

Abstract
The intestinal lumen is a host place for a wide range of microbiota and sets a unique interplay between local immune system, inflammatory cells and intestinal epithelium, forming a physical barrier against microbial invaders and toxins. Bacterial translocation is the migration of viable or nonviable microorganisms or their pathogen-associated molecular patterns, such as lipopolysaccharide, from the gut lumen to the mesenteric lymph nodes, systemic circulation and other normally sterile extraintestinal sites. A series of studies have shown that translocation of bacteria and their products across the intestinal barrier is a commonplace in patients with liver disease. The deterioration of intestinal barrier integrity and the consulting increased intestinal permeability in cirrhotic patients play a pivotal pathophysiological role in the development of severe complications as high rate of infections, spontaneous bacterial peritonitis, hepatic encephalopathy, hepatorenal syndrome, variceal bleeding, progression of liver injury and hepatocellular carcinoma. Nevertheless, the exact cellular and molecular mechanisms implicated in the phenomenon of microbial translocation in liver cirrhosis have not been fully elucidated yet.
Keywords: Cirrhosis, Intestinal barrier, Tight junction, Bacterial translocation, Intestinal bacterial overgrowth
Core tip: Intestinal barrier function is impaired in patients with cirrhosis and this derangement seems to be associated with liver disease severity. This phenomenon is multifactorial and the exact pathophysiological mechanisms which are implicated in this deterioration have not been fully elucidated yet. The disruption of intestinal barrier integrity and the subsequent increased intestinal permeability in cirrhotic patients promote bacterial translocation and play a major role in the development of severe clinical complications affecting natural history of liver disease and patients’ survival.
INTRODUCTION
Cirrhosis and portal hypertension associated complications are a common cause of mortality worldwide. Increased intestinal permeability and subsequent bacterial translocation to the mesenteric lymph nodes and extraintestinal sites are well established in these patients[1,2]. Endotoxemia seems to be a key factor and results in a cascade of immunomodulatory, cellular and molecular events. Potential mechanisms that can promote BT are intestinal bacterial overgrowth (IBO) and gut flora disturbances, increased intestinal permeability via the paracellular and intracellular route and local as well as systemic immune dysfunction[3-7]. Cirrhosis is also associated with increased oxidative stress in the systematic circulation, the intestinal and liver tissue, which in turn acts as a harmful agent to the intestinal epithelial cells, affects apoptosis and cellular proliferation, deteriorates the expression of tight junction (TJ) proteins and favors bacterial translocation[8-12]. Endotoxemia plays a critical role in the exacerbation of host and acquired immune responses, activation of cells to release cytokines, which can promote intestinal and liver tissue damage[13-16]. Furthermore, bacterial translocation (BT) is associated with severity of liver disease and provokes serious clinical events and complications[17].
THE INTESTINAL BARRIER STRUCTURAL AND FUNCTIONAL ELEMENTS
The intestinal tract represents the body’s largest interface between the host and the external environment. The complexity of its function is obvious when thinking that the intestine has to serve simultaneously two distinct functions; the absorption and transport of necessary nutrients from the intestinal lumen into the circulation and the internal milieu in general and, on the other hand, the prevention of the penetration of harmful entities including microorganisms, luminal antigens and proinflammatory factors. The latter function is known as barrier function. Gut barrier function depends on both the immune barrier, composed of locally acting factors, such as the secretory IgA, intramucosal lymphocytes, Payer’s nodules, mesenteric lymph nodes and of the systemic host defense, the latter represented mainly by the reticuloendothelial system, the biological barrier-made up of normal intestinal flora responsible for colonization resistance - the mechanical barrier as well, consisted of the closed-lining intestinal epithelial cells and by the capillary endothelial cells. All these components of gut barrier integrity can be majorly affected by liver cirrhosis[12,18,19].
The intestinal mechanical barrier in cirrhosis
The intestinal mucosal barrier consists of the mucus layer and intestinal epithelial cells. The epithelium prevents translocation of pathogens via transcellular and paracellular route[20]. The enterocytes are connected to each other by junctional complexes consisting of TJs, adherens junctions, desmosomes, and gap junctions forming a selective physical barrier that regulates paracellular transport[21,22]. The main transmembrane protein families in tight junctions are members of the occludin, claudins, and junctional adhesion molecules, which are linked to the actin cytoskeleton regulating paracellular movement of micromolecules, bacteria and macromolecules such as lipopolysaccharide[22-24]. TJs regulate transport via two distinct pathways: a charge selective, claudin-based pores that are 4 Å in radius for small ions and uncharged molecules, and a second one pathway, regardless of molecules charge and size[25,26]. Liver cirrhosis induces prominent changes in enterocytes’ tight junction proteins, representing a cellular mechanism for intestinal barrier disruption and hyperpermeability[19,27]. In cholestatic liver injury, increased myosin light-chain kinase activation and diminished expression of occludin and zonula occludens-1 (ZO-1) have been reported in colonic epithelium with a concomitant increased intestinal permeability[4]. Reduced expression of duodenal occludin and Claudin-1 has been found in patients with cirrhosis compared to controls. Also, these alterations were more apparent in decompensated patients as compared to compensated ones. Negative regression was proved between occludin and claudin-1 expression, Child-Pugh score, the size of esophageal varices and serum endotoxin levels. These data support the view that there is a dynamic relationship between portal hypertension, bacterial translocation and TJs expression in intestinal epithelial cells[19]. In patients with nonalcoholic steatohepatitis and alcoholic decompensated cirrhosis, increased Claudin-2 was proved and could comprise a pivotal factor inducing intestinal barrier disruption. Conflicting are the findings about TJ proteins ZO, occludin and claudin-1 and the gap junction protein Connexin expression[28]. In cirrhosis, one of the main contributing factors to TJ alterations is the increased production of tumor necrosis factor-α (TNF-α) by monocytes in mesenteric lymph nodes[29,30]. TNF-α increases miR-122a expression in Caco-2 enterocytes and in vivo in a mouse model. miR-122a binds to the noncoding region three prime untranslated region of occludin mRNA and impacts on occludin mRNA downregulation and subsequent occludin diminished expression, as well as upregulates claudin-2 and -8 expression but does not induct any alteration of claudins-1, -3, -5. Moreover, a linear relationship between TNF-α induced reduction of occludin and a higher inulin flux has been observed, indicating an increased Caco-2 permeability to inulin[31].
Histopathological changes of intestinal mucosa: Specific ultrastructural alterations of intestinal mucosa have been observed in cirrhotic patients that may be related to increased BT. In a case control study of cirrhotic patients using electron microscopy, dilated extracellular space between adjacent enterocytes, more prominent in the lower portion of the intestinal epithelial cells and reduced number of shorter and thicker microvilli were observed[32]. In experimental models of cirrhotic rats the intestinal mucosa was presented with atrophic, shorter, fractured villi and infiltration of inflammatory cells into the lamina propria and the muscular layer. The glandular epithelia resembled as irregular structures after the loss of their cylindrical shape. Excessive villi swell and loose structure of mucous membrane were correlated positively to endotoxemia[33].
Mucus: The mucus layer overlying the intestinal mucosa provides a first line defense mechanism against harmful antigens, and prevents bacteria and their byproducts from invading the microvillus environment. Mucus consists of glycoproteins secreted by goblet cells called mucins. Mucin (MUC) secretion is affected by transcription factors [nuclear factor-κB (NF-κB)], growth factors, lipopolysaccharide (LPS), microbes presence, inflammatory cytokines[34,35]. NF-κB is activated during gastrointestinal tract inflammation and binds to specific sites in the promoter of MUC2[36]. Chronic alcohol feeding increases the mucus content in the small intestine in rats. Furthermore, increased mucus thickness has been observed in the duodenum of alcoholic patients as a concomitant protective modification[37,38]. Increased MUC2 and MUC3 mRNA expression has been found in the ileum of rats with liver cirrhosis compared to those without cirrhosis[2]. Intestinal mucus modulates bacterial adherence to the intestinal mucosal surface and is associated with a loss of intestinal barrier function[39].
Intestinal oxidative stress: Oxidative stress is a mediator of intestinal mucosal barrier damage in patients with liver cirrhosis, affecting intestinal epithelial cell apoptosis and proliferation, and enhances BT and endotoxemia[12]. Portal hypertension results in intestinal mucosa hypoperfusion and hypoxia, which exacerbate oxidative damage in the gut mucosa by the increased xanthine oxidase activity and oxygen free radicals release[11]. Xanthine oxidase found in the liver and intestinal mucosa catalyzes the oxidation of hypoxanthine to xanthine, the conversion of xanthine to uric acid and is an important source of free radicals in the intestinal epithelium. Increased xanthine oxidase and decreased xanthine dehydrogenase activity have been observed in the intestinal mucosa and enterocyte mitochondria in the state of liver cirrhosis. Oxidative stress causes tissue damage at the subcellular level by lipid peroxidation affecting mitochondrial function. Reactive oxygen species break down the cellular membrane stability and induct cell death by lipid peroxidation in the cirrhotic rats[9,11]. Increased levels of malondialdehyde, a product of the lipid peroxidation, have been found in ileal and cecal mucosa in cirrhotic rats with ascites when compared to control rats, and in cirrhotic rats with BT compared to those without BT[8,40]. Experimental cirrhotic rats received pentoxifylline treatment, a regimen which exerts anti-inflammatory and antioxidant effects, appeared to have lower malondialdehyde levels in the cecal mucosa compared to placebo-treated ones. Pentoxifylline administration attenuates bacterial overgrowth, BT to cecal lymph nodes and impacts on elimination of spontaneous bacterial peritonitis[40]. Free radicals can also affect viscosity of the mucus in the gastric mucosa, enhance bacterial adherence ability to the epithelial cells and facilitate the translocation across the mucosa, resulting in complications such as spontaneous bacterial peritonitis (SBP)[10,41].
The intestinal immunological barrier in cirrhosis
Gut-associated lymphoid tissue alterations: The host innate immune system is the first line defense mechanism which is activated against bacteria and other toxins. The intestinal immune system consists of the gut-associated lymphoid tissue, which comprises four lymphoid compartments: Peyer’s patches, lamina propria lymphocytes, including dendritic cells (DCs), intraepithelial lymphocytes and mesenteric lymph nodes, which are implicated in both the adaptive and innate immune defense mechanism[42]. The interaction between the host immune system and the microbiota inducts the activation of the intestinal immune system and the gut-associated lymphoid tissue that in turn modifies the microbiota environment[43]. DCs induce the development of Th1/Th17 T cells, regulatory T cells and promote TNF-α production[44]. Dendritic cells of the lamina propria induct tight junction alterations and sample microbes from the intestinal lumen[45]. An increased count of activated monocytes, dendritic cells and T lymphocytes in the intestinal mucosa and mesenteric lymph nodes (MLNs) coincided with specific alterations of cytokine expression in the intestinal mucosa as well as increased phagocytosis by intestinal dendritic cells in cirrhosis as a response to intestinal bacteria and other pathogens. Increased activated macrophages in the duodenal lamina propria, augmented intestinal permeability and altered intestinal tight junction protein expression have been demonstrated in patients with decompensated cirrhosis[28,31,46-48]. In response to BT, intestinal epithelial cells release chemokines, which exert chemoattractant effects and induce the recruitment of DCs to the mucosa as well as in MLNs[47]. IgA is one of the most important molecules in the regulation of intestinal homeostasis. Peyer’s patches and isolated lymphoid follicles are implicated in commensal-specific IgA production that aids to prevent the commensals from invading the gut mucosa[49]. Mice deficient in the toll-like receptors (TLR)-adapter molecule MyD88 on B cells lack commensal-specific immunoglobulin-response that results in impaired epithelial integrity and enables commensal bacteria to function as highly pathogenic organisms[50]. A pronounced reduction in CD27+ memory B-cells count and functional capacity as well as a reduced ability to recruit T-cells, have been observed in cirrhotic patients. These B-cell defects may explain the susceptibility to bacterial infection. Also blockade of TLR4 and TLR9 signaling abrogates the activation of normal donor B-cells by cirrhotic plasma, suggesting a role for bacterial translocation in cirrhosis[51]. T cells are critical in host defense against the translocation of enteric bacteria since their depletion has been correlated with augmented BT and spreading of bacteria to extraintestinal sites and MLNs[37,52,53].
Antimicrobial peptides: Deficiency in antimicrobial peptides (AMPs) leads to disruption of the mucosal barrier, a shift in the bacterial composition, bacterial overgrowth and increase in BT. Antimicrobial peptides, also called host defense peptides, are part of the innate immune response and act as broad spectrum antibiotics killing Gram negative and Gram positive bacteria, viruses and fungi. AMPs include defensins, cathelcidins, lysozyme, resistin-like molecules and lectins. Defensins have a broad range of antimicrobial activity by binding to the microbial cell membrane and forming pore-like membrane defects. Human a-defensins that are expressed by neutrophils and Paneth cells located at the base of Lieberkuhn crypts, in response to bacteria and LPS exposure, regulate and maintain microbial balance in the intestinal lumen[54-56]. Reduced expression of Paneth cell defensins and diminished in vitro antibacterial activity of a-defensins against Enterobacteriacea have been observed in ascitic cirrhotic rats with BT to MLNs[57]. Regenerating islet derived proteins RegIII, produced by Paneth cells via activation of TLRs by pathogen-associated molecular patterns, bind to cell wall peptidoglycans of Gram-positive bacteria, and maintain a physical barrier between the epithelial cell surface and intestinal microbes[58,59]. Chronic alcohol intake has been shown to diminish RegIII expression in the small intestine of mice as well as in humans[3]. IgA antibodies released into the intestinal lumen, bind and aggregate bacteria, preventing mucosal adherence and colonization[60]. Reduced fecal IgA content as well as diminished secretion of mucosal IgA into the jejunum have been reported, suggesting a potential relationship between IgA, BT and development of infections in cirrhosis[37,61].
Cytokine alterations in cirrhosis and immune dysfunction: Endotoxemia as a result of intestinal barrier dysfunction, triggers the activation of the innate immune system and the release of proinflammatory cytokines[62]. The increased proinflammatory cytokine production (TNF-α, IFN, IL-6) and reduced anti-inflammatory cytokines (IL-10), in state of liver cirrhosis, by intestinal immune cells, affect the intestinal epithelial barrier integrity disrupting the epithelial tight Junctions and favour the increase of bacterial translocation[29,33,63,64]. Insulin-like growth factor I therapy in cirrhotic rats has been found to promote portal pressure, bacterial translocation and endotoxemia reduction through diminished TNF-α expression[65].
The intestinal biological barrier in cirrhosis
Gut microbiota alterations: Intestinal bacterial overgrowth is common in cirrhosis and it has been shown to be particularly frequent in those with more severe liver disease and in those with a prior history of SBP and/or hepatic encephalopathy[66-71]. Reduced gastric acid secretion, intestinal dysmotility, lack of bile salts and reduced antimicrobial peptides killing capacity as well as portal hypertension have been recognized as contributory factors to IBO[3,72,73]. Changes in the gut microflora favor bacterial translocation and promote endotoxemia in patients with cirrhosis and experimental models of cirrhosis[67,74,75]. A direct relationship between the density and composition of cecal bacteria and the number of viable bacteria of this strain, present in MLNs, has been demonstrated in mouse models[76]. Intestinal bacterial overgrowth promotes the development of SBP by increasing bacterial translocation. Aerobic bacteria in cecal stool are increased in cirrhotic rats with bacterial translocation with or without spontaneous bacterial peritonitis compared to cirrhotic rats without bacterial translocation and SBP[72]. The impaired motility of the small intestine is a common feature in cirrhosis and may be a crucial factor in the pathophysiology of intestinal bacterial overgrowth, increased intestinal permeability and subsequent bacterial translocation[77]. The small intestinal transit is delayed in cirrhotic rats and the cecal aerobic bacteria count is higher compared to healthy controls[78].
CLINICAL IMPLICATIONS
Liver injury
Intestinal inflammation and bacterial translocation play a major role in the progression of liver fibrosis via TLR2, the receptor for products from Gram-positive bacteria such as peptidoglycan which in turn promotes a cascade of signals on monocytes in the lamina propria and tumor necrosis factor receptor type I (TNFRI) on intestinal epithelial cells. TLR2-/- mice have shown significantly less positive mesenteric lymph node cultures and lower endotoxin levels in the systematic circulation as a marker of bacterial translocation compared to wild type mice. TNFRI-/- mice are protected from liver fibrosis by a decreased collagen α (I) gene expression and deposition of extracellular matrix proteins, suggesting that TNFRI on intestinal epithelial cells enhances the paracellular leakage and favors bacterial translocation and liver fibrogenesis[46]. LPS leads to host immune activation and enhances plasma sCD14 as a response. In patients with severe fibrosis higher plasma levels of sCD14 and more hepatic CD14+ cells have been documented compared to patients with minimal fibrosis. LPS-mediated activation of both circulating monocytes and hepatic Kupffer cells induces liver fibrosis and progression to end-stage liver disease[79]. Seki et al[80] demonstrated that the intestinal bacterial microflora and a functional TLR4 are required for hepatic fibrogenesis. Hepatic stellate cells (HSCs) are the target through which TLR4 ligands such as lipopolysacharide promote fibrogenesis. In quiescent HSCs, TLR4 activation triggers chemokine secretion, induces chemotaxis of Kupffer cells, downregulates the transforming growth factor (TGF)-b, sensitizes HSCs to TGF-b - induced signals and allows unrestricted activation by Kupffer cells. LPS-induced HSCs sensitization to TGF-b leads to collagen production and deposition and seems to be mediated by a MyD88-NF-κB-dependent pathway[80].
Hepatocellular cancer
The majority of hepatocellular cancer (HCC) cases are generated in the state of chronic liver inflammation. Increased intestinal permeability, bacterial translocation and LPS accumulation activating the NF-κB pathway, suggest a hallmark of chronic liver disease and contribute to hepatic inflammation, proinflammatory cytokines TNF-α, IL-6 and IL-1 release, oxidative damage and fibrosis. The deterioration of normal equilibrium in the intestinal microbiota and NF-κB activation through upregulation of TNF-α exert promotional properties in HCC development[81]. Decreased hepatocarcinogenesis has been found in mice lacking IKK-b, a kinase required for NF-κB activation, in both hepatocytes and hematopoietic-derived Kupffer cells, suggesting that IKK-b orchestrates inflammatory crosstalk between hepatocytes and Kupffer cells and promotes liver cancer induction[82]. Infusion of LPS, which is an agonist of Toll-Like Receptor, increases hepatocarcinogenesis, tumor number and size in experimental animal model of mice intoxicated with DEN/CCl4. In advanced liver disease HCC development is mediated by TLR4-dependent secretion of growth factors such as epiregulin hepatomitogen by hepatic stellate cells, leading to EGFR and HER2 activation during the first stages of carcinogenesis, whereas it reduces hepatocyte apoptosis by NF-κB nuclear translocation[83-85]. TLR4 deficiency and antibiotic-induced gut sterilization decrease hepatic proliferation and fibrogenesis and could prevent HCC in patients with chronic liver injury, suggesting that the intestinal microbiota and TLR4 overexpression represent a possible molecular mechanism for the induction of HCC promotion[84]. These data suggest that disturbances of intestinal microflora, endotoxemia, and subsequent TLR4 mediated hepatic stellate cell activation might provide a dynamic interplay between endotoxemia, hepatic fibrosis and HCC promotion by increasing growth factors[83,85,86]. The hepatic expression of the glutathione S-transferase placental form, a marker for cellular alteration in the early stage of HCC development, has decreased in rats treated with probiotic MIYAIRI 588 compared to the choline deficient amino acids - diet-fed rats. The number and the size of the HCC lesion reduction in the MIYAIRI 588-treated rats have been correlated with endotoxemia elimination and increased ZO-1 and occludin expression, suggesting that bacterial translocation enhancement may constitute a promoting factor in hepatocarcinogenesis[87].
Hepatic encephalopathy
Intestinal dysbiosis and bacterial infections are precipitating factors for the induction of hepatic encephalopathy overt or subclinical. In previous studies cognitive impairment was recorded in 42% of cirrhotics without infection, in 79% of those with infection and without SIRS and in 90% of septic patients[88,89]. Altered flora, increased endotoxin levels, and excessive inflammation (IL-6, TNF-α, IL-2, and IL-13) have been found in cirrhotics with HE compared with those without hepatic encephalopathy (HE)[90]. Streptococcus salivarius is more prominent in cirrhotic patients with minimal hepatic encephalopathy (MHE) in comparison to those without HE, and is significantly associated with ammonia concentration[91]. Bacterial overgrowth with abundance of Gram-negative [Escherichia coli (E. coli)] and Gram-positive (Staphylococcus spp.) has been associated with cirrhosis complicated with MHE[92]. A higher incidence of previous hepatic encephalopathy episodes has been revealed in patients with TLR4 D299G and/or T399I polymorphisms, which are associated with intestinal barrier dysfunction, compared to wild-type patients (78% vs 20%)[93].
Gastrointestinal bleeding
Bacterial infection might increase the risk of variceal hemorrhage[94,95]. Cirrhotic patients with impaired intestinal permeability, high lipopolysaccharide binding protein and IL-6 levels represent a higher risk of variceal bleeding[96]. Bacterial infection is responsible for early rebleeding[95]. In a prospective study by Bernard et al[97], early rebleeding, defined as recurrence of bleeding within 7 d after admission, was observed in 43.5% of patients with bacterial infection compared to 9.8% in those without infection. Furthermore, the mean number of blood units transfused and the 4-wk mortality were significantly higher in patients with infection[97]. Bacterial infection was independently associated with failure to control bleeding in a previous study[98]. Patients with hepatocellular carcinoma and variceal bleeding tend to have a greater rebleeding rate due to a higher infection rate. Antibiotic prophylaxis can prevent infection and rebleeding, improving survival rate as well as decreasing the amount of blood transfused in patients with acute gastroesophageal variceal bleeding following endoscopic treatment[99,100]. A retrospective study suggested that administration of antibiotics prior to endoscopy or up to 8 h following endoscopy, if this is initially missed, reduces rebleeding and improves 28-d survival[101,102].
Hepatopulmonary syndrome
Bacterial translocation and subsequent endotoxemia in cirrhotic rats may be a pathogenetic mechanism implicated in hepatopulmonary syndrome (HPS) progression. Endotoxin mediated stimulation of Kupffer cells via mitogen-activated protein kinase pathway upregulates TNF-α production and constitutes a key step in the induction of hepatopulmonary syndrome[103]. In cirrhotic rats endotoxemia, severity of liver disease and portal vein pressure are strongly correlated with the expression of eNOs, inducible nitric oxide synthase (iNOS), HO-1, histological changes in lung tissue, such as an increased number of dilated capillaries, infiltration of phagocytes and neutrophils and play a central role in the development of hepatopulmonary syndrome by inducing NO and CO[104]. In cirrhotic rats treated with norfloxacin, elimination of Gram-negative bacterial translocation, reduced count of pulmonary microvessels containing more than 10 macrophages, decreased expression and activity of lung iNOS have been observed, suggesting that bacterial translocation may be a major mechanism for the pathogenesis of HPS[105].
Hepatorenal syndrome
Hepatorenal syndrome is a specific type of renal failure that affects individuals suffering from liver cirrhosis[106]. Hepatorenal syndrome (HRS) is due to constriction of the blood vessels of the kidneys and dilation of the splachnic vessels which supplies the intestine[107]. Portal hypertension in cirrhosis has been associated with circulatory disturbances, arterial splanchnic vasodilatation and subsequent reduction in systemic vascular resistance, which results in reduced blood volume. Compensatory mechanisms such as vasoconstrictor systems and sodium retention in the kidneys are activated. However, increased cardiac output and hyperdynamic circulation, in advanced cirrhosis are insufficient to retain ideal intravascular effective volume resulting in hypoperfusion of kidneys[108,109]. The markedly decreased renal blood flow in decompensated cirrhosis, leads to hepatorenal syndrome that is frequently triggered from infections[110-112]. Patients with SBP without shock who exhibit high proinflammatory response are at high risk of developing kidney failure[111]. Renal failure occurs in approximately one third of patients with cirrhosis and bacterial infections and is irreversible or progressive in two-thirds of patients with treatment of infection only. The presence of a nosocomial infection, the absence of infection resolution with antibiotics and the peak count of neutrophil leukocytes in blood have been demonstrated as significant predictive factors of irreversibility of HRS[112-115]. Cirrhotic patients with culture-negative, non-neutrocytic ascites and bacterial DNA presence in ascitic fluid have a significantly higher TNF-α level in serum and ascitic fluid and a major risk of HRS compared to those without bacterial DNA, suggesting that bacterial translocation, subsequent inflammation and bacterial DNA presence are implicated in HRS induction[17]. Supportive to previous data are the results of Kalambokis et al[116] study, according to which intestinal decontamination with rifaximin therapy improves systemic circulation and renal function in patients with advanced alcoholic cirrhosis. Additionally, gut sterilization reduces CO and plasma renin activity, and inducts systemic vascular resistance increase. Rifaximin administration significantly improves the glomerular filtration rate and natriuresis while attenuates endotoxemia and reduces IL-6 and TNF-α production, suggesting that the prevention of infection in cirrhotic patients with renal failure seems to be a beneficial approach[116,117].
Infections
The intestinal permeability index (IPI) is increased in patients with advanced liver cirrhosis and active gastrointestinal hemorrhage, especially in those with proven or possible infections. IPI is an independent factor for the prediction of infection incidence in cirrhotic patients, suggesting that intestinal barrier dysfunction inducts bacterial translocation and affects the patient susceptibility to infections[118]. Patients with a bacterial infection suffer from a more severe liver disease with lower serum albumin and prolonged prothrombin time compared to cirrhotics without signs of infection[119]. Rimola et al[120] demonstrated that decompensated cirrhotics with a depressed reticuloendothelial system phagocytic activity have a higher risk of bacteremia affecting the survival rate.
Spontaneous bacterial peritonitis
Spontaneous bacterial peritonitis is a common complication of cirrhosis. Bacterial contamination of ascites fluid leading to SBP is caused by bacterial translocation. In cirrhotic rats identical bacterial species are cultivated in both mesenteric lymph nodes and ascitic fluid[121]. Among the patients with liver cirrhosis and culture-negative, non-neutrocytic ascites has been documented that the presence of ascitic bacterial DNA coincides with a higher relative risk of spontaneous bacterial peritonitis, suggesting a distinct association of SBP with impaired intestinal barrier function and increased bacterial translocation[17]. Patients with decompensated cirrhosis carrying Nucleotide-binding oligomerization domain containing 2 (NOD2) risk alleles (1007fs, G908R, R702W) which have been linked with impaired intestinal barrier or a history of prior SBP are at significant risk for development of spontaneous bacterial peritonitis and bacterascites[122,123]. It remains controversial whether proton-pump inhibitors use increases bacterial translocation and the risk of SBP[124-126]. On the other hand, treatment with b-blockers may prevent spontaneous bacterial peritonitis[127].
MORTALITY
Patients with liver cirrhosis and bacterial DNA in ascites as molecular evidence of intestinal bacterial translocation have an increased risk of death compared to those without bacterial DNA[17]. NOD2 gene variants in patients with advanced liver cirrhosis linked to impaired mucosal barrier function may be genetic risk factors for death. NOD2 risk alleles and spontaneous bacterial peritonitis are independent predictive factors of death[122,123]. In a prospective study of fifty-three patients with cirrhosis, univariate Kaplan Meier analysis showed that Child-Pugh group, serum bilirubin, serum albumin, plasma endotoxin, and prothrombin time were associated with mortality[67].
CONCLUSION
In conclusion, intestinal barrier function is impaired in patients with cirrhosis and this derangement seems to be more pronounced in advanced cirrhosis. The disruption of mucosal barrier integrity is multifactorial, depends on a series of cellular and immune-mediated events, and affects the natural history of liver disease and patients’ survival as illustrated in the Figure 1. Therefore, there is an open field for clinical investigations intending new customized treatment interventions at a molecular level and the modification of bacterial translocation events.
Figure 1.
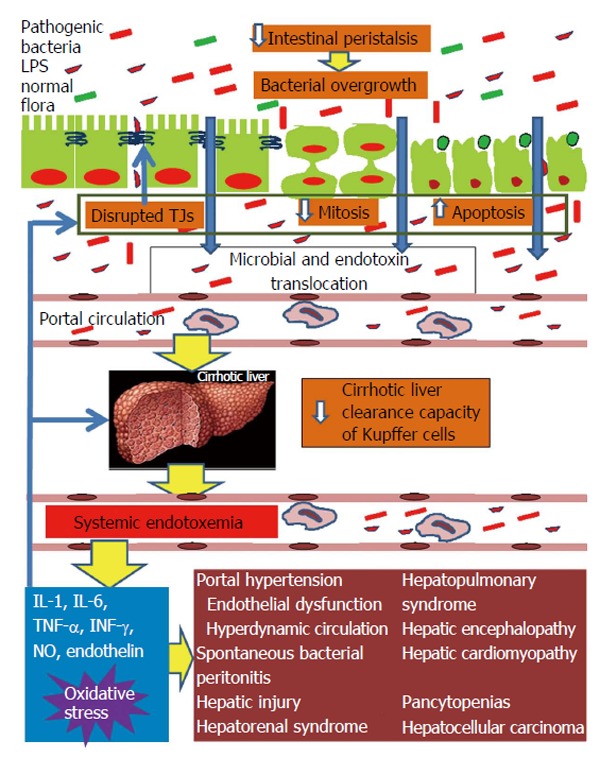
Pathophysiological overview of intestinal barrier dysfunction in liver cirrhosis and its interconnection with cirrhosis’ complications from diverse organs. Liver cirrhosis delays intestinal motility thus changing the indigenous gut microecology and promoting intraluminal bacterial and endotoxin overgrowth. In parallel, the structural and functional integrity of the intestinal mucosa is disrupted leading to increased gut permeability. Important factors implicated in increased intestinal permeability are the disruption of the tight junctions structural complex and altered epithelial homeostasis, with decreased mitotic activity and increased apoptosis of enterocytes. Systemic cytokinemia and oxidative stress are pivotal promoters of these intestinal alterations. Increased gut permeability permits the escape of intraluminal bacteria and endotoxins initially into portal blood and subsequently, through a decreased clearance capacity of the cirrhotic liver, into systemic circulation. Systemic endotoxemia activates a systemic inflammatory response with release of interleukin-1 (IL-1), IL-6, tumor necrosis factor-alpha, interferon-γ, nitric oxide and endothelin-1, which can induce circulatory and remote organ dysfunction, partially through promotion of reactive oxygen species formation in the endothelium, lung, kidney, brain, heart and bone marrow. At the same time, the endotoxin-induced increased systemic levels of proinflammatory cytokines and oxidative stress aggravate intestinal and hepatic injury, further promoting bacterial translocation and endotoxemia, thus, maintaining the vicious cycle of gut barrier dysfunction, bacterial and endotoxin translocation, systemic release of proinflammatory cytokines and oxidative stress, complications of cirrhosis from diverse organs. TJ: Tight junction; LPS: Lipopolysaccharide.
Footnotes
P- Reviewer: Decorti G, Zoller M S- Editor: Ji FF L- Editor: A E- Editor: Liu SQ
Conflict-of-interest statement: None of the authors have any conflict of interest related to this work.
Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Peer-review started: February 26, 2015
First decision: May 14, 2015
Article in press: July 2, 2015
References
- 1.Llamas MA, Aller MA, Marquina D, Nava MP, Arias J. Bacterial translocation to mesenteric lymph nodes increases in chronic portal hypertensive rats. Dig Dis Sci. 2010;55:2244–2254. doi: 10.1007/s10620-009-1001-3. [DOI] [PubMed] [Google Scholar]
- 2.Xie YR, Liu SL, Liu X, Luo ZB, Zhu B, Li ZF, Li LJ, He Y, Jiang L, Li H, et al. Intestinal microbiota and innate immunity-related gene alteration in cirrhotic rats with liver transplantation. Transplant Proc. 2011;43:3973–3979. doi: 10.1016/j.transproceed.2011.08.113. [DOI] [PubMed] [Google Scholar]
- 3.Yan AW, Fouts DE, Brandl J, Stärkel P, Torralba M, Schott E, Tsukamoto H, Nelson KE, Brenner DA, Schnabl B. Enteric dysbiosis associated with a mouse model of alcoholic liver disease. Hepatology. 2011;53:96–105. doi: 10.1002/hep.24018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fouts DE, Torralba M, Nelson KE, Brenner DA, Schnabl B. Bacterial translocation and changes in the intestinal microbiome in mouse models of liver disease. J Hepatol. 2012;56:1283–1292. doi: 10.1016/j.jhep.2012.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen Y, Yang F, Lu H, Wang B, Chen Y, Lei D, Wang Y, Zhu B, Li L. Characterization of fecal microbial communities in patients with liver cirrhosis. Hepatology. 2011;54:562–572. doi: 10.1002/hep.24423. [DOI] [PubMed] [Google Scholar]
- 6.Chen Y, Qin N, Guo J, Qian G, Fang D, Shi D, Xu M, Yang F, He Z, Van Nostrand JD, et al. Functional gene arrays-based analysis of fecal microbiomes in patients with liver cirrhosis. BMC Genomics. 2014;15:753. doi: 10.1186/1471-2164-15-753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rogers GB, van der Gast CJ, Bruce KD, Marsh P, Collins JE, Sutton J, Wright M. Ascitic microbiota composition is correlated with clinical severity in cirrhosis with portal hypertension. PLoS One. 2013;8:e74884. doi: 10.1371/journal.pone.0074884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chiva M, Guarner C, Peralta C, Llovet T, Gómez G, Soriano G, Balanzó J. Intestinal mucosal oxidative damage and bacterial translocation in cirrhotic rats. Eur J Gastroenterol Hepatol. 2003;15:145–150. doi: 10.1097/00042737-200302000-00007. [DOI] [PubMed] [Google Scholar]
- 9.Marnett LJ. Oxy radicals, lipid peroxidation and DNA damage. Toxicology. 2002;181-182:219–222. doi: 10.1016/s0300-483x(02)00448-1. [DOI] [PubMed] [Google Scholar]
- 10.Natarajan SK, Ramamoorthy P, Thomas S, Basivireddy J, Kang G, Ramachandran A, Pulimood AB, Balasubramanian KA. Intestinal mucosal alterations in rats with carbon tetrachloride-induced cirrhosis: changes in glycosylation and luminal bacteria. Hepatology. 2006;43:837–846. doi: 10.1002/hep.21097. [DOI] [PubMed] [Google Scholar]
- 11.Ramachandran A, Prabhu R, Thomas S, Reddy JB, Pulimood A, Balasubramanian KA. Intestinal mucosal alterations in experimental cirrhosis in the rat: role of oxygen free radicals. Hepatology. 2002;35:622–629. doi: 10.1053/jhep.2002.31656. [DOI] [PubMed] [Google Scholar]
- 12.Assimakopoulos SF, Tsamandas AC, Tsiaoussis GI, Karatza E, Zisimopoulos D, Maroulis I, Kontogeorgou E, Georgiou CD, Scopa CD, Thomopoulos KC. Intestinal mucosal proliferation, apoptosis and oxidative stress in patients with liver cirrhosis. Ann Hepatol. 2013;12:301–307. [PubMed] [Google Scholar]
- 13.Úbeda M, Muñoz L, Borrero MJ, Díaz D, Francés R, Monserrat J, Lario M, Lledó L, Such J, Álvarez-Mon M, et al. Critical role of the liver in the induction of systemic inflammation in rats with preascitic cirrhosis. Hepatology. 2010;52:2086–2095. doi: 10.1002/hep.23961. [DOI] [PubMed] [Google Scholar]
- 14.Francés R, Zapater P, González-Navajas JM, Muñoz C, Caño R, Moreu R, Pascual S, Bellot P, Pérez-Mateo M, Such J. Bacterial DNA in patients with cirrhosis and noninfected ascites mimics the soluble immune response established in patients with spontaneous bacterial peritonitis. Hepatology. 2008;47:978–985. doi: 10.1002/hep.22083. [DOI] [PubMed] [Google Scholar]
- 15.Guarner C, González-Navajas JM, Sánchez E, Soriando G, Francés R, Chiva M, Zapater P, Benlloch S, Muñoz C, Pascual S, et al. The detection of bacterial DNA in blood of rats with CCl4-induced cirrhosis with ascites represents episodes of bacterial translocation. Hepatology. 2006;44:633–639. doi: 10.1002/hep.21286. [DOI] [PubMed] [Google Scholar]
- 16.Attar BM, Moore CM, George M, Ion-Nedelcu N, Turbay R, Zachariah A, Ramadori G, Fareed J, Van Thiel DH. Procalcitonin, and cytokines document a dynamic inflammatory state in non-infected cirrhotic patients with ascites. World J Gastroenterol. 2014;20:2374–2382. doi: 10.3748/wjg.v20.i9.2374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.El-Naggar MM, Khalil el-SA, El-Daker MA, Salama MF. Bacterial DNA and its consequences in patients with cirrhosis and culture-negative, non-neutrocytic ascites. J Med Microbiol. 2008;57:1533–1538. doi: 10.1099/jmm.0.2008/001867-0. [DOI] [PubMed] [Google Scholar]
- 18.Assimakopoulos SF, Scopa CD, Vagianos CE. Pathophysiology of increased intestinal permeability in obstructive jaundice. World J Gastroenterol. 2007;13:6458–6464. doi: 10.3748/wjg.v13.i48.6458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Assimakopoulos SF, Tsamandas AC, Tsiaoussis GI, Karatza E, Triantos C, Vagianos CE, Spiliopoulou I, Kaltezioti V, Charonis A, Nikolopoulou VN, et al. Altered intestinal tight junctions’ expression in patients with liver cirrhosis: a pathogenetic mechanism of intestinal hyperpermeability. Eur J Clin Invest. 2012;42:439–446. doi: 10.1111/j.1365-2362.2011.02609.x. [DOI] [PubMed] [Google Scholar]
- 20.Groschwitz KR, Hogan SP. Intestinal barrier function: molecular regulation and disease pathogenesis. J Allergy Clin Immunol. 2009;124:3–20; quiz 21-22. doi: 10.1016/j.jaci.2009.05.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schneeberger EE, Lynch RD. The tight junction: a multifunctional complex. Am J Physiol Cell Physiol. 2004;286:C1213–C1228. doi: 10.1152/ajpcell.00558.2003. [DOI] [PubMed] [Google Scholar]
- 22.Turner JR. Intestinal mucosal barrier function in health and disease. Nat Rev Immunol. 2009;9:799–809. doi: 10.1038/nri2653. [DOI] [PubMed] [Google Scholar]
- 23.Van Itallie CM, Anderson JM. The molecular physiology of tight junction pores. Physiology (Bethesda) 2004;19:331–338. doi: 10.1152/physiol.00027.2004. [DOI] [PubMed] [Google Scholar]
- 24.Nusrat A, Turner JR, Madara JL. Molecular physiology and pathophysiology of tight junctions. IV. Regulation of tight junctions by extracellular stimuli: nutrients, cytokines, and immune cells. Am J Physiol Gastrointest Liver Physiol. 2000;279:G851–G857. doi: 10.1152/ajpgi.2000.279.5.G851. [DOI] [PubMed] [Google Scholar]
- 25.Shen L, Weber CR, Raleigh DR, Yu D, Turner JR. Tight junction pore and leak pathways: a dynamic duo. Annu Rev Physiol. 2011;73:283–309. doi: 10.1146/annurev-physiol-012110-142150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pijls KE, Jonkers DM, Elamin EE, Masclee AA, Koek GH. Intestinal epithelial barrier function in liver cirrhosis: an extensive review of the literature. Liver Int. 2013;33:1457–1469. doi: 10.1111/liv.12271. [DOI] [PubMed] [Google Scholar]
- 27.Assimakopoulos SF, Charonis AS. Uncovering the molecular events associated with increased intestinal permeability in liver cirrhosis: the pivotal role of enterocyte tight junctions and future perspectives. J Hepatol. 2013;59:1144–1146. doi: 10.1016/j.jhep.2013.06.031. [DOI] [PubMed] [Google Scholar]
- 28.Du Plessis J, Vanheel H, Janssen CE, Roos L, Slavik T, Stivaktas PI, Nieuwoudt M, van Wyk SG, Vieira W, Pretorius E, et al. Activated intestinal macrophages in patients with cirrhosis release NO and IL-6 that may disrupt intestinal barrier function. J Hepatol. 2013;58:1125–1132. doi: 10.1016/j.jhep.2013.01.038. [DOI] [PubMed] [Google Scholar]
- 29.Genescà J, Martí R, Rojo F, Campos F, Peribáñez V, Gónzalez A, Castells L, Ruiz-Marcellán C, Margarit C, Esteban R, et al. Increased tumour necrosis factor alpha production in mesenteric lymph nodes of cirrhotic patients with ascites. Gut. 2003;52:1054–1059. doi: 10.1136/gut.52.7.1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Muñoz L, Albillos A, Nieto M, Reyes E, Lledó L, Monserrat J, Sanz E, de la Hera A, Alvarez-Mon M. Mesenteric Th1 polarization and monocyte TNF-alpha production: first steps to systemic inflammation in rats with cirrhosis. Hepatology. 2005;42:411–419. doi: 10.1002/hep.20799. [DOI] [PubMed] [Google Scholar]
- 31.Ye D, Guo S, Al-Sadi R, Ma TY. MicroRNA regulation of intestinal epithelial tight junction permeability. Gastroenterology. 2011;141:1323–1333. doi: 10.1053/j.gastro.2011.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Such J, Guardiola JV, de Juan J, Casellas JA, Pascual S, Aparicio JR, Solá-Vera J, Pérez-Mateo M. Ultrastructural characteristics of distal duodenum mucosa in patients with cirrhosis. Eur J Gastroenterol Hepatol. 2002;14:371–376. doi: 10.1097/00042737-200204000-00006. [DOI] [PubMed] [Google Scholar]
- 33.Wen JB, Zhu FQ, Chen WG, Jiang LP, Chen J, Hu ZP, Huang YJ, Zhou ZW, Wang GL, Lin H, et al. Oxymatrine improves intestinal epithelial barrier function involving NF-κB-mediated signaling pathway in CCl4-induced cirrhotic rats. PLoS One. 2014;9:e106082. doi: 10.1371/journal.pone.0106082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kim YS, Ho SB. Intestinal goblet cells and mucins in health and disease: recent insights and progress. Curr Gastroenterol Rep. 2010;12:319–330. doi: 10.1007/s11894-010-0131-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Andrianifahanana M, Moniaux N, Batra SK. Regulation of mucin expression: mechanistic aspects and implications for cancer and inflammatory diseases. Biochim Biophys Acta. 2006;1765:189–222. doi: 10.1016/j.bbcan.2006.01.002. [DOI] [PubMed] [Google Scholar]
- 36.Li JD, Feng W, Gallup M, Kim JH, Gum J, Kim Y, Basbaum C. Activation of NF-kappaB via a Src-dependent Ras-MAPK-pp90rsk pathway is required for Pseudomonas aeruginosa-induced mucin overproduction in epithelial cells. Proc Natl Acad Sci USA. 1998;95:5718–5723. doi: 10.1073/pnas.95.10.5718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wiest R, Lawson M, Geuking M. Pathological bacterial translocation in liver cirrhosis. J Hepatol. 2014;60:197–209. doi: 10.1016/j.jhep.2013.07.044. [DOI] [PubMed] [Google Scholar]
- 38.Hartmann P, Chen P, Wang HJ, Wang L, McCole DF, Brandl K, Stärkel P, Belzer C, Hellerbrand C, Tsukamoto H, et al. Deficiency of intestinal mucin-2 ameliorates experimental alcoholic liver disease in mice. Hepatology. 2013;58:108–119. doi: 10.1002/hep.26321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Katayama M, Xu D, Specian RD, Deitch EA. Role of bacterial adherence and the mucus barrier on bacterial translocation: effects of protein malnutrition and endotoxin in rats. Ann Surg. 1997;225:317–326. doi: 10.1097/00000658-199703000-00012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Corradi F, Brusasco C, Fernández J, Vila J, Ramirez MJ, Seva-Pereira T, Fernández-Varo G, Mosbah IB, Acevedo J, Silva A, et al. Effects of pentoxifylline on intestinal bacterial overgrowth, bacterial translocation and spontaneous bacterial peritonitis in cirrhotic rats with ascites. Dig Liver Dis. 2012;44:239–244. doi: 10.1016/j.dld.2011.10.014. [DOI] [PubMed] [Google Scholar]
- 41.Mojzis J, Hegedüsová R, Mirossay L. Role of mucus in ischemia/reperfusion-induced gastric mucosal injury in rats. Physiol Res. 2000;49:441–446. [PubMed] [Google Scholar]
- 42.Bellot P, Francés R, Such J. Pathological bacterial translocation in cirrhosis: pathophysiology, diagnosis and clinical implications. Liver Int. 2013;33:31–39. doi: 10.1111/liv.12021. [DOI] [PubMed] [Google Scholar]
- 43.Round JL, Mazmanian SK. The gut microbiota shapes intestinal immune responses during health and disease. Nat Rev Immunol. 2009;9:313–323. doi: 10.1038/nri2515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hase K, Kawano K, Nochi T, Pontes GS, Fukuda S, Ebisawa M, Kadokura K, Tobe T, Fujimura Y, Kawano S, et al. Uptake through glycoprotein 2 of FimH(+) bacteria by M cells initiates mucosal immune response. Nature. 2009;462:226–230. doi: 10.1038/nature08529. [DOI] [PubMed] [Google Scholar]
- 45.Rescigno M, Urbano M, Valzasina B, Francolini M, Rotta G, Bonasio R, Granucci F, Kraehenbuhl JP, Ricciardi-Castagnoli P. Dendritic cells express tight junction proteins and penetrate gut epithelial monolayers to sample bacteria. Nat Immunol. 2001;2:361–367. doi: 10.1038/86373. [DOI] [PubMed] [Google Scholar]
- 46.Hartmann P, Haimerl M, Mazagova M, Brenner DA, Schnabl B. Toll-like receptor 2-mediated intestinal injury and enteric tumor necrosis factor receptor I contribute to liver fibrosis in mice. Gastroenterology. 2012;143:1330–1340.e1. doi: 10.1053/j.gastro.2012.07.099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Muñoz L, José Borrero M, Ubeda M, Lario M, Díaz D, Francés R, Monserrat J, Pastor O, Aguado-Fraile E, Such J, et al. Interaction between intestinal dendritic cells and bacteria translocated from the gut in rats with cirrhosis. Hepatology. 2012;56:1861–1869. doi: 10.1002/hep.25854. [DOI] [PubMed] [Google Scholar]
- 48.Albillos A, Lario M, Álvarez-Mon M. Cirrhosis-associated immune dysfunction: distinctive features and clinical relevance. J Hepatol. 2014;61:1385–1396. doi: 10.1016/j.jhep.2014.08.010. [DOI] [PubMed] [Google Scholar]
- 49.Knoop KA, Newberry RD. Isolated Lymphoid Follicles are Dynamic Reservoirs for the Induction of Intestinal IgA. Front Immunol. 2012;3:84. doi: 10.3389/fimmu.2012.00084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kirkland D, Benson A, Mirpuri J, Pifer R, Hou B, DeFranco AL, Yarovinsky F. B cell-intrinsic MyD88 signaling prevents the lethal dissemination of commensal bacteria during colonic damage. Immunity. 2012;36:228–238. doi: 10.1016/j.immuni.2011.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Doi H, Iyer TK, Carpenter E, Li H, Chang KM, Vonderheide RH, Kaplan DE. Dysfunctional B-cell activation in cirrhosis resulting from hepatitis C infection associated with disappearance of CD27-positive B-cell population. Hepatology. 2012;55:709–719. doi: 10.1002/hep.24689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gautreaux MD, Deitch EA, Berg RD. T lymphocytes in host defense against bacterial translocation from the gastrointestinal tract. Infect Immun. 1994;62:2874–2884. doi: 10.1128/iai.62.7.2874-2884.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Choudhry MA, Fazal N, Goto M, Gamelli RL, Sayeed MM. Gut-associated lymphoid T cell suppression enhances bacterial translocation in alcohol and burn injury. Am J Physiol Gastrointest Liver Physiol. 2002;282:G937–G947. doi: 10.1152/ajpgi.00235.2001. [DOI] [PubMed] [Google Scholar]
- 54.Vaishnava S, Behrendt CL, Ismail AS, Eckmann L, Hooper LV. Paneth cells directly sense gut commensals and maintain homeostasis at the intestinal host-microbial interface. Proc Natl Acad Sci USA. 2008;105:20858–20863. doi: 10.1073/pnas.0808723105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ayabe T, Satchell DP, Wilson CL, Parks WC, Selsted ME, Ouellette AJ. Secretion of microbicidal alpha-defensins by intestinal Paneth cells in response to bacteria. Nat Immunol. 2000;1:113–118. doi: 10.1038/77783. [DOI] [PubMed] [Google Scholar]
- 56.Ogle CK, Noel JG, Guo X, Wells DA, Valente JF, Ogle JD, Alexander JW. The ability of endotoxin-stimulated enterocytes to produce bactericidal factors. Crit Care Med. 2002;30:428–434. doi: 10.1097/00003246-200202000-00027. [DOI] [PubMed] [Google Scholar]
- 57.Teltschik Z, Wiest R, Beisner J, Nuding S, Hofmann C, Schoelmerich J, Bevins CL, Stange EF, Wehkamp J. Intestinal bacterial translocation in rats with cirrhosis is related to compromised Paneth cell antimicrobial host defense. Hepatology. 2012;55:1154–1163. doi: 10.1002/hep.24789. [DOI] [PubMed] [Google Scholar]
- 58.Cash HL, Whitham CV, Behrendt CL, Hooper LV. Symbiotic bacteria direct expression of an intestinal bactericidal lectin. Science. 2006;313:1126–1130. doi: 10.1126/science.1127119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mukherjee S, Partch CL, Lehotzky RE, Whitham CV, Chu H, Bevins CL, Gardner KH, Hooper LV. Regulation of C-type lectin antimicrobial activity by a flexible N-terminal prosegment. J Biol Chem. 2009;284:4881–4888. doi: 10.1074/jbc.M808077200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Spaeth G, Gottwald T, Specian RD, Mainous MR, Berg RD, Deitch EA. Secretory immunoglobulin A, intestinal mucin, and mucosal permeability in nutritionally induced bacterial translocation in rats. Ann Surg. 1994;220:798–808. doi: 10.1097/00000658-199412000-00014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Saitoh O, Sugi K, Lojima K, Matsumoto H, Nakagawa K, Kayazawa M, Tanaka S, Teranishi T, Hirata I. Increased prevalence of intestinal inflammation in patients with liver cirrhosis. World J Gastroenterol. 1999;5:391–396. doi: 10.3748/wjg.v5.i5.391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ewaschuk J, Endersby R, Thiel D, Diaz H, Backer J, Ma M, Churchill T, Madsen K. Probiotic bacteria prevent hepatic damage and maintain colonic barrier function in a mouse model of sepsis. Hepatology. 2007;46:841–850. doi: 10.1002/hep.21750. [DOI] [PubMed] [Google Scholar]
- 63.Coant N, Simon-Rudler M, Gustot T, Fasseu M, Gandoura S, Ragot K, Abdel-Razek W, Thabut D, Lettéron P, Ogier-Denis E, et al. Glycogen synthase kinase 3 involvement in the excessive proinflammatory response to LPS in patients with decompensated cirrhosis. J Hepatol. 2011;55:784–793. doi: 10.1016/j.jhep.2010.12.039. [DOI] [PubMed] [Google Scholar]
- 64.Gómez-Hurtado I, Moratalla A, Moya-Pérez Á, Peiró G, Zapater P, González-Navajas JM, Giménez P, Such J, Sanz Y, Francés R. Role of interleukin 10 in norfloxacin prevention of luminal free endotoxin translocation in mice with cirrhosis. J Hepatol. 2014;61:799–808. doi: 10.1016/j.jhep.2014.05.031. [DOI] [PubMed] [Google Scholar]
- 65.Lorenzo-Zúñiga V, Rodríguez-Ortigosa CM, Bartolí R, Martínez-Chantar ML, Martínez-Peralta L, Pardo A, Ojanguren I, Quiroga J, Planas R, Prieto J. Insulin-like growth factor I improves intestinal barrier function in cirrhotic rats. Gut. 2006;55:1306–1312. doi: 10.1136/gut.2005.079988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Jalan R, Fernandez J, Wiest R, Schnabl B, Moreau R, Angeli P, Stadlbauer V, Gustot T, Bernardi M, Canton R, et al. Bacterial infections in cirrhosis: a position statement based on the EASL Special Conference 2013. J Hepatol. 2014;60:1310–1324. doi: 10.1016/j.jhep.2014.01.024. [DOI] [PubMed] [Google Scholar]
- 67.Bauer TM, Schwacha H, Steinbrückner B, Brinkmann FE, Ditzen AK, Aponte JJ, Pelz K, Berger D, Kist M, Blum HE. Small intestinal bacterial overgrowth in human cirrhosis is associated with systemic endotoxemia. Am J Gastroenterol. 2002;97:2364–2370. doi: 10.1111/j.1572-0241.2002.05791.x. [DOI] [PubMed] [Google Scholar]
- 68.Morencos FC, de las Heras Castaño G, Martín Ramos L, López Arias MJ, Ledesma F, Pons Romero F. Small bowel bacterial overgrowth in patients with alcoholic cirrhosis. Dig Dis Sci. 1995;40:1252–1256. doi: 10.1007/BF02065533. [DOI] [PubMed] [Google Scholar]
- 69.Yang CY, Chang CS, Chen GH. Small-intestinal bacterial overgrowth in patients with liver cirrhosis, diagnosed with glucose H2 or CH4 breath tests. Scand J Gastroenterol. 1998;33:867–871. doi: 10.1080/00365529850171549. [DOI] [PubMed] [Google Scholar]
- 70.Chang CS, Yang SS, Kao CH, Yeh HZ, Chen GH. Small intestinal bacterial overgrowth versus antimicrobial capacity in patients with spontaneous bacterial peritonitis. Scand J Gastroenterol. 2001;36:92–96. doi: 10.1080/00365520150218110. [DOI] [PubMed] [Google Scholar]
- 71.Jun DW, Kim KT, Lee OY, Chae JD, Son BK, Kim SH, Jo YJ, Park YS. Association between small intestinal bacterial overgrowth and peripheral bacterial DNA in cirrhotic patients. Dig Dis Sci. 2010;55:1465–1471. doi: 10.1007/s10620-009-0870-9. [DOI] [PubMed] [Google Scholar]
- 72.Chang CS, Chen GH, Lien HC, Yeh HZ. Small intestine dysmotility and bacterial overgrowth in cirrhotic patients with spontaneous bacterial peritonitis. Hepatology. 1998;28:1187–1190. doi: 10.1002/hep.510280504. [DOI] [PubMed] [Google Scholar]
- 73.Pérez-Paramo M, Muñoz J, Albillos A, Freile I, Portero F, Santos M, Ortiz-Berrocal J. Effect of propranolol on the factors promoting bacterial translocation in cirrhotic rats with ascites. Hepatology. 2000;31:43–48. doi: 10.1002/hep.510310109. [DOI] [PubMed] [Google Scholar]
- 74.Gómez-Hurtado I, Santacruz A, Peiró G, Zapater P, Gutiérrez A, Pérez-Mateo M, Sanz Y, Francés R. Gut microbiota dysbiosis is associated with inflammation and bacterial translocation in mice with CCl4-induced fibrosis. PLoS One. 2011;6:e23037. doi: 10.1371/journal.pone.0023037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Guarner C, Runyon BA, Young S, Heck M, Sheikh MY. Intestinal bacterial overgrowth and bacterial translocation in cirrhotic rats with ascites. J Hepatol. 1997;26:1372–1378. doi: 10.1016/s0168-8278(97)80474-6. [DOI] [PubMed] [Google Scholar]
- 76.Steffen EK, Berg RD. Relationship between cecal population levels of indigenous bacteria and translocation to the mesenteric lymph nodes. Infect Immun. 1983;39:1252–1259. doi: 10.1128/iai.39.3.1252-1259.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Thalheimer U, De Iorio F, Capra F, del Mar Lleo M, Zuliani V, Ghidini V, Tafi MC, Caburlotto G, Gennari M, Burroughs AK, et al. Altered intestinal function precedes the appearance of bacterial DNA in serum and ascites in patients with cirrhosis: a pilot study. Eur J Gastroenterol Hepatol. 2010;22:1228–1234. doi: 10.1097/MEG.0b013e32833b4b03. [DOI] [PubMed] [Google Scholar]
- 78.Sánchez E, Casafont F, Guerra A, de Benito I, Pons-Romero F. Role of intestinal bacterial overgrowth and intestinal motility in bacterial translocation in experimental cirrhosis. Rev Esp Enferm Dig. 2005;97:805–814. doi: 10.4321/s1130-01082005001100005. [DOI] [PubMed] [Google Scholar]
- 79.Sandler NG, Koh C, Roque A, Eccleston JL, Siegel RB, Demino M, Kleiner DE, Deeks SG, Liang TJ, Heller T, et al. Host response to translocated microbial products predicts outcomes of patients with HBV or HCV infection. Gastroenterology. 2011;141:1220–130, 1220-130. doi: 10.1053/j.gastro.2011.06.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Seki E, De Minicis S, Osterreicher CH, Kluwe J, Osawa Y, Brenner DA, Schwabe RF. TLR4 enhances TGF-beta signaling and hepatic fibrosis. Nat Med. 2007;13:1324–1332. doi: 10.1038/nm1663. [DOI] [PubMed] [Google Scholar]
- 81.Pikarsky E, Porat RM, Stein I, Abramovitch R, Amit S, Kasem S, Gutkovich-Pyest E, Urieli-Shoval S, Galun E, Ben-Neriah Y. NF-kappaB functions as a tumour promoter in inflammation-associated cancer. Nature. 2004;431:461–466. doi: 10.1038/nature02924. [DOI] [PubMed] [Google Scholar]
- 82.Maeda S, Kamata H, Luo JL, Leffert H, Karin M. IKKbeta couples hepatocyte death to cytokine-driven compensatory proliferation that promotes chemical hepatocarcinogenesis. Cell. 2005;121:977–990. doi: 10.1016/j.cell.2005.04.014. [DOI] [PubMed] [Google Scholar]
- 83.Darnaud M, Faivre J, Moniaux N. Targeting gut flora to prevent progression of hepatocellular carcinoma. J Hepatol. 2013;58:385–387. doi: 10.1016/j.jhep.2012.08.019. [DOI] [PubMed] [Google Scholar]
- 84.Dapito DH, Mencin A, Gwak GY, Pradere JP, Jang MK, Mederacke I, Caviglia JM, Khiabanian H, Adeyemi A, Bataller R, et al. Promotion of hepatocellular carcinoma by the intestinal microbiota and TLR4. Cancer Cell. 2012;21:504–516. doi: 10.1016/j.ccr.2012.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Zhang HL, Yu LX, Yang W, Tang L, Lin Y, Wu H, Zhai B, Tan YX, Shan L, Liu Q, et al. Profound impact of gut homeostasis on chemically-induced pro-tumorigenic inflammation and hepatocarcinogenesis in rats. J Hepatol. 2012;57:803–812. doi: 10.1016/j.jhep.2012.06.011. [DOI] [PubMed] [Google Scholar]
- 86.Fox JG, Feng Y, Theve EJ, Raczynski AR, Fiala JL, Doernte AL, Williams M, McFaline JL, Essigmann JM, Schauer DB, et al. Gut microbes define liver cancer risk in mice exposed to chemical and viral transgenic hepatocarcinogens. Gut. 2010;59:88–97. doi: 10.1136/gut.2009.183749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Endo H, Niioka M, Kobayashi N, Tanaka M, Watanabe T. Butyrate-producing probiotics reduce nonalcoholic fatty liver disease progression in rats: new insight into the probiotics for the gut-liver axis. PLoS One. 2013;8:e63388. doi: 10.1371/journal.pone.0063388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Merli M, Lucidi C, Pentassuglio I, Giannelli V, Giusto M, Di Gregorio V, Pasquale C, Nardelli S, Lattanzi B, Venditti M, et al. Increased risk of cognitive impairment in cirrhotic patients with bacterial infections. J Hepatol. 2013;59:243–250. doi: 10.1016/j.jhep.2013.03.012. [DOI] [PubMed] [Google Scholar]
- 89.Chavarria L, Oria M, Romero-Giménez J, Alonso J, Lope-Piedrafita S, Cordoba J. Brain magnetic resonance in experimental acute-on-chronic liver failure. Liver Int. 2013;33:294–300. doi: 10.1111/liv.12032. [DOI] [PubMed] [Google Scholar]
- 90.Bajaj JS, Ridlon JM, Hylemon PB, Thacker LR, Heuman DM, Smith S, Sikaroodi M, Gillevet PM. Linkage of gut microbiome with cognition in hepatic encephalopathy. Am J Physiol Gastrointest Liver Physiol. 2012;302:G168–G175. doi: 10.1152/ajpgi.00190.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Zhang Z, Zhai H, Geng J, Yu R, Ren H, Fan H, Shi P. Large-scale survey of gut microbiota associated with MHE Via 16S rRNA-based pyrosequencing. Am J Gastroenterol. 2013;108:1601–1611. doi: 10.1038/ajg.2013.221. [DOI] [PubMed] [Google Scholar]
- 92.Liu Q, Duan ZP, Ha DK, Bengmark S, Kurtovic J, Riordan SM. Synbiotic modulation of gut flora: effect on minimal hepatic encephalopathy in patients with cirrhosis. Hepatology. 2004;39:1441–1449. doi: 10.1002/hep.20194. [DOI] [PubMed] [Google Scholar]
- 93.Nieto JC, Sánchez E, Román E, Vidal S, Oliva L, Guarner-Argente C, Poca M, Torras X, Juárez C, Guarner C, et al. Cytokine production in patients with cirrhosis and TLR4 polymorphisms. World J Gastroenterol. 2014;20:17516–17524. doi: 10.3748/wjg.v20.i46.17516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Goulis J, Patch D, Burroughs AK. Bacterial infection in the pathogenesis of variceal bleeding. Lancet. 1999;353:139–142. doi: 10.1016/S0140-6736(98)06020-6. [DOI] [PubMed] [Google Scholar]
- 95.Bleichner G, Boulanger R, Squara P, Sollet JP, Parent A. Frequency of infections in cirrhotic patients presenting with acute gastrointestinal haemorrhage. Br J Surg. 1986;73:724–726. doi: 10.1002/bjs.1800730916. [DOI] [PubMed] [Google Scholar]
- 96.Reiberger T, Ferlitsch A, Payer BA, Mandorfer M, Heinisch BB, Hayden H, Lammert F, Trauner M, Peck-Radosavljevic M, Vogelsang H. Non-selective betablocker therapy decreases intestinal permeability and serum levels of LBP and IL-6 in patients with cirrhosis. J Hepatol. 2013;58:911–921. doi: 10.1016/j.jhep.2012.12.011. [DOI] [PubMed] [Google Scholar]
- 97.Bernard B, Cadranel JF, Valla D, Escolano S, Jarlier V, Opolon P. Prognostic significance of bacterial infection in bleeding cirrhotic patients: a prospective study. Gastroenterology. 1995;108:1828–1834. doi: 10.1016/0016-5085(95)90146-9. [DOI] [PubMed] [Google Scholar]
- 98.Goulis J, Armonis A, Patch D, Sabin C, Greenslade L, Burroughs AK. Bacterial infection is independently associated with failure to control bleeding in cirrhotic patients with gastrointestinal hemorrhage. Hepatology. 1998;27:1207–1212. doi: 10.1002/hep.510270504. [DOI] [PubMed] [Google Scholar]
- 99.Bernard B, Grangé JD, Khac EN, Amiot X, Opolon P, Poynard T. Antibiotic prophylaxis for the prevention of bacterial infections in cirrhotic patients with gastrointestinal bleeding: a meta-analysis. Hepatology. 1999;29:1655–1661. doi: 10.1002/hep.510290608. [DOI] [PubMed] [Google Scholar]
- 100.Hou MC, Lin HC, Liu TT, Kuo BI, Lee FY, Chang FY, Lee SD. Antibiotic prophylaxis after endoscopic therapy prevents rebleeding in acute variceal hemorrhage: a randomized trial. Hepatology. 2004;39:746–753. doi: 10.1002/hep.20126. [DOI] [PubMed] [Google Scholar]
- 101.Brown MR, Jones G, Nash KL, Wright M, Guha IN. Antibiotic prophylaxis in variceal hemorrhage: timing, effectiveness and Clostridium difficile rates. World J Gastroenterol. 2010;16:5317–5323. doi: 10.3748/wjg.v16.i42.5317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Lee YY, Tee HP, Mahadeva S. Role of prophylactic antibiotics in cirrhotic patients with variceal bleeding. World J Gastroenterol. 2014;20:1790–1796. doi: 10.3748/wjg.v20.i7.1790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Zhang HY, Han DW, Wang XG, Zhao YC, Zhou X, Zhao HZ. Experimental study on the role of endotoxin in the development of hepatopulmonary syndrome. World J Gastroenterol. 2005;11:567–572. doi: 10.3748/wjg.v11.i4.567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Zhang HY, Han de W, Su AR, Zhang LT, Zhao ZF, Ji JQ, Li BH, Ji C. Intestinal endotoxemia plays a central role in development of hepatopulmonary syndrome in a cirrhotic rat model induced by multiple pathogenic factors. World J Gastroenterol. 2007;13:6385–6395. doi: 10.3748/wjg.v13.i47.6385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Rabiller A, Nunes H, Lebrec D, Tazi KA, Wartski M, Dulmet E, Libert JM, Mougeot C, Moreau R, Mazmanian M, et al. Prevention of gram-negative translocation reduces the severity of hepatopulmonary syndrome. Am J Respir Crit Care Med. 2002;166:514–517. doi: 10.1164/rccm.200201-027OC. [DOI] [PubMed] [Google Scholar]
- 106.Ng CK, Chan MH, Tai MH, Lam CW. Hepatorenal syndrome. Clin Biochem Rev. 2007;28:11–17. [PMC free article] [PubMed] [Google Scholar]
- 107.Ginès P, Arroyo V. Hepatorenal syndrome. J Am Soc Nephrol. 1999;10:1833–1839. doi: 10.1681/ASN.V1081833. [DOI] [PubMed] [Google Scholar]
- 108.Ginès P, Schrier RW. Renal failure in cirrhosis. N Engl J Med. 2009;361:1279–1290. doi: 10.1056/NEJMra0809139. [DOI] [PubMed] [Google Scholar]
- 109.Newby DE, Hayes PC. Hyperdynamic circulation in liver cirrhosis: not peripheral vasodilatation but ‘splanchnic steal’. QJM. 2002;95:827–830. doi: 10.1093/qjmed/95.12.827. [DOI] [PubMed] [Google Scholar]
- 110.Tristani FE, Cohn JN. Systemic and renal hemodynamics in oliguric hepatic failure: effect of volume expansion. J Clin Invest. 1967;46:1894–1906. doi: 10.1172/JCI105679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Navasa M, Follo A, Filella X, Jiménez W, Francitorra A, Planas R, Rimola A, Arroyo V, Rodés J. Tumor necrosis factor and interleukin-6 in spontaneous bacterial peritonitis in cirrhosis: relationship with the development of renal impairment and mortality. Hepatology. 1998;27:1227–1232. doi: 10.1002/hep.510270507. [DOI] [PubMed] [Google Scholar]
- 112.Fasolato S, Angeli P, Dallagnese L, Maresio G, Zola E, Mazza E, Salinas F, Donà S, Fagiuoli S, Sticca A, et al. Renal failure and bacterial infections in patients with cirrhosis: epidemiology and clinical features. Hepatology. 2007;45:223–229. doi: 10.1002/hep.21443. [DOI] [PubMed] [Google Scholar]
- 113.Terra C, Guevara M, Torre A, Gilabert R, Fernández J, Martín-Llahí M, Baccaro ME, Navasa M, Bru C, Arroyo V, et al. Renal failure in patients with cirrhosis and sepsis unrelated to spontaneous bacterial peritonitis: value of MELD score. Gastroenterology. 2005;129:1944–1953. doi: 10.1053/j.gastro.2005.09.024. [DOI] [PubMed] [Google Scholar]
- 114.Barreto R, Fagundes C, Guevara M, Solà E, Pereira G, Rodríguez E, Graupera I, Martín-Llahí M, Ariza X, Cárdenas A, et al. Type-1 hepatorenal syndrome associated with infections in cirrhosis: natural history, outcome of kidney function, and survival. Hepatology. 2014;59:1505–1513. doi: 10.1002/hep.26687. [DOI] [PubMed] [Google Scholar]
- 115.Wong F, O’Leary JG, Reddy KR, Patton H, Kamath PS, Fallon MB, Garcia-Tsao G, Subramanian RM, Malik R, Maliakkal B, et al. New consensus definition of acute kidney injury accurately predicts 30-day mortality in patients with cirrhosis and infection. Gastroenterology. 2013;145:1280–8.e1. doi: 10.1053/j.gastro.2013.08.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Kalambokis GN, Mouzaki A, Rodi M, Pappas K, Fotopoulos A, Xourgia X, Tsianos EV. Rifaximin improves systemic hemodynamics and renal function in patients with alcohol-related cirrhosis and ascites. Clin Gastroenterol Hepatol. 2012;10:815–818. doi: 10.1016/j.cgh.2012.02.025. [DOI] [PubMed] [Google Scholar]
- 117.Bruns T, Zimmermann HW, Stallmach A. Risk factors and outcome of bacterial infections in cirrhosis. World J Gastroenterol. 2014;20:2542–2554. doi: 10.3748/wjg.v20.i10.2542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Kim BI, Kim HJ, Park JH, Park DI, Cho YK, Sohn CI, Jeon WK, Kim HS, Kim DJ. Increased intestinal permeability as a predictor of bacterial infections in patients with decompensated liver cirrhosis and hemorrhage. J Gastroenterol Hepatol. 2011;26:550–557. doi: 10.1111/j.1440-1746.2010.06490.x. [DOI] [PubMed] [Google Scholar]
- 119.van Vlerken LG, Huisman EJ, van Hoek B, Renooij W, de Rooij FW, Siersema PD, van Erpecum KJ. Bacterial infections in cirrhosis: role of proton pump inhibitors and intestinal permeability. Eur J Clin Invest. 2012;42:760–767. doi: 10.1111/j.1365-2362.2011.02643.x. [DOI] [PubMed] [Google Scholar]
- 120.Rimola A, Soto R, Bory F, Arroyo V, Piera C, Rodes J. Reticuloendothelial system phagocytic activity in cirrhosis and its relation to bacterial infections and prognosis. Hepatology. 1984;4:53–58. doi: 10.1002/hep.1840040109. [DOI] [PubMed] [Google Scholar]
- 121.Llovet JM, Bartolí R, Planas R, Cabré E, Jimenez M, Urban A, Ojanguren I, Arnal J, Gassull MA. Bacterial translocation in cirrhotic rats. Its role in the development of spontaneous bacterial peritonitis. Gut. 1994;35:1648–1652. doi: 10.1136/gut.35.11.1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Appenrodt B, Grünhage F, Gentemann MG, Thyssen L, Sauerbruch T, Lammert F. Nucleotide-binding oligomerization domain containing 2 (NOD2) variants are genetic risk factors for death and spontaneous bacterial peritonitis in liver cirrhosis. Hepatology. 2010;51:1327–1333. doi: 10.1002/hep.23440. [DOI] [PubMed] [Google Scholar]
- 123.Bruns T, Peter J, Reuken PA, Grabe DH, Schuldes SR, Brenmoehl J, Schölmerich J, Wiest R, Stallmach A. NOD2 gene variants are a risk factor for culture-positive spontaneous bacterial peritonitis and monomicrobial bacterascites in cirrhosis. Liver Int. 2012;32:223–230. doi: 10.1111/j.1478-3231.2011.02561.x. [DOI] [PubMed] [Google Scholar]
- 124.Bajaj JS, Zadvornova Y, Heuman DM, Hafeezullah M, Hoffmann RG, Sanyal AJ, Saeian K. Association of proton pump inhibitor therapy with spontaneous bacterial peritonitis in cirrhotic patients with ascites. Am J Gastroenterol. 2009;104:1130–1134. doi: 10.1038/ajg.2009.80. [DOI] [PubMed] [Google Scholar]
- 125.Trikudanathan G, Israel J, Cappa J, O’Sullivan DM. Association between proton pump inhibitors and spontaneous bacterial peritonitis in cirrhotic patients - a systematic review and meta-analysis. Int J Clin Pract. 2011;65:674–678. doi: 10.1111/j.1742-1241.2011.02650.x. [DOI] [PubMed] [Google Scholar]
- 126.Terg R, Casciato P, Garbe C, Cartier M, Stieben T, Mendizabal M, Niveyro C, Benavides J, Marino M, Colombato L, et al. Proton pump inhibitor therapy does not increase the incidence of spontaneous bacterial peritonitis in cirrhosis: a multicenter prospective study. J Hepatol. 2015;62:1056–1060. doi: 10.1016/j.jhep.2014.11.036. [DOI] [PubMed] [Google Scholar]
- 127.Senzolo M, Cholongitas E, Burra P, Leandro G, Thalheimer U, Patch D, Burroughs AK. beta-Blockers protect against spontaneous bacterial peritonitis in cirrhotic patients: a meta-analysis. Liver Int. 2009;29:1189–1193. doi: 10.1111/j.1478-3231.2009.02038.x. [DOI] [PubMed] [Google Scholar]