

Abstract
CELF1 is a multifunctional RNA-binding protein that controls several aspects of RNA fate. The targeted disruption of the Celf1 gene in mice causes male infertility due to impaired spermiogenesis, the postmeiotic differentiation of male gametes. Here, we investigated the molecular reasons that underlie this testicular phenotype. By measuring sex hormone levels, we detected low concentrations of testosterone in Celf1-null mice. We investigated the effect of Celf1 disruption on the expression levels of steroidogenic enzyme genes, and we observed that Cyp19a1 was upregulated. Cyp19a1 encodes aromatase, which transforms testosterone into estradiol. Administration of testosterone or the aromatase inhibitor letrozole partly rescued the spermiogenesis defects, indicating that a lack of testosterone associated with excessive aromatase contributes to the testicular phenotype. In vivo and in vitro interaction assays demonstrated that CELF1 binds to Cyp19a1 mRNA, and reporter assays supported the conclusion that CELF1 directly represses Cyp19a1 translation. We conclude that CELF1 downregulates Cyp19a1 (Aromatase) posttranscriptionally to achieve high concentrations of testosterone compatible with spermiogenesis completion. We discuss the implications of these findings with respect to reproductive defects in men, including patients suffering from isolated hypogonadotropic hypogonadism and myotonic dystrophy type I.
INTRODUCTION
Spermatogenesis is a complex process that can be divided into three phases: mitotic proliferation of spermatogonia, meiosis, and postmeiotic differentiation. Differentiation, or spermiogenesis, transforms round spermatids into elongated spermatids and spermatozoa. It is morphologically characterized by the formation of the acrosome and flagellum, nucleus condensation, and cell elongation (1). The mature gametes are then released by Sertoli cells into the lumen of the seminiferous tubules by spermiation (2).
Transcription ceases during spermiogenesis upon nuclear condensation. At this stage, proteins are translated from mRNAs transcribed at earlier stages and stored in a translationally inactive state (3). Gene expression is therefore regulated only posttranscriptionally in differentiating spermatids, but in general, mechanisms of posttranscriptional controls are of key importance in the testis. Small noncoding RNAs, and especially microRNAs (miRNAs) and Piwi-interacting RNAs (piRNAs), downregulate specific transcripts. This regulation is crucial, since spermatogenesis is severely impaired in mice lacking the enzymes that synthesize this class of regulatory RNAs (4). Other key actors in posttranscriptional control are RNA-binding proteins (RBPs). Collectively, they control several aspects of RNA life: nuclear maturation, transport, storage in cellular compartments, translation, and stability or decay. Several RBPs are involved in spermatogenesis, and the inactivation in mice of some genes encoding RBPs is associated with infertility due to spermatogenesis defects (3, 5–7). In most cases, the molecular reasons for infertility have not been identified yet.
CELF1 (CUGBP, Elav-like family member 1; also named CUGBP1) is among the RBPs that are involved in spermatogenesis. The Celf1 gene is largely expressed in mouse testis, and mice with a targeted disruption of Celf1 (referred to here as Celf1−/− mice) display an arrest of spermiogenesis just before the round spermatids start elongating (8). This blockage is observed in adults but also during the first wave of spermatogenesis (9). CELF1 is a multifunctional RBP, with a nuclear and cytoplasmic localization (10). In the nucleus, it controls pre-mRNA alternative splicing (11, 12). In the cytoplasm, it targets bound mRNAs for rapid decay (13, 14) and/or modulates their translation (15–19). The cytoplasmic functions are probably mediated by interactions with molecular partners, like the poly(A) nuclease PARN (20) or the translation initiation complex eIF2 (21). It is highly probable that CELF1 controls several RNAs in the testis, which are misregulated in Celf1−/− mice. However, we still lack the link between the RNA targets of CELF1 and spermiogenesis completion.
The goal of the present work was to understand the molecular bases for impaired spermatid elongation in Celf1-null mice, and consequently, the function of CELF1 during spermiogenesis. Because spermiogenesis is largely under hormonal control (22, 23), we investigated the hormonal status of Celf1−/− males, and we found that the testosterone level was severely reduced. This was the result of increased aromatase activity, the enzyme that aromatizes testosterone into estradiol. Treatments with aromatase inhibitors or with exogenous testosterone demonstrated a causal relationship between lack of testosterone associated with excessive aromatase activity and spermiogenesis defects in these mice. Furthermore, we showed that CELF1 directly downregulates the expression of Cyp19a1, the gene encoding aromatase. Our results reveal that the RBP CELF1 represses aromatase to achieve sufficient concentrations of testosterone for round spermatid differentiation. We discuss the implications of this finding with respect to reproductive troubles in men.
MATERIALS AND METHODS
Ethics statement.
The animals were housed in the animal facilities of Biosit Rennes (Arche) as approved by the French animal care agency (Direction des Services Vétérinaires, approval number A3523840). Experiments were carried out according to standard procedures after acceptance by the local ethics committee (approval number R-2011-CGC-01).
Animal experiments and histology.
The targeted disruption of Celf1 has been described (8). We crossed heterozygous mice to obtain homozygous Celf1−/− mice, and we kept the Celf1+/+ littermates (bred separately after weaning) as controls. For testosterone treatments, males (1.7 ± 1 month) were anesthetized and implanted with a subdermal Silastic implant (1 cm; Dow Corning, Midland, MI) containing testosterone propionate (5 mg; 86541-5G; Fluka) or a placebo implant as described previously (24). For letrozole treatments, males (5.5 ± 2.5 months) underwent daily subcutaneous (s.c.) injections of 40 μg letrozole dissolved in 0.3% hydroxypropyl cellulose (both from Sigma). We killed the adult mice by cervical dislocation and collected intracardiac blood (stored as serum at −80°C), seminal vesicles, testes, and epididymides. For each animal, we used one testis for RNA and protein extraction, microsomal fraction preparation, and steroid extraction and the contralateral testis with its epididymis for histological examination as described previously (8).
We detected Leydig cells on 5-μm-thick transverse sections of testis using an antibody directed against HSD3B6 (1:3,000 in phosphate-buffered saline [PBS]; generously supplied by Ian Mason) (25). Staining was carried out with the Vectastain Elite ABC kit (Vector Laboratories, Burlingame, CA) and diaminobenzidine substrate kit (Vector Laboratories), and counterstaining was done with hematoxylin. To count Leydig cells, we used a BX-50 Olympus microscope with Cast-Grid VI.10 (Olympus, Munich, Germany) to generate unbiased counting fields by systematic uniform random sampling scheme. An area of six testicular cross sections was measured to estimate the number of Leydig cells per square millimeter.
Hormone measurement.
We assayed circulating testosterone by extracting 150 μl of serum with ethyl ether, which was evaporated to leave a dry residue. The testosterone in dry residues was quantified using the testosterone radioimmunoassay (RIA) kit (IM1087; Immunotech, Beckman), following the manufacturer's instructions.
For intratesticular steroids, one testis was weighed and homogenized in phosphate buffer (pH 7.4) with a Dounce homogenizer, and steroids were extracted twice with dichloromethane. The dry residues were dissolved in phosphate buffer (pH 7.25), and testosterone and estradiol were quantified by RIA (26).
The other hormones were measured from sera at the University of Virginia Center for Research in Reproduction Ligand Assay and Analysis Core.
Biochemical methods.
For protein-RNA immunoprecipitations, wild-type adult testes were dissociated in ice-cold PBS, UV irradiated (5 times, 4,000 μJ/cm2, 254 nm), and stored at −80°C. The testes were homogenized, and CELF1 was immunoprecipitated (3B1; Santa Cruz) or pseudoimmunoprecipitated (nonimmune IgG) as previously described (27). The coimmunoprecipitated RNAs were treated with proteinase K (2 μg/μl) for 30 min at 37°C and 60°C for 30 min, extracted with phenol-chloroform, precipitated, and dissolved in water. The residual genomic DNA was removed by DNase treatment (Ambion). For the expression profiling, total RNAs were extracted with TRI Reagent (Euromedex) (1 ml TRI Reagent/testis), and the residual genomic DNA was removed by DNase treatment. RNAs were purified from cultured cells with the NucleoSpin RNA kit (Macherey-Nagel).
The cDNAs were prepared from the purified RNAs with random primers and Superscript II reverse transcriptase (Invitrogen). We carried out real-time PCR using Power SYBR green PCR master mix (Applied Biosystems) in the ABI Prism 7900 (Applied Biosystems). The primer sequences for Star (L36062) and Cyp17a1 (NM_007809) were published previously (28). The primer sequences are as follows: Celf1, GCAGCAAGTTATTATCATTGGC and TTCTTTGAGACAGGACTGGA; Cyp17a1, GGGCACTGCATCACGATAAA and GATCTAAGAAGCGCTCAGGCA; Cyp19a1, GAGAGTTCATGAGAGTCTGGATCA and CATGGAACATGCTTGAGGACT; Cyp19a1 (pre-mRNA, E3-I3), GCATGAGAACGGCATCATATT and AGTTGGGGCTGCCAGTGTAT; Cyp19a1 (pre-mRNA, I5-E6), GAATCTGTTTTGCTGGAAGTCTAA and TCAAATCAGTTGCAAAATCCATAC; Cyp19a1 (pre-mRNA, E8-I8), AAGAATGCACAGGCTCGAGTA and GAAGGGCAGATGAAGCTTTG; Hprt, CTGGTGAAAAGGACCTCTCG and TCAAGGGCATATCCAACAACAAAC; B2m, TGGTGCTTGTCTCACTGACC and CCGTTCTTCAGCATTTGGAT; Star, CCGGAGCAGAGTGGTGTCA and CAGTGGATGAAGCACCATGC; Cyp11a1, CCAGTGTCCCCATGCTCAA and CAGCTGCATGGTCCTTCCA; Hsd17b3, CAAGCTCTTTCCTGCGATCAAT and TTGAGTCCATGTCTGGCCAAC; Srd5a GCGTAGTCTACCTGGAGGGT and GAAGAGCCCACCATCTGGAG; firefly luciferase, GCTCAGCAAGGAGGTAGGTG and TCTTACCGGTGTCCAAGTCC; Renilla luciferase, AATGGCTCATATCGCCTCCTGGAT and TGGACGATGGCCTTGATCTTGTCT. The relative amount of RNA was calculated as 2−ΔCT, where ΔCT = CT − CT(reference). For RNA immunoprecipitation experiments, the reference was the total extract before immunoprecipitation. For expression profiling, the reference was the geometrical mean for two reference genes, Hprt and B2m (29).
We prepared microsomal fractions to measure aromatase activity. One half testis was weighed and homogenized in (0.05 mM NaP [pH 7.5], 0.15 M KCl, 21 mM NaCl, 20 mM sucrose, 5 mM dithiothreitol [DTT], protease inhibitor cocktail [P8340; Sigma]). The total extract was cleared (500 × g, 10 min, 4°C) and ultracentrifuged (100,000 × g, 2 h, 4°C). We measured aromatase activity in the dissolved pellet with the tritiated-water release assay (30).
For Western blot analyses of testicular extracts, one half testis was lysed in radioimmunoprecipitation assay (RIPA) buffer (50 mM Tris-HCl [pH 7.4], 150 mM NaCl, 5 mM EDTA, 1% deoxycholate, 5 mM DTT, 0.1% SDS, 0.5% NP-40, protease inhibitor cocktail [P8340; Sigma]), digested with DNase, and sonicated. We loaded 100 μg of proteins per lane. For Western blot analyses of HeLa cells, we used protein extracts from ∼105 cells. The primary antibodies are as follows: HSD3B6 (25), TUBB (tubulin; Sigma), CELF1 (3B1; sc20003; Santa Cruz), PCNA (P8825; Sigma).
For electrophoretic mobility shift assays (EMSAs), cDNAs corresponding to the different parts of the Cyp19a1 3′ untranslated region (UTR) were amplified from testis cDNA with the following PCR primers: fragment a, taatacgactcactatagggGTTGGGGACCAGTGAAGAAA and ACATGCTGGGATGTTGACCT; fragment b, taatacgactcactatagggAGGTCAACATCCCAGCATGT and TATTTCACTTTTGCCCCCAA. Lowercase indicates the nucleotides corresponding to the T7 RNA polymerase promoter. RNA was obtained from the PCR products by T7 in vitro transcription using [32P]UTP, and the resulting RNA was purified and quantified. We carried out EMSAs as described previously (31), with 2-fold dilutions of CELF1 starting from 400 nM or with a constant concentration of 100 nM in the presence of 400 nM competitor oligonucleotides (1, ACACACACACACACATATGAAAGTC; 2, GCGCGCGCACACACACACACACACA; 3, GCATGCACAAGTACACACACAGACT; 4, CACACACACACACACTATGTATTCA; 5, AGAGTCTACCACACACACACACACA).
Cell manipulations and luciferase reporter assays.
To construct plasmids for reporter 3′ UTR assays, we inserted the wild-type Cyp19a1 3′ UTR downstream of the firefly luciferase gene in the pmirGlo vector (Promega), using the Gibson Assembly master mix kit (NEBE2611S/L) with the following oligonucleotides: forward, ctagttgtttaaacgagctgtcgacAGAAGTGTGGCCTATCTAC and reverse gcctgcaggtcgactAGAAAGACTTCTTTATTTGAAATGC. The nucleotides corresponding to the target vector for the Gibson assembly are in lowercase. For the mutant Cyp19a1 3′ UTR, we separately amplified the region upstream of the UG stretches with the same forward primer as above and a novel reverse primer (ATGTAGAAAGAGTCTTATGAAAGTCCTGTTGTTC) and the region downstream of the UG stretches with the same reverse primer as above and a novel forward primer (AACAGGACTTTCATAAGACTCTTTCTACATTAAACC). We simultaneously inserted both fragments in the pmirGlo vector.
We cultured HeLa cells in Dulbecco's modified Eagle medium (DMEM) with 10% fetal bovine serum and antibiotics. For the experiments that did not request cotransfection, we obtained nonclonal populations of stably transfected cells by adding 800 μg/ml Geneticin 72 h after transfection (reduced to 400 μg/ml after 2 weeks of selection). For RNA interference (RNAi) experiments, we cotransfected the cells with 50 ng of reporter plasmid and 25 pmol of anti-CELF1 or control small interfering RNA (siRNA) (anti-CELF1, a mix of 5′-GAGCCAACCUGUUCAUCUA-3′, 5′-GCUGUUUAUUGGUAUGAUU-3′ and 5′-GCUGCAUUAGAAGCUCAGA-3′; control, 5′-GUCUAGACGAGUGUGACAU-3′) using JetPrime (PolyPlus). For the overexpression experiments, we cotransfected cells in 24-well plates with 80 ng reporter plasmid and 150 ng mCherry-H2B or mCherry-CELF1. The firefly and Renilla luciferase activities were measured 30 to 48 h later using the dual-luciferase reporter assay kit (Promega).
Statistics.
All data are expressed as means ± standard errors of the means or strip charts with the median. We assessed the statistical significance by Kruskal-Wallis tests, Wilcoxon rank sum tests with continuity correction, or Wilcoxon signed-rank tests with continuity correction when both wild-type and Celf1−/− littermates were available. All statistical analyses were performed with R.
RESULTS
Hypogonadism in Celf1−/− males.
Celf1−/− mice suffer from defective spermiogenesis (8), which is largely dependent on testosterone. We therefore assayed the serum testosterone concentrations in Celf1−/− and wild-type adult mice (4 months). The testosterone concentrations in sera were much lower in Celf1−/− mice than in control mice (Table 1). The same was true for the concentrations in testicular extracts (Table 1). Hence, Celf1−/− males are hypogonadal.
TABLE 1.
Hormone measurements in wild-type and Celf1−/− males
| Hormone | Sample type (unit) | Concn in mice with genotypea: |
Pb | |
|---|---|---|---|---|
| Celf1+/+ | Celf1−/− | |||
| Testosterone | Serum (ng/ml) | 7.41 ± 1.59 (17) | 0.55 ± 0.10 (17) | 1.7 × 10−5 |
| Intratestis (ng/mg) | 0.123 ± 0.040 (7) | 0.032 ± 0.010 (7) | 3.1 × 10−2 | |
| Estradiol | Serum (pg/ml) | 2.67 ± 0.22 (10) | 3.11 ± 0.11 (10) | 6.4 × 10−2 |
| Intratestis (pg/mg) | 0.200 ± 0.045 (7) | 0.630 ± 0.180 (7) | 1.8 × 10−2 | |
| FSH | Serum (ng/ml) | 37.2 ± 2.6 (10) | 41.0 ± 2.1 (10) | 0.14 |
| LH | Serum (ng/ml) | 0.813 ± 0.320 (13) | 0.581 ± 0.278 (14) | 0.13 |
Hormone concentrations in the sera or testicular extracts of wild-type and Celf1−/− mice were quantified by radioimmunoassays or ELISA. Data are means ± standard errors of the means. Numbers of mice are in parentheses.
We compared the Celf1+/+ and Celf1−/− data by a Wilcoxon rank sum test.
A possible cause of male hypogonadism is a defective production of luteinizing hormone (LH) and follicle-stimulating hormone (FSH), since these pituitary hormones stimulate directly or indirectly testosterone production. We therefore measured their concentrations in serum (Table 1). The FSH levels were nearly identical in Celf1−/− and wild-type mice. The LH levels were possibly lower in Celf1−/− mice, but the dispersion of the LH concentrations makes this difference nonsignificant. Hence, the concentrations of the pituitary hormones FSH and LH in serum in Celf1−/− males do not differ or differ only slightly from those in Celf1+/+ mice, suggesting that their hypogonadism stems from testicular failure.
Genital tract development.
We next weighed the testes of Celf1−/− and control adult mice. Celf1−/− mice are overall smaller (8), and so are their genital organs. However, after normalization by the total body weight, the testes from Celf1−/− mice had weights similar to those of testes from Celf1+/+ mice (Fig. 1A). Hence, hypotestosteronemia in these mice is not a consequence of small testes.
FIG 1.
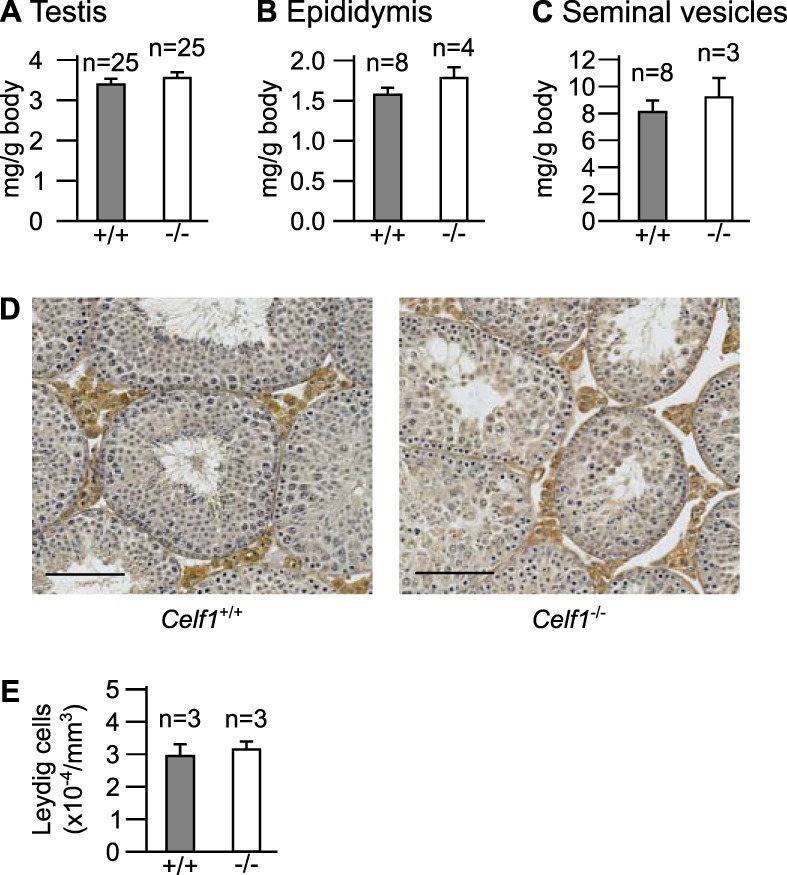
Characterization of the male genital tract. We weighed Celf1−/− or Celf1+/+ adult mice, as well as their genital organs. We show here the weights of a single testis (A), a single epididymis (B), and both seminal vesicles (C) for each animal, divided by the total body weight. n, number of wild-type and Celf1−/− males. (D) Representative histological sections of wild-type and Celf1−/− testes. The Leydig cells were stained brown by immunohistochemistry with an anti-HSD3B6 antibody (25). Bars, 100 μm. (E) Number of Leydig cells per square millimeter of testicular cross sections, from wild-type and Celf1−/− littermates. In panels A to C and E, the differences between Celf1−/− and Celf1+/+ organs are not significant (P > 0.1; Wilcoxon rank sum test).
We also found the weights of the epididymides and of the seminal vesicles to be very similar in wild-type and Celf1−/− mice (Fig. 1B and C). Together with their normal appearance in Celf1−/− mice (8), this shows that these organs develop correctly. Furthermore, cryptorchidism does not occur, while it is commonly associated with severe forms of hypogonadism (32). The normal internal genitalia and the normal testicular descent, which are testosterone dependent, suggest that the concentrations of testosterone in Celf1−/− mice, albeit low, are sufficient to drive differentiation and development of the genital tract.
Finally, we analyzed Leydig cells, because they are the major sites of testosterone synthesis. Their morphology and their distribution in testicular interstitial tissue were similar in Celf1−/− and control mice (Fig. 1D). Systematically counting the number of Leydig cells also failed to reveal any difference between the two genotypes (Fig. 1E). Hence, hypogonadism in Celf1−/− mice is not due to a defective proliferation of Leydig cells.
Expression of steroidogenic enzyme genes.
Although Leydig cells are apparently normal in size and number in Celf1−/− mice, they may be deficient in steroidogenesis. Steroidogenesis is a multistep process that involves several enzymatic reactions taking cholesterol as a substrate to synthesize testosterone, which is converted to dihydrotestosterone or estradiol (Fig. 2A) (33). We compared the levels of expression of all the genes encoding steroidogenic enzymes in testes from Celf1−/− and control mice. The abundances of five mRNAs out of seven (Star, Cyp11a1, Hsd17b3, Cyp17a1, and Srd5a) were not different between the two genotypes (Fig. 2B), and we focused on the two remaining mRNAs. At the mRNA level, Hsd3b6 (the only Hsd3b gene expressed in adult mouse testis [28]) was downregulated in Celf1−/− mice by 40% (Fig. 2B). However, Western blots carried out with several animals revealed only a low, nonsignificant decrease in levels of HSD3B6 protein in Celf1−/− testes (Fig. 2C). Hence, Hsd3b6 is not or only weakly downregulated in Celf1−/− mice. Conversely, the abundance of Cyp19a1 mRNA, encoding aromatase, was increased by 45% (Fig. 2B). We were unable to directly measure the amount of CYP19A1/aromatase protein due to the lack of appropriate antibodies, but we measured aromatase activity. As shown in Fig. 2D, aromatase activity was 80% higher in microsomal fractions from Celf1−/− testes than in microsomal fractions from control testes. Hence, Cyp19a1 is upregulated in Celf1−/− males, which may contribute to reduced testosterone concentrations.
FIG 2.

Expression of steroidogenic enzyme genes in wild-type and Celf1−/− males. (A) Pathway of sex steroid synthesis. The CYP17A1 protein has two sequential enzymatic activities, but the intermediate products were omitted for clarity. (B) Quantification of the indicated mRNAs by RT-qPCR in the testes of wild-type and Celf1−/− littermates. (C) (Left) Representative Western blot of testis extracts from Celf1−/− or Celf1+/+ mice with antibodies against HSD3B6 and TUBB (loading control). (Right) Quantification of the HSD3B6/TUBB ratios from Western blots performed with 7 wild-type and Celf1−/− testis extracts. (D) Aromatase activities in microsomal fractions of testes measured by a tritiated-water release assay. (E) Quantification of the abundance of Cyp19a1 pre-mRNA using primers directed against the indicated intron (I)-exon (E) junctions. In all panels, gray bars represent wild-type mice, and white bars represent Celf1−/− mice. The P values of Wilcoxon signed rank tests below 0.1 are shown. n, number of pairs of wild-type and Celf1−/− littermates.
We measured the abundance of Cyp19a1 pre-mRNA by specifically amplifying intron-exon junctions by reverse transcription-quantitative PCR (RT-qPCR). This experiment, which we carried out with three different pairs of primers, revealed similar amounts of Cyp19a1 pre-mRNA in Celf1−/− and wild-type testes (Fig. 2E). We conclude that the targeted disruption of Celf1 does not affect Cyp19a1 transcription and that Cyp19a1 overexpression in Celf1−/− mice is associated with changes to posttranscriptional regulations.
Aromatase converts testosterone into estradiol. Therefore, hyperactive aromatase is expected not only to reduce testosterone levels but also to increase estradiol levels. We verified this prediction by measuring estradiol concentrations in sera and testicular extracts (Table 1). Estradiol levels were increased 3-fold in Celf1−/− mouse testes. There was also a trend for increased serum estradiol concentration. Altogether, these results show that the high aromatase activity in Celf1−/− mice results in imbalanced levels of testosterone and estradiol.
The high aromatase activity is a major cause of defective spermiogenesis in Celf1−/− males.
We next investigated a putative causal relationship between defective spermatogenesis (8) and high aromatase activity, resulting in hypogonadism in Celf1−/−. We can raise Celf1−/− mice only on a mixed genetic background because the mutation is homozygous lethal on a pure background (9), resulting in variable expressivity of spermatogenesis defects. As previously observed (8), Celf1−/− testes could be grouped into three classes. The unaffected testes display apparently normal seminiferous tubules (Fig. 3A, left). The strongly affected testes display seminiferous tubules with vacuoles and giant multinuclear cells that arise from the fusion of several round spermatids through cytoplasmic bridges (34) and are virtually depleted of elongated spermatids and spermatozoa (Fig. 3A, right). The moderately affected testes have heterogeneous seminiferous tubules, with normal tubules and tubules containing fewer spermatozoa or elongated spermatids (Fig. 3A, middle). We sorted 17 Celf1−/− mice by the phenotype of their testes, and we measured their serum testosterone. The strongly affected mice had low levels of testosterone (∼400 pg/ml) (Fig. 3B). There was a tendency toward higher levels of testosterone from strongly affected to moderately affected and unaffected mice (Fig. 3B), but this did not reach statistical significance (Kruskal-Wallis rank sum test), and even the unaffected mice had low concentrations of testosterone (∼650 pg/ml) compared with wild-type mice (see Table 1). We also measured aromatase activities (Fig. 3B). Aromatase activity was high for all three classes of mice, and, although the strongly affected mice had even stronger aromatase activities than the other two classes, the differences were again nonsignificant. These data show that all the Celf1−/− mice are characterized by low testosterone and high aromatase, irrespective of their spermatogenesis defects. This suggests that other genetic factors shape the testicular phenotype of hypogonadal Celf1−/− males. Hence, hypotestosteronemia in Celf1−/− males can either play no role in the appearance of spermatogenesis defects or define a specific endocrine background that is permissive for these defects.
FIG 3.

Changes to testosterone concentration are a cause of defective spermiogenesis in Celf1−/− mice. (A) Histological sections of seminiferous tubules from Celf1−/− mice were stained with hematoxylin and eosin. Three representative sections of testes classified as unaffected, moderately affected, and strongly affected are shown from left to right. (B) Seventeen mice were sorted by the phenotype of their seminiferous tubules (n = 6, 6, and 5 for unaffected, moderately affected, and strongly affected tubules, respectively). The mean serum testosterone concentrations of these three classes of mice are shown as black bars. The white bars show the mean aromatase activities in testes of 12 other mice (n = 3 for unaffected, 6 for moderately affected, and 3 for strongly affected). (C) Representative sections of the cauda epididymis from Celf1−/− males, showing a graduation of phenotypes from left to right. The inset shows a high magnification. (D) Serum testosterone concentrations in wild-type males, testosterone-implanted Celf1−/− males, and letrozole-treated Celf1−/− males. To make comparisons easier, we show here the results for Celf1−/− males presented in Table 1. ***, P = 5.4 × 10−4 (−/− mice versus +/+ mice); #, P = 0.029 (testosterone-treated −/− mice versus −/− mice) or 0.012 (letrozole-treated −/− mice versus −/− mice). The other P values (Wilcoxon rank sum tests) are above 0.1. (E) Representative sections of epididymides from untreated, testosterone-treated, and letrozole-treated Celf1−/− mice. (F) Distribution of the number of round cells per section of cauda epididymal tubule in untreated wild-type mice, untreated Celf1−/− mice, testosterone-treated Celf1−/− mice, and letrozole-treated Celf1−/− mice. ***, P = 6.4 × 10−7 (−/− mice versus +/+ mice); **, P = 2.3 × 10−3 (testosterone-treated −/− mice versus +/+ mice); *, P = 0.036 (letrozole-treated −/− mice versus +/+ mice); ##, P = 1.8 × 10−3 (testosterone-treated −/− mice versus −/− mice); #, P = 0.032 (letrozole-treated −/− mice versus −/− mice). The other P values (Wilcoxon rank sum tests) are above 0.1. Bars, 100 μm (A, C, and E) and 30 μm (inset). V, vacuole. Asterisks indicate multinuclear cells, the arrow indicates a round spermatid prematurely released by the Sertoli cells, and the arrowhead indicates a degenerating cell.
We needed a quantitative assessment of spermatogenesis defects to discriminate between these possibilities. We reasoned that, since spermatozoa are stored in the cauda epididymides before ejaculation, the contents of the cauda epididymis reflect the overall activity of numerous seminiferous tubules. We analyzed histological sections of the cauda epididymis from several Celf1−/− mice. The epididymides of unaffected testes essentially contained spermatozoa (Fig. 3C, left), like those of wild-type males. The epididymides of strongly affected testes contained only round cells (Fig. 3C, right), corresponding to round spermatids prematurely released by Sertoli cells, to degenerating cells, or to multinuclear cells. Finally, the epididymides of moderately affected testes contained variable numbers of round cells (Fig. 3C, middle), probably reflecting different degrees of severity. Hence, the number of round cells in sections of cauda epididymis objectifies the defective spermatogenesis of Celf1−/− mice with good sensitivity.
We carried out rescue experiments to analyze the links between spermiogenesis defects, testosterone, and aromatase. We treated Celf1−/− males with testosterone implants for 45 days. This raised their serum testosterone level almost up to the wild-type level (Fig. 3D). This also reduced the number of round cells per section of cauda epididymis tubules (Fig. 3E). Systematically counting the number of round cells revealed that this number was much higher in nonimplanted Celf1−/− mice than in wild-type mice. Importantly, this number was significantly reduced in testosterone-treated Celf1−/− mice (Fig. 3F), demonstrating a role for hypotestosteronemia in spermatogenesis defects. Together with the poor correlation between testosterone concentrations and spermatogenesis defects (Fig. 3B), this observation reveals that hypotestosteronemia in Celf1−/− mice is necessary but not sufficient to cause spermatogenesis defects. We also treated Celf1−/− males with the aromatase inhibitor letrozole for 45 days. This restored a normal testosterone concentration (Fig. 3D), indicating that Cyp19a1 overexpression is the major cause of reduced testosterone concentrations in Celf1−/− males. This also significantly reduced the number of round cells in epididymides (Fig. 3E and F), confirming the link between low testosterone and spermatogenesis defects. Together, these results demonstrate that the reduced testosterone levels arising from high Cyp19a1 expression strongly contribute to spermatogenesis defects of Celf1−/− males.
CELF1 directly binds to Cyp19a1 mRNA.
We have shown that Cyp19a1 is upregulated posttranscriptionally in Celf1−/− testes (Fig. 2). This raised the hypothesis that CELF1 directly controls Cyp19a1 expression in wild-type testes. We first tested if CELF1 interacts with Cyp19a1 mRNA in vivo. We irradiated freshly dilacerated testes with UV light to induce covalent bridges between RNAs and their associated proteins. We immunoprecipitated CELF1 with a specific antibody. A high-stringency wash allowed us to recover only RNAs directly interacting with CELF1, as shown for cross-linking and immunoprecipitation (CLIP) experiments (35). Western blots confirmed the efficiency of the immunoprecipitations (Fig. 4A). We assessed coimmunoprecipitated RNAs by RT-qPCR (Fig. 4B). Because CELF1 interacts with its own mRNA (27), we used Celf1 mRNA as a positive control for coimmunoprecipitations. Celf1 mRNA was efficiently immunoprecipitated with the anti-CELF1 antibody but not with nonimmune IgG. Cyp17a1 mRNA, which is not differentially regulated in Celf1−/− testes (Fig. 2B), was not immunoprecipitated by either antibody. Cyp19a1 mRNA was coimmunoprecipitated with CELF1, demonstrating that CELF1 protein interacts with Cyp19a1 mRNA in mouse testes.
FIG 4.

CELF1 directly binds to Cyp19a1 mRNA. (A and B) We irradiated freshly dilacerated testes with UV light and carried out immunoprecipitations with either anti-CELF1 or nonimmune immunoglobulins (IgG, mock). (A) Western blots with anti-CELF1 antibodies of total testis extracts and of immunoprecipitates obtained with nonimmune and anti-CELF1 antibodies. The results of three representative independent immunoprecipitation experiments are shown. Except where indicated (−/−), all the testes were from wild-type mice. The positions of CELF1 and of the endogenous immunoglobulins detected by the secondary antibody are indicated. (B) Quantification by RT-qPCR of the coimmunoprecipitated mRNAs. The abundance of Cyp17a1 (negative control), Celf1 (positive control), and Cyp19a1 mRNAs in the immunoprecipitates relative to the inputs is shown. n, number of independent immunoprecipitation experiments. (C) Schematic drawing of the 3′ untranslated region of murine Cyp19a1 mRNA. The positions of the two UG stretches potentially able to interact with CELF1 and the positions of the fragments and the competitor oligonucleotides for electrophoretic mobility shift assays (EMSAs) are shown. (D and E) EMSAs using increasing amounts of recombinant CELF1 protein and the labeled RNA fragment b (D) or a (E). The fragment names are those used in panel C. (F) EMSA using recombinant CELF1 protein and the RNA fragment b, plus the indicated competitor oligonucleotides (Comp. 1 to 5).
We next sought to identify the region of Cyp19a1 mRNA bound by CELF1. We described a motif-scoring algorithm that predicts UGU-rich RNA elements potentially able to interact with human CELF1 protein (27). Taking advantage of the strong conservation between human and mouse CELF1, we used this algorithm to identify candidate regions of Cyp19a1 transcripts associated with CELF1. This approach allowed us to identify only one candidate region within Cyp19a1 mRNA and pre-mRNA, which consists of a fragment of the 3′ untranslated region (3′ UTR) 150 nucleotides (nt) long and containing two UG stretches. We used an electrophoretic mobility shift assay (EMSA) to confirm that CELF1 interacts with an ∼200-nucleotide probe containing both UG stretches (fragment b) (Fig. 4C). The addition of increasing amounts of recombinant CELF1 progressively shifted fragment b from a position corresponding to the free probe to a position corresponding to a complex (Fig. 4D). The dissociation constant (Kd) for this interaction is 23 ± 4 nM, which is similar to the Kd between CELF1 and other RNA-binding partners of this protein (31). The addition of CELF1 protein did not affect the migration of the control fragment a (Fig. 4C), which corresponds to another region of the Cyp19a1 3′ UTR that is not predicted to contain CELF1 binding sites (Fig. 4E). Hence, in EMSA, CELF1 specifically interacts with a fragment of the Cyp19a1 3′ UTR.
We then sought to confirm that CELF1 interacts with fragment b through the UG stretches. We tested the capacity of five different antisense competitor oligonucleotides (numbered 1 to 5; Fig. 4C) to inhibit the interaction between CELF1 and fragment b in an EMSA. As expected, competitor 3, which is complementary to a sequence located outside the UG stretches, had no effect on the interaction between fragment b and CELF1 (Fig. 4F, lane 5). Conversely, all the competitor oligonucleotides complementary to UG stretches and to anchoring sequences flanking either of the UG stretches abolished the interaction of CELF1 with Cyp19a1 3′ UTR (Fig. 4F, lanes 3 and 4 and lanes 6 and 7). This suggests that CELF1 requires two different sites to efficiently bind to the Cyp19a1 3′ UTR, possibly as a consequence of the capacity of that protein to oligomerize (36). Altogether, the above-described experiments demonstrate the capacity of CELF1 to interact with Cyp19a1 mRNA in vivo and in vitro.
CELF1 posttranscriptionally controls Cyp19a1 expression.
The above results indicate that CELF1 protein interacts with the Cyp19a1 3′ UTR and that Cyp19a1 is upregulated in the absence of CELF1. This suggests that CELF1 directly acts on Cyp19a1 mRNA to reduce the abundance of the encoded protein. We carried out luciferase reporter assays in HeLa cells to test this hypothesis. We transfected the cells with a plasmid carrying the firefly luciferase gene associated with the Cyp19a1 3′ UTR (Fig. 5A). This plasmid also encodes Renilla luciferase as an internal control. Compared with the control (vector) 3′ UTR, the Cyp19a1 3′ UTR repressed luciferase expression ∼5-fold (Fig. 5B). Importantly, a mutant Cyp19a1 3′ UTR with a deletion of the UG stretches that were shown to mediate CELF1 binding (Fig. 4) failed to repress luciferase (Fig. 5B). This suggests that CELF1 mediates the repression conferred by Cyp19a1 3′ UTR. We measured luciferase mRNA levels in the same cells (Fig. 5C). The mRNA containing the wild-type Cyp19a1 3′ UTR was as abundant as the other mRNAs, consistent with wild-type Cyp19a1 3′ UTR essentially exerting its repressive effect at a translational level.
FIG 5.
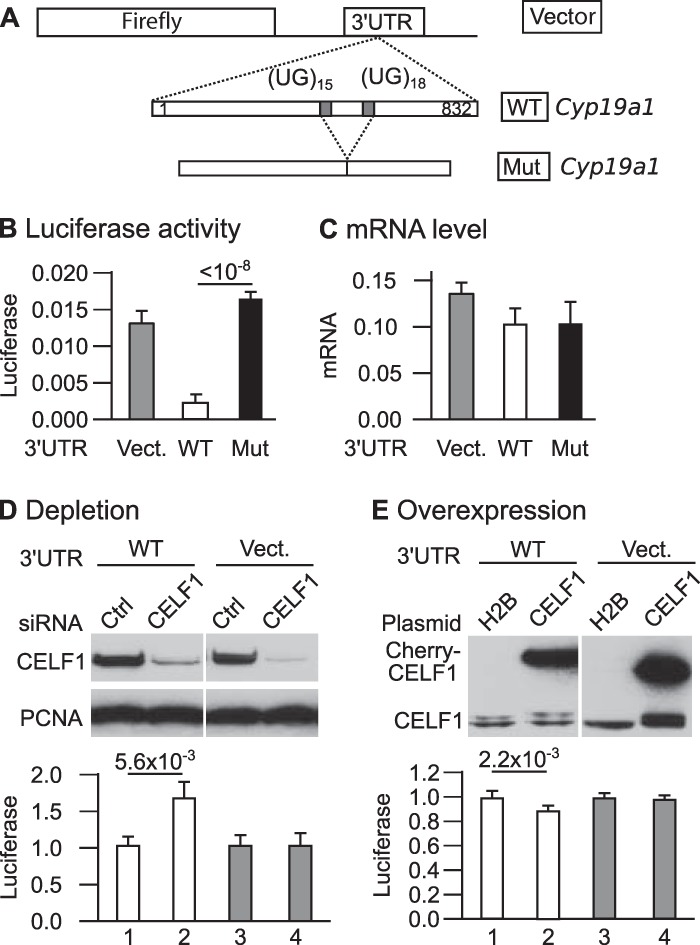
Luciferase reporter assays with the Cyp19a1 3′ UTR. (A) Structure of the plasmids. The firefly gene is coupled to a vector (control) 3′ UTR, the complete wild-type Cyp19a1 3′ UTR (WT Cyp19a1), or a mutant Cyp19a1 3′ UTR with the region containing the UG stretches deleted (Mut Cyp19a1). (B) Firefly luciferase activities measured in populations of HeLa cells stably transfected with the indicated plasmids (n = 16 independent experiments; P = 4.5 × 10−8, Kruskal-Wallis rank sum test). (C) Firefly luciferase mRNA levels (relative to Renilla luciferase levels) in populations of HeLa cells stably transfected with the indicated plasmids (n = 5 independent experiments; P = 0.2753, Kruskal-Wallis rank sum test). (D) Cells were cotransfected with the indicated reporter plasmids and anti-CELF1 or control siRNA. (Top) Western blots to assess CELF1 abundance; (bottom) luciferase activity (n = 9 independent transfection experiments). (E) Cells were cotransfected with the indicated reporter plasmids and expression vector encoding Cherry-tagged CELF1 or Cherry-tagged histone 2B. (Top) Western blots to assess CELF1 abundance; (bottom) luciferase activity (n = 6 independent transfection experiments). The P values from Wilcoxon rank sum tests are shown (B, D, and E; Bonferroni adjusted in panel B). In all experiments, firefly luciferase activities were normalized to Renilla luciferase activities measured in the same extracts.
The fact that the deletion of CELF1 binding sites abolishes the translational repression exerted by Cyp19a1 3′ UTR suggests that this repression is mediated by CELF1. We carried out depletion and overexpression experiments to confirm this hypothesis. As previously shown (27), transfecting HeLa cells with specific siRNAs efficiently depletes them in CELF1 (Fig. 5D). This depletion upregulated by 64% the expression of the reporter with Cyp19a1 3′ UTR (Fig. 5D, lanes 1 and 2) but had no effect on the expression of the reporter with the vector 3′ UTR (lanes 3 and 4). Conversely, overexpressing CELF1 weakly but significantly repressed the expression of the reporter with the Cyp19a1 but not the vector 3′ UTR (Fig. 5E). This confirms that CELF1 represses the expression of a reporter gene with Cyp19a1 3′ UTR. Altogether, these data reveal that CELF1 is a direct, posttranscriptional regulator of Cyp19a1 expression in mouse testes.
DISCUSSION
In the present work, we looked for the molecular mechanisms responsible for the spermiogenesis blockage in Celf1−/− males. We show that Cyp19a1 is upregulated posttranscriptionally in Celf1−/− testes, leading to high aromatase activity. The finding that pharmacological inhibition of aromatase improves spermiogenesis in Celf1−/− males indicates that excessive aromatase is at least in part responsible for spermiogenesis defects. Furthermore, CELF1 interacts with Cyp19a1 mRNA, and luciferase reporter assays in HeLa cells support the interpretation that it represses Cyp19a1 expression directly. Interestingly, the Cyp19a1 3′ UTR represses the expression of the reporter gene at the protein level in a CELF1-dependent manner, but not at the mRNA level. This suggests that CELF1 represses the translation of Cyp19a1, in accordance with known properties of CELF1 (10, 16–19). These results in HeLa cells seem to contradict the in vivo results, as we detected high levels of Cyp19a1 mRNA in Celf1−/− testes (Fig. 2B). However, Cyp19a1 upregulation is stronger at the protein level (aromatase activity) than at the mRNA level (compare Fig. 2B and D), which is consistent with CELF1-dependent translational control of Cyp19a1 expression in testes. Together, these results identify a novel physiological function for CELF1, in supporting spermatogenesis completion by translationally downregulating Cyp19a1.
What is the link between high aromatase activity and testicular defects in Celf1−/− males? A salient feature of Celf1−/− males is their low concentrations of testosterone in sera and testicular extracts (Table 1). The role of androgen signaling on spermiogenesis has been clarified using genetically modified mice. Serum testosterone concentrations are low in mice in which the luteinizing hormone receptor (LurKO) is constitutively inactivated (37–39) or the androgen receptor in Leydig cells is conditionally inactivated (40). In both mouse models, hypotestosteronemia is associated with a blockage of spermatid elongation at stage 7, like in Celf1−/− mice (8). Supplementation of testosterone, which was carried out in LurKO mice, restores correct spermatid elongation, demonstrating the function of testosterone in the progression of spermatid elongation (41). High concentrations of testosterone support the appearance of ectoplasmic specializations between late spermatids and Sertoli cells, allowing spermiogenesis completion (42–46). Our observation that Celf1−/− epididymides accumulate round cells (Fig. 3C) is consistent with a premature release of round spermatids by Sertoli cells arising from low testosterone concentrations.
In addition to low testosterone, the hormonal characterization of Celf1−/− males revealed increased estradiol concentrations, at least in testicular extracts (Table 1). The administration of estradiol to adult rats or mice affects spermatogenesis (47–49). At first glance, this suggests that high estradiol concentrations may contribute to spermiogenesis defects in Celf1−/− males. However, exogenous estradiol represses the hypothalamus pituitary gonadal axis to reduce the circulating concentrations of pituitary hormones, which strongly reduces the intratesticular testosterone concentrations (48). Hence, spermatogenesis defects in rodents supplemented with exogenous estradiol may arise from decreased testosterone, and it is not certain whether excessive estradiol directly acts on spermatogenesis. Regarding Celf1−/− males, exogenous testosterone partly restores correct spermiogenesis (Fig. 3F), which would not be the case if spermiogenesis defects arose mainly from high estradiol concentrations. For these reasons, we think that lack of testosterone is the main cause of defective spermiogenesis in Celf1−/− mice, with excessive estradiol being a putative secondary cause. Noteworthily, estradiol-mediated repression of the hypothalamus pituitary gonadal axis may explain why LH levels are not increased in Celf1−/− males despite reduced testosterone (Table 1).
The hormonal status of Celf1−/− males is evocative of human isolated hypogonadotropic hypogonadism (IHH), with low testosterone and low to normal LH (50). IHH is frequent in obese men, possibly as a consequence of enhanced aromatization of testosterone by aromatase, which is present in adipocytes in human (50). Treating obese men with an aromatase inhibitor restores normal testosterone levels (51). Some nonobese men also have excessive aromatase activity, possibly for genetic reasons, which results in nonobstructive azoospermia and low testosterone. Aromatase inhibitors seem to be efficient treatments, though this represents an off-label use (52). Rodent models of impaired spermatogenesis associated with high aromatase activity would provide insights into this pathology and allow testing of different treatment options (52). Mouse males with a Cyp19a1 transgene develop testicular defects with low testosterone concentrations (53–55). However, Cyp19a1 is probably much more strongly upregulated in these mouse lines than in human patients. Male mice with a targeted disruption of Dax1 are sterile and have high aromatase activity (56). However, increased estradiol is the major endocrine consequence of high aromatase in Dax1-disrupted males, with essentially no impact on testosterone (56). This differs from Celf1−/− male mice and from human patients. Hence, Celf1−/− mice might be the first rodent model for human IHH with excessive aromatase activity.
The results presented here may also have significant consequences for another human pathology, myotonic dystrophy type 1 (DM1). DM1 is the most frequent adult myopathy, with a mean prevalence of 1 case/8,000 individuals. The main symptoms of DM1 are myotonia and muscle weakness, but many other clinical signs are associated with this disease, including hypogonadism. Most men with DM1 display abnormal testicular functions, with testicular atrophy, oligospermia or azoospermia, and small amounts of testosterone (57–59). The underlying molecular defect of DM1 involves an unstable CTG expansion in the 3′ UTR of the DMPK gene (60, 61). This expansion has only limited consequences for DMPK expression, and most DM1 symptoms arise in trans from the DMPK RNA harboring the CUG expansion in the 3′ UTR. It is well recognized that this “toxic” RNA upregulates the expression of CELF1 and reduces the availability of another RBP, MBLN1, through pathways that are not fully understood. The resulting imbalance between the two RBPs eventually causes most cardiac and muscular signs of DM1 (62). However, in DM1, CELF1 seems to be translocated from the cytoplasm to the nucleus (63) and to be concentrated in stress granules (64). It also seems to lose its capacity to interact with its cytoplasmic partner, eIF2 (21). This suggests that, in DM1, CELF1 is less active in the cytoplasm though more abundant overall (65). Accordingly, mice with null or low CELF1 activity display several traits evocative of DM1. These include muscle weakness, cardiac defects, and cataracts (66–68) but also male hypogonadism (this article). During the course of this work, we also observed that Celf1−/− mice are highly sensitive to anesthesia (data not shown), similar to DM1 patients (69). Therefore, it is possible that DM1 is accompanied by a decrease of CELF1 activity in the cytosol, together with the increase of CELF1 activity in the nucleus. Hence, it is not unreasonable to suppose that CYP19A1 is overexpressed in men with DM1 due to a reduced cytoplasmic CELF1 and that this overexpression contributes to their hormonal defects. Aromatase inhibitors would then be candidate drugs to alleviate hypogonadism in DM1 men.
ACKNOWLEDGMENTS
This work was supported by the Agence Nationale pour la Recherche (ANR-07-JCJC-0097-01) and the Ligue régionale contre le Cancer to L.P. Hormone measurements were carried out at the University of Virginia Center for Research in Reproduction Ligand Assay and Analysis Core, which is supported by Eunice Kennedy Shriver NICHD/NIH (SCCPIR) grant U54-HD28934.
We thank Ian Mason for the generous gift of the HSD3B6 antibody, Virginie Rouiller-Fabre, Olivier Kah, Bernard Jégou, and Thierry Charlier for stimulating discussions, Erwan Watrin for opportune suggestions regarding stable transfections, and Bernard Jégou for giving us access to the CastGrid. We thank the Biosit Structure and its members for access to their facilities, including the mouse facility Arche and Histo-Pathologie H2P2. We thank David Bec for invaluable help with mouse implantation.
REFERENCES
- 1.Jan SZ, Hamer G, Repping S, de Rooij DG, van Pelt AM, Vormer TL. 2012. Molecular control of rodent spermatogenesis. Biochim Biophys Acta 1822:1838–1850. doi: 10.1016/j.bbadis.2012.02.008. [DOI] [PubMed] [Google Scholar]
- 2.O'Donnell L, Nicholls PK, O'Bryan MK, McLachlan RI, Stanton PG. 2011. Spermiation: the process of sperm release. Spermatogenesis 1:14–35. doi: 10.4161/spmg.1.1.14525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Idler RK, Yan W. 2012. Control of messenger RNA fate by RNA-binding proteins: an emphasis on mammalian spermatogenesis. J Androl 33:309–337. doi: 10.2164/jandrol.111.014167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.de Mateo S, Sassone-Corsi P. 2014. Regulation of spermatogenesis by small non-coding RNAs: role of the germ granule. Semin Cell Dev Biol 29:84–92. doi: 10.1016/j.semcdb.2014.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Venables JP, Cooke HJ. 2000. Lessons from knockout and transgenic mice for infertility in men. J Endocrinol Invest 23:584–591. doi: 10.1007/BF03343780. [DOI] [PubMed] [Google Scholar]
- 6.Paronetto MP, Sette C. 2010. Role of RNA-binding proteins in mammalian spermatogenesis. Int J Androl 33:2–12. doi: 10.1111/j.1365-2605.2009.00959.x. [DOI] [PubMed] [Google Scholar]
- 7.Iguchi N, Tobias JW, Hecht NB. 2006. Expression profiling reveals meiotic male germ cell mRNAs that are translationally up- and down-regulated. Proc Natl Acad Sci U S A 103:7712–7717. doi: 10.1073/pnas.0510999103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kress C, Gautier-Courteille C, Osborne HB, Babinet C, Paillard L. 2007. Inactivation of CUG-BP1/CELF1 causes growth, viability, and spermatogenesis defects in mice. Mol Cell Biol 27:1146–1157. doi: 10.1128/MCB.01009-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cibois M, Boulanger G, Audic Y, Paillard L, Gautier-Courteille C. 2012. Inactivation of the Celf1 gene that encodes an RNA-binding protein delays the first wave of spermatogenesis in mice. PLoS One 7:e46337. doi: 10.1371/journal.pone.0046337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dasgupta T, Ladd AN. 2012. The importance of CELF control: molecular and biological roles of the CUG-BP, Elav-like family of RNA-binding proteins. Wiley Interdiscip Rev RNA 3:104–121. doi: 10.1002/wrna.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Philips AV, Timchenko LT, Cooper TA. 1998. Disruption of splicing regulated by a CUG-binding protein in myotonic dystrophy. Science 280:737–741. doi: 10.1126/science.280.5364.737. [DOI] [PubMed] [Google Scholar]
- 12.Savkur RS, Philips AV, Cooper TA. 2001. Aberrant regulation of insulin receptor alternative splicing is associated with insulin resistance in myotonic dystrophy. Nat Genet 29:40–47. doi: 10.1038/ng704. [DOI] [PubMed] [Google Scholar]
- 13.Rattenbacher B, Beisang D, Wiesner DL, Jeschke JC, von Hohenberg M, St Louis-Vlasova IA, Bohjanen PR. 2010. Analysis of CUGBP1 targets identifies GU-repeat sequences that mediate rapid mRNA decay. Mol Cell Biol 30:3970–3980. doi: 10.1128/MCB.00624-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Beisang D, Rattenbacher B, Vlasova-St Louis IA, Bohjanen PR. 2012. Regulation of CUG-binding protein 1 (CUGBP1) binding to target transcripts upon T cell activation. J Biol Chem 287:950–960. doi: 10.1074/jbc.M111.291658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ezzeddine N, Paillard L, Capri M, Maniey D, Bassez T, Aït-Ahmed O, Osborne B. 2002. EDEN dependent translational repression of maternal mRNAs is conserved between Xenopus and Drosophila. Proc Natl Acad Sci U S A 99:257–262. doi: 10.1073/pnas.012555499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cibois M, Gautier-Courteille C, Vallee A, Paillard L. 2010. A strategy to analyze the phenotypic consequences of inhibiting the association of an RNA-binding protein with a specific RNA. Rna 16:10–15. doi: 10.1261/rna.1742610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Iakova P, Wang GL, Timchenko L, Michalak M, Pereira-Smith OM, Smith JR, Timchenko NA. 2004. Competition of CUGBP1 and calreticulin for the regulation of p21 translation determines cell fate. EMBO J 23:406–417. doi: 10.1038/sj.emboj.7600052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu L, Ouyang M, Rao JN, Zou T, Xiao L, Chung HK, Wu J, Donahue JM, Gorospe M, Wang JY. 2015. Competition between RNA-binding proteins CELF1 and HuR modulates MYC translation and intestinal epithelium renewal. Mol Biol Cell 26:1797–1810. doi: 10.1091/mbc.E14-11-1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yu TX, Rao JN, Zou T, Liu L, Xiao L, Ouyang M, Cao S, Gorospe M, Wang JY. 2013. Competitive binding of CUGBP1 and HuR to occludin mRNA controls its translation and modulates epithelial barrier function. Mol Biol Cell 24:85–99. doi: 10.1091/mbc.E12-07-0531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Moraes KC, Wilusz CJ, Wilusz J. 2006. CUG-BP binds to RNA substrates and recruits PARN deadenylase. Rna 12:1084–1091. doi: 10.1261/rna.59606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Salisbury E, Sakai K, Schoser B, Huichalaf C, Schneider-Gold C, Nguyen H, Wang GL, Albrecht JH, Timchenko LT. 2008. Ectopic expression of cyclin D3 corrects differentiation of DM1 myoblasts through activation of RNA CUG-binding protein, CUGBP1. Exp Cell Res 314:2266–2278. doi: 10.1016/j.yexcr.2008.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Borg CL, Wolski KM, Gibbs GM, O'Bryan MK. 2010. Phenotyping male infertility in the mouse: how to get the most out of a ‘non-performer’. Hum Reprod Update 16:205–224. doi: 10.1093/humupd/dmp032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.O'Shaughnessy PJ. 2014. Hormonal control of germ cell development and spermatogenesis. Semin Cell Dev Biol 29:55–65. doi: 10.1016/j.semcdb.2014.02.010. [DOI] [PubMed] [Google Scholar]
- 24.Singh J, O'Neill C, Handelsman DJ. 1995. Induction of spermatogenesis by androgens in gonadotropin-deficient (hpg) mice. Endocrinology 136:5311–5321. doi: 10.1210/endo.136.12.7588276. [DOI] [PubMed] [Google Scholar]
- 25.Lo YC, Brett L, Kenyon CJ, Morley SD, Mason JI, Williams BC. 1998. StAR protein is expressed in both medulla and cortex of the bovine and rat adrenal gland. Endocr Res 24:559–563. doi: 10.3109/07435809809032645. [DOI] [PubMed] [Google Scholar]
- 26.Guiguen Y, Jalabert B, Thouard E, Fostier A. 1993. Changes in plasma and gonadal steroid hormones in relation to the reproductive cycle and the sex inversion process in the protandrous seabass, Lates calcarifer. Gen Comp Endocrinol 92:327–338. doi: 10.1006/gcen.1993.1170. [DOI] [PubMed] [Google Scholar]
- 27.Le Tonqueze O, Gschloessl B, Namanda-Vanderbeken A, Legagneux V, Paillard L, Audic Y. 2010. Chromosome wide analysis of CUGBP1 binding sites identifies the tetraspanin CD9 mRNA as a target for CUGBP1-mediated down-regulation. Biochem Biophys Res Commun 394:884–889. doi: 10.1016/j.bbrc.2010.03.020. [DOI] [PubMed] [Google Scholar]
- 28.O'Shaughnessy PJ, Willerton L, Baker PJ. 2002. Changes in Leydig cell gene expression during development in the mouse. Biol Reprod 66:966–975. doi: 10.1095/biolreprod66.4.966. [DOI] [PubMed] [Google Scholar]
- 29.Vandesompele J, De Preter K, Pattyn F, Poppe B, Van Roy N, De Paepe A, Speleman F. 2002. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol 3:RESEARCH0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gohin M, Bodinier P, Fostier A, Bobe J, Chesnel F. 2011. Aromatase expression in Xenopus oocytes: a three cell-type model for the ovarian estradiol synthesis. J Mol Endocrinol 47:241–250. doi: 10.1530/JME-11-0080. [DOI] [PubMed] [Google Scholar]
- 31.Marquis J, Paillard L, Audic Y, Cosson B, Danos O, Le Bec C, Osborne HB. 2006. CUG-BP1/CELF1 requires UGU-rich sequences for high-affinity binding. Biochem J 400:291–301. doi: 10.1042/BJ20060490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pitteloud N, Hayes FJ, Boepple PA, DeCruz S, Seminara SB, MacLaughlin DT, Crowley WF Jr. 2002. The role of prior pubertal development, biochemical markers of testicular maturation, and genetics in elucidating the phenotypic heterogeneity of idiopathic hypogonadotropic hypogonadism. J Clin Endocrinol Metab 87:152–160. doi: 10.1210/jcem.87.1.8131. [DOI] [PubMed] [Google Scholar]
- 33.Payne AH, Hales DB. 2004. Overview of steroidogenic enzymes in the pathway from cholesterol to active steroid hormones. Endocr Rev 25:947–970. doi: 10.1210/er.2003-0030. [DOI] [PubMed] [Google Scholar]
- 34.Russell L, Ettlin R, SinhaHikim A, Clegg E. 1990. Histological and histopathological evaluation of the testis. Cache River Press, St. Louis, MO. [Google Scholar]
- 35.Ule J, Jensen K, Mele A, Darnell RB. 2005. CLIP: a method for identifying protein-RNA interaction sites in living cells. Methods 37:376–386. doi: 10.1016/j.ymeth.2005.07.018. [DOI] [PubMed] [Google Scholar]
- 36.Cosson B, Gautier-Courteille C, Maniey D, Ait-Ahmed O, Lesimple M, Osborne HB, Paillard L. 2006. Oligomerization of EDEN-BP is required for specific mRNA deadenylation and binding. Biol Cell 98:653–665. doi: 10.1042/BC20060054. [DOI] [PubMed] [Google Scholar]
- 37.Lei ZM, Mishra S, Zou W, Xu B, Foltz M, Li X, Rao CV. 2001. Targeted disruption of luteinizing hormone/human chorionic gonadotropin receptor gene. Mol Endocrinol 15:184–200. doi: 10.1210/mend.15.1.0586. [DOI] [PubMed] [Google Scholar]
- 38.Zhang FP, Poutanen M, Wilbertz J, Huhtaniemi I. 2001. Normal prenatal but arrested postnatal sexual development of luteinizing hormone receptor knockout (LuRKO) mice. Mol Endocrinol 15:172–183. doi: 10.1210/mend.15.1.0582. [DOI] [PubMed] [Google Scholar]
- 39.Zhang FP, Pakarainen T, Zhu F, Poutanen M, Huhtaniemi I. 2004. Molecular characterization of postnatal development of testicular steroidogenesis in luteinizing hormone receptor knockout mice. Endocrinology 145:1453–1463. doi: 10.1210/en.2003-1049. [DOI] [PubMed] [Google Scholar]
- 40.Xu Q, Lin HY, Yeh SD, Yu IC, Wang RS, Chen YT, Zhang C, Altuwaijri S, Chen LM, Chuang KH, Chiang HS, Yeh S, Chang C. 2007. Infertility with defective spermatogenesis and steroidogenesis in male mice lacking androgen receptor in Leydig cells. Endocrine 32:96–106. doi: 10.1007/s12020-007-9015-0. [DOI] [PubMed] [Google Scholar]
- 41.Pakarainen T, Zhang FP, Makela S, Poutanen M, Huhtaniemi I. 2005. Testosterone replacement therapy induces spermatogenesis and partially restores fertility in luteinizing hormone receptor knockout mice. Endocrinology 146:596–606. doi: 10.1210/en.2004-0913. [DOI] [PubMed] [Google Scholar]
- 42.O'Donnell L, McLachlan RI, Wreford NG, Robertson DM. 1994. Testosterone promotes the conversion of round spermatids between stages VII and VIII of the rat spermatogenic cycle. Endocrinology 135:2608–2614. doi: 10.1210/en.135.6.2608. [DOI] [PubMed] [Google Scholar]
- 43.O'Donnell L, McLachlan RI, Wreford NG, de Kretser DM, Robertson DM. 1996. Testosterone withdrawal promotes stage-specific detachment of round spermatids from the rat seminiferous epithelium. Biol Reprod 55:895–901. doi: 10.1095/biolreprod55.4.895. [DOI] [PubMed] [Google Scholar]
- 44.Wong CH, Xia W, Lee NP, Mruk DD, Lee WM, Cheng CY. 2005. Regulation of ectoplasmic specialization dynamics in the seminiferous epithelium by focal adhesion-associated proteins in testosterone-suppressed rat testes. Endocrinology 146:1192–1204. doi: 10.1210/en.2004-1275. [DOI] [PubMed] [Google Scholar]
- 45.Zhang J, Wong CH, Xia W, Mruk DD, Lee NP, Lee WM, Cheng CY. 2005. Regulation of Sertoli-germ cell adherens junction dynamics via changes in protein-protein interactions of the N-cadherin-beta-catenin protein complex which are possibly mediated by c-Src and myotubularin-related protein 2: an in vivo study using an androgen suppression model. Endocrinology 146:1268–1284. doi: 10.1210/en.2004-1194. [DOI] [PubMed] [Google Scholar]
- 46.Verhoeven G, Willems A, Denolet E, Swinnen JV, De Gendt K. 2010. Androgens and spermatogenesis: lessons from transgenic mouse models. Philos Trans R Soc Lond B Biol Sci 365:1537–1556. doi: 10.1098/rstb.2009.0117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Toyama Y, Hosoi I, Ichikawa S, Maruoka M, Yashiro E, Ito H, Yuasa S. 2001. beta-estradiol 3-benzoate affects spermatogenesis in the adult mouse. Mol Cell Endocrinol 178:161–168. doi: 10.1016/S0303-7207(01)00419-1. [DOI] [PubMed] [Google Scholar]
- 48.D'Souza R, Gill-Sharma MK, Pathak S, Kedia N, Kumar R, Balasinor N. 2005. Effect of high intratesticular estrogen on the seminiferous epithelium in adult male rats. Mol Cell Endocrinol 241:41–48. doi: 10.1016/j.mce.2005.04.011. [DOI] [PubMed] [Google Scholar]
- 49.D'Souza R, Pathak S, Upadhyay R, Gaonkar R, D'Souza S, Sonawane S, Gill-Sharma M, Balasinor NH. 2009. Disruption of tubulobulbar complex by high intratesticular estrogens leading to failed spermiation. Endocrinology 150:1861–1869. doi: 10.1210/en.2008-1232. [DOI] [PubMed] [Google Scholar]
- 50.Hofstra J, Loves S, van Wageningen B, Ruinemans-Koerts J, Jansen I, de Boer H. 2008. High prevalence of hypogonadotropic hypogonadism in men referred for obesity treatment. Neth J Med 66:103–109. [PubMed] [Google Scholar]
- 51.Loves S, Ruinemans-Koerts J, de Boer H. 2008. Letrozole once a week normalizes serum testosterone in obesity-related male hypogonadism. Eur J Endocrinol 158:741–747. doi: 10.1530/EJE-07-0663. [DOI] [PubMed] [Google Scholar]
- 52.Schlegel PN. 2012. Aromatase inhibitors for male infertility. Fertil Steril 98:1359–1362. doi: 10.1016/j.fertnstert.2012.10.023. [DOI] [PubMed] [Google Scholar]
- 53.Fowler KA, Gill K, Kirma N, Dillehay DL, Tekmal RR. 2000. Overexpression of aromatase leads to development of testicular leydig cell tumors: an in vivo model for hormone-mediated testicular cancer. Am J Pathol 156:347–353. doi: 10.1016/S0002-9440(10)64736-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tekmal RR, Ramachandra N, Gubba S, Durgam VR, Mantione J, Toda K, Shizuta Y, Dillehay DL. 1996. Overexpression of int-5/aromatase in mammary glands of transgenic mice results in the induction of hyperplasia and nuclear abnormalities. Cancer Res 56:3180–3185. [PubMed] [Google Scholar]
- 55.Li X, Nokkala E, Yan W, Streng T, Saarinen N, Warri A, Huhtaniemi I, Santti R, Makela S, Poutanen M. 2001. Altered structure and function of reproductive organs in transgenic male mice overexpressing human aromatase. Endocrinology 142:2435–2442. doi: 10.1210/endo.142.6.8211. [DOI] [PubMed] [Google Scholar]
- 56.Wang ZJ, Jeffs B, Ito M, Achermann JC, Yu RN, Hales DB, Jameson JL. 2001. Aromatase (Cyp19) expression is up-regulated by targeted disruption of Dax1. Proc Natl Acad Sci U S A 98:7988–7993. doi: 10.1073/pnas.141543298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Udd B, Krahe R. 2012. The myotonic dystrophies: molecular, clinical, and therapeutic challenges. Lancet Neurol 11:891–905. doi: 10.1016/S1474-4422(12)70204-1. [DOI] [PubMed] [Google Scholar]
- 58.Cruz Guzman Odel R, Chavez Garcia AL, Rodriguez-Cruz M. 2012. Muscular dystrophies at different ages: metabolic and endocrine alterations. Int J Endocrinol 2012:485376. doi: 10.1155/2012/485376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Antonini G, Clemenzi A, Bucci E, De Marco E, Morino S, Di Pasquale A, Latino P, Ruga G, Lenzi A, Vanacore N, Radicioni AF. 2011. Hypogonadism in DM1 and its relationship to erectile dysfunction. J Neurol 258:1247–1253. doi: 10.1007/s00415-011-5914-3. [DOI] [PubMed] [Google Scholar]
- 60.Mahadevan M, Tsilfidis C, Sabourin L, Shutler G, Amemiya C, Jansen G, Neville C, Narang M, Barcelo J, O'Hoy K, et al. . 1992. Myotonic dystrophy mutation: an unstable CTG repeat in the 3′ untranslated region of the gene. Science 255:1253–1255. doi: 10.1126/science.1546325. [DOI] [PubMed] [Google Scholar]
- 61.Brook JD, McCurrach ME, Harley HG, Buckler AJ, Church D, Aburatani H, Hunter K, Stanton VP, Thirion JP, Hudson T, et al. . 1992. Molecular basis of myotonic dystrophy: expansion of a trinucleotide (CTG) repeat at the 3′ end of a transcript encoding a protein kinase family member. Cell 68:799–808. doi: 10.1016/0092-8674(92)90154-5. [DOI] [PubMed] [Google Scholar]
- 62.Lee JE, Cooper TA. 2009. Pathogenic mechanisms of myotonic dystrophy. Biochem Soc Trans 37:1281–1286. doi: 10.1042/BST0371281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Roberts R, Timchenko N, Miller J, Reddy S, Caskey C, Swanson M, Timchenko L. 1997. Altered phosphorylation and intracellular distribution of a (CUG)n triplet repeat RNA-binding protein in patients with myotonic dystrophy and in myotonin protein kinase knockout mice. Proc Natl Acad Sci U S A 94:13221–13226. doi: 10.1073/pnas.94.24.13221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Huichalaf C, Sakai K, Jin B, Jones K, Wang GL, Schoser B, Schneider-Gold C, Sarkar P, Pereira-Smith OM, Timchenko N, Timchenko L. 2010. Expansion of CUG RNA repeats causes stress and inhibition of translation in myotonic dystrophy 1 (DM1) cells. FASEB J 24:3706–3719. doi: 10.1096/fj.09-151159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Jones K, Wei C, Iakova P, Bugiardini E, Schneider-Gold C, Meola G, Woodgett J, Killian J, Timchenko NA, Timchenko LT. 2012. GSK3beta mediates muscle pathology in myotonic dystrophy. J Clin Invest 122:4461–4472. doi: 10.1172/JCI64081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ladd AN, Taffet G, Hartley C, Kearney DL, Cooper TA. 2005. Cardiac tissue-specific repression of CELF activity disrupts alternative splicing and causes cardiomyopathy. Mol Cell Biol 25:6267–6278. doi: 10.1128/MCB.25.14.6267-6278.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Berger DS, Moyer M, Kliment GM, van Lunteren E, Ladd AN. 2011. Expression of a dominant negative CELF protein in vivo leads to altered muscle organization, fiber size, and subtype. PLoS One 6:e19274. doi: 10.1371/journal.pone.0019274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kim YK, Mandal M, Yadava RS, Paillard L, Mahadevan MS. 2014. Evaluating the effects of CELF1 deficiency in a mouse model of RNA toxicity. Hum Mol Genet 23:293–302. doi: 10.1093/hmg/ddt419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Mathieu J, Allard P, Gobeil G, Girard M, De Braekeleer M, Begin P. 1997. Anesthetic and surgical complications in 219 cases of myotonic dystrophy. Neurology 49:1646–1650. doi: 10.1212/WNL.49.6.1646. [DOI] [PubMed] [Google Scholar]