

Abstract
Ezrin is a key regulator of cancer metastasis that links the extracellular matrix to the actin cytoskeleton and regulates cell morphology and motility. We discovered a small-molecule inhibitor, NSC305787, that directly binds to ezrin and inhibits its function. In this study, we used a nano-liquid chromatography-tandem mass spectrometry (nano-LC–MS-MS)-based proteomic approach to identify ezrin-interacting proteins that are competed away by NSC305787. A large number of the proteins that interact with ezrin were implicated in protein translation and stress granule dynamics. We validated direct interaction between ezrin and the RNA helicase DDX3, and NSC305787 blocked this interaction. Downregulation or long-term pharmacological inhibition of ezrin led to reduced DDX3 protein levels without changes in DDX3 mRNA. Ectopic overexpression of ezrin in low-ezrin-expressing osteosarcoma cells caused a notable increase in DDX3 protein levels. Ezrin inhibited the RNA helicase activity of DDX3 but increased its ATPase activity. Our data suggest that ezrin controls the translation of mRNAs preferentially with a structured 5′ untranslated region, at least in part, by sustaining the protein level of DDX3 and/or regulating its function. Therefore, our findings suggest a novel function for ezrin in regulation of gene translation that is distinct from its canonical role as a cytoskeletal scaffold at the cell membrane.
INTRODUCTION
Ezrin is a prototype member of the ERM (ezrin-radixin-moesin) family of proteins that functions as a scaffold between the plasma membrane and the underlying cortical actin cytoskeleton (1, 2). Ezrin regulates cytoskeletal dynamics in response to both internal and external stimuli through its intracellular localization and protein binding activities; thus, it plays an important role in the maintenance of cell shape, cell polarity, adhesion, and movement (3). All members of the ERM family are characterized by the presence of a shared FERM domain at the amino terminus, which can bind to transmembrane proteins, including CD43, CD44, CD95, ICAMs, syndecan 2, EBP50/NHERF1, and E3KARP/NHERF2. The carboxy termini of ERM proteins contain an F-actin binding domain, which both regulates intramolecular interactions with amino-terminal FERM domains and promotes F-actin organization. The pleiotropic functions of ezrin in a wide range of cellular processes can be explained through its association with numerous proteins with diverse functions (4). Several lines of evidence have indicated that ezrin can oscillate between various “open/active” and “closed/dormant” states, which are regulated by self-association of N-terminal and C-terminal regions. Head-to-tail folding of the molecule likely masks the respective protein binding sites and leads to the localization of ezrin in the cytoplasm in its monomeric form. The conformational switch to an open state requires direct interaction with the plasma membrane phospholipid phosphatidylinositol 4,5-bisphosphate [PI(4,5)P2] and phosphorylation of a conserved threonine (T567) located in the C terminus (5–9). To date, all ezrin-related cellular phenotypes, including its involvement in different signal transduction pathways, have been attributed to this conformational shift in tertiary structure, followed by membrane localization.
Osteosarcoma (OS) is the most common primary bone cancer in children and adolescents (10). Respiratory failure due to pulmonary metastasis is the main cause of mortality in patients suffering from OS (11). Evidence from animal and human studies suggests that ezrin contributes to tumor metastasis. High ezrin expression is closely associated with poor survival in OS, as well as in pancreatic cancer, ovarian cancer, soft tissue sarcomas, gliomas, breast cancer, and rhabdomyosarcoma (12–21). However, the underlying molecular mechanisms of ezrin-mediated metastasis are largely unknown.
DDX3 is a member of the DEAD-box family of putative helicases, which are characterized by the presence of a conserved Asp-Glu-Ala-Asp motif (22). All DEAD-box proteins contain a highly conserved catalytic core domain with ATP-binding and RNA-binding sites that mediates the ATPase and RNA-unwinding activities. In addition to their established unwinding activities on RNA duplexes, DEAD-box proteins participate in a large variety of cellular processes that require manipulation of the RNA structure (23, 24). The lack of identification of the physiological RNA substrates or consensus sequences for the vast majority of DEAD-box proteins has limited our understanding of these enzymes (22). Because DEAD-box helicases are components of multiprotein complexes, their interaction with other proteins likely influences their RNA substrate specificities.
Human DDX3 (also named DDX3X and DBX) is a ubiquitously expressed protein that constantly shuttles between the cytoplasm and the nucleus. DDX3 regulates gene expression at different steps from transcription to translation, including both splicing and mRNA export. DDX3 interacts with poly(A)-binding protein 1 (PABP1) and several translation initiation factors, including eukaryotic translation initiation factor 4E (eIF4E), eIF4G, eIF4A, eIF2α, and eIF3 (25–29). DDX3 is a critical factor for stress granule (SG) assembly and cell survival under stress conditions (26, 28). DDX3 effects upon protein translation include both positive and negative regulatory roles (23, 24, 30). DDX3 functions as a repressor of cap-dependent translation by acting as an eIF4E-inhibitory protein in a helicase-independent manner (25, 26). There is also evidence supporting a role of DDX3 RNA helicase in promoting the translation initiation of a specific subset of mRNAs that include complex secondary structure in their 5′ untranslated regions (5′ UTRs) (27, 31–33). DDX3 is also implicated in cancer; however, as for its role in protein translation, the results are mixed, suggesting both oncogenic and tumor suppressor functions (25, 33–42).
Here, we demonstrate that ezrin interacts with proteins involved in the translation initiation process and SG assembly and function. Among these proteins, we identified DDX3 as a novel direct binding partner of ezrin; this interaction regulates the DDX3 protein level and activity and is inhibited by the antiezrin compound NSC305787. Our findings suggest that ezrin preferentially regulates translation of mRNAs with a structured 5′ UTR, at least in part, through maintaining the intracellular protein level of DDX3. In particular, ezrin blocks the RNA helicase activity of DDX3 while increasing its ATPase activity. Since DDX3 is involved in multiple aspects of RNA metabolism with a prominent role in translation initiation, the results presented here suggested that there might be novel functions of ezrin in regulating gene translation and SG dynamics that are independent of its canonical role as a cytoskeletal scaffolding protein at the plasma membrane.
MATERIALS AND METHODS
Cell lines.
Human MG63.3 and mouse K7M2, K12, and AS 1.46 OS cell lines were kindly provided by Chand Khanna from the National Cancer Institute (NCI) (NIH, Bethesda, MD). The MG63.3, K7M2, and K12 cells were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS). The K7M2/ezrin antisense clone AS 1.46 was cultured in DMEM with 10% FBS containing 500 μg/ml G418 (Invitrogen, Carlsbad, CA). The human hepatoma cell line HepG2 (ATCC; HB-8065) was grown in DMEM with 10% FBS and 1% nonessential amino acids. The human lung adenocarcinoma cell line H1944 (ATCC; CRL-5907) was grown in RPMI medium with 10% FBS.
Transient DDX3 and ezrin overexpression.
MG63.3 OS cells were transfected with a pCMV-entry plasmid expressing the human DDX3X transcript variant 1 cDNA with a Myc-FLAG tag (Origene, Rockville, MD; RC204171). K12 mouse OS cells were transfected with a pcDNA 3.1 plasmid expressing the human ezrin cDNA with a Myc-His tag. Transfections of cells were done using the X-tremeGene 9 DNA transfection reagent (Roche, San Francisco, CA) according to the manufacturer's recommendations. Transient expression of DDX3X and ezrin from the vectors was assessed after 48 h.
Gene silencing with siRNA.
The expression of DDX3 was suppressed by using two prevalidated small interfering RNA (siRNA) duplexes targeting different coding regions of DDX3 (Invitrogen, Carlsbad, CA; s4004 and s4005). The prevalidated siRNA sequence used for silencing human ezrin was ordered from Invitrogen (Carlsbad, CA) (s14796). siGENOME nontargeting siRNA pool 2 (Dharmacon, Lafayette, CO; siGENOME nontargeting siRNA control pools/pool 2 D-001206-14-50, which is a pool of 4 nontargeting siRNAs) was used as the control. MG63.3 cells were transfected with DDX3 or ezrin siRNA oligonucleotides using X-tremeGene siRNA transfection reagent (Roche, San Francisco, CA) according to the manufacturer's protocol. The cells were analyzed for DDX3 and ezrin knockdown after 48 or 72 h by immunoblotting.
Luciferase reporter assays.
The pCMV-Luciferase (pCMV-LUC) and pCMV-Stem Loop-Luciferase (pCMV-SL-LUC) reporter constructs were kindly provided by Hsin-Sheng Yang from the University of Kentucky (43). Firefly luciferase expression from pCMV-LUC and pCMV-SL-LUC was measured after 24 h using a luciferase assay kit (Promega, Madison, WI) on an LMax II 384 luminometer (Molecular Devices, Union City, CA) according the manufacturer's protocol. The luciferase activities were normalized to the total protein concentration. Transfection of reporter constructs was carried out using X-tremeGene 9 DNA transfection reagent (Roche, San Francisco, CA) according to the manufacturer's instructions.
RT-PCR.
Total RNA was extracted from cell lines using an RNeasy minikit (Qiagen, Valencia, CA) and reverse transcribed using a transcriptor first-strand cDNA synthesis kit (Roche, San Francisco, CA) according to the manufacturer's protocols. Quantitative reverse transcription (qRT)-PCR was done on a LightCycler 480 II system using SYBR green mix (Sigma-Aldrich, St. Louis, MO) in a 20-μl volume (10 μl of 2× master mix, 1.0 μl of 10 μM forward and reverse primer mix, and 2.0 μl cDNA). Reactions were performed in triplicate on a 96-multiwell plate. qRT-PCR was performed under the following operating conditions: preincubation at 95°C for 10 min and 40 amplification cycles, including denaturation at 95°C for 30 s, annealing at 55°C for 30 s, and extension at 72°C for 45 s. Finally, melting curves were determined to check primer specificity. Changes in relative gene expression were calculated using the comparative threshold cycle (CT) method with 18S rRNA as the reference. Primer pairs were as follows: ezrin, sense (5′-CATCACTGAGGCAGAGAAGAAC-3′) and antisense (5′-TGTCATTGTGGGTCCTCTTATTC-3′); DDX3, sense (5′-GGTATTAGCACCAACGAGAGAG-3′) and antisense (5′-TATCGGCACCACCATAAACC-3′); 18S rRNA, sense (5′-CTTAGAGGGACAAGTGGCG-3′) and antisense (5′-ACGCTGAGCCAGTCAGTGTA-3′).
IP and Western blotting.
Immunoprecipitation (IP) experiments were done with protein lysates from mouse and human OS cells, as described previously (44). Total cell lysates were prepared by lysing the cells on ice for 30 min with phospho-lysis buffer (50 mM HEPES, pH 7.9, 100 mM NaCl, 4.0 mM sodium pyrophosphate, 10 mM EDTA, 10 mM sodium fluoride, and 1% Triton X-100) containing 2.0 mM sodium vanadate, 1.0 mM phenylmethylsulfonyl fluoride (PMSF), 4.0 μg/ml aprotinin, 4.0 μg/ml leupeptin, and 1.0 μg/ml calyculin A. IP experiments were performed using approximately 1,000 to 1,500 μg total cellular protein in 1.0 ml phospho-lysis buffer. After preclearing, the lysates were incubated with 2.0 μl of antiezrin antibody (Sigma-Aldrich, St. Louis, MO; E8897) or an equal amount of control mouse IgG (Santa Cruz Biotechnology, Santa Cruz, CA; sc-2025) overnight at 4°C on a rotating axis. For RNase A treatment, immunoprecipitates were treated with 0.5 mg/ml RNase A at 37°C for 30 min. In the morning, a 40-μl resuspended volume of bovine serum albumin (BSA)-blocked protein A/G Plus agarose beads (Santa Cruz Biotechnology, Santa Cruz, CA) was added to the samples, and the mixtures were rotated for 1 h at 4°C. The pelleted beads were washed three times with 0.3 ml (each) IP wash buffer (10 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1.0 mM EGTA, 1.0 mM EDTA, 1% Triton X-100, 0.5% Nonidet P-40, 0.2 mM sodium vanadate, and 0.2 mM phenylmethylsulfonyl fluoride [PMSF]), and all the proteins bound to the beads were eluted by boiling samples in SDS-PAGE sample buffer for 5 min. Immunoblotting was performed as previously described (45). Briefly, samples were subjected to SDS-PAGE and then transferred to an Immobilon-P membrane (Millipore, Billerica, MA). Nonspecific binding sites were blocked upon incubation in 5% nonfat dry milk diluted in 1× TTBS (20 mM Tris-HCl, pH 7.5, 150 mM NaCl, 0.05% Tween 20) for 1 h at room temperature. The membranes were incubated with primary antibodies diluted in either 5% nonfat dry milk or 5% BSA in 1× TTBS at room temperature for 2 h or at 4°C overnight, according to the manufacturer's recommendations. The dilutions for primary antibodies were as follows: antiezrin, 1:2,500 (Sigma-Aldrich, St. Louis, MO; E8897); anti-DDX3, 1:500 (Santa Cruz Biotechnology, Santa Cruz, CA; sc-81247); anti-Flag, 1:1,000 (Sigma-Aldrich, St. Louis, MO; F1804); anti-caprin-1, 1:4,000 (Bethyl Laboratories; A303-882A), anti-SND1, 1:3,000 (Bethyl Laboratories; A302-883A); anti-PABP1, 1:1,000 (Cell Signaling, Danvers, MA; 4992); anti-MTDH, 1:1,000 (R&D Systems, Minneapolis, MN; MAB7180) and antiactin-horseradish peroxidase (HRP), 1:5,000 (Santa Cruz Biotechnology, Santa Cruz, CA; sc-1615). After rinsing three times with 1× TTBS, the membranes were incubated for 1 h at room temperature in HRP-conjugated anti-rabbit (GE Healthcare Bio-Sciences, Pittsburgh, PA; NA934V) or anti-mouse (GE Healthcare Bio-Sciences, Pittsburgh, PA; NA931V) secondary antibody diluted 1:5,000 in 1× TTBS or anti-rat secondary antibody (R&D Systems, Minneapolis, MN; HAF005) diluted 1:1,000 in 5% nonfat dry milk in 1× TTBS. The blots were then washed three times in 1× TTBS and then developed using Millipore Immobilon Western chemiluminescent HRP substrate according to the manufacturer's instructions (Millipore Corporation, Billerica, MA). Chemiluminescence was detected using a Fujifilm LAS-3000 imaging system.
SPR.
Surface plasmon resonance (SPR) studies for testing direct interaction between ezrin and DDX3 were performed on a Biacore T-200 instrument at room temperature. Purified wild-type ezrin or a phosphomimicking ezrin mutant was immobilized on the second flow cell of a CM5 sensor chip (GE Healthcare Bio-Sciences, Pittsburgh, PA) by the amine-coupling method in sodium acetate buffer, pH 4.5 (both at ∼7,600 resonance units [RU]). The first flow cell was left empty for background signal subtraction. HBS-P (10 mM HEPES, pH 7.4, 150 mM NaCl, 0.05% [vol/vol] nonionic polysorbate surfactant P20 [GE Healthcare Bio-Sciences, Pittsburgh, PA]) was used as the running buffer. Kinetic analysis was done by injecting six different concentrations of DDX3 (1.25, 2.5, 5, 10, 20, and 40 nM) over ezrin-immobilized and control surfaces in triplicate. Each injection was done with 90 s of association time and 300 s of dissociation time. For testing of the ability of ezrin to directly bind MTDH, purified ezrin was immobilized on the second flow of a CM5 sensor chip under the same conditions as described above (∼6,500 RU). In order to study the binding kinetics of ezrin and the MTDH interaction, HBS-P buffer containing 1% glycerol was used as the running buffer. Purified recombinant human MTDH was purchased from Origene (Rockville, MD) (TP307238). Kinetic analysis was done by injecting four different concentrations of MTDH (0.63, 1.25, 2.5, and 5 nM) over ezrin-captured and control surfaces in triplicate. Each injection was done with 60 s of association time and 300 s of dissociation time. KD (equilibrium dissociation constant) values for ezrin-DDX3 and ezrin-MTDH interactions were obtained using BiaEvaluation software (version 1.0).
ELISA.
Enzyme-linked immunosorbent assays (ELISAs) were performed on MaxiSorp 96-well plates in triplicate wells. The wells were coated with either recombinant wild-type ezrin or an ezrin T567D phosphomimicking mutant (300 ng/well) in 100 μl PBS overnight at 4°C and then incubated with 4% BSA in PBS at room temperature for 2 h to block uncoated sites and prevent nonspecific binding. After washing of the wells five times with 200 μl of PBS containing 0.1% Tween 20 to remove unbound protein, NSC305787 was added at a concentration range of 0.012 to 12.0 μM. After 1 h of incubation and washing as described above, DDX3 (300 ng/well) in 100 μl PBS was added as a ligand in the presence of NSC305787 at the same concentrations. The plate was incubated overnight at 4°C, followed by extensive washing of the wells. The protein binding was detected using anti-DDX3 antiserum, followed by a secondary antibody coupled to horseradish peroxidase and using a 3,3′,5,5′-tetramethylbenzidine (TMB) peroxidase substrate kit (Bio-Rad, Irvine, CA) according to the manufacturer's instructions. The dilutions of primary and secondary antibody used were as follows: DDX3 antibody (Santa Cruz Biotechnology, Santa Cruz, CA; sc-81247), 1:500 dilution in washing buffer; HRP-conjugated anti-mouse secondary antibody (GE Healthcare Bio-Sciences, Pittsburgh, PA; NA931V), 1:2,000 in washing buffer. Color development was monitored, and the absorbances were measured at 450 nm with a microplate reader.
Affinity pulldown of NSC305787-competed ezrin-interacting proteins.
Recombinant ezrin was first coupled to a CNBr-activated Sepharose 4B matrix (Sigma-Aldrich, St. Louis, MO) according to the manufacturer's recommendations. Briefly, 4.7 mg of purified ezrin in 0.1 M NaHCO3 buffer, pH 8.3, containing 0.5 M NaCl was added to 1.0 ml of swollen and washed CNBr-activated resin, and the mixture was rotated overnight at 4°C. The unreacted ligand was washed away using NaHCO3-NaCl coupling buffer. The unreacted groups on the resin were then blocked with 0.2 M glycine, pH 8.0, overnight on a rotating axis at 4°C. The beads were washed first with basic coupling buffer, pH 8.5, and then with 0.1 M acetate buffer, pH 4.0, containing 0.5 M NaCl to remove the blocking solution. This wash cycle with high- and low-pH buffer solutions was repeated five times. The efficiency of coupling was checked by SDS-PAGE, using samples of the starting ezrin preparation and the postcoupling supernatant. The ezrin-coupled resin was stored as a 25% slurry in phospho-lysis buffer (50 mM HEPES, pH 7.9, 100 mM NaCl, 4.0 mM sodium pyrophosphate, 10 mM EDTA, 10 mM sodium fluoride, and 1% Triton) in the presence of 0.2% sodium azide at 4°C. Total cell lysates from K7M2 cells were prepared as described above. One hundred microliters of a 25% slurry of ezrin-coated beads was incubated overnight with 2.0 mg total cellular protein in 2.0 ml phospho-lysis buffer in the presence of either vehicle (dimethyl sulfoxide [DMSO]) or NSC305787 at a 50.0 μM concentration at 4°C on a rotating axis. The resin was washed four times with 0.75 ml (each time) phospho-lysis buffer containing 2.0 mM sodium vanadate, 1.0 mM PMSF, 4.0 μg/ml aprotinin, 4.0 μg/ml leupeptin, and 1.0 μg/ml calyculin A. The proteins bound to the beads were eluted by boiling samples in 2× SDS-PAGE sample buffer for 5 min and then run on a 12% SDS-PAGE gel. The protein bands were stained with a GelCode Coomassie blue staining kit (Pierce, Rockford, IL). The protein bands corresponding to the control- and NSC305787-treated pulldown lanes that differed in staining intensity were excised from the gel and subsequently subjected to in-gel tryptic digestion to extract peptides for further nano-liquid chromatography-tandem mass spectrometry (nano-LC–MS-MS) analysis.
Preparation of recombinant proteins.
Full-length wild-type ezrin and the phosphomimicking ezrin mutant (T567D) were cloned into the pQE16 vector and expressed in Escherichia coli strain M15(pREP4) (Qiagen, Valencia, CA). Cultures were inoculated at 1:20 dilution in LB medium from saturated overnight cultures containing 100 μg/ml ampicillin and 25 μg/ml kanamycin and were grown for 90 to 120 min until the optical density at 600 nm (OD600) reached 0.6 at 37°C. To induce the expression of ezrin, 1.0 mM isopropyl-β-d-thiogalactopyranoside was added, and the cells were grown for an additional 4 h. Cells were harvested by centrifugation at 8,000 × g for 15 min. To purify bacterium-expressed ezrin, the induced cells were resuspended in 20 mM 2-(N-morpholino)ethanesulfonic acid (MES), pH 6.8, containing 100 mM NaCl, 10% glycerol, 0.4% Tween 20, 5.0 mM β-mercaptoethanol, 50 μg/ml PMSF, 75 μg/ml benzamidine, and 1 Complete protease inhibitor tablet/50 ml (Roche, San Francisco, CA) and lysed by sonication. The lysate was then clarified by centrifugation at 13,500 × g for 10 min and applied to a HiTrap SP Sepharose Fast Flow (GE Healthcare Bio-Sciences, Pittsburgh, PA) cation-exchange column preequilibrated with 25 mM HEPES, pH 6.8, containing 100 mM NaCl, 10% glycerol, and 1.0 mM tris(2-carboxyethyl)phosphine (TCEP) in an AKTA Explorer chromatography system (GE Healthcare Bio-Sciences, Pittsburgh, PA). The column was developed using a linear gradient of 100 to 1,000 mM NaCl, and fractions were monitored by SDS-PAGE on 10% gels. Fractions rich in ezrin were pooled, diluted appropriately to lower the salt concentration to 100 mM, and applied to a HiTrap heparin-Sepharose high-performance column (GE Healthcare Bio-Sciences, Pittsburgh, PA) preequilibrated with 25 mM HEPES, pH 7.5, containing 100 mM NaCl, 10% glycerol, and 1.0 mM TCEP. The column was developed with a linear gradient of 100 to 1,000 mM NaCl, and the protein purity was determined by SDS-PAGE followed by Coomassie staining. The eluted fractions were dialyzed against 25 mM HEPES, pH 7.5, 100 mM NaCl, 10% glycerol, and 1.0 mM TCEP and stored at −80°C for further use. Recombinant DDX3 protein was expressed in insect cells using the baculovirus expression system. Baculovirus stock was generated using a DDX3 bacmid vector in adherent SF9 cells (Invitrogen, Carlsbad, CA). A Rapid Titer kit (Clontech Laboratories Inc., Palo Alto, CA) was used to determine the multiplicity of infection (MOI) of the virus stock. Virus stock at an MOI of 2.5 was used to infect 1 liter of a serum-independent suspension of Sf9 cells (2.5 × 106 cells/ml). Cells were harvested after 42 to 44 h of infection, and all subsequent purification steps were carried out at 4°C. A whole-cell extract was prepared in ice-cold lysis buffer containing 25 mM HEPES, pH 7.5, 500 mM NaCl, 5.0 mM MgCl2, 1% Triton X-100, 1% sodium deoxycholate, 10% glycerol, 5.0 mM β-mercaptoethanol, 25 mM imidazole, 1.0 mM PMSF, and 1 Complete protease inhibitor tablet/50 ml (Roche, San Francisco, CA). The lysate was incubated on ice for 30 min and then centrifuged for 10 min at 13,500 × g. To remove the nucleic acids, the lysate was treated with 2.0 mg/ml protamine sulfate (Sigma-Aldrich, St. Louis, MO) and spun again at 13,500 × g for 10 min. DDX3 was purified using immobilized metal affinity chromatography (IMAC) on a nickel-charged Hi-Trap chelating high-performance column (GE Healthcare Bio-Sciences, Pittsburgh, PA) in an AKTA Explorer chromatography system (GE Healthcare Bio-Sciences, Pittsburgh, PA). The binding buffer was composed of 25 mM HEPES, pH 7.5, 300 mM NaCl, 10% glycerol, 5.0 mM MgCl2, 0.1% Tween 20, 25 mM imidazole, and 5.0 mM β-mercaptoethanol. Protein fractions were eluted in the same buffer using a continuous gradient of 25 to 600 mM imidazole. The purity of the protein was analyzed by SDS-PAGE. The eluted fractions were dialyzed against 25 mM HEPES, pH 7.5, 150 mM NaCl, 50% glycerol, 5.0 mM MgCl2, and 2.0 mM TCEP and stored at −80°C for further use.
Peptide analysis by nano-LC–MS-MS.
Selected peptide bands were excised and destained using 50% acetonitrile (ACN) in 25 mM ammonium bicarbonate, pH 7.8, dehydrated with 200 μl of ACN for 5 min at 30°C using a thermoshaker (Eppendorf AG, Hamburg, Germany), alkylated with 200 μl 15 mM iodoacetamide (IAA) for 15 min, washed twice with 200 μl 25 mM ammonium bicarbonate, pH 7.8, at 30°C, and then vacuum dried in a Speed-Vac (Labconco, Kansas City, MO). Gel pieces were rehydrated, and proteins were digested for 8 h at 37°C in a thermoshaker with 40 μl of 25-ng/μl trypsin (trypsin gold, mass spectrometry grade; Promega, Madison, WI) in 25 mM ammonium bicarbonate, pH 7.8. After digestion, the peptides were extracted from the gel pieces using step-by-step extraction with a gradient of ACN (15 to 60% ACN with 1% trifluoroacetic acid [TFA]). The extraction was performed using a sonicator (Bransonic Ultrasonics Corporation, Danbury, CT) with ice cubes. Peptide separation was achieved on a Tempo Capillary LC device coupled with HiPLC-nanoflex (Eksigent, Framingham, MA) using a nano-cHiPLC trap column (200 μm by 500 μm; ChromXP C18-CL; 3 μm; 300 Å) (Eksigent, Framingham, MA) interfaced with a 5600 TripleTOF mass spectrometer (AB Sciex, Framingham, MA). The tryptic digest was separated with a 10-min trapping step using 2% ACN and 0.1% formic acid at a 3-μl/min flow rate, and chromatographic separation was performed at a 0.3-μl/min flow rate as follows: starting conditions, 2% ACN, 0.1% formic acid; 1 to 35 min, 5 to 50% ACN, 0.1% formic acid; 35 to 37 min, 50 to 95% ACN, 0.1% formic acid; 37 to 40 min, 95% ACN, 0.1% formic acid, followed by equilibration with 2% ACN, 0.1% formic acid for an additional 20 min. For all runs, 1 μl of sample was injected directly after enzymatic digestion. Analysis was performed using an information-dependent acquisition workflow with one full scan (400 to 1,500 m/z) and 50 MS-MS fragmentations of major multiply charged precursor ions with rolling collision energy. Mass spectra were recorded in the MS range of 400 to 1,500 m/z and MS-MS spectra in the range of 100 to 1,800 m/z, with a resolution of 30,000 and mass accuracy up to 2 ppm using the following experimental parameters: declustering potential, 80 V; curtain gas, 15; ion spray voltage, 2,300 V; ion source gas 1, 20; interface heater, 180°C; entrance potential, 10 V; collision exit potential, 11 V; exclusion time, 5 s. The collision energy was set automatically according to the m/z of the precursor (rolling collision energy). The data were processed using ProteinPilot 4.0 software (AB Sciex, Framingham, MA).
DDX3 helicase assay.
The helicase activity of recombinant human DDX3 was determined by measuring the extent of unwinding of the double-stranded RNA (dsRNA) with a 3′ overhang. dsRNA was prepared by annealing the two complementary nucleic acid strands, 5′-/56-FAM/ACGUCGAUCCGAAACUAUACUUAAUUUUAA-3′ (30-mer reporter strand) and 5′-GUUUCGGAUCGACGU/3IABkFQ/-3′ (15-mer quenching strand), which were purchased from IDTDNA (Coralville, IA). The longer strand was fluorescence (6-carboxyfluorescein [FAM]) labeled at its 5′ end. The shorter strand was labeled with a 3′ quencher (3′ Iowa Black FQ [3IABkFQ]), which quenches the fluorescence when bound to the reporter strand. The helicase assays were performed using 96-well plates in 50-μl reaction volumes containing 25 mM HEPES, pH 7.5, 1.0 mM MgCl2, 1.0 mM dithiothreitol (DTT), 4.0 nM purified DDX3, and 20 nM dsRNA at 37°C. Reactions were initiated by the addition of 1.0 mM ATP to the incubation mixture with the help of an injector pump. Reactions were monitored for 15 min in a BioTek Synergy H4 hybrid reader (Winooski, VT) using excitation and emission wavelengths of 490 nm and 519 nm, respectively. The specificity of the enzyme reaction was confirmed by including control reactions, such as a helicase reaction with heat-inactivated DDX3 or helicase reactions in the absence of ATP and Mg2+. To assess the effect of ezrin on the DDX3-helicase reaction, increasing concentrations of purified recombinant human ezrin from 4.0 to 160 nM, which corresponds to a range of 1- to 40-fold molar excess over the amount of DDX3, were added to the reaction mixture. To confirm the specificity of enzyme inhibition experiments, a helicase assay was performed using an unrelated protein, myoglobin.
DDX3 ATPase assay.
The ATPase activity of DDX3 was determined using a fixed-type assay by measuring the amount of free phosphate formed upon ATP hydrolysis using a PiColorLock ALS assay kit (Innova Biosciences, Cambridge, United Kingdom) according to the manufacturer's instructions. Reactions were performed using a 96-well plate in 25 mM HEPES, pH 7.5, 1.0 mM DTT, 1.0 mM MgCl2, 1.0 mM ATP, 200 nM purified DDX3, and 400 nM dsRNA with a 3′ overhang in a final volume of 25 μl. The sequence of the dsRNA was the same as that of the RNA used in the DDX3 RNA helicase assay. The reaction was started by the addition of ATP with the help of an injector pump, and then the mixture was incubated for 20 min at 37°C. After adding 100 μl phosphate detection reagent according to the instructions of the kit supplier, the absorbance values were measured at 625 nm after a 25-min incubation period using a BioTek Synergy H4 hybrid reader (Winooski, VT). A phosphate standard curve was also prepared by using known concentrations of free phosphate under the conditions described above. The effect of ezrin on DDX3 ATPase activity was tested by adding increasing concentrations of purified recombinant human ezrin from 1.0 to 8.0 μM, which corresponds to a range of 5- to 40-fold molar excess of the amount of DDX3, to the reaction mixture. Negative-control reactions, including heat-inactivated DDX3 with the indicated molar excess of recombinant ezrin, were also carried out along with the experimental reactions under the same conditions. Experiments were also performed using myoglobin as a negative-control protein under the same reaction conditions.
In vitro translation assay.
The effects of recombinant ezrin and DDX3 on the translation of nonstructured and stem-loop-structured luciferase mRNAs were determined by using the T7 Quick-Coupled transcription/translation system (Promega, Madison, WI) according to the manufacturer's instructions. Each 50-μl reaction mixture contained 40 μl TNT Quick master mix (Promega, Madison, WI), 1.0 μl of 1.0 mM methionine, 2.0 μl of 0.50-μg/μl pCMV-LUC or pCMV-SL-LUC reporter construct, and appropriate amounts of purified recombinant proteins. The reaction mixtures were incubated at 30°C for 90 min, and the results of protein expression were analyzed by measuring the luciferase activities as described above. To assess whether recombinant ezrin or DDX3 affects the transcription of luciferase, translation reaction mixtures were assembled as described above with 1.0 μg of each protein and appropriate buffer controls. After a 90-min incubation period at 30°C, 35-μl aliquots of 50-μl incubation mixtures were used for total RNA isolation. RNA was purified using MegaClear columns (Ambion, Austin, TX; AM1908), and reverse transcribed using random-hexamer primers as described above. qRT-PCR was done under the same conditions as described above, using primer pairs specific for luciferase (sense, 5′-TGTGGATTTCGAGTCGTCTTAAT-3′; antisense, 5′-CGAAGAAGGAGAATAGGGTTGG-3′) and 18S rRNA (see above).
Statistical analysis.
The statistical significance of differences between groups was evaluated by an unpaired Student t test using Prism version 6.0c (GraphPad Software, La Jolla, CA). Statistical significance was defined as a P value of <0.05.
RESULTS
Proteomics identifies novel binding partners of ezrin.
The small molecule NSC305787 directly binds to ezrin and inhibits the metastatic phenotype both in vitro and in vivo (46). Since ezrin lacks intrinsic enzymatic activity and exerts its biological functions through protein-protein interactions, we sought protein partners of ezrin that would be disrupted by NSC305787. We used an affinity pulldown strategy with recombinant ezrin covalently linked to Sepharose beads. Total cell lysates from mouse K7M2 OS cells were incubated with ezrin-coated beads in the presence of either NSC305787 or vehicle control, followed by elution and resolution with PAGE (Fig. 1A). Coomassie-stained protein bands that were differentially present in the NSC305787-treated sample were cut from the gel and analyzed by nano-LC–MS-MS. The candidate proteins were prioritized to eliminate those that were not identified by at least two unique peptides with a confidence level of ≥95%, a ProteinPilot Unused ProtScore of ≥2 (99% confidence level), and a correct molecular weight (Fig. 1B). These filtering steps yielded 240 proteins that were expected to compete with NSC305787 for binding to ezrin (see Table S1 in the supplemental material). Nine of the 240 proteins were previously identified as ezrin-binding partners, including ezrin itself (47), other ERM proteins, radixin and moesin (48), PABP1 (49), calpain 1 and calpain 2 (50–52), protein kinase C iota (53), focal adhesion kinase (54), and Na+/H+ exchange regulatory cofactor (EBP50/NHERF1) (55). Moreover, a recent study demonstrated that the ELMO-DOCK1 (docking protein 1) complex, a bipartite guanine nucleotide exchange factor complex for the small GTPase Rac1, orchestrates ciliary basal body migration, docking, and positioning by interacting with ezrin and that the ELMO-DOCK1-Rac1 complex influences ezrin phosphorylation (56). Among the proteins, we identified DOCK1, as well as the other regulators of Rac1 function, Rho guanine nucleotide exchange factor (GEF-H1), regulator of chromosome condensation 2 (RCC2), and Rac GTPase activating protein 1 (RacGap1), in our MS-MS data (see Table S1 in the supplemental material). The presence of the above-mentioned ezrin-binding proteins in the MS screen confirmed the validity of our experimental approach.
FIG 1.

Identification of candidate NSC305787-competed ezrin-binding proteins. (A) Schematic diagram describing an affinity pulldown coupled with an MS-MS approach for identification of ezrin-interacting proteins that can be competed away by NSC305787. Recombinant ezrin was purified and coupled with CNBr-activated Sepharose 4B beads. The ezrin-coated beads were then used as bait on total cell lysates from K7M2 mouse OS cells in the presence of either NSC305787 or vehicle control. The proteins bound to the beads were eluted by boiling samples in SDS-PAGE sample buffer. Proteins were then run on an SDS-PAGE gel and stained with Coomassie brilliant blue. Protein bands that were competed away with NSC305787 were analyzed by nano-LC–MS-MS. (B) Schematic flow diagram for analysis of the raw data obtained from MS. Through stepwise filtering, a total of 240 potential ezrin-interacting proteins that can be competed away by NSC305787 were identified. Proteins identified by MS-MS were first filtered based on the elimination of proteins that were not identified by at least two unique peptides with a confidence level of ≥95% and with a ProteinPilot Unused ProtScore of ≥2 (99% confidence level). Further subtraction of proteins was then based on the removal of proteins whose predicted molecular weights differed by more than 20% from the molecular weights of the corresponding bands on the gel.
Ezrin interacts with proteins involved in the translation initiation process and stress granule dynamics.
When we analyzed the 240 potential ezrin-interacting proteins, we identified 21 key ezrin-binding proteins associated with translation initiation and SG dynamics, including DDX3, PABP1, caprin-1, staphylococcal nuclease domain-containing 1 (SND1), several heat shock/stress proteins, eIF3 subunits, heterogeneous nuclear ribonucleoprotein Q (hnRNP Q), fragile X mental retardation syndrome-related protein 1 (FXR1), heterogeneous nuclear ribonucleoprotein K (hnRNP K), lyric/metadherin (MTDH), argonaute-2/eIF2C2, fragile X mental retardation protein (FMRP), and RNA-binding motif protein 42 (RBM42) (see Table S1 in the supplemental material). Among these proteins, DDX3 had the top ranking, with 199 peptide matches, 71% coverage, and an Unused ProtScore of 128.70 in the MS-MS data (see Table S1 in the supplemental material). We first confirmed the endogenous ezrin interaction with DDX3 in K7M2 mouse and MG63.3 human OS cell lines by coimmunoprecipitation (co-IP) (Fig. 2A). When mouse K7M2 OS cells were treated with NSC305787, the amount of coprecipitated DDX3 with antiezrin antibody decreased significantly (Fig. 2B). Surface plasmon resonance (SPR) confirmed a direct interaction between purified recombinant ezrin and DDX3, since IP experiments do not distinguish between direct and indirect protein complex interactions (Fig. 2C and D). The role of ezrin Thr567 phosphorylation upon its binding to DDX3 was assessed using a phosphomimicking ezrin mutant in which the C-terminal threonine, T567, was replaced by aspartate (T567D). We observed that DDX3 directly interacts with both wild-type ezrin and phosphomimicking mutant ezrin at low nanomolar concentrations, with comparable KD values of 2.81 nM and 4.68 nM, respectively (Fig. 2C and D). These findings establish that ezrin and DDX3 are direct binding partners and that the interaction is inhibited by NSC305787. In order to evaluate if ezrin interaction with DDX3 was dependent on the presence of RNA in the complex, we immunoprecipitated ezrin from cell lysates, followed by RNase A treatment. The amount of coprecipitated DDX3 diminished, but it did not completely disappear from ezrin immunoprecipitates, when RNase A was added, suggesting that RNA has a role in, but is not required for, ezrin-DDX3 interaction (Fig. 2E).
FIG 2.

Ezrin interacts with DDX3, and the antiezrin compound NSC305787 inhibits this interaction. (A) Interaction of ezrin with DDX3 in K7M2 mouse (left) and MG63.3 human (right) OS total cell lysates (TCL) by co-IP experiments. Protein complexes were immunoprecipitated from the cell lysates with antiezrin antibody (Ab) or negative-control total mouse IgG and immunoblotted (IB) with anti-DDX3 antibody. (B) K7M2 cells were treated with 3.0 μM NSC305787 for 8 h. Total cell lysates were immunoprecipitated with control IgG or an ezrin antibody, which was followed by Western blotting for ezrin and DDX3. (C) (Top) Recombinant wild-type ezrin and the phosphomimicking ezrin T567D mutant were expressed in bacteria and purified by cation-exchange chromatography on an SP Sepharose column, followed by adsorption chromatography on a heparin column. DDX3 protein was expressed in baculovirus-infected insect cells and purified by Ni2+ affinity column chromatography. Approximately 1.5 μg of each protein was run on a gel and stained with Coomassie blue. (Bottom) A purified DDX3 sample (1.5 μg) was run on a gel and stained with Coomassie blue (Coom.) (left), or 250 ng of purified DDX3 was run on a gel and transferred to a membrane for Western blot analysis using anti-DDX3 antibody (right). (D) Recombinant wild-type ezrin or the phosphomimicking ezrin mutant was immobilized on a CM5 chip in Biacore T-200. DDX3 was injected over the chip surface at six different concentrations (1.25, 2.5, 5.0, 10, 20, and 40 nM) in triplicate. The colored lines show real data, and the black lines represent curves fitted for a 1:1 binding model. (E) Equal amounts of lysates from K7M2 mouse OS cells were subjected to co-IP with antiezrin antibody. The resulting coprecipitates were treated with 0.5 mg/ml RNase A at 37°C for 30 min. −RNase A and no treat., samples that were incubated in the absence of RNase A for 30 min at 37°C or at 4°C, respectively. Immunoblotting was performed with anti-DDX3 and antiezrin antibodies.
To further validate that NSC305787 can interrupt the interaction between ezrin and DDX3, we performed ELISAs using purified proteins, wild-type ezrin, a T567D phosphomimicking mutant of ezrin, and DDX3 (Fig. 3). The ELISA involved binding of DDX3 to ezrin immobilized on a polystyrene surface, which was detected using an anti-DDX3 antiserum followed by a secondary antibody coupled to horseradish peroxidase. Negative controls (wells coated with BSA alone, ezrin alone, or BSA alone followed by incubation with DDX3 protein) and a positive control (wells coated with DDX3 protein alone) were included to verify the specificity of the ELISA. The inhibitory activity of NSC305787 was monitored by a concentration-dependent reduction in the binding signal due to disruption of the complex. The 50% inhibitory concentrations (IC50s) of NSC305787 required to disrupt wild-type ezrin-DDX3 and ezrin T567D-DDX3 interactions were calculated as 182.4 nM and 2,096 nM, respectively, which correspond to 4.6-fold and 52.4-fold molar excess over the amount of DDX3 protein used as a ligand in solution (Fig. 3). These findings suggest that NSC305787 is much more effective in preventing the binding of DDX3 to wild-type ezrin than to the ezrin T567D mutant.
FIG 3.

ELISA demonstrating inhibition of the ezrin-DDX3 protein interaction by NSC305787. NSC305787 was much more effective in competing away the binding of DDX3 to wild-type ezrin than that to the ezrin T567D phosphomimicking mutant. (A) The surface of an ELISA plate was coated with wild-type ezrin protein (300 ng/well). Binding of DDX3 to the immobilized ezrin protein was detected using anti-DDX3 antiserum, followed by a secondary antibody coupled to horseradish peroxidase and a chromogenic substrate. ELISA wells coated with BSA served as negative controls (far-left bar). Additional negative controls included wells coated with ezrin protein alone (second bar) and wells coated with BSA alone followed by incubation with DDX3 protein (third bar). For the positive control, ELISA wells were coated with DDX3 protein alone (far-right bar). The inhibitory activity of NSC305787 was tested using a range of concentrations from 0.012 μM to 12.0 μM. Inhibition of the ezrin-DDX3 interaction led to a reduction in the color signal. The graph below shows the IC50 curve. (B) ELISA performed as for panel A, except that the purified ezrin T567D phosphomimicking mutant was used in place of the wild-type protein.
Apart from DDX3, we performed co-IP experiments to verify the interaction of ezrin with caprin-1, MTDH, PABP1, and SND1 in cell lysates from K7M2 mouse OS cells. IP of endogenous ezrin with antiezrin antibody resulted in coprecipitation of caprin-1, MTDH, and PABP1 but not SND1, suggesting that ezrin interacts with the caprin-1, MTDH, and PABP1 proteins in OS cells (Fig. 4A). We were also able to demonstrate the direct binding of ezrin to MTDH by SPR (Fig. 4B).
FIG 4.
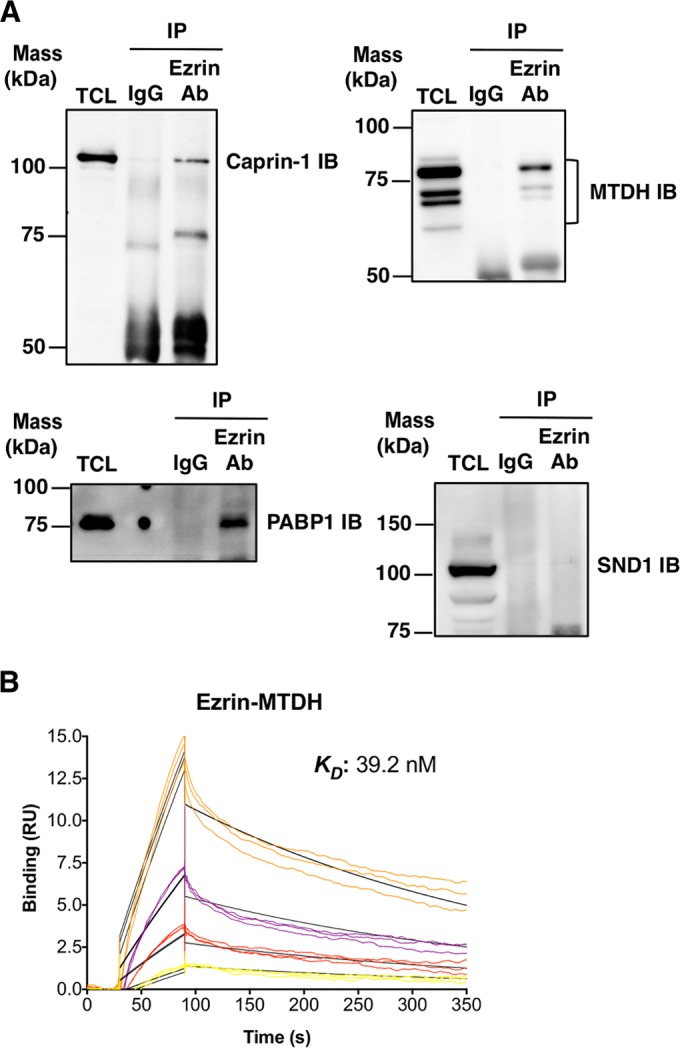
Ezrin interacts with caprin-1, MTDH, and PABP1. (A) The ability of ezrin to interact with caprin-1, MTDH, PABP1, and SND1 was tested by co-IP experiments in mouse K7M2 OS total cell lysates. Protein complexes were immunoprecipitated from the cell lysates with antiezrin antibody or negative-control total mouse IgG, followed by immunoblotting with anti-caprin-1, anti-MTHD, anti-PABP1, and anti-SND1 antibodies. (B) For SPR studies, recombinant human ezrin was immobilized on a CM5 chip in Biacore T-200. Recombinant human MTDH was injected over the chip surface at four different concentrations (0.63, 1.25, 2.50, and 5.0 nM) in triplicate. The colored lines show real data, and the black lines represent curves fitted for a 1:1 binding model.
Ezrin regulates DDX3 protein levels in cancer cells.
We used RNA interference (RNAi) to silence ezrin expression in OS cells. In MG63.3 cells transfected with siRNA targeting ezrin, DDX3 protein levels were also reduced significantly (Fig. 5A, left). This unexpected drop in DDX3 protein levels in response to ezrin siRNA was validated in human hepatocellular (HepG2) and lung (H1944) cancer cell lines (Fig. 5A, middle and right, respectively). To rule out the possibility that the observed changes in DDX3 protein levels were due to nonspecific off-target effects of ezrin siRNA, we checked protein expression in the K7M2 and K12 mouse OS cell lines. K7M2 was derived from K12 cells following multiple cycles of orthotopic implantation of K12 cells into mouse tibias and recovery of metastatic cells from the lung. Highly metastatic K7M2 clones express significantly higher levels of ezrin protein than K12 cells. The AS 1.46 line was generated through suppression of ezrin expression by stable transfection of an antisense oligonucleotide in K7M2 cells, which resulted in reduced metastatic potential (13, 57). DDX3 protein expression levels were found to be significantly lower in both K12 and AS 1.46 cells than in K7M2 cells (Fig. 5B). There was no change in DDX3 mRNA levels in MG63.3 cells following ezrin reduction compared to the control (Fig. 5C). Ectopic overexpression of ezrin in K12 cells also resulted in higher expression of the DDX3 protein than in the cells transfected with the empty vector (Fig. 5D).
FIG 5.

Ezrin regulates the DDX3 protein level. (A) Expression of ezrin protein was inhibited by siRNA in human OS (MG63.3), hepatocellular cancer (HepG2), and lung cancer (H1944) cell lines, which resulted in reduced DDX3 expression. Protein levels were detected by Western blotting of total cell lysates. (B) K12 is a mouse OS cell line, and K7M2 is a subclone of K12 with high ezrin expression and high metastatic potential. Ezrin expression in K7M2 was inhibited by stable antisense oligonucleotides (labeled as AS1.46 cells). The expression levels of ezrin, DDX3, and actin proteins were determined by Western blotting. (C) Ezrin and DDX3 mRNA levels were measured by real-time PCR in MG63.3 cells from panel A, and the results were normalized to 18S rRNA levels and expressed as fold change over the control group (*, P < 0.0001; Student's t test). The values are presented as the means and standard deviations of the results of triplicate experiments. (D) Low-ezrin-expressing K12 mouse OS cells were transfected with either an empty vector (EV) or a cDNA coding for wild-type ezrin containing a Myc-His tag. The protein levels of ezrin, DDX3, and actin were detected by Western blotting after 48 h. (E) MG63.3 cells were transfected with a cDNA coding for DDX3 containing a FLAG tag or a negative-control EV. Ezrin expression was inhibited by siRNA, and the level of expression of DDX3 from the vector was determined by Western blotting using anti-FLAG antibody. (F) K7M2 cells were treated with 2.0 μM NSC305787 or vehicle control for 4 days, and the expression levels of ezrin, DDX3, and actin were determined by Western blotting. (G) The expression levels of DDX3 in MG63.3 cells were determined at 0 h, 4 h, 8 h, 18 h, 24 h, 48 h, and 72 h after transfection of cells with siRNA for ezrin. (H) MG63.3 cells were transfected with either an siRNA targeting endogenous DDX3 (left) or a cDNA coding for DDX3 containing a FLAG tag (right). The expression level of ezrin was determined by Western blotting using antiezrin antibody. The asterisk indicates the Flag-tagged exogenous DDX3.
We next investigated whether inhibition of endogenous ezrin expression could lead to a reduction in DDX3 protein levels expressed ectopically. MG63.3 cells were transfected with a mammalian expression vector containing cDNA for human DDX3 with a FLAG tag. Similar to the previous experiments, siRNA targeting of ezrin reduced expression levels of the FLAG-tagged DDX3 protein (Fig. 5E). Each of these results supported the conclusion that the alterations in the DDX3 protein level in response to ezrin depletion or overexpression were due to the changes either in the half-life or in the translational rate of the DDX3 protein; the data did not support alterations in transcription. In order to address the question of whether pharmacological inhibition of ezrin binding could lead to a reduction in DDX3 protein levels, we treated K7M2 cells with low concentrations of NSC305787 for 4 days. This long-term exposure of the cells caused a significant decrease in DDX3 protein levels compared to the vehicle-treated cells (Fig. 5F). We performed a time course analysis of RNAi-mediated ezrin knockdown to identify how soon the decrease in DDX3 protein expression becomes evident (Fig. 5G). The analysis of ezrin and DDX3 expression in MG63.3 OS cells at various time points after transfection with siRNA for ezrin demonstrated that the level of DDX3 started to decline after 24 h, with the most significant decrease seen at 72 h. The reduction in DDX3 protein levels was correlated with the ezrin protein levels in the cells. To further test whether a similar inhibitory effect on ezrin protein expression could be detected by modulating DDX3 levels, we both transiently reduced and ectopically overexpressed DDX3 in MG63.3 cells (Fig. 5H). Neither approach altered ezrin protein levels.
Ezrin regulates translation of mRNAs containing a structured 5′ untranslated region through DDX3.
DDX3 regulates protein translation from mRNAs containing a structured 5′ UTR (27, 31–33). A specific complex 5′ UTR contains a 24-bp stem-loop structure that is only 2% efficient in translation of upstream protein-coding genes compared to a non-stem-loop-structured mRNA (43). We used this 24-bp stem-loop in cis with a firefly luciferase expression reporter to test if ezrin-altered translation is based upon secondary mRNA structure (Fig. 6A).
FIG 6.

Ezrin modulates translation of structured mRNAs through DDX3 in transfected cells, and DDX3 preferentially inhibits translation of 5′ UTR structured mRNA in vitro. (A) MG63.3 human OS cells were transfected with constructs designed to represent a nonstructured (pCMV-LUC) or a weakly translated gene with a structured (pCMV-SL-LUC) 5′ untranslated region adjacent to the reporter fLUC gene. Cells were also transfected with anti-DDX3 or antiezrin siRNAs. (B) Fold changes in normalized firefly luciferase activities from both structured and nonstructured luciferase mRNAs in ezrin- and DDX3-depleted cells with respect to mock-transfected cells. The firefly luciferase data were normalized to total protein (*, P < 0.0001; Student's t test). The values are presented as the means and standard deviations of the results of triplicate experiments. The graph shows a representative of three independent experiments. (C) Western blots showing ezrin and DDX3 protein levels from panel B. (D) Rabbit reticulocyte lysates containing pCMV-LUC or pCMV-SL-LUC reporter constructs carrying luciferase mRNAs with a nonstructured or stem-loop-structured 5′ UTR, respectively, were incubated with increasing amounts of DDX3 (1 and 2 μg, corresponding to 0.27 and 0.53 μM, respectively) or ezrin (1 and 2 μg, corresponding to 0.25 and 0.50 μM, respectively) under conditions for coupled transcription-translation. The translation was performed in a 50-μl reaction volume at 30°C for 90 min, as described in Materials and Methods. The amount of functionally active luciferase protein synthesized from a non-stem-loop- or stem-loop-structured mRNA was measured in a luminometer. The values are presented as the means ± standard deviations of triplicate determinations. (E) Reactions were set up as for panel D, except the effect of ezrin on luciferase expression was tested in the presence of DDX3. The graph on the left shows the translation reactions for pCMV-LUC and pCMV-SL-LUC performed in the presence of 1.0 μg ezrin alone (two left bars), 1.0 μg DDX3 alone (two middle bars), and both 1.0 μg ezrin and 1.0 μg DDX3 (two right bars). The graph on the right shows translation reactions for pCMV-LUC and pCMV-SL-LUC performed in the presence of 10.0 μg ezrin alone (two left bars), 1.0 μg DDX3 alone (two middle bars), and both 10.0 μg ezrin and 1.0 μg DDX3 (two right bars). (F) In vitro-coupled transcription-translation reactions were set up as for panel D with either recombinant DDX3 (1 μg) or ezrin (1 μg). After a 90-min incubation period at 30°C, total RNA was isolated as described in Materials and Methods. Luciferase mRNA levels were measured by real-time PCR, and the results were normalized to the 18S rRNA levels. The values are presented as the means and standard deviations of triplicate determinations. (G) Total RNA was isolated as described for panel E from in vitro-coupled transcription-translation reactions with rabbit reticulocyte lysates containing pCMV-LUC or pCMV-SL-LUC reporter constructs in the absence of any recombinant protein. Luciferase mRNA levels were measured by real-time PCR, and the results were normalized to 18S rRNA levels. The values are presented as the means and standard deviations of triplicate determinations.
The 24-bp stem-loop dramatically inhibited protein synthesis from the cis-luciferase gene in our transfected cells, which was only 2% of the luciferase activity from a nonstructured mRNA. The same structure showed a 2.5-fold increase in luciferase activity following reduction of DDX3 protein expression in MG63.3 cells (Fig. 6B). However, we observed no difference in luciferase activity from a nonmodified mRNA between DDX3-depleted and control cells. When the same experiments were performed with siRNA targeting ezrin, similar correlations in both stem-loop and nonstructured luciferase activities were seen in MG63.3 cells (Fig. 6B). Since siRNA-mediated knockdown of ezrin expression resulted in reduced protein levels of DDX3 (Fig. 6C), these results suggested that the effect of ezrin depletion on luciferase reporter activities was most likely mediated through the loss of DDX3 protein.
We used an in vitro translation assay to examine the effect of DDX3 on translation of mRNA with a stem-loop structure in the 5′ UTR and whether ezrin has any role in this process. The effects of ezrin and DDX3 on translation were measured by adding purified recombinant proteins to a rabbit reticulocyte lysate containing a pcDNA-LUC or a pcDNA-SL-LUC template under conditions for coupled transcription-translation. As expected, translation of luciferase mRNA with a stable secondary structure in the 5′ UTR was approximately 25-fold less efficient than that of the nonstructured luciferase mRNA in the absence of any added recombinant protein. Addition of 1 μg (0.27 μM) and 2 μg (0.53 μM) purified DDX3 to the reticulocyte lysate inhibited luciferase activity from both stem-loop and non-stem-loop luciferases in a concentration-dependent manner, whereas addition of ezrin did not produce any inhibitory effect on either luciferase activity (Fig. 6D). DDX3 inhibited the translation of stem-loop-structured luciferase more effectively than that of the non-stem-loop luciferase. Addition of 1 μg DDX3 to the reticulocyte lysate inhibited stem-loop-structured luciferase expression by 64%. In contrast, when the same amount of DDX3 was added to the reaction mixture, inhibition of non-stem-loop luciferase expression was only 30% (Fig. 6D). We then tested the effect of ezrin on luciferase expression in the presence of DDX3 (Fig. 6E). However, we observed no change in the amount of inhibition of luciferase expression caused by DDX3 when we added 1.0 μg and 10.0 μg of purified ezrin to the reticulocyte lysates containing 1.0 μg of DDX3, which corresponds to 1:1 and 10:1 molar excess ratios. Since the experiments were performed under conditions for coupled transcription-translation, we verified that the DDX3-dependent decrease in protein expression was not due to decreased levels of luciferase mRNA (Fig. 6F). We also did not observe significant differences in luciferase mRNA levels in nonmodified or structured 5′ UTRs (Fig. 6G). Thus, all the above-mentioned findings supported the idea that a secondary structure in the 5′ UTR of the mRNA enhances its susceptibility to inhibition of translation by DDX3.
Ezrin inhibits helicase activity but stimulates ATPase activity of DDX3.
We investigated whether ezrin affects the RNA helicase or ATPase function of DDX3, which might also have a role in translation regulation by ezrin. We evaluated both wild-type ezrin and phosphomimicking mutant ezrin (ezrin T567D) to explore the possible influence of ezrin Thr-567 phosphorylation on the modulation of DDX3 RNA helicase or ATPase activity. A fluorescence resonance energy transfer (FRET)-based reporter assay that permits monitoring of the enzyme activity in real time was employed to study the helicase activity of recombinant DDX3 protein. The assay utilizes a dsRNA substrate with a 3′ overhang consisting of a fluorophore-labeled reporter strand and a complementary strand modified with a spectrally paired quencher dye. When the dsRNA substrate is unwound by the helicase, the dye emits fluorescence upon its release from the quencher (58).
Both purified recombinant wild-type ezrin and the ezrin T567D mutant inhibited DDX3 helicase activity in a concentration-dependent manner (Fig. 7A and B). However, the amount of inhibition was significantly reduced by the phosphomimic. The concentrations of wild-type ezrin and ezrin T567D required to elicit a 50% inhibition in the fluorescence signal (IC50) were determined to be 44.0 ± 7.1 nM and 117.1 ± 9.0 nM, respectively (Fig. 7A and B, right). This corresponds to almost 11- and 29-fold molar excesses of wild-type ezrin and ezrin T567D over DDX3, respectively. A negative-control protein, myoglobin, was added to reaction mixtures at a high molar ratio, yet there was no change in the DDX3 enzyme activity (Fig. 7C). Neither purified wild-type ezrin nor myoglobin alone had any effect on fluorescence (Fig. 7C). Control incubation mixtures in which either Mg2+ or ATP was omitted or heat-inactivated DDX3 was used did not produce any detectable single-stranded RNA (ssRNA) (Fig. 7D).
FIG 7.

Wild-type ezrin inhibits RNA helicase activity of DDX3 more effectively than the phosphomimicking ezrin T567D mutant in a concentration-dependent manner. (A) The effect of ezrin on DDX3 helicase activity was tested by a FRET-based reporter assay in real time. Double-stranded RNA with a 3′ overhang consisting of a 5′ fluorophore-labeled reporter strand and a 3′ quencher-labeled strand was used as the substrate. Reaction mixtures containing 4 nM recombinant DDX3 and 20 nM dsRNA with a 3′ overhang were incubated with increasing amounts of recombinant wild-type ezrin (4 to 160 nM, corresponding to 1- to 40-fold molar excesses of ezrin over DDX3). The measured fluorescence intensity indicates the amount of single-stranded RNA product formation. (Right) Nonlinear curve fitting of the concentration-response data to calculate the IC50 of wild-type ezrin on helicase activity. (B) The RNA duplex-unwinding activity of DDX3 was measured under the same conditions as described above, except instead of wild-type ezrin, the phosphomimicking ezrin T567D mutant (4 to 400 nM, corresponding to 1- to 100-fold molar excesses of ezrin over DDX3) was used in the reaction mixtures. Both graphs are representative of the results of three independent experiments. The calculated IC50 data for both wild-type ezrin and the phosphomimicking ezrin mutant represent the means ± standard deviations from three independent experiments. (C) Ezrin at 40-fold molar excess concentration over DDX3 caused almost complete inhibition of DDX3 helicase activity, whereas myoglobin as a negative-control protein at 80-fold molar excess did not inhibit the helicase activity. Ezrin or myoglobin itself did not produce any change in fluorescence readings over the time course of the experiment. (D) When ATP and Mg2+ ions were removed from the reaction mixture, no helicase activity was observed. Heat-inactivated DDX3 was also unable to catalyze the unwinding of dsRNA. The graphs show representatives of three independent experiments.
We determined the ATPase activity of DDX3 in the presence of dsRNA with a 3′ overhang and increasing amounts of recombinant ezrin. The sequence of the dsRNA was the same as that of the RNA used in the DDX3 RNA helicase assay. We found that wild-type ezrin actually stimulated ATPase in a concentration-dependent manner. Ezrin T567D was significantly less effective in enhancing the DDX3 ATPase activity than wild-type ezrin (Fig. 8A). Addition of wild-type ezrin to the reaction mixture at a concentration of 4 μM, corresponding to 20-fold molar excess over DDX3, stimulated the ATPase activity by approximately 5-fold, with further increases in ezrin leading to saturation. In contrast, the ezrin T567D mutant at 8 μM concentration (40-fold molar excess) minimally enhanced the DDX3 ATPase (1.7-fold). Myoglobin added to the reaction mixture in 30- and 40-fold excess over the amount of DDX3 did not alter the rate of ATP hydrolysis (Fig. 8B). Although low-level basal ATPase activity was reported for DDX3 in the absence of RNA (59), we were unable to detect it under our assay conditions without any RNA substrate or in the presence of only a complementary longer single-stranded template of dsRNA (Fig. 8C). We also did not observe any increase in ATPase activity when DDX3 was incubated with wild-type ezrin at 6 μM concentration (30-fold molar excess over DDX3) without any RNA or in the presence of ssRNA (Fig. 8C).
FIG 8.

Wild-type ezrin stimulates ATPase activity of DDX3 more effectively than the phosphomimicking ezrin T567D mutant in a concentration-dependent manner. (A) The effect of ezrin on DDX3 ATPase activity was determined using a fixed-type assay by measuring the amount of free phosphate generated during the hydrolysis of ATP, as described in Materials and Methods. Reaction mixtures containing 200 nM DDX3 and 400 nM dsRNA with a 3′ overhang were incubated with increasing amounts of either recombinant wild-type ezrin or the phosphomimicking ezrin T567D mutant (1 to 8 μM, corresponding to 5- to 40-fold molar excesses of ezrin over DDX3). The graphs are representative of the results of three independent experiments. (B) When myoglobin was added to the reaction mixtures as a negative-control protein at increasing concentrations corresponding to 30- and 40-fold molar excesses over the amount of DDX3, no apparent change in enzyme activity was observed, whereas ezrin at the same concentrations significantly stimulated DDX3 ATPase activity. The results are expressed as means and standard deviations of duplicate determinations. (C) Reaction mixtures containing 200 nM DDX3 were incubated with 6 μM recombinant wild-type ezrin (corresponding to a 30-fold molar excess of ezrin over DDX3) without any RNA substrate or in the presence of 400 nM either dsRNA with a 3′ overhang or its complementary, longer ssRNA. The sequence of the dsRNA is given in Materials and Methods.
DISCUSSION
Ezrin lacks intrinsic enzyme activity and shows its pleiotropic functions by interacting with different protein binding partners that are involved in diverse cellular processes. We hypothesized that the antimetastatic effect of NSC305787 occurs putatively by preventing specific ezrin protein-protein interactions. We identified a subset of proteins in our ezrin interactome list, including DDX3, PABP1, caprin-1, SND1, several heat shock/stress proteins, eIF3 subunits, hnRNP Q, FXR1, hnRNP K, MTDH, argonaute 2/eIF2C2, FMRP, and RBM42, that are known to be components and/or modulators of protein translation initiation and SG assembly (26, 60–71). SGs are dynamic, dense aggregates of proteins, including translation initiation factors, and RNAs that allow the cell to survive under stress conditions by serving as a reservoir for nontranslated mRNAs (60). DDX3 has been characterized as a pivotal SG-nucleating protein conferring a cell survival advantage under stress conditions (26). As RNA-binding proteins, both caprin-1 and MTDH have been shown to regulate the translation of multiple mRNAs and the formation of SGs in response to stress (62, 63). Our previous findings based on polysome profile analysis suggested that ezrin associates with ribonucleoprotein complexes and has a role in the early phases of translation initiation rather than active and ongoing translation. Ezrin was detected in the same polysomal fractions containing the RNA-binding proteins YB-1 and PABP1, as well as the cap binding proteins eIF4E and RACK1 (49). We have validated through co-IP experiments that ezrin interacts with PABP1 (Fig. 4A), which is a core component of SGs and an SG marker protein. PABP1 stimulates translation initiation by forming a complex with the poly(A) tail of mRNAs and interacting with eIF4G, a large scaffolding protein that provides a platform for binding of several initiation factors, which gives rise to a “closed-loop” model for translation initiation. In light of the above-mentioned evidence, we propose a model demonstrating that ezrin is a component of the messenger ribonucleoprotein (mRNP) complex associated with translation initiation machinery and stress granule aggregates, which includes DDX3, PABP1, and other RNA-binding proteins, such as caprin-1 and metadherin (Fig. 9). This model suggests a role for ezrin in regulation of the mRNP homeostasis required for transitions between translationally active and inactive states under normal or oncogenic/stress conditions. Ezrin might perform this task by modulating protein-protein and protein-RNA interactions and/or regulating enzymatic functions of the helicase components of the mRNP complex required for its remodeling. In support of our model, recently, two different studies analyzing the mRNA interactome using quantitative proteomics have provided evidence that ezrin is an mRNA binding protein (72, 73). The present findings suggest that ezrin allows tumor cells to endure specific stress conditions encountered during metastatic progression via integrating translational regulation with the cellular stress response and regulating SG dynamics. Our findings also suggest that NSC305787 interferes with the ability of ezrin to perform its delicate task of coordinating or orchestrating those biological processes by inhibiting protein-protein interactions.
FIG 9.

Working model for ezrin and DDX3 functions. The model demonstrates a novel role for ezrin in regulating transcription and translation independently of its membrane-localized open conformation. We propose that ezrin and DDX3 are the components of an mRNP complex associated with translation initiation machinery and stress granule aggregates. In this model, ezrin functions as a regulator of mRNP homeostasis required for transitions between translationally active (translation initiation complex) and inactive (stress granule) states under normal or oncogenic/stress conditions.
Several lines of evidence have demonstrated reciprocal regulation between ERM proteins and Rho GTPases in cytoskeletal modeling, cell polarity, and migration. Although, little is known about signaling cascades coupling the Rho family of GTPases with ERM proteins, different studies suggest that members of the Rho GTPases, including Rac1, might either stimulate or inactivate ERM proteins (56, 74, 75). Recently, Chen et al. (33) identified several factors in the Rac signaling pathway, including Rac1, as potential translation targets of DDX3. They have shown that DDX3 modulates cell migration and invasion via its activity in promoting Rac1 mRNA translation, possibly by interacting with the 5′ UTR, which is required for Wnt–β-catenin signaling. Although in our experiments DDX3 preferentially inhibited the translation of mRNAs with a complex 5′ UTR, these findings nevertheless suggest that the cross talk between ezrin-, DDX3-, and Rac1-mediated signaling pathways could cooperatively regulate the metastatic phenotype of cancer cells. These findings also highlight the importance of identification of the endogenous mRNAs associated with metastatic progression, the translation of which is regulated by ezrin-DDX3 interaction.
It has been demonstrated that caprin-1 promotes OS metastasis by conferring resistance to apoptosis and via the activation of Akt and extracellular signal-regulated kinase 1/2 (ERK1/2) signaling pathways (76). MTDH has also recently been shown to mediate OS metastasis by upregulating MMP-2 via endothelin-1/endothelin A receptor and Jun N-terminal protein kinase (JNK)/c-Jun/MMP-2 signaling pathways (77–79), as well as by regulating epithelial-mesenchymal transition (80). Consequently, these novel interactions between ezrin, caprin-1, and MTDH might also have a substantial effect on determining the ability of cells to acquire metastatic capability. MTDH also interacts with SND1, and this interaction has been shown to be critical for cell survival under oncogenic/stress conditions (81, 82). Although, we demonstrated direct interaction between ezrin and MTDH through co-IP and SPR experiments, we were unable to detect the SND1 protein in the same coprecipitates with ezrin (Fig. 4A).
Recently, Viswanatha et al. (83) identified 38 ezrin-binding proteins by expressing various mutant forms of ezrin corresponding to its closed, open, and hyperactivated states in Jeg-3 epithelial cells and performing affinity pulldown followed by MS. This study has revealed that the ezrin interactome might change depending on the conformational state of ezrin, and an unexpectedly significant fraction of all ezrin-binding proteins bind preferentially to ezrin in its closed form. In the current understanding, the closed conformation of ezrin is considered the inactive form, which exists in the cytoplasm mainly as dormant monomers.
Our findings suggested that ezrin preferentially modulates translation of a specific subset of mRNAs with a complex 5′ UTR by posttranscriptionally upregulating the intracellular protein level of DDX3. Ezrin likely sustains the intracellular protein level of DDX3 either by stabilizing it or by stimulating its translational expression. Taking into account that long-term exposure of OS cells to NSC305787 resulted in lower DDX3 protein levels (Fig. 5F), it might be ezrin binding that is critical to stabilize DDX3. In addition, the results shown in Fig. 5E suggest that the ezrin-mediated effects on the DDX3 protein level are not exerted at the stage of translation initiation, as the cDNA encoding the full-length human DDX3 did not contain the 5′ UTR. We also determined whether ezrin has any role in maintaining the intracellular protein levels of caprin-1 and MTDH, similar to DDX3. Contrary to what we observed for DDX3 in the K12, K7M2, and K7M2-ezrin antisense AS 1.46 cell lines (Fig. 5B), the protein levels of caprin-1 and MTDH were not associated with changes in the expression of ezrin (data not shown).
Although, the role of DDX3 in translation initiation is ambiguous, several studies have indicated that DDX3 acts as a translational repressor rather than an activator (25, 26, 28). Our results were consistent with those findings in that in our experimental systems, DDX3 had an inhibitory effect on translation. In addition, here, we provide biochemical evidence that ezrin-mediated modulation of DDX3 enzyme activity seems not to be involved in repression of translation by DDX3, at least in isolated systems using purified proteins (Fig. 6E). However, we cannot rule out the possibility that ezrin regulates mRNA homeostasis by modulating enzymatic activities of DDX3 in cells under normal and oncogenic/stress conditions, and therefore, further detailed experimentation is needed in order to clarify the biological role of ezrin-DDX3 interaction in translation initiation. It should be noted that it is still unclear how DDX3 regulates translation—as an enzyme possessing both helicase and ATPase functions or as an interacting partner that binds other components of the translation initiation machinery. A study on the role of the Saccharomyces cerevisiae DDX3 homolog, Ded1, has suggested that it can act both as a repressor of translation initiation through its ability to interact with other components of the translation initiation factors and as an activator via its ATP-dependent activity (28). The situation is even further complicated given that the eukaryotic translation initiation complex contains other proteins with helicase and ATPase functions, such as eIF4A (DDX2A/B), which is also a DEAD-box RNA helicase and unwinds RNA secondary structures in the 5′ UTR of mRNAs. Even though it is well known that formation of an eIF4G, eIF4E, eIF4B, and eIF4H multiprotein complex greatly stimulates the ATPase and helicase activities of free eIF4A (84), how other accessory factors present in the translation initiation complex, such as DDX3 and possibly ezrin, modulate the biochemical functions of eIF4A remains elusive.
Although, we demonstrated that wild-type (closed-conformation) and constitutively active mutant (open-conformation) ezrin proteins bind to DDX3 with comparable affinities (Fig. 2D), wild-type ezrin modulates the enzymatic activity of DDX3 better than the mutant (Fig. 7 and 8). Recombinant wild-type and mutant ezrin proteins were expressed in E. coli. The biochemical purity of the wild-type and mutant ezrin proteins was established (Fig. 2C); however, the conformational purity of the proteins (i.e., what percentage of the total protein is in open versus closed form) is unknown. Binding of wild-type and mutant ezrin proteins to DDX3 with comparable affinities may lead to different conformational changes in DDX3 and its enzymatic functions. Therefore, similar binding affinities might not reflect similar modulations of DDX3 enzyme activity. In addition, although in our previous study, we demonstrated that NSC305787 inhibits ezrin T567D phosphorylation, we believe that it is not the sole mechanism involved and that inhibition of ezrin function is still possible without inhibiting its phosphorylation. Computational prediction of the potential binding sites of NSC305787 was based on the only available X-ray crystal structure of the N-terminal FERM domain of ezrin (46). The analysis produced several potential binding sites that are not at C-terminal sites, including the T567 residue. Binding of NSC305787 might occur at several sites on ezrin, including C-terminal sites, which can lead to conformational changes resulting in modulation of the interaction of ezrin with other proteins without directly interfering with T567 phosphorylation.
Since we were unable to measure the ATPase activity of DDX3 without RNA or in the presence of ssRNA (Fig. 8C), the observed stimulation by ezrin may not be a direct effect on the ATPase function of DDX3 but rather an indirect effect of the inhibition of its RNA helicase activity. Ezrin might increase the nonproductive unwinding events by uncoupling the ATP hydrolysis cycle from the strand separation function of DDX3. Given that ATP hydrolysis is necessary for dissociation of DEAD-box proteins from the RNA and multiple substrate turnovers and thus for enzyme recycling (85), our observations support a model in which inhibition of the RNA helicase activity of DDX3 by ezrin might have resulted in stimulation of its ATPase activity in order to compensate for and minimize the nonproductive unwinding events at the expense of more ATP.
An understanding of the molecular events accompanying ezrin-DDX3 interaction will certainly have implications in cancer biology; however, more studies need to be undertaken to establish its biological significance. Studies concerning the role of DDX3 in cancer have shown ambiguous results, suggesting both oncogenic and tumor suppressor functions, depending on the cancer type (23, 34). This dual role of DDX3 could be related to its precise function in regulation of translation, which might be differentially modulated in different types of cells and under different conditions (e.g., stress conditions). To our knowledge, no study has yet demonstrated the role of DDX3 in OS. We hypothesize that direct interaction between ezrin and DDX3 has biological significance in the metastatic phenotype of cancer cells, including OS, which needs to be further clarified. In addition, although our preliminary data show that the antiezrin compound NSC305787 disrupts the interaction between ezrin and DDX3, it remains uncertain whether its antimetastatic effects are dependent on ezrin-DDX3 interaction.
In conclusion, in this study, we identified DDX3 as a novel binding partner of ezrin, which suggests a novel molecular function for ezrin in integrating environmental stress with translational regulation. This novel function of ezrin is distinct from its known functions at the plasma membrane as a cytoskeletal scaffolding protein. Our findings also suggest that the antiezrin compound NSC305787 exerts its antimetastatic effects, at least in part, by blocking specific protein-protein interactions involving ezrin that otherwise enhance the ability of tumor cells to endure specific stresses encountered during metastatic progression. Finally, the novel protein-protein interaction involving ezrin and DDX3 opens new windows of opportunity, which can link the helicase field with cancer researchers and collective knowledge about ezrin from cell biology to virology.
Supplementary Material
ACKNOWLEDGMENTS
We are grateful to Eckhard Jankowsky from Case Western Reserve University for critical discussions and helpful comments on the manuscript. We thank Hsin-Sheng Yang from the University of Kentucky for the pCMV-LUC and pCMV-SL-LUC plasmids and Chand Khanna from the National Cancer Institute, NIH, for the OS cell lines.
This work was supported by U.S. Department of Defense grant W81XWH-10-1-0137 (to A.Ü.). We also thank the Proteomics and Metabolomics and Biacore Molecular Interaction shared resources at the Lombardi Comprehensive Cancer Center (Georgetown University), which are supported by grant P30 CA51008 from the National Cancer Institute.
Georgetown University has filed a patent application for the use of NSC305787 and related compounds in cancer therapy in which J.A.T. and A.Ü. are listed as inventors.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/MCB.00332-15.
REFERENCES
- 1.Takeuchi K, Kawashima A, Nagafuchi A, Tsukita S. 1994. Structural diversity of band 4.1 superfamily members. J Cell Sci 107:1921–1928. [DOI] [PubMed] [Google Scholar]
- 2.Tsukita S, Yonemura S. 1997. ERM (ezrin/radixin/moesin) family: from cytoskeleton to signal transduction. Curr Opin Cell Biol 9:70–75. doi: 10.1016/S0955-0674(97)80154-8. [DOI] [PubMed] [Google Scholar]
- 3.Bretscher A, Edwards K, Fehon RG. 2002. ERM proteins and merlin: integrators at the cell cortex. Nat Rev Mol Cell Biol 3:586–599. doi: 10.1038/nrm882. [DOI] [PubMed] [Google Scholar]
- 4.Fehon RG, McClatchey AI, Bretscher A. 2010. Organizing the cell cortex: the role of ERM proteins. Nat Rev Mol Cell Biol 11:276–287. doi: 10.1038/nrm2866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Matsui T, Maeda M, Doi Y, Yonemura S, Amano M, Kaibuchi K, Tsukita S, Tsukita S. 1998. Rho-kinase phosphorylates COOH-terminal threonines of ezrin/radixin/moesin (ERM) proteins and regulates their head-to-tail association. J Cell Biol 140:647–657. doi: 10.1083/jcb.140.3.647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gautreau A, Louvard D, Arpin M. 2000. Morphogenic effects of ezrin require a phosphorylation-induced transition from oligomers to monomers at the plasma membrane. J Cell Biol 150:193–203. doi: 10.1083/jcb.150.1.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fievet BT, Gautreau A, Roy C, Del Maestro L, Mangeat P, Louvard D, Arpin M. 2004. Phosphoinositide binding and phosphorylation act sequentially in the activation mechanism of ezrin. J Cell Biol 164:653–659. doi: 10.1083/jcb.200307032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhu L, Zhou R, Mettler S, Wu T, Abbas A, Delaney J, Forte JG. 2007. High turnover of ezrin T567 phosphorylation: conformation, activity, and cellular function. Am J Physiol Cell Physiol 293:C874–C884. doi: 10.1152/ajpcell.00111.2007. [DOI] [PubMed] [Google Scholar]
- 9.Bosk S, Braunger JA, Gerke V, Steinem C. 2011. Activation of F-actin binding capacity of ezrin: synergism of PIP(2) interaction and phosphorylation. Biophys J 100:1708–1717. doi: 10.1016/j.bpj.2011.02.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Marina N, Gebhardt M, Teot L, Gorlick R. 2004. Biology and therapeutic advances for pediatric osteosarcoma. Oncologist 9:422–441. doi: 10.1634/theoncologist.9-4-422. [DOI] [PubMed] [Google Scholar]
- 11.Gordon N, Kleinerman ES. 2009. The role of Fas/FasL in the metastatic potential of osteosarcoma and targeting this pathway for the treatment of osteosarcoma lung metastases. Cancer Treat Res 152:497–508. doi: 10.1007/978-1-4419-0284-9_29. [DOI] [PubMed] [Google Scholar]
- 12.Khanna C, Khan J, Nguyen P, Prehn J, Caylor J, Yeung C, Trepel J, Meltzer P, Helman L. 2001. Metastasis-associated differences in gene expression in a murine model of osteosarcoma. Cancer Res 61:3750–3759. [PubMed] [Google Scholar]
- 13.Khanna C, Wan X, Bose S, Cassaday R, Olomu O, Mendoza A, Yeung C, Gorlick R, Hewitt SM, Helman LJ. 2004. The membrane-cytoskeleton linker ezrin is necessary for osteosarcoma metastasis. Nat Med 10:182–186. doi: 10.1038/nm982. [DOI] [PubMed] [Google Scholar]
- 14.Park HR, Jung WW, Bacchini P, Bertoni F, Kim YW, Park YK. 2006. Ezrin in osteosarcoma: comparison between conventional high-grade and central low-grade osteosarcoma. Pathol Res Pract 202:509–515. doi: 10.1016/j.prp.2006.01.015. [DOI] [PubMed] [Google Scholar]
- 15.Akisawa N, Nishimori I, Iwamura T, Onishi S, Hollingsworth MA. 1999. High levels of ezrin expressed by human pancreatic adenocarcinoma cell lines with high metastatic potential. Biochem Biophys Res Commun 258:395–400. doi: 10.1006/bbrc.1999.0653. [DOI] [PubMed] [Google Scholar]
- 16.Yu Y, Khan J, Khanna C, Helman L, Meltzer PS, Merlino G. 2004. Expression profiling identifies the cytoskeletal organizer ezrin and the developmental homeoprotein Six-1 as key metastatic regulators. Nat Med 10:175–181. doi: 10.1038/nm966. [DOI] [PubMed] [Google Scholar]
- 17.Tynninen O, Carpen O, Jaaskelainen J, Paavonen T, Paetau A. 2004. Ezrin expression in tissue microarray of primary and recurrent gliomas. Neuropathol Appl Neurobiol 30:472–477. doi: 10.1111/j.1365-2990.2004.00562.x. [DOI] [PubMed] [Google Scholar]
- 18.Weng WH, Ahlen J, Astrom K, Lui WO, Larsson C. 2005. Prognostic impact of immunohistochemical expression of ezrin in highly malignant soft tissue sarcomas. Clin Cancer Res 11:6198–6204. doi: 10.1158/1078-0432.CCR-05-0548. [DOI] [PubMed] [Google Scholar]
- 19.Hustinx SR, Fukushima N, Zahurak ML, Riall TS, Maitra A, Brosens L, Cameron JL, Yeo CJ, Offerhaus GJ, Hruban RH, Goggins M. 2005. Expression and prognostic significance of 14-3-3sigma and ERM family protein expression in periampullary neoplasms. Cancer Biol Ther 4:596–601. doi: 10.4161/cbt.4.5.1748. [DOI] [PubMed] [Google Scholar]
- 20.Kobel M, Gradhand E, Zeng K, Schmitt WD, Kriese K, Lantzsch T, Wolters M, Dittmer J, Strauss HG, Thomssen C, Hauptmann S. 2006. Ezrin promotes ovarian carcinoma cell invasion and its retained expression predicts poor prognosis in ovarian carcinoma. Int J Gynecol Pathol 25:121–130. doi: 10.1097/01.pgp.0000185410.39050.ac. [DOI] [PubMed] [Google Scholar]
- 21.Sarrio D, Rodriguez-Pinilla SM, Dotor A, Calero F, Hardisson D, Palacios J. 2006. Abnormal ezrin localization is associated with clinicopathological features in invasive breast carcinomas. Breast Cancer Res Treat 98:71–79. doi: 10.1007/s10549-005-9133-4. [DOI] [PubMed] [Google Scholar]
- 22.Linder P, Jankowsky E. 2011. From unwinding to clamping—the DEAD box RNA helicase family. Nat Rev Mol Cell Biol 12:505–516. doi: 10.1038/nrm3154. [DOI] [PubMed] [Google Scholar]
- 23.Schroder M. 2010. Human DEAD-box protein 3 has multiple functions in gene regulation and cell cycle control and is a prime target for viral manipulation. Biochem Pharmacol 79:297–306. doi: 10.1016/j.bcp.2009.08.032. [DOI] [PubMed] [Google Scholar]
- 24.Rocak S, Linder P. 2004. DEAD-box proteins: the driving forces behind RNA metabolism. Nat Rev Mol Cell Biol 5:232–241. doi: 10.1038/nrm1335. [DOI] [PubMed] [Google Scholar]
- 25.Shih JW, Tsai TY, Chao CH, Wu Lee YH. 2008. Candidate tumor suppressor DDX3 RNA helicase specifically represses cap-dependent translation by acting as an eIF4E inhibitory protein. Oncogene 27:700–714. doi: 10.1038/sj.onc.1210687. [DOI] [PubMed] [Google Scholar]
- 26.Shih JW, Wang WT, Tsai TY, Kuo CY, Li HK, Wu Lee YH. 2012. Critical roles of RNA helicase DDX3 and its interactions with eIF4E/PABP1 in stress granule assembly and stress response. Biochem J 441:119–129. doi: 10.1042/BJ20110739. [DOI] [PubMed] [Google Scholar]
- 27.Soto-Rifo R, Rubilar PS, Limousin T, de Breyne S, Decimo D, Ohlmann T. 2012. DEAD-box protein DDX3 associates with eIF4F to promote translation of selected mRNAs. EMBO J 31:3745–3756. doi: 10.1038/emboj.2012.220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hilliker A, Gao Z, Jankowsky E, Parker R. 2011. The DEAD-box protein Ded1 modulates translation by the formation and resolution of an eIF4F-mRNA complex. Mol Cell 43:962–972. doi: 10.1016/j.molcel.2011.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lee CS, Dias AP, Jedrychowski M, Patel AH, Hsu JL, Reed R. 2008. Human DDX3 functions in translation and interacts with the translation initiation factor eIF3. Nucleic Acids Res 36:4708–4718. doi: 10.1093/nar/gkn454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tarn WY, Chang TH. 2009. The current understanding of Ded1p/DDX3 homologs from yeast to human. RNA Biol 6:17–20. doi: 10.4161/rna.6.1.7440. [DOI] [PubMed] [Google Scholar]
- 31.Lai MC, Lee YH, Tarn WY. 2008. The DEAD-box RNA helicase DDX3 associates with export messenger ribonucleoproteins as well as tip-associated protein and participates in translational control. Mol Biol Cell 19:3847–3858. doi: 10.1091/mbc.E07-12-1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lai MC, Chang WC, Shieh SY, Tarn WY. 2010. DDX3 regulates cell growth through translational control of cyclin E1. Mol Cell Biol 30:5444–5453. doi: 10.1128/MCB.00560-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen HH, Yu HI, Cho WC, Tarn WY. 21 July 2014. DDX3 modulates cell adhesion and motility and cancer cell metastasis via Rac1-mediated signaling pathway. Oncogene doi: 10.1038/onc.2014.190. [DOI] [PubMed] [Google Scholar]
- 34.Fuller-Pace FV. 2013. DEAD box RNA helicase functions in cancer. RNA Biol 10:121–132. doi: 10.4161/rna.23312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Botlagunta M, Vesuna F, Mironchik Y, Raman A, Lisok A, Winnard P Jr, Mukadam S, Van Diest P, Chen JH, Farabaugh P, Patel AH, Raman V. 2008. Oncogenic role of DDX3 in breast cancer biogenesis. Oncogene 27:3912–3922. doi: 10.1038/onc.2008.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sun M, Song L, Zhou T, Gillespie GY, Jope RS. 2011. The role of DDX3 in regulating Snail. Biochim Biophys Acta 1813:438–447. doi: 10.1016/j.bbamcr.2011.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Huang JS, Chao CC, Su TL, Yeh SH, Chen DS, Chen CT, Chen PJ, Jou YS. 2004. Diverse cellular transformation capability of overexpressed genes in human hepatocellular carcinoma. Biochem Biophys Res Commun 315:950–958. doi: 10.1016/j.bbrc.2004.01.151. [DOI] [PubMed] [Google Scholar]
- 38.Bol GM, Raman V, van der Groep P, Vermeulen JF, Patel AH, van der Wall E, van Diest PJ. 2013. Expression of the RNA helicase DDX3 and the hypoxia response in breast cancer. PLoS One 8:e63548. doi: 10.1371/journal.pone.0063548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chang PC, Chi CW, Chau GY, Li FY, Tsai YH, Wu JC, Wu Lee YH. 2006. DDX3, a DEAD box RNA helicase, is deregulated in hepatitis virus-associated hepatocellular carcinoma and is involved in cell growth control. Oncogene 25:1991–2003. doi: 10.1038/sj.onc.1209239. [DOI] [PubMed] [Google Scholar]
- 40.Chao CH, Chen CM, Cheng PL, Shih JW, Tsou AP, Lee YH. 2006. DDX3, a DEAD box RNA helicase with tumor growth-suppressive property and transcriptional regulation activity of the p21waf1/cip1 promoter, is a candidate tumor suppressor. Cancer Res 66:6579–6588. doi: 10.1158/0008-5472.CAN-05-2415. [DOI] [PubMed] [Google Scholar]
- 41.Wu DW, Liu WS, Wang J, Chen CY, Cheng YW, Lee H. 2011. Reduced p21(WAF1/CIP1) via alteration of p53-DDX3 pathway is associated with poor relapse-free survival in early-stage human papillomavirus-associated lung cancer. Clin Cancer Res 17:1895–1905. doi: 10.1158/1078-0432.CCR-10-2316. [DOI] [PubMed] [Google Scholar]
- 42.Wu DW, Lee MC, Wang J, Chen CY, Cheng YW, Lee H. 2014. DDX3 loss by p53 inactivation promotes tumor malignancy via the MDM2/Slug/E-cadherin pathway and poor patient outcome in non-small-cell lung cancer. Oncogene 33:1515–1526. doi: 10.1038/onc.2013.107. [DOI] [PubMed] [Google Scholar]
- 43.Yang HS, Cho MH, Zakowicz H, Hegamyer G, Sonenberg N, Colburn NH. 2004. A novel function of the MA-3 domains in transformation and translation suppressor Pdcd4 is essential for its binding to eukaryotic translation initiation factor 4A. Mol Cell Biol 24:3894–3906. doi: 10.1128/MCB.24.9.3894-3906.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Toretsky JA, Erkizan V, Levenson A, Abaan OD, Parvin JD, Cripe TP, Rice AM, Lee SB, Uren A. 2006. Oncoprotein EWS-FLI1 activity is enhanced by RNA helicase A. Cancer Res 66:5574–5581. doi: 10.1158/0008-5472.CAN-05-3293. [DOI] [PubMed] [Google Scholar]
- 45.Beauchamp E, Bulut G, Abaan O, Chen K, Merchant A, Matsui W, Endo Y, Rubin JS, Toretsky J, Uren A. 2009. GLI1 is a direct transcriptional target of EWS-FLI1 oncoprotein. J Biol Chem 284:9074–9082. doi: 10.1074/jbc.M806233200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bulut G, Hong SH, Chen K, Beauchamp EM, Rahim S, Kosturko GW, Glasgow E, Dakshanamurthy S, Lee HS, Daar I, Toretsky JA, Khanna C, Uren A. 2012. Small molecule inhibitors of ezrin inhibit the invasive phenotype of osteosarcoma cells. Oncogene 31:269–281. doi: 10.1038/onc.2011.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bhartur SG, Goldenring JR. 1998. Mapping of ezrin dimerization using yeast two-hybrid screening. Biochem Biophys Res Commun 243:874–877. doi: 10.1006/bbrc.1998.8196. [DOI] [PubMed] [Google Scholar]
- 48.Gary R, Bretscher A. 1993. Heterotypic and homotypic associations between ezrin and moesin, two putative membrane-cytoskeletal linking proteins. Proc Natl Acad Sci U S A 90:10846–10850. doi: 10.1073/pnas.90.22.10846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Briggs JW, Ren L, Nguyen R, Chakrabarti K, Cassavaugh J, Rahim S, Bulut G, Zhou M, Veenstra TD, Chen Q, Wei JS, Khan J, Uren A, Khanna C. 2012. The ezrin metastatic phenotype is associated with the initiation of protein translation. Neoplasia 14:297–310. doi: 10.1593/neo.11518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yao X, Thibodeau A, Forte JG. 1993. Ezrin-calpain I interactions in gastric parietal cells. Am J Physiol 265:C36–C46. [DOI] [PubMed] [Google Scholar]
- 51.Wang H, Guo Z, Wu F, Long F, Cao X, Liu B, Zhu Z, Yao X. 2005. PKA-mediated protein phosphorylation protects ezrin from calpain I cleavage. Biochem Biophys Res Commun 333:496–501. doi: 10.1016/j.bbrc.2005.05.143. [DOI] [PubMed] [Google Scholar]
- 52.McRobert EA, Young AN, Bach LA. 2012. Advanced glycation end-products induce calpain-mediated degradation of ezrin. FEBS J 279:3240–3250. doi: 10.1111/j.1742-4658.2012.08710.x. [DOI] [PubMed] [Google Scholar]
- 53.Wald FA, Oriolo AS, Mashukova A, Fregien NL, Langshaw AH, Salas PJ. 2008. Atypical protein kinase C (iota) activates ezrin in the apical domain of intestinal epithelial cells. J Cell Sci 121:644–654. doi: 10.1242/jcs.016246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Poullet P, Gautreau A, Kadare G, Girault JA, Louvard D, Arpin M. 2001. Ezrin interacts with focal adhesion kinase and induces its activation independently of cell-matrix adhesion. J Biol Chem 276:37686–37691. doi: 10.1074/jbc.M106175200. [DOI] [PubMed] [Google Scholar]
- 55.Reczek D, Berryman M, Bretscher A. 1997. Identification of EBP50: a PDZ-containing phosphoprotein that associates with members of the ezrin-radixin-moesin family. J Cell Biol 139:169–179. doi: 10.1083/jcb.139.1.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Epting D, Slanchev K, Boehlke C, Hoff S, Loges NT, Yasunaga T, Indorf L, Nestel S, Lienkamp SS, Omran H, Kuehn EW, Ronneberger O, Walz G, Kramer-Zucker A. 2015. The Rac1 regulator ELMO controls basal body migration and docking in multiciliated cells through interaction with ezrin. Development 142:174–184. doi: 10.1242/dev.112250. [DOI] [PubMed] [Google Scholar]
- 57.Mendoza A, Hong SH, Osborne T, Khan MA, Campbell K, Briggs J, Eleswarapu A, Buquo L, Ren L, Hewitt SM, Dakir el H, Garfield S, Walker R, Merlino G, Green JE, Hunter KW, Wakefield LM, Khanna C. 2010. Modeling metastasis biology and therapy in real time in the mouse lung. J Clin Invest 120:2979–2988. doi: 10.1172/JCI40252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tani H, Fujita O, Furuta A, Matsuda Y, Miyata R, Akimitsu N, Tanaka J, Tsuneda S, Sekiguchi Y, Noda N. 2010. Real-time monitoring of RNA helicase activity using fluorescence resonance energy transfer in vitro. Biochem Biophys Res Commun 393:131–136. doi: 10.1016/j.bbrc.2010.01.100. [DOI] [PubMed] [Google Scholar]
- 59.Garbelli A, Beermann S, Di Cicco G, Dietrich U, Maga G. 2011. A motif unique to the human DEAD-box protein DDX3 is important for nucleic acid binding, ATP hydrolysis, RNA/DNA unwinding and HIV-1 replication. PLoS One 6:e19810. doi: 10.1371/journal.pone.0019810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Buchan JR, Parker R. 2009. Eukaryotic stress granules: the ins and outs of translation. Mol Cell 36:932–941. doi: 10.1016/j.molcel.2009.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gao X, Ge L, Shao J, Su C, Zhao H, Saarikettu J, Yao X, Yao Z, Silvennoinen O, Yang J. 2010. Tudor-SN interacts with and co-localizes with G3BP in stress granules under stress conditions. FEBS Lett 584:3525–3532. doi: 10.1016/j.febslet.2010.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Meng X, Zhu D, Yang S, Wang X, Xiong Z, Zhang Y, Brachova P, Leslie KK. 2012. Cytoplasmic metadherin (MTDH) provides survival advantage under conditions of stress by acting as RNA-binding protein. J Biol Chem 287:4485–4491. doi: 10.1074/jbc.C111.291518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Solomon S, Xu Y, Wang B, David MD, Schubert P, Kennedy D, Schrader JW. 2007. Distinct structural features of caprin-1 mediate its interaction with G3BP-1 and its induction of phosphorylation of eukaryotic translation initiation factor 2alpha, entry to cytoplasmic stress granules, and selective interaction with a subset of mRNAs. Mol Cell Biol 27:2324–2342. doi: 10.1128/MCB.02300-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Napoli I, Mercaldo V, Boyl PP, Eleuteri B, Zalfa F, De Rubeis S, Di Marino D, Mohr E, Massimi M, Falconi M, Witke W, Costa-Mattioli M, Sonenberg N, Achsel T, Bagni C. 2008. The fragile X syndrome protein represses activity-dependent translation through CYFIP1, a new 4E-BP. Cell 134:1042–1054. doi: 10.1016/j.cell.2008.07.031. [DOI] [PubMed] [Google Scholar]
- 65.Say E, Tay HG, Zhao ZS, Baskaran Y, Li R, Lim L, Manser E. 2010. A functional requirement for PAK1 binding to the KH(2) domain of the fragile X protein-related FXR1. Mol Cell 38:236–249. doi: 10.1016/j.molcel.2010.04.004. [DOI] [PubMed] [Google Scholar]
- 66.Didiot MC, Subramanian M, Flatter E, Mandel JL, Moine H. 2009. Cells lacking the fragile X mental retardation protein (FMRP) have normal RISC activity but exhibit altered stress granule assembly. Mol Biol Cell 20:428–437. doi: 10.1091/mbc.E08-07-0737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Pillai RS, Bhattacharyya SN, Artus CG, Zoller T, Cougot N, Basyuk E, Bertrand E, Filipowicz W. 2005. Inhibition of translational initiation by Let-7 microRNA in human cells. Science 309:1573–1576. doi: 10.1126/science.1115079. [DOI] [PubMed] [Google Scholar]
- 68.Fukuda T, Naiki T, Saito M, Irie K. 2009. hnRNP K interacts with RNA binding motif protein 42 and functions in the maintenance of cellular ATP level during stress conditions. Genes Cells 14:113–128. doi: 10.1111/j.1365-2443.2008.01256.x. [DOI] [PubMed] [Google Scholar]
- 69.Bomsztyk K, Denisenko O, Ostrowski J. 2004. hnRNP K: one protein multiple processes. Bioessays 26:629–638. doi: 10.1002/bies.20048. [DOI] [PubMed] [Google Scholar]
- 70.Kim DY, Kim W, Lee KH, Kim SH, Lee HR, Kim HJ, Jung Y, Choi JH, Kim KT. 2013. hnRNP Q regulates translation of p53 in normal and stress conditions. Cell Death Differ 20:226–234. doi: 10.1038/cdd.2012.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lynch M, Chen L, Ravitz MJ, Mehtani S, Korenblat K, Pazin MJ, Schmidt EV. 2005. hnRNP K binds a core polypyrimidine element in the eukaryotic translation initiation factor 4E (eIF4E) promoter, and its regulation of eIF4E contributes to neoplastic transformation. Mol Cell Biol 25:6436–6453. doi: 10.1128/MCB.25.15.6436-6453.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Baltz AG, Munschauer M, Schwanhausser B, Vasile A, Murakawa Y, Schueler M, Youngs N, Penfold-Brown D, Drew K, Milek M, Wyler E, Bonneau R, Selbach M, Dieterich C, Landthaler M. 2012. The mRNA-bound proteome and its global occupancy profile on protein-coding transcripts. Mol Cell 46:674–690. doi: 10.1016/j.molcel.2012.05.021. [DOI] [PubMed] [Google Scholar]
- 73.Castello A, Fischer B, Eichelbaum K, Horos R, Beckmann BM, Strein C, Davey NE, Humphreys DT, Preiss T, Steinmetz LM, Krijgsveld J, Hentze MW. 2012. Insights into RNA biology from an atlas of mammalian mRNA-binding proteins. Cell 149:1393–1406. doi: 10.1016/j.cell.2012.04.031. [DOI] [PubMed] [Google Scholar]
- 74.Ivetic A, Ridley AJ. 2004. Ezrin/radixin/moesin proteins and Rho GTPase signalling in leucocytes. Immunology 112:165–176. doi: 10.1111/j.1365-2567.2004.01882.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Valderrama F, Thevapala S, Ridley AJ. 2012. Radixin regulates cell migration and cell-cell adhesion through Rac1. J Cell Sci 125:3310–3319. doi: 10.1242/jcs.094383. [DOI] [PubMed] [Google Scholar]
- 76.Sabile AA, Arlt MJ, Muff R, Husmann K, Hess D, Bertz J, Langsam B, Aemisegger C, Ziegler U, Born W, Fuchs B. 2013. Caprin-1, a novel Cyr61-interacting protein, promotes osteosarcoma tumor growth and lung metastasis in mice. Biochim Biophys Acta 1832:1173–1182. doi: 10.1016/j.bbadis.2013.03.014. [DOI] [PubMed] [Google Scholar]
- 77.Wang F, Ke ZF, Sun SJ, Chen WF, Yang SC, Li SH, Mao XP, Wang LT. 2011. Oncogenic roles of astrocyte elevated gene-1 (AEG-1) in osteosarcoma progression and prognosis. Cancer Biol Ther 12:539–548. doi: 10.4161/cbt.12.6.16301. [DOI] [PubMed] [Google Scholar]
- 78.Liu B, Wu Y, Peng D. 2013. Astrocyte elevated gene-1 regulates osteosarcoma cell invasion and chemoresistance via endothelin-1/endothelin A receptor signaling. Oncol Lett 5:505–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wang F, Ke ZF, Wang R, Wang YF, Huang LL, Wang LT. 6 September 2014. Astrocyte elevated gene-1 (AEG-1) promotes osteosarcoma cell invasion through the JNK/c-Jun/MMP-2 pathway. Biochem Biophys Res Commun doi: 10.1016/j.bbrc.2014.09.009. [DOI] [PubMed] [Google Scholar]
- 80.Tang J, Shen L, Yang Q, Zhang C. 2014. Overexpression of metadherin mediates metastasis of osteosarcoma by regulating epithelial-mesenchymal transition. Cell Prolif 47:427–434. doi: 10.1111/cpr.12129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wan L, Lu X, Yuan S, Wei Y, Guo F, Shen M, Yuan M, Chakrabarti R, Hua Y, Smith HA, Blanco MA, Chekmareva M, Wu H, Bronson RT, Haffty BG, Xing Y, Kang Y. 2014. MTDH-SND1 interaction is crucial for expansion and activity of tumor-initiating cells in diverse oncogene- and carcinogen-induced mammary tumors. Cancer Cell 26:92–105. doi: 10.1016/j.ccr.2014.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Guo F, Wan L, Zheng A, Stanevich V, Wei Y, Satyshur KA, Shen M, Lee W, Kang Y, Xing Y. 2014. Structural insights into the tumor-promoting function of the MTDH-SND1 complex. Cell Rep 8:1704–1713. doi: 10.1016/j.celrep.2014.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Viswanatha R, Wayt J, Ohouo PY, Smolka MB, Bretscher A. 2013. Interactome analysis reveals ezrin can adopt multiple conformational states. J Biol Chem 288:35437–35451. doi: 10.1074/jbc.M113.505669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Marintchev A, Edmonds KA, Marintcheva B, Hendrickson E, Oberer M, Suzuki C, Herdy B, Sonenberg N, Wagner G. 2009. Topology and regulation of the human eIF4A/4G/4H helicase complex in translation initiation. Cell 136:447–460. doi: 10.1016/j.cell.2009.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Liu F, Putnam A, Jankowsky E. 2008. ATP hydrolysis is required for DEAD-box protein recycling but not for duplex unwinding. Proc Natl Acad Sci U S A 105:20209–20214. doi: 10.1073/pnas.0811115106. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.