) Immunoblots of whole-cell extracts from wild-type HeLa cells (HeLa WT), or HeLa cells stably expressing short hairpin RNA against luciferase (ctrl KD) or XPG (KD #1 and KD #2), probed with the antibodies indicated on the left. (B) Real-time PCR analysis for FOS and GAPDH mRNAs. Total RNA was prepared from cells stimulated for the indicated times with 0.1 μg/ml EGF. The results are normalized to the unstimulated condition at time zero and expressed as the mean ± standard error of the mean (SEM) from at least three independent experiments. (C) Real-time PCR analysis of EGF-inducible and housekeeping gene expression. Total RNA prepared from cells stimulated for 30 min with 0.1 μg/ml EGF was examined as described for panel B. (D) XPG knockdown has little effect on the EGF signal transduction pathway. Whole extracts from cells that were stimulated for 7.5 min with 0.1 μg/ml EGF or left untreated (−) were analyzed by immunoblotting with the antibodies indicated on the left.
EGF also induces the consecutive phosphorylation of mitogen-activated protein kinase/ERK and the transcription factor Elk-1. Therefore, we examined the effects of XPG knockdown on the EGF signal transduction pathway. Immunoblot analysis showed that EGF-induced ERK and Elk-1 phosphorylation levels were similar in WT, shRNA control, and XPG knockdown cells, indicating that XPG has little effect on the EGF signal transduction pathway (Fig. 1D).
The XPG-TFIIH complex interacts with the transcription elongation complex.
EGF-induced FOS transcription is regulated at the elongation phase, and elongation factor knockdown is shown to reduce transcription (15, 17). These observations, coupled with our results showing that XPG knockdown decreases EGF-induced FOS transcription, suggest that XPG plays a role in transcription elongation. Therefore, we examined XPG interaction with transcription elongation factors. Because few proteins other than TFIIH subunits immunoprecipitate with epitope-tagged XPG under normal conditions (8), we assessed XPG protein interactions under DSP cross-linking conditions (Fig. 2A). HEK293 cells stably expressing GFP- and V5-tagged XPG were treated with DSP, followed by immunoprecipitation and Western blotting of whole-cell homogenates. Consistent with previous observations (8, 18), immunoprecipitation with the anti-GFP or anti-V5 antibody coprecipitated the TFIIH p62 subunit and Rpb1 (Fig. 2B and C). In addition to p62 and Rpb1, immunoblot analysis revealed that XPG interacts with a DSIF subunit (Spt5), Tat-SF1, Paf1 complex subunits (CTR9 and Paf1), and a P-TEFb subunit (Cdk9), all of which are involved in transcription elongation (15, 17). To confirm the interaction between XPG and transcription elongation factors, the cell extracts were incubated with an anti-Spt5 antibody (Fig. 2D). This reaction coprecipitated Cdk7 and XPG (V5) along with Rpb1, Tat-SF1, and Spt5. In contrast, two proteins that are not involved in transcription elongation, MMS19 and retinoblastoma (Rb), were not detected in either immunoprecipitation experiment. These results suggest that the XPG-TFIIH complex interacts specifically with the transcription elongation complex.
FIG 2.
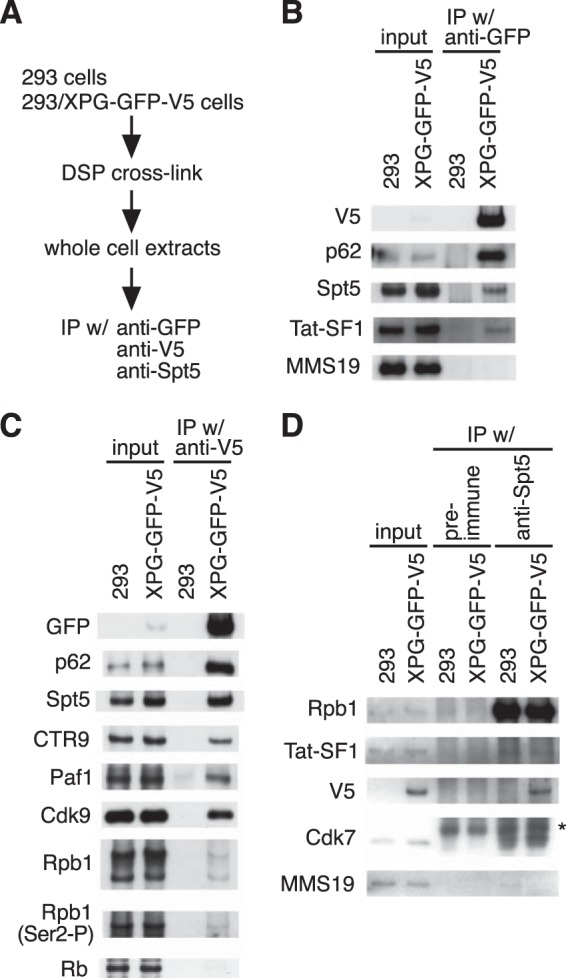
The XPG-TFIIH complex interacts with the transcription elongation complex. (A) Workflow summary of experimental procedures for cross-linking and immunoprecipitation (IP). (B to D) DSP cross-linked whole-cell extracts from untransfected HEK293 cells (293) or HEK293 cells stably expressing XPG-GFP-V5 were immunoprecipitated using anti-GFP antibody (B), anti-V5 antibody (C), and preimmune serum (ctrl) or anti-Spt5 serum (D). Immunoprecipitated materials and 0.5% of the cross-linked samples taken prior to immunoprecipitation (input) were analyzed by immunoblotting with the indicated antibodies. The asterisk denotes nonspecific bands.
The XPG-TFIIH complex physically associates with the entire FOS gene after EGF stimulation.
To better understand the role of XPG in EGF-induced FOS expression, we examined the distribution of XPG, RNAPII, Spt5, and other general transcription factors across the FOS gene (Fig. 3A) by ChIP analysis. Prior to and following EGF induction, TBP and TFIIB associated only with the FOS promoter region (Fig. 3B); this association pattern was similar in XPG knockdown and control cells. Consistent with earlier reports (17), RNAPII and Spt5 associated with the FOS promoter region in the absence of EGF stimulation, while following induction, their associations extended to include downstream regions (Fig. 3B). Prior to induction, RNAPII and Spt5 associations with the FOS gene were similar in XPG knockdown cells and control cells. In contrast, EGF induction reduced RNAPII association with both promoter and downstream regions of FOS in XPG knockdown cells, compared to control cells (Fig. 3B and C). These results suggest that XPG contributes to transcription elongation as well as initiation.
FIG 3.

The XPG-TFIIH complex physically associates with the entire FOS gene after EGF stimulation. (A) Diagram of FOS gene. The transcription start site is indicated by an arrow. The boxes represent open reading frames. The four amplicons (a, b, c, and u) used for ChIP are indicated under the diagram. (B) Soluble chromatin from cells stimulated for 10 min with EGF or left unstimulated was subjected to ChIP with anti-Rpb1, anti-Spt5, anti-TBP, or anti-TFIIB antibody. Real-time PCR was performed using primers amplifying a control intergenic region on chromosome 2 (labeled i) and the four regions of FOS. Each bar represents a mean ± SEM for at least three independent experiments. Asterisks indicate a statistically significant difference between corresponding ctrl KD and XPG KD #2 values (*, P < 0.05; Student's t test). In the graphs for RNAPII and Spt5, the left y axis shows the percentage of ChIPed DNA for amplicon a, and the right y axis shows the percentage of ChIPed DNA for the other amplicons. (C) Comparison of RNAPII ChIP signals of XPG KD cells with those of control KD cells in panel B. (D) Soluble chromatin from cells stimulated for 10 min with EGF or unstimulated cells were subjected to two-step cross-linking ChIP with control rabbit IgG or anti-XPG, anti-p62, anti-XPD, or anti-MAT1 antibody. Real-time PCR was performed as described for panel B. Each bar represents a mean ± SEM for at least three independent experiments. Asterisks indicate a statistically significant difference between corresponding ctrl KD and XPG KD #2 values (*, P < 0.05; **, P < 0.01; Student's t test). In the graphs of XPG, MAT1, and XPD, the left y axis shows the percentage of ChIPed DNA for amplicon a, and the right y axis shows the percentage of ChIPed DNA for the other amplicons. (E) Comparison of p62 and XPD ChIP signals of XPG KD cells with those of control KD cells in panel D. The results are represented as fold enrichment relative to the enrichment under unstimulated conditions.
We also examined the distribution of XPG and TFIIH on FOS; however, there was poor signal resolution using conventional ChIP analysis. Instead, we used a recently published two-step DSG cross-linking ChIP method (19) to detect transcription factors binding to their target loci. Similar to our previous results, the two-step ChIP assay showed that TBP and TFIIB bound specifically to the promoter region of the EEF1A1 gene (see below), indicating that this method retains the same binding specificity as the original ChIP assay. Using the two-step ChIP assay, we found XPG to be associated with multiple regions of the FOS gene both before and after EGF stimulation (Fig. 3D). However, in XPG knockdown cells, XPG ChIP signals were reduced to the same levels as those of rabbit IgG. Therefore, XPG is distributed throughout the genome and is further recruited to the FOS gene upon EGF stimulation (Fig. 3D). In addition, in shRNA control cells, TFIIH subunits (XPD, p62, and MAT1) were also recruited to the FOS gene upon EGF stimulation (Fig. 3D and E), suggesting that XPG and TFIIH track together along FOS. In contrast, in XPG knockdown cells, ChIP signals for MAT1 were markedly reduced (Fig. 3D) and ChIP signals for p62 and XPD were less enriched on the FOS gene upon EGF stimulation (Fig. 3D and E), suggesting that XPG is required for EGF-induced TFIIH recruitment to FOS. Although MAT1, p62, and XPD are TFIIH components, they exhibited different distributions on FOS. This may be due to the existence of complexes other than TFIIH that contain a TFIIH subunit(s), such as the MMXD (MMS19-MIP18-XPD) complex (20). Together, these results indicate that upon EGF stimulation, the XPG-TFIIH complex is recruited to the entire FOS gene and participates in FOS transcription.
The XPG-TFIIH complex also associates with housekeeping gene loci.
To further demonstrate that the XPG-TFIIH complex contributes to transcription elongation, we used ChIP analysis to examine the distribution of these same factors on the housekeeping gene EEF1A1 (Fig. 4A). We found that RNAPII and Spt5 associate with both the promoter and downstream regions, whereas TBP and TFIIB associate only with the promoter region of EEF1A1 (Fig. 4B). Similar to the ChIP findings for FOS (Fig. 3D), substantial amounts of XPG and TFIIH subunits were associated with both the promoter and downstream regions of EEF1A1. These results indicate that the XPG-TFIIH complex associates with the transcribed regions of constitutively active genes as well as those of stimulus-induced genes.
FIG 4.

The XPG-TFIIH complex is associated with a housekeeping gene (EEF1A1). (A) Diagram of the EEF1A1 gene. The four amplicons (d, e, f, and g) used for ChIP are indicated (diagram notation is identical to that shown in the legend for Fig. 3A). (B) ChIP assays for XPG-TFIIH complex proteins involved in EEF1A1 gene transcription. ChIP was performed using a two-step cross-linking protocol with soluble chromatin from HeLa cells and immunoprecipitated with the indicated antibodies. Real-time PCR was performed using primers amplifying a control intergenic region (i), a region upstream of FOS (u), and the four regions of the EEF1A1 gene (d to g). Each bar represents the mean ± SEM for at least three independent experiments. Asterisks indicate a statistically significant difference with respect to values for the u and/or i regions (*, P < 0.05; **, P < 0.01; Student's t test).
EGF-induced FOS transcription is compromised in the cells of XP-G/CS patients.
The above findings prompted us to investigate whether EGF-induced FOS transcription is affected in primary fibroblasts from human patients harboring mutations in XPG. For this, we used two XP-G (XP125LO and XP65BE) and two XP-G/CS (XP82DC and XP20BE) cell lines (Fig. 5A). Consistent with previous reports (21), immunoblot analysis revealed that full-length XPG was not present in XP-G/CS cells, whereas small but significant amounts of full-length XPG were detected in XP-G cells (Fig. 5B).
FIG 5.

EGF-induced FOS transcription is compromised in the cells of XP-G/CS patients. (A) Schematic representation of wild-type XPG and mutations predicted to be expressed in XP125LO, XP65BE, XP82DC, and XP20BE mutant cells. The two nuclease motifs (N and I regions) and mutations are indicated as gray and black boxes, respectively. (B) Whole-cell extracts of primary fibroblasts from normal (FS3), XP-G (XP125LO and XP65BE), and XP-G/CS (XP82DC and XP20BE) patients were analyzed by immunoblotting with the indicated antibodies. The asterisk denotes nonspecific bands. (C) Real-time PCR analysis of FOS expression was performed at the indicated times following EGF (0.1 μg/ml) treatment. FOS expression values are normalized to GAPDH expression. In each experiment, the total FOS expression was estimated after 60 min of EGF stimulation (yellow area in the graphs) and is shown as the mean ± SEM from at least three independent experiments (lower right panel). The differences between normal and XP-G/CS cells were statistically significant (*, P < 0.05; **, P < 0.01; Student's t test).
Next, we used real-time PCR to examine FOS transcription in XP-G, XP-G/CS, and normal WT control (FS3) cells at 0, 15, 30, and 60 min after EGF stimulation (Fig. 5C). At 30 min after EGF stimulation (peak response), FOS transcription increased in FS3, XP125LO, XP65BE, XP82DC, and XP20BE cells by 53-, 40-, 26-, 22-, and 22-fold, respectively. In addition, the estimated level of FOS expression over 60 min of EGF stimulation was significantly reduced in XP-G/CS cells compared to normal control cells, while FOS expression was markedly less attenuated in XP-G cells. The extent of the effect of XPG deficiency on FOS transcription shown here was lesser than that shown in Fig. 1B, probably because different cells were used in the two figures (HeLa cells and primary cells from patients). Note that while inducible FOS transcription at 30 min was similar in XP65BE and XP-G/CS cells, the cumulative FOS transcription over 60 min was higher in XP65BE than in XP-G/CS cells. Taken together, these results indicate that the level of EGF-induced FOS transcription is significantly lower in XP-G/CS cells than in normal control cells but is at an intermediate level in XP-G cells, although in XP-G cells, reduced transcription is likely due to markedly decreased XPG protein expression (Fig. 5B). It was also suggested that XPG endonuclease activity is not essential for EGF-induced FOS transcription.
The XPG C-terminal region is required for EGF-induced association with FOS.
To further understand the basis for differences in EGF-induced FOS transcription, we used ChIP to examine the FOS gene for distribution of WT XPG and mutant XPG proteins derived from XP-G and XP-G/CS patients. The XPG mutants included an A792V missense mutation identified from an XP-G (XP125LO) patient and a C-terminal truncation (ΔC) mutation mimicking that of an XP-G/CS (XPCS1RO) patient (Fig. 6A). WT and mutant XPG proteins were fused to GFP and V5 tags and stably expressed in HEK293 cells (Fig. 6B). ChIP analysis was subsequently performed using these cells and parental HEK293 cells, with an anti-Rpb1 or anti-GFP antibody (Fig. 6C). With the Rpb1 antibody, ChIP signals were similar in magnitude and distribution across the FOS gene in all cells examined. Using the anti-GFP antibody, after induction in XPG (WT) cells, ChIP signals increased across the entire FOS gene, consistent with the results we obtained using an anti-XPG antibody (Fig. 3D). Similar inducible XPG distribution patterns were observed in XPG A792V cells, yet no significant signal increases were detected in XPG ΔC or parental HEK293 cells. Taken together, these results indicate that the C-terminal region of XPG is crucial for EGF-stimulated recruitment to FOS.
FIG 6.

The C-terminal region of XPG is required for association with the FOS gene after EGF induction. (A) Schematic structure of recombinant XPG proteins expressed in HEK293 cells. (B) Whole-cell extracts from HEK293 cells or HEK293 cells stably expressing GFP-V5-tagged recombinant XPG were analyzed by immunoblotting with the indicated antibodies. (C) ChIP using anti-Rpb1 or anti-GFP antibody and real-time PCR was performed as described in the legend for Fig. 3D. Asterisks indicate a statistically significant difference between the value before EGF stimulation and the values after EGF stimulation (*, P < 0.05; **, P < 0.01; Student's t test).
The XPG C-terminal truncation mutant binds to transcription elongation factors but not to TFIIH in vivo.
To examine whether WT and mutant XPG constructs interact with TFIIH and transcription elongation factors in vivo, extracts of HEK293 cells stably expressing exogenous WT or mutant XPG (A792V or ΔC) were cross-linked with DSP and then immunoprecipitated with an anti-V5 antibody. Subsequent immunoblot analysis of the immunoprecipitate revealed that WT XPG and XPG A792V bound both TFIIH and transcription elongation factors, whereas XPG ΔC bound to transcription elongation factors but not to TFIIH (Fig. 7A). While XPG ΔC could be included in the elongation complex via interactions with transcription elongation factors, XPG ΔC did not bind to FOS in vivo (Fig. 6C). Moreover, FOS transcription was induced to a significantly lower level in XP-G/CS cells (Fig. 5C). Taken together, these results suggest that the interaction between the XPG C terminus and TFIIH plays an important role in FOS transcription.
FIG 7.

Mutant XPG bearing a C-terminal truncation binds to transcription elongation factors but not to TFIIH. (A) DSP cross-linked extracts from whole HEK293 cells stably expressing recombinant XPG were immunoprecipitated with an anti-V5 antibody. Bound materials were analyzed by immunoblotting with the indicated antibodies. (B) Schematic model for nucleotide excision repair, ligand-induced transactivation, and transcription elongation in normal, XP-G, and XP-G/CS cells (NR, nuclear receptor; RE, response element; GTFs, general transcription factors; CTD, C-terminal domain).
DISCUSSION
The XPG-TFIIH complex associates with transcription elongation factors.
Interaction between XPG and TFIIH has been reported by many groups (18, 22–26). In addition, we reported that XPG forms a complex with TFIIH, stabilizing it and allowing the phosphorylation and transactivation of nuclear receptors (8). Therefore, XPG could be considered to be the eleventh subunit of TFIIH (27). In this report, we confirmed that XPG (WT) and XPG A792V, but not XPG ΔC, interact with TFIIH under cross-linking conditions (Fig. 7A) and found that XPG interacts with transcription elongation factors, including Spt5, Tat-SF1, Cdk9, and Paf1 complex subunits, along with TFIIH (Fig. 2 and 7A). This result is consistent with the previous findings that XPG binds to phosphorylated RNAPII (18) and that rad2 (a yeast homolog of the XPG gene) interacts genetically with dst1 (a yeast homolog of the TFIIS gene) (28). XPG ΔC did not bind to TFIIH but still interacted with transcription elongation factors, suggesting that XPG is important for TFIIH to participate in the transcription elongation complex. This idea is supported by our results showing that XPG knockdown attenuates recruitment of TFIIH subunits to FOS upon EGF stimulation (Fig. 3D). Although the C-terminally truncated XPG mutant bound to transcription elongation factors in an immunoprecipitation assay, the intact XPG-TFIIH complex is required for transcription elongation in vivo. This is demonstrated in that XPG (WT) and XPG A792V were recruited to FOS after EGF stimulation but XPG ΔC was not (Fig. 6C).
Role of the XPG-TFIIH complex in transcription elongation.
We showed that the XPG-TFIIH complex associates with the actively transcribed regions of not only stimulus-induced genes but also housekeeping genes. However, knockdown of XPG did not change the expression levels of various proteins (Fig. 1A and D) or RNA synthesis as measured by the incorporation of [3H]uridine (T. Narita and M. Saijo, unpublished data), suggesting that the XPG-TFIIH complex is not crucial for the expression of the genes. However, the XPG-TFIIH complex still has the potential to be involved in transcription elongation of some genes on the basis of the following observations: (i) the XPG-TFIIH complex was enriched at the promoters and coding regions of actively transcribed genes (Fig. 3D and 4B), and (ii) the XPG-TFIIH complex interacted with the transcription elongation complex regardless of EGF stimulation (Fig. 2). Other factors may compensate for the function of the XPG-TFIIH complex in the transcription of housekeeping genes.
The XPG-TFIIH complex has four enzymatic activities: endonuclease (XPG), helicase (XPB and XPD), ubiquitin ligase (p44), and protein kinase (Cdk7) (27). Most likely, Cdk7 kinase activity coordinates the phosphorylation state of the RNAPII C-terminal domain (CTD) during transcription elongation. Although most prior studies analyzed the phosphorylation of Ser2 and Ser5 in the CTD, recent studies reveal that Ser7 is also phosphorylated in both yeast and mammalian cells (29, 30). Ser7 phosphorylation is detected when RNAPII is located between the promoter region and the 3′ end of both protein-coding and noncoding genes (31, 32). Cdk7 (Kin28 in yeast) phosphorylates Ser5 and plays a role in Ser7 phosphorylation in both yeast and human cells (31, 33–36). Genome-wide analyses using a Kin28 analog-sensitive mutant (37) revealed that (i) the phosphorylation levels of both Ser5 and Ser7 increase throughout the open reading frame to levels far beyond those normally observed in WT cells in the absence of Kin28 catalytic activity and (ii) this abnormal phosphorylation is partially, but significantly, suppressed by the deletion of Bur2 (the human homolog of the Bur1-Bur2 complex is P-TEFb). These results suggest that the kinase activity of Kin28 (Cdk7) maintains the proper phosphorylation levels of Ser5 and Ser7 by preventing opportunistic phosphorylation by other kinases. We found that the XPG-TFIIH complex interacts with the transcription elongation complex, including P-TEFb, and is recruited to the coding regions of both primary response and housekeeping genes in this study. In addition, XPG knockdown affects Ser5 phosphorylation of the RNAPII CTD at the coding region as well as at the promoter region of the FOS gene after EGF stimulation (T. Narita and K. Tanaka, unpublished data). Therefore, the XPG-TFIIH complex may regulate the phosphorylation of elongating RNAPII on the CTD at Ser5 and Ser7.
Links between CS features and aberrant transcription in CS and XP/CS patients.
The CS-specific features in CS and XP/CS patients are unlikely to be due solely to a defect in NER, based on the observations that (i) CS-A and CS-B patients are deficient in TCR but proficient in GGR, whereas mutations in XPA, which is indispensable for both TCR and GGR, do not cause CS, and (ii) neither Xpa−/− nor Xpg−/− mice are capable of NER, yet only Xpg−/− mice show severe CS-like features (38). We found that both WT XPG and an A792V mutant derived from an XP-G patient form stable complexes with TFIIH, allowing the phosphorylation and transactivation of nuclear receptors, whereas a mutant XPG harboring a C-terminal deletion (derived from an XP-G/CS patient) did not form a complex with TFIIH, resulting in reduced transactivation of nuclear receptors (8). In addition, XP-D/CS, but not XP-D, cells were unable to restart DHFR transcription after UV irradiation, due to Sirt1-mediated heterochromatinization of the promoter (11). Moreover, CSB is known to regulate RNAPI and RNAPII transcription (14, 39). These results suggest that the pathological features in patients with CS and XP/CS are caused by dysregulation of transcriptional networks.
From the data presented in this study, we propose a model for the function of the XPG-TFIIH complex in cells and to explain the link between mutations in XPG and clinical features (Fig. 7B). The XPG-TFIIH complex has at least three functions: (i) NER, by interacting with NER factors; (ii) ligand-induced transactivation through the phosphorylation of nuclear receptors; and (iii) control of transcriptional elongation by interacting with the transcription elongation complex. Point mutations in XPG that impair endonuclease activity only affect NER, leading to the pathogenesis typical of XP. In contrast, destabilization of the XPG-TFIIH complex due to XPG C-terminal truncation affects not only NER but also ligand-induced transactivation and transcription elongation, leading to the pathogenesis observed in XP-G/CS. Because dysregulation of transcriptional elongation causes developmental defects (40–42), our results indicate that the CS features in XP-G/CS patients are due in part to defects in regulation of transcriptional elongation.
ACKNOWLEDGMENTS
We thank Hiroshi Handa for providing antibodies. We also thank Yuki Yamaguchi and Shinsuke Ito for helpful discussion.
This work was supported in part by a Grant-in-Aid for Scientific Research on Innovative Areas (to K.T., no. 22131009) from the Ministry of Education, Cultures, Sports, Science and Technology (MEXT). This work was also supported by Health and Labor Sciences Research Grants for Research on Intractable Diseases (to K.T.), Grants-in-Aid for Young Scientists (B) (to T.N., no. 19770146 and 22770171), a Grant-in-Aid for Scientific Research (C) (to T.N., no. 24570156), a Grant-in-Aid for Scientific Research on Priority Areas (to T.N., no. 20052015) from MEXT, a grant from the Circle for the Promotion of Science and Engineering (to T.N.), and a grant from the Uehara Memorial Foundation (to T.N.).
REFERENCES
- 1.Kamileri I, Karakasilioti I, Garinis GA. 2012. Nucleotide excision repair: new tricks with old bricks. Trends Genet 28:566–573. doi: 10.1016/j.tig.2012.06.004. [DOI] [PubMed] [Google Scholar]
- 2.Lehmann AR. 2003. DNA repair-deficient diseases, xeroderma pigmentosum, Cockayne syndrome and trichothiodystrophy. Biochimie 85:1101–1111. doi: 10.1016/j.biochi.2003.09.010. [DOI] [PubMed] [Google Scholar]
- 3.Brooks PJ. 2013. Blinded by the UV light: how the focus on transcription-coupled NER has distracted from understanding the mechanisms of Cockayne syndrome neurologic disease. DNA Repair (Amst) 12:656–671. doi: 10.1016/j.dnarep.2013.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fisher RP. 2005. Secrets of a double agent: CDK7 in cell-cycle control and transcription. J Cell Sci 118:5171–5180. doi: 10.1242/jcs.02718. [DOI] [PubMed] [Google Scholar]
- 5.Zurita M, Merino C. 2003. The transcriptional complexity of the TFIIH complex. Trends Genet 19:578–584. doi: 10.1016/j.tig.2003.08.005. [DOI] [PubMed] [Google Scholar]
- 6.O'Donovan A, Scherly D, Clarkson SG, Wood RD. 1994. Isolation of active recombinant XPG protein, a human DNA repair endonuclease. J Biol Chem 269:15965–15968. [PubMed] [Google Scholar]
- 7.Scharer OD. 2008. XPG: its products and biological roles. Adv Exp Med Biol 637:83–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ito S, Kuraoka I, Chymkowitch P, Compe E, Takedachi A, Ishigami C, Coin F, Egly JM, Tanaka K. 2007. XPG stabilizes TFIIH, allowing transactivation of nuclear receptors: implications for Cockayne syndrome in XP-G/CS patients. Mol Cell 26:231–243. doi: 10.1016/j.molcel.2007.03.013. [DOI] [PubMed] [Google Scholar]
- 9.Barreto G, Schafer A, Marhold J, Stach D, Swaminathan SK, Handa V, Doderlein G, Maltry N, Wu W, Lyko F, Niehrs C. 2007. Gadd45a promotes epigenetic gene activation by repair-mediated DNA demethylation. Nature 445:671–675. doi: 10.1038/nature05515. [DOI] [PubMed] [Google Scholar]
- 10.Le May N, Mota-Fernandes D, Velez-Cruz R, Iltis I, Biard D, Egly JM. 2010. NER factors are recruited to active promoters and facilitate chromatin modification for transcription in the absence of exogenous genotoxic attack. Mol Cell 38:54–66. doi: 10.1016/j.molcel.2010.03.004. [DOI] [PubMed] [Google Scholar]
- 11.Velez-Cruz R, Zadorin AS, Coin F, Egly JM. 2013. Sirt1 suppresses RNA synthesis after UV irradiation in combined xeroderma pigmentosum group D/Cockayne syndrome (XP-D/CS) cells. Proc Natl Acad Sci U S A. 110:E212-220. doi: 10.1073/pnas.1213076110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schmitz KM, Schmitt N, Hoffmann-Rohrer U, Schafer A, Grummt I, Mayer C. 2009. TAF12 recruits Gadd45a and the nucleotide excision repair complex to the promoter of rRNA genes leading to active DNA demethylation. Mol Cell 33:344–353. doi: 10.1016/j.molcel.2009.01.015. [DOI] [PubMed] [Google Scholar]
- 13.Zhou Q, Li T, Price DH. 2012. RNA polymerase II elongation control. Annu Rev Biochem 81:119–143. doi: 10.1146/annurev-biochem-052610-095910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Selby CP, Sancar A. 1997. Cockayne syndrome group B protein enhances elongation by RNA polymerase II. Proc Natl Acad Sci U S A 94:11205–11209. doi: 10.1073/pnas.94.21.11205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen Y, Yamaguchi Y, Tsugeno Y, Yamamoto J, Yamada T, Nakamura M, Hisatake K, Handa H. 2009. DSIF, the Paf1 complex, and Tat-SF1 have nonredundant, cooperative roles in RNA polymerase II elongation. Genes Dev 23:2765–2777. doi: 10.1101/gad.1834709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Oshima S, Nakamura T, Namiki S, Okada E, Tsuchiya K, Okamoto R, Yamazaki M, Yokota T, Aida M, Yamaguchi Y, Kanai T, Handa H, Watanabe M. 2004. Interferon regulatory factor 1 (IRF-1) and IRF-2 distinctively up-regulate gene expression and production of interleukin-7 in human intestinal epithelial cells. Mol Cell Biol 24:6298–6310. doi: 10.1128/MCB.24.14.6298-6310.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yamada T, Yamaguchi Y, Inukai N, Okamoto S, Mura T, Handa H. 2006. P-TEFb-mediated phosphorylation of hSpt5 C-terminal repeats is critical for processive transcription elongation. Mol Cell 21:227–237. doi: 10.1016/j.molcel.2005.11.024. [DOI] [PubMed] [Google Scholar]
- 18.Sarker AH, Tsutakawa SE, Kostek S, Ng C, Shin DS, Peris M, Campeau E, Tainer JA, Nogales E, Cooper PK. 2005. Recognition of RNA polymerase II and transcription bubbles by XPG, CSB, and TFIIH: insights for transcription-coupled repair and Cockayne syndrome. Mol Cell 20:187–198. doi: 10.1016/j.molcel.2005.09.022. [DOI] [PubMed] [Google Scholar]
- 19.Nowak DE, Tian B, Brasier AR. 2005. Two-step cross-linking method for identification of NF-kappaB gene network by chromatin immunoprecipitation. Biotechniques 39:715–725. doi: 10.2144/000112014. [DOI] [PubMed] [Google Scholar]
- 20.Ito S, Tan LJ, Andoh D, Narita T, Seki M, Hirano Y, Narita K, Kuraoka I, Hiraoka Y, Tanaka K. 2010. MMXD, a TFIIH-independent XPD-MMS19 protein complex involved in chromosome segregation. Mol Cell 39:632–640. doi: 10.1016/j.molcel.2010.07.029. [DOI] [PubMed] [Google Scholar]
- 21.Nouspikel T, Lalle P, Leadon SA, Cooper PK, Clarkson SG. 1997. A common mutational pattern in Cockayne syndrome patients from xeroderma pigmentosum group G: implications for a second XPG function. Proc Natl Acad Sci U S A 94:3116–3121. doi: 10.1073/pnas.94.7.3116. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 22.Araujo SJ, Nigg EA, Wood RD. 2001. Strong functional interactions of TFIIH with XPC and XPG in human DNA nucleotide excision repair, without a preassembled repairosome. Mol Cell Biol 21:2281–2291. doi: 10.1128/MCB.21.7.2281-2291.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dunand-Sauthier I, Hohl M, Thorel F, Jaquier-Gubler P, Clarkson SG, Scharer OD. 2005. The spacer region of XPG mediates recruitment to nucleotide excision repair complexes and determines substrate specificity. J Biol Chem 280:7030–7037. doi: 10.1074/jbc.M412228200. [DOI] [PubMed] [Google Scholar]
- 24.Gervais V, Lamour V, Jawhari A, Frindel F, Wasielewski E, Dubaele S, Egly JM, Thierry JC, Kieffer B, Poterszman A. 2004. TFIIH contains a PH domain involved in DNA nucleotide excision repair. Nat Struct Mol Biol 11:616–622. doi: 10.1038/nsmb782. [DOI] [PubMed] [Google Scholar]
- 25.Mu D, Park CH, Matsunaga T, Hsu DS, Reardon JT, Sancar A. 1995. Reconstitution of human DNA repair excision nuclease in a highly defined system. J Biol Chem 270:2415–2418. doi: 10.1074/jbc.270.6.2415. [DOI] [PubMed] [Google Scholar]
- 26.Thorel F, Constantinou A, Dunand-Sauthier I, Nouspikel T, Lalle P, Raams A, Jaspers NG, Vermeulen W, Shivji MK, Wood RD, Clarkson SG. 2004. Definition of a short region of XPG necessary for TFIIH interaction and stable recruitment to sites of UV damage. Mol Cell Biol 24:10670–10680. doi: 10.1128/MCB.24.24.10670-10680.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Egly JM, Coin F. 2011. A history of TFIIH: two decades of molecular biology on a pivotal transcription/repair factor. DNA Repair (Amst) 10:714–721. doi: 10.1016/j.dnarep.2011.04.021. [DOI] [PubMed] [Google Scholar]
- 28.Lee SK, Yu SL, Prakash L, Prakash S. 2002. Requirement of yeast RAD2, a homolog of human XPG gene, for efficient RNA polymerase II transcription. Implications for Cockayne syndrome. Cell 109:823–834. [DOI] [PubMed] [Google Scholar]
- 29.Chapman RD, Heidemann M, Albert TK, Mailhammer R, Flatley A, Meisterernst M, Kremmer E, Eick D. 2007. Transcribing RNA polymerase II is phosphorylated at CTD residue serine-7. Science 318:1780–1782. doi: 10.1126/science.1145977. [DOI] [PubMed] [Google Scholar]
- 30.Schwer B, Shuman S. 2011. Deciphering the RNA polymerase II CTD code in fission yeast. Mol Cell 43:311–318. doi: 10.1016/j.molcel.2011.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tietjen JR, Zhang DW, Rodriguez-Molina JB, White BE, Akhtar MS, Heidemann M, Li X, Chapman RD, Shokat K, Keles S, Eick D, Ansari AZ. 2010. Chemical-genomic dissection of the CTD code. Nat Struct Mol Biol 17:1154–1161. doi: 10.1038/nsmb.1900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mayer A, Lidschreiber M, Siebert M, Leike K, Soding J, Cramer P. 2010. Uniform transitions of the general RNA polymerase II transcription complex. Nat Struct Mol Biol 17:1272–1278. doi: 10.1038/nsmb.1903. [DOI] [PubMed] [Google Scholar]
- 33.Glover-Cutter K, Larochelle S, Erickson B, Zhang C, Shokat K, Fisher RP, Bentley DL. 2009. TFIIH-associated Cdk7 kinase functions in phosphorylation of C-terminal domain Ser7 residues, promoter-proximal pausing, and termination by RNA polymerase II. Mol Cell Biol 29:5455–5464. doi: 10.1128/MCB.00637-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Akhtar MS, Heidemann M, Tietjen JR, Zhang DW, Chapman RD, Eick D, Ansari AZ. 2009. TFIIH kinase places bivalent marks on the carboxy-terminal domain of RNA polymerase II. Mol Cell 34:387–393. doi: 10.1016/j.molcel.2009.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kim M, Suh H, Cho EJ, Buratowski S. 2009. Phosphorylation of the yeast Rpb1 C-terminal domain at serines 2, 5, and 7. J Biol Chem 284:26421–26426. doi: 10.1074/jbc.M109.028993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Boeing S, Rigault C, Heidemann M, Eick D, Meisterernst M. 2010. RNA polymerase II C-terminal heptarepeat domain Ser-7 phosphorylation is established in a mediator-dependent fashion. J Biol Chem 285:188–196. doi: 10.1074/jbc.M109.046565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bataille AR, Jeronimo C, Jacques PE, Laramee L, Fortin ME, Forest A, Bergeron M, Hanes SD, Robert F. 2012. A universal RNA polymerase II CTD cycle is orchestrated by complex interplays between kinase, phosphatase, and isomerase enzymes along genes. Mol Cell 45:158–170. doi: 10.1016/j.molcel.2011.11.024. [DOI] [PubMed] [Google Scholar]
- 38.Shiomi N, Mori M, Kito S, Harada YN, Tanaka K, Shiomi T. 2005. Severe growth retardation and short life span of double-mutant mice lacking Xpa and exon 15 of Xpg. DNA Repair (Amst) 4:351–357. doi: 10.1016/j.dnarep.2004.10.009. [DOI] [PubMed] [Google Scholar]
- 39.Bradsher J, Auriol J, de Santis LP, Iben S, Vonesch JL, Grummt I, Egly JM. 2002. CSB is a component of RNA pol I transcription. Mol Cell 10:819–829. doi: 10.1016/S1097-2765(02)00678-0. [DOI] [PubMed] [Google Scholar]
- 40.Guo S, Yamaguchi Y, Schilbach S, Wada T, Lee J, Goddard A, French D, Handa H, Rosenthal A. 2000. A regulator of transcriptional elongation controls vertebrate neuronal development. Nature 408:366–369. doi: 10.1038/35042590. [DOI] [PubMed] [Google Scholar]
- 41.Barboric M, Lenasi T, Chen H, Johansen EB, Guo S, Peterlin BM. 2009. 7SK snRNP/P-TEFb couples transcription elongation with alternative splicing and is essential for vertebrate development. Proc Natl Acad Sci U S A 106:7798–7803. doi: 10.1073/pnas.0903188106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bai X, Kim J, Yang Z, Jurynec MJ, Akie TE, Lee J, LeBlanc J, Sessa A, Jiang H, DiBiase A, Zhou Y, Grunwald DJ, Lin S, Cantor AB, Orkin SH, Zon LI. 2010. TIF1gamma controls erythroid cell fate by regulating transcription elongation. Cell 142:133–143. doi: 10.1016/j.cell.2010.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]