

Abstract
In recent years, mGlu4 has received great attention and research effort because of the potential benefits of mGlu4 activation in treating numerous brain disorders, such as Parkinson’s disease (PD). Many positive allosteric modulators of mGlu4 have been developed. To better understand the role of mGlu4 in healthy and disease conditions, we are interested in developing an mGlu4 selective radioligand for in vivo studies. Thus, we had synthesized and studied [11C]2 as a PET tracer for mGlu4, which demonstrated some promising features as a PET radioligand as well as the limitation need to be improved. In order to develop an mGlu4 ligand with enhanced affinity and improved metabolic stability, we have modified, synthesized and evaluated a series of new N-phenylpicolinamide derivatives. The SAR study has discovered a number of compounds with low nM affinity to mGlu4. The dideuteriumfluoromethoxy modified compound 24 is identified as a very promising mGlu4 ligand, which has demonstrated enhanced affinity, improved in vitro microsomal stability, good selectivity and good permeability.
Keywords: metabotropic glutamate receptor subtype 4 (mGlu4), positive allosteric modulator (PAM), positron emission tomography (PET), affinity, metabolic stability, structure-affinity relationship (SAR)
Graphical Abstract
L-Glutamate is the most abundant excitatory neurotransmitter in the CNS (Central nerve system) of vertebrates and probably mediates more than 50% of all synapses.1,2 Two major classes of receptors, mGlu and iGlu, are involved in glutamate signal transfer. The mGlu belong to Class C of the GPCR (G protein-coupled receptor) super family, which are thought to exist as dimers and have a distinct large extracellular N-terminus. This extracellular N-terminal domain contains two hinged globular domains referred as the Venus Flytrap Domain (VFD), which is the orthosteric binding site for the endogenous ligand, L-glutamate.3 The mGlu can be further divided into three subgroups including eight known receptor subtypes (group I: mGlu1 and mGlu5, group II: mGlu2 and mGlu3, and group III: mGlu4, mGlu6, mGlu7 and mGlu8) based on their structural similarity, ligand specificity, and preferred coupling mechanisms.4 The mGlu are involved in glutamate signaling in almost every excitatory synapse in CNS, and they have distinctive biodistribution in CNS depending on subtypes and subgroups.5 In recent years, mGlu4 has received great attention and research effort because of the potential benefits of mGlu4 activation in treating numerous brain disorders, such as Parkinson’s disease (PD).6,7 As a group III mGlu, mGlu4 interacts with the Gαi/o subunit of G-protein which negatively couples with adenylate cyclase to inhibit cAMP dependent signal pathways.8,9 The mGlu4 is expressed at multiple synapses throughout the basal ganglia, mainly localized presynaptically and expressed in the striatum, hippocampus, thalamus, and cerebellum.4,10,11 Its activation reduces neurotransmitter release, a mechanism implicated in the pathophysiology of PD. The activation of the mGlu4 receptor can be accomplished by two different mechanisms: orthosteric agonists (competing with L-glutamate) or noncompetitive positive allosteric modulators (PAMs). Most orthosteric ligands of mGlu4 made in the past lack clear subtype selectivity and BBB (Blood-brain barrier) penetration, but notable examples exist of selective and brain penetrant orthosteric agonists, such as LSP4-2022.12,13 Much recent effort has been focused on the development of allosteric modulators, which target the seven-transmembrane spanning domain. In particular, the allosteric modulation of mGlu4 has spurred intense interest after (−)-PHCCC (1, N-phenyl-7-(hydroxyimino)-cyclopropa[b]chromen-1a-carboxamide), a partially selective mGlu4 PAM, was discovered and demonstrated activity in models of neuroprotection and PD. Since then there has been substantial progress in identifying PAMs for mGlu4.6,14,15 Figure 1 shows some representative mGlu4 PAMs.6,14,16,17,18,19,20 Subsequent results with PAMs of mGlu4 have further validated the antiparkinsonian activity in animal models of PD,11,17,21,22,23,24 in which this approach has opened a new avenue for developing nondopaminergic treatments for PD and for identifying a novel disease modifying therapeutics.
Figure 1.

Some representative mGlu4 PAMs.
To better understand the role of mGlu4 in healthy and disease conditions, we are interested in developing an mGlu4 selective radioligand for in vivo study. As a noninvasive medical imaging technique and a powerful tool in neurological research, positron emission tomography (PET) offers a possibility to visualize and analyze the target receptor expression under physiological and pathophysiological conditions. PET is being applied more often to detect disease-related biochemical changes before the disease-associated anatomical changes could be found by standard medical imaging modalities. Moreover, PET tracers serve as invaluable biomarkers during the development of potential therapeutic drugs. Thus, extensive research efforts have been directed toward the development of PET radioligands suitable for probing mGlu such as mGlu1 and mGlu5.15
Recently, we have reported a carbon-11 labeled PET ligand [11C]2 (N-(4-Chloro-3-[11C]methoxyphenyl)picolinamide)25, which was based on a reported mGlu4 PAM 216. In 2009, two research groups at Addex Pharma26 and Vanderbilt University16 have independently disclosed a series of small arylamide compounds as a new class of mGlu4 PAMs. Engers et al. found from a high-throughput screening there were a number of small arylamide compounds having mGlu4 PAM activity. They reported the SAR study, in vitro pharmacokinetic (PK) parameters and in vivo rat PK, which included the SAR results for sixteen N-phenylpicolinamide derivatives.16 Compounds 2 and 3 were the most potent mGlu4 PAMs in this series and showed some potentially suitable properties for PET tracer development, which include: 1) Rapid penetration into rat brain following intraperitoneal injection (Tmax for brain: 0.5 h); 2) High brain:plasma (B/P) partition coefficients for both compounds (B/P=4.1 for 2 and 9.9 for 3), in which B/P was determined by AUC0–8h, Brain/AUC0–8h, Plasma; 3) Good in vitro potency and efficacy for both human and rat mGlu4 compared to previous reported mGlu4 PAM; 4) Good selectivity over other mGlu subtypes; 5) Compound 2 was the first mGlu4 PAM to demonstrate efficacy in a preclinical rodent model of motor impairments associated with PD.6 Thus, we had synthesized and studied [11C]2 as a PET tracers for mGlu4. This compound demonstrated some promising features as a PET radioligand such as the fast uptake into brain and the specific accumulation in mGlu4-rich regions of the brain. However, in comparison to one of the best mGlu5 PET tracer [18F]FPEB (3-[18F]fluoro-5-(2-pyridinylethynyl)benzonitrile)27,28, [11C]2 showed the decreased retention time in the brain, which may affect the quality of the imaging. The results indicate that the affinity and metabolic stability of this class of tracers need further optimization. We report here the synthesis and structure-affinity relationship study of new N-phenylpicolinamide derivatives to develop mGlu4 ligands with improved affinity and metabolic stability.
We have modified and synthesized a series of new N-phenylpicolinamide derivatives for SAR study, in which the syntheses are shown in Scheme 1 – 3 (see Supplementary data). Three most active known compounds in this series (2, 3, and 10) were also synthesized and evaluated as the reference compounds for optimization. On the basis of previous SAR results16, we modified compound 2 at three positions (3- or 4-phenyl, 6-pyrindyl) as illustrated in Figure 2. It is known that the SAR of this series was tight,6,16 so we started with minor modifications based on the reported data. As shown in Table 1, the modifications include the isosteric replacement of hydrogen by fluorine or deuterium, oxygen by sulfur, methoxy by cyano group and change for different halogen atoms. The radiolabeling strategy was also considered in lead optimization design to generate the facile labeling positions for either C-11 or F-18 tracer.
Scheme 1.
Synthesis of the N-phenylpicolinamide derivatives.
Reagents and conditions: (a) for carboxylic acids, EDC.HCl, HOBt.H2O, DIPEA, dioxane; (b) for carboxylic acids, 1. thionyl chloride, benzene, reflux for 2 h; 2. TEA, THF, 40 °C, 1 h; (c) for acid chloride, DIPEA, CH2Cl2, 4h.
Scheme 3.
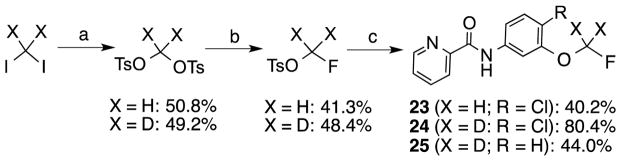
Synthesis of the N-(3-fluoromethoxyphenyl)picolinamides 23 – 25.
Reagents and conditions: (a) Ag(OTs), MeCN, reflux, overnight; (b) CsF, HO(CH2O)6H, reflux, 3.5 h; (c) N-(4-R-3-hydroxyphenyl)picolinamide, K2CO3, 40–50 °C, 3 days.
Figure 2.
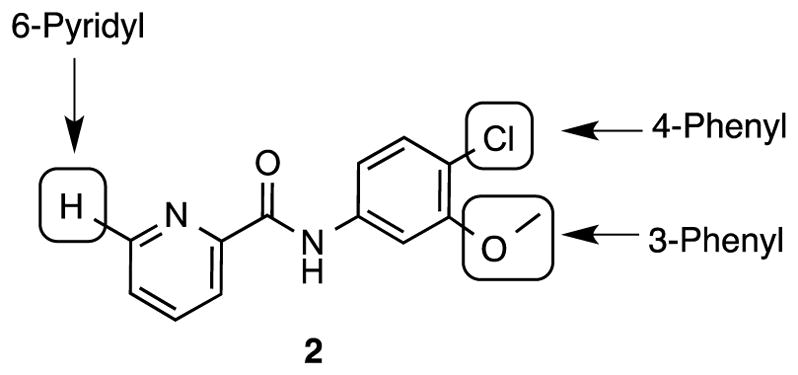
Modificatons on compound 2
Table 1.
SAR of N-phenylpicolinamide derivatives.
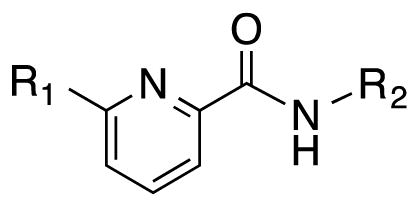
| |||||||
|---|---|---|---|---|---|---|---|
| Compd | R1 | R2 | MW | CLogP | tPSA | Affinity IC50 (nM) | Log(IC50 ± SE) |
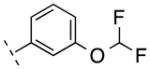
| 2 | H |
|
262.69 | 2.80 | 50.69 | 5.1 | −8.29 ± 0.09 |
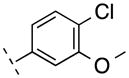
| 3 | H |
|
264.23 | 2.67 | 50.69 | 4.6 | −8.34 ± 0.08 |
| 10 | H |
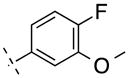
|
246.24 | 2.26 | 50.69 | 31.6 | −7.50 ± 0.09 |
| 11 | H |
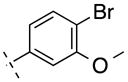
|
307.15 | 2.95 | 50.69 | 322 | −6.49 ± 0.08 |
| 12 | H |

|
354.15 | 3.16 | 50.69 | 146 | −6.84 ± 0.10 |
| 13 | H |
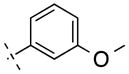
|
228.25 | 2.22 | 50.69 | 13.7 | −7.86 ± 0.10 |
| 14 | H |
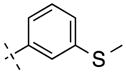
|
244.31 | 2.81 | 41.46 | 4.9 | −8.31 ± 0.07 |
| 15 | F |
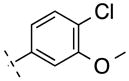
|
280.68 | 2.96 | 50.69 | 7.3 | −8.13 ± 0.11 |
| 16 | F |
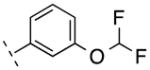
|
282.22 | 2.85 | 50.69 | 6.7 | −8.18 ± 0.10 |
| 17 | F |
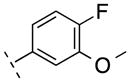
|
264.23 | 2.42 | 50.69 | 89.2 | −7.05 ± 0.10 |
| 18 | H |
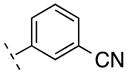
|
223.23 | 1.89 | 65.25 | 10.4 | −7.98 ± 0.05 |
| 19 | H |
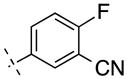
|
241.23 | 2.06 | 65.25 | 7.4 | −8.13 ± 0.04 |
| 20 | H |
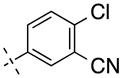
|
257.68 | 2.50 | 65.25 | 5.3 | −8.28 ± 0.04 |
| 21 | H |
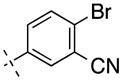
|
302.13 | 2.78 | 65.25 | 47 | −7.33 ± 0.08 |
| 22 | H |
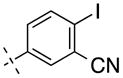
|
349.13 | 3.04 | 65.25 | 172 | −6.77 ± 0.08 |
| 23 | H |

|
280.68 | 2.97 | 50.69 | 3.2 | −8.49 ± 0.07 |
| 24 | H |
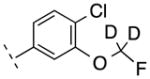
|
282.70 | 2.97 | 50.69 | 3.2 | −8.49 ± 0.11 |
| 25 | H |
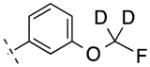
|
248.25 | 2.39 | 50.69 | 3.7 | −8.43 ± 0.03 |
Since poor BBB permeability and high nonspecific binding (NSB) are among the most frequent causes for failure in CNS PET ligand development, it is necessary to consider some important physicochemical parameter such as MW, ClogP and tPSA at the design stage. It has been recently proposed that more desirable ranges for CNS drugs are ClogP <3, MW < 360 and 40 < tPSA < 90.29 As shown in Table 1, all compounds except 12 and 22 possess the favorable physicochemical parameters, making them ideal candidates for CNS ligand development.
The lead compounds 2 and 3 were identified as mGlu4 PAMs by using functional assays (calcium mobilization assays for human mGlu4 and thallium flux assays for rat mGlu4) and characterized with EC50, the maximum response and the fold shift values.16 It is known that the EC50 value may not always correlated closely to the affinity value for PAM.30 It is very important to study the binding affinity for developing PET ligands. Thus, we prepared the tritium-labeled compound 2 ([3H]2, N-(4-chloro-3-(methoxy-t3)phenyl)picolinamide) for competitive binding assay.31 The synthesized compounds were characterized with competitive binding studies using mGlu4 transfected CHO cells by increasing the concentration of test materials from 0.01 nM to 10 μM in presence of 2 nM of [3H]2, in which the binding affinities to mGlu4 were described as IC50 values (Table 1).32
In structure-affinity study, we first evaluated the substitutions at the 4-phenyl position by keeping the 3-methoxy group constant. The 4-phenyl position of N-phenylpicolinamide was tolerated with some substitutions as demonstrated in known compounds 6–8, in which compounds 6 and 7 were reported very potent but poor brain penetration.20 Thus we limited the 4-phenyl substitutions for different halogens. The results show that the 4-chloro substitution give the best affinity, in which the affinity values of 2 and 10–13 are in the following order: Cl < H < F < I < Br. Larger halogen substitutions such as iodine and bromine led to substantial loss in affinity. It was then found that the 3-methylthio group was superior to the 3-methoxy group by comparing compounds 13 and 14, showing a 2.8 fold enhancement in affinity.
On the other hand, compounds 15–17 had been incorporated a fluorine atom at 6-pyrindyl position of N-phenylpicolinamide, which can have a relatively facile fluorine-18 labeling. Compared to 2 and 3, the affinity of 15 and 16 was not significantly reduced.
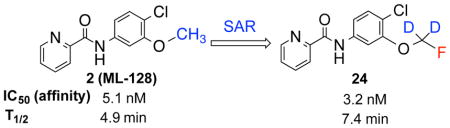
Next we turned our attention to the 3-phenyl position. It is considered that the metabolic stability was one of major issues for ML-128 (2), in which the 3-methoxy group was identified as the soft group. The 3-phenyl position was also very sensitive with substitutions. It was reported a simple change of 3-difluoromethoxy in compound 3 to 3-trifluoromethoxy group imparted a more than 10 fold loss of activity.16 Our initial effort was directed at 3-cyano substitution, in which 11C-cyanation may be carried out through a palladium-mediated cyanation or the Rousenmund-von Braun reaction.33 Five 3-cyanophenyl compounds (18–22) with different 4-phenyl substitutions were evaluated. The results show that the 3-cyano-4-chloro-analog 20 give a similar affinity compared to the 3-methoxy-4-chloro-analog 2. The affinity values of 18–22 are depending on 4-phenyl substitution and in the following order: Cl < F < H < Br < I, which shows different substitution effect compared to 3-methoxy analogs 2 and 10–13. We then replaced 3-methoxy with 3-fluoromethoxy for two reasons: first, since both 3-methoxy- and 3-difluoromethoxy-analogs exhibited the activity, fluoromethoxy should be also active; second, it generates a position for fluorine-18 labeling. Fluorine-18 is often the radionuclide of choice for both its physical and nuclear characteristics. Its half-life is long enough to carry out relatively extended imaging protocols when compared to what is possible with carbon-11. This facilitates kinetic studies and high-quality metabolic and plasma analysis. However, fluorine-18 labeling is normally limited to chemical structures already containing a fluorine atom and the possible labeling strategies are limited for the preparation of radiotracers of high specific radioactivity. The result shows that 3-fluoromethoxy compound 23 has an improved affinity (3.2 nM) compared to that (5.1 nM) of 2, which improves 1.6 fold. However, 3-fluoromethoxy group may not be metabolically stable, since the 3-methoxy and 3-difluoromethoxy groups were metabolically unstable in compounds 2 and 3. On the other hand, 3-trifluoromethoxy analog of 3 was significantly more stable but lack activity.16 Hence, we had applied a 3-dideuteriumfluoromethoxy group to replace 3-fluoromethoxy group as shown in compounds 24 and 25. Deuterium isotope effects have been used to reduce in vivo metabolic rates. For example, Zhang et al. reported that a deuterium-substituted analog (with 18FD2CO) as a radioligand for peripheral benzodiazepine receptor (PBR) had remarkably prolonged the half-life (T1/2) in mice brain.34 The deuterium substitution may reduce the rate of defluorination initiated by cleavage of the C–H bond without altering the binding affinity to mGlu4. The result shows that the 3-dideuteriumfluoromethoxy modified compounds 24 and 25 have excellent affinity.
On the basis of the affinity of these picolinamide derivatives, we subsequently determined the in vitro microsomal stability of the selected compounds that include 2–4 and 23–24 (Table 2). Compound 4 (ADX88178) is one of a most potent mGlu4 PAM to date and was shown to be orally active in a number of preclinical in vivo PD models.14,22 As Table 2 shows, the dideuteriumfluoromethoxy-compound 24 (T1/2 = 7.4 min) is more stable than the corresponding fluoromethoxy-analog 23 (T1/2 = 5.8 min) and the methoxy-analog 2(T1/2 = 4.9 min). It was reported that the cleaving rate of the C–H bond wasabout 6.7 times faster than that of C–D bond at 25 °C.34 On the other hand, the half time and the difference of the metabolic rates of the dideuteriumfluoromethoxy analog and the fluoromethoxy analog depended on the level of the enzyme. In developing the PET ligand for PBR, Zhang et al. found that the half time (T1/2) in the plasma was 2.575 min for the deuterium-substituted analog (with 18FD2CO) and 2.367 min for the non-deuterated analog. However, the half time (T1/2) of the deuterium-substituted analog in the brain was >60 min, whereas that of for the non-deuterated analog was only 2.227 min.34 We anticipate that the difference of the half times in the brain between compounds 24 and 23 as well as 2 could be more significant. Compared to 4, compound 24 has the same affinity and a similar in vitro microsomal stability. It is clear that compound 24 has both enhanced affinity and improved in vitro microsomal stability compared to 2.
Table 2.
In Vitro properties of the selected compounds.
| Compound | Affinity IC50 (nM) (n=3) | κa | SEM(κ) (n=2) | T1/2 (min) | Avg. Pe (10−6 cm/s) |
|---|---|---|---|---|---|
| 2 | 5.1 | 0.141 | 0.010 | 4.9 | 256 |
| 3 | 4.6 | 0.132 | 0.008 | 5.2 | 214 |
| 4 | 3.2 | 0.099 | 0.005 | 7.0 | 257 |
| 23 | 3.2 | 0.120 | 0.010 | 5.8 | 272 |
| 24 | 3.2 | 0.093 | 0.005 | 7.4 |
The decay constant that is slope of log concentration vs time profile (T1/2=Ln2/κ).
The selectivity of compound 24 was also determined among the various mGlu subtypes, in which the functional assays were carried out on mGlu1, mGlu2, mGlu5, mGlu6 and mGlu8. Compound 24 showed little activity against these mGlu (Supporting Information).
In addition, the permeability values of 2–4 and 23 were measured using BBB PAMPA model at pH 7.4, which characterized the rate across the BBB due to passive diffusion. The determined effective permeability (Pe) values are summarized in Table 2, in which the Pe results for internal highly and low permeable standards are 160 for propranolol and <2.8 for atenolol, respectively. This result indicates that compounds 23 and 24 have good BBB permeability. Although high BBB passive permeability does not necessary translate to sufficient unbound drug concentration in the brain because of potential intrinsic clearance and efflux transport, it is beneficial for CNS drug candidates.
In summary, we have modified, synthesized and evaluated a series of new N-phenylpicolinamide derivatives. Our research further demonstrated that N-phenylpicolinamide is a good template to develop mGlu4 ligands, which has offered extensive SAR results by us and other labs.16,26 The SAR study has discovered a number of compounds with good affinities (<10 nM) to mGlu4. The dideuteriumfluoromethoxy modified compound 24 is identified as a very promising mGlu4 ligand, which has demonstrated enhanced affinity, improved in vitro microsomal stability, good selectivity and good permeability. Compound 24 is considered as an attractive candidate for future labeling with fluorine-18 as an mGlu4 PET tracer. Since a number of compounds have good affinity we are studying their PAM activity to mGlu4 and potential therapeutic applications.
Supplementary Material
Scheme 2.
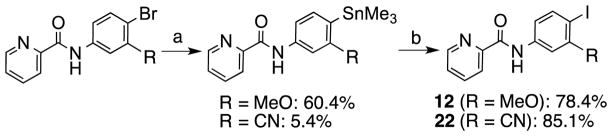
Synthesis of the N-phenylpicolinamides 12 and 22.
Reagents and conditions: (a) (SnMe3)2, Pd(PPh3)4, Toluene, reflux, 8.5 h; (b) I2, CH2Cl2, 2 h.
Acknowledgments
Funding was provided by NIBIB-R01EB012864 and NIMH-R01MH91684 to A.-L. B. Authors would like to acknowledge supporting grants for the instrumentation 1S10RR029495-01, 1S10RR026666-01, and 1S10RR023452-01. The mGlu functional data was generously provided by the National Institute of Mental Health’s Psychoactive Drug Screening Program, Contract # HHSN-271-2013-00017-C (NIMH PDSP). The NIMH PDSP is Directed by Bryan L. Roth MD, PhD at the University of North Carolina at Chapel Hill and Project Officer Jamie Driscoll at NIMH, Bethesda MD, USA. Financial support for PP from The Orion Farmos Research Foundation, Kuopio University Foundation, and Sigrid Juselius Foundation is gratefully acknowledged.
Footnotes
Supplementary data (experimental procedures and spectroscopic characterization of all new compounds) associated with this article can be found in the online version, at http://dx.doi.org/10.1016/j.bmcl.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Conn PJ. Physiological roles and therapeutic potential of metabotropic glutamate receptors. Annals of the New York Academy Sciences. 2003;1003:12–21. doi: 10.1196/annals.1300.002. [DOI] [PubMed] [Google Scholar]
- 2.Watkins JC, Jane DE. The glutamate story. British Journal of Pharmacology. 2006;147(S1):S100–S108. doi: 10.1038/sj.bjp.0706444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hampson DR, Rose EM, Antflick JE. The structure of metabotropic glutamate receptors. In: Gereau RWSGT, editor. The glutamate receptors. Human Press; Totowa: 2008. pp. 363–386. [Google Scholar]
- 4.Conn PJ, Pin JP. Pharmacology and functions of metabotropic glutamate receptors. Annual Review of Pharmacology and Toxicology. 1997;37:205–237. doi: 10.1146/annurev.pharmtox.37.1.205. [DOI] [PubMed] [Google Scholar]
- 5.Riedel G, Platt B, Micheau J. Glutamate receptor function in learning and memory. Behavioral Brain Research. 2003;140:1–47. doi: 10.1016/s0166-4328(02)00272-3. [DOI] [PubMed] [Google Scholar]
- 6.Robichaud AJ, Engers DW, Lindsley CW, Hopkins CR. Recent progress on the identification of metabotropic glutamate 4 receptor ligands and their potential utility as CNS therapeutics. ACS Chemical Neuroscience. 2011;17:433–449. doi: 10.1021/cn200043e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Amalric M, Lopez S, Goudet C, Fisone G, Battaglia G, Nicoletti F, Pin JP, Acher FC. Group III and subtype 4 metabotropic glutamate receptor agonists: Discovery and pathophysiological applications in Parkinson’s disease. Neuropharmacology. 2013;66:53–64. doi: 10.1016/j.neuropharm.2012.05.026. [DOI] [PubMed] [Google Scholar]
- 8.Fettagutti F, Balani-Guerra B, Corsi M, Nakanishi S, Corti C. Activation of the extracellular signal regulated kinase 2 by metabotropic glutamate receptors. European Journal of Neuroscience. 1999;11:2073–2082. doi: 10.1046/j.1460-9568.1999.00626.x. [DOI] [PubMed] [Google Scholar]
- 9.Niswender CM, Conn PJ. Metabotropic glutamate receptors: physiology, pharmacology, and disease. Annual Review of Pharmacology and Toxicology. 2010;50:295–322. doi: 10.1146/annurev.pharmtox.011008.145533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Corti C, Aldegheri L, Somogyi P, Ferraguti F. Distribution and synaptic localization of the metabotropic glutamate receptor 4 (mGluR4) in the rodent CNS. Neuroscience. 2002;110:403–420. doi: 10.1016/s0306-4522(01)00591-7. [DOI] [PubMed] [Google Scholar]
- 11.Valenti O, Mannaioni G, Seabrook GR, Conn PJ, Marino MJ. Group III metabotropic glutamate-receptor-mediated modulation of excitatory transmission in rodent substantia nigra pars compacta dopamine neurons. Journal of Pharmcology and Experimental Therapeutics. 2005;313:1296–1304. doi: 10.1124/jpet.104.080481. [DOI] [PubMed] [Google Scholar]
- 12.Goudet C, Vilar B, Courtiol T, Deltheil T, Bessiron T, Brabet I, Oueslati N, Rigault D, Bertrand HO, McLean H, Daniel H, Amalric M, Acher F, Pin JP. A novel selective metabotropic glutamate receptor 4 agonist reveals new possibilities for developing subtype selective ligands with therapeutic potential. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2012;26(4):1682–93. doi: 10.1096/fj.11-195941. [DOI] [PubMed] [Google Scholar]
- 13.Cajina M, Nattini M, Song D, Smagin G, Joergensen EB, Chandrasena G, Bundgaard C, Toft DB, Huang X, Acher F, Doller D. Qualification of LSP1-2111 as a Brain Penetrant Group III Metabotropic Glutamate Receptor Orthosteric Agonist. ACS Medicinal Chemistry Letters. 2014;5(2):119–123. doi: 10.1021/ml400338f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lindsley CW, Hopkins CR. Metabotropic glutamate receptor 4 (mGlu4)-positive allosteric modulators for the treatment of Parkinson’s disease: historical perspective and review of the patent literature. Expert Opinion on Therapeutic Patents. 2012;22(5):461–481. doi: 10.1517/13543776.2012.679437. [DOI] [PubMed] [Google Scholar]
- 15.Zhang Z, Brownell A-L. Imaging of Metabotropic Glutamate Receptors (mGluR)s. In: Bright P, editor. Neuroimaging - Clinical applications. InTech - Open Access Publisher; Rijeka, Croatia: 2012. pp. 499–532. [Google Scholar]
- 16.Engers DW, Niswender CM, Weaver CD, Jadhav S, Menon UN, Zamorano R, Conn PJ, Lindsley CW, Hopkins CR. Synthesis and evaluation of a series of heterobiaryl amides that are centrally penetrant metabotropic glutamate receptor 4 (mGluR4) positive allosteric modulators (PAMs) Journal of Medicinal Chemistry. 2009;52:4115–4118. doi: 10.1021/jm9005065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jones CK, Engers DW, Thompson AD, Field JR, Blobaum AL, Lindsley SR, Zhou Y, Gogliotti RD, Jadhav S, Zamorano R, Bogenpohl J, Smith Y, Morrison R, Daniels JS, Weaver CD, Conn PJ, Lindsley CW, Niswender CM, Hopkins CR. Discovery, Synthesis, and Structure-Activity Relationship Development of a Series of N-4-(2,5-Dioxopyrrolidin-1-yl)phenylpicolinamides (VU0400195, ML182): Characterization of a Novel Positive Allosteric Modulator of the Metabotropic Glutamate Receptor 4 (mGlu4) with Oral Efficacy in an Antiparkinsonian Animal Model. Journal of Medicinal Chemistry. 2011;54(21):7639–7647. doi: 10.1021/jm200956q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kalinichev M, Le Poul E, Bolea C, Girard F, Campo B, Fonsi M, Royer-Urios I, Browne SE, Uslaner JM, Davis MJ, Raber J, Duvoisin R, Bate ST, Reynolds IJ, Poli S, Celanire S. Characterization of the novel positive allosteric modulator of the metabotropic glutamate receptor 4 ADX88178 in rodent models of neuropsychiatric disorders. Journal of Pharmacology and Experimental Therapeutics. 2014;350(3):495–505. 11. doi: 10.1124/jpet.114.214437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hong SP, Liu KG, Ma G, Sabio M, Uberti MA, Bacolod MD, Peterson J, Zou ZZ, Robichaud AJ, Doller D. Tricyclic Thiazolopyrazole Derivatives as Metabotropic Glutamate Receptor 4 Positive Allosteric Modulators. Journal of Medicinal Chemistry. 2011;54(14):5070–5081. doi: 10.1021/jm200290z. [DOI] [PubMed] [Google Scholar]
- 20.Lindsley CW, Niswender CM, Engers DW, Hopkins CR. Recent progress in the development of mGluR4 positive allosteric modulators for the treatment of Parkinson’s disease. Current Topics in Medicinal Chemistry (Sharjah, United Arab Emirates) 2009;9(10):949–963. [PubMed] [Google Scholar]
- 21.Valenti O, Marino MJ, Wittmann M, Lis E, DiLella AG, Kinney GG, Conn PJ. Group III metabotropic glutamate receptor-mediated modulation of the striatopallidal synapse. Journal of Neuroscience. 2003;23:7218–7226. doi: 10.1523/JNEUROSCI.23-18-07218.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Le Poul E, Bolea C, Girard F, Poli S, Charvin D, Campo B, Bortoli J, Bessif A, Luo B, Koser AJ, Hodge LM, Smith KM, DiLella AG, Liverton N, Hess F, Browne SE, Reynolds IJ. A potent and selective metabotropic glutamate receptor 4 positive allosteric modulator improves movement in rodent models of Parkinson’s disease. Journal of Pharmacology and Experimental Therapeutics. 2012;343(1):167–177. doi: 10.1124/jpet.112.196063. [DOI] [PubMed] [Google Scholar]
- 23.Bennouar KE, Uberti MA, Melon C, Bacolod MD, Jimenez HN, Cajina M, Kerkerian-Le Goff L, Doller D, Gubellini P. Synergy between L-DOPA and a novel positive allosteric modulator of metabotropic glutamate receptor 4: Implications for Parkinson’s disease treatment and dyskinesia. Neuropharmacology. 2013;66:158–169. doi: 10.1016/j.neuropharm.2012.03.022. [DOI] [PubMed] [Google Scholar]
- 24.Iderberg H, Maslava N, Thompson AD, Bubser M, Niswender CM, Hopkins CR, Lindsley CW, Conn PJ, Jones CK, Cenci MA. Pharmacological stimulation of metabotropic glutamate receptor type 4 in a rat model of Parkinson’s disease and L-DOPA-induced dyskinesia: Comparison between a positive allosteric modulator and an orthosteric agonist. Neuropharmacology. 2015 doi: 10.1016/j.neuropharm.2015.02.023. Ahead of Print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kil KE, Zhang Z, Jokivarsi K, Gong C, Choi JK, Kura S, Brownell AL. Radiosynthesis of N-(4-chloro-3-[11C]methoxyphenyl)-2-picolinamide ([11C]ML128) as a PET radiotracer for metabotropic glutamate receptor subtype 4 (mGlu4) Bioorganic and Medicinal Chemistry. 2013;21:5955–5962. doi: 10.1016/j.bmc.2013.07.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bolea C. Preparation of amido derivatives and their use as positive allosteric modulators of metabotropic glutamate receptors. 2009 2008-EP59043,2009010454, 20080710. [Google Scholar]
- 27.Patel S, Hamill T, Connolly B, Jagoda E, Li W, Gibson R. Species differences in mGluR5 binding sites in mammalian central nervous system determined using in vitro binding with [18F]F-PEB. Nuclear Medicine and Biology. 2007;34:1009–17. doi: 10.1016/j.nucmedbio.2007.07.009. [DOI] [PubMed] [Google Scholar]
- 28.Wang J, Tueckmantel W, Zhu A. Pellegrino D, Brownell A-L, Synthesis and preliminary biological evaluation of 3-[(18)F]fluoro-5-(2-pyridinylethynyl)benzonitrile as a PET radiotracer for imaging metabotropic glutamate receptor subtype 5. Synapse. 2007;61(12):951–61. doi: 10.1002/syn.20445. [DOI] [PubMed] [Google Scholar]
- 29.Zhang L, Villalobos A, Beck EM, Bocan T, Chappie TA, Chen L, Grimwood S, Heck SD, Helal CJ, Hou X, Humphrey JM, Lu J, Skaddan MB, McCarthy TJ, Verhoest PR, Wager TT, Zasadny K. Design and Selection Parameters to Accelerate the Discovery of Novel Central Nervous System Positron Emission Tomography (PET) Ligands and Their Application in the Development of a Novel Phosphodiesterase 2A PET Ligand. Journal of Medicinal Chemistry. 2013;56(11):4568–4579. doi: 10.1021/jm400312y. [DOI] [PubMed] [Google Scholar]
- 30.Kenakin T, Onaran O. The ligand paradox between affinity and efficacy: can you be there and not make a difference? Trends Pharmacol Sci. 2002;23(6):275–280. doi: 10.1016/s0165-6147(02)02036-9. [DOI] [PubMed] [Google Scholar]
- 31.Kil KE, Poutiainen P, Zhang Z, Zhu A, Choi JK, Jokivarsi K, Brownell AL. Radiosynthesis and Evaluation of an 18F-Labeled Positron Emission Tomography (PET) Radioligand for Metabotropic Glutamate Receptor Subtype 4 (mGlu4) Journal of Medicinal Chemistry. 2014;57(21):9130–9138. doi: 10.1021/jm501245b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Poutiainen P, Kil K-E, Zhang Z, Kuruppu D, Tannous B, Brownell A-L. Co-operative binding assay for the characterization of mGlu4 allosteric modulators. Neuropharmacology. 2015 doi: 10.1016/j.neuropharm.2015.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Miller PW, Long NJ, Vilar R, Gee AD. Synthesis of 11C, 18F, 15O, and 13N radiolabels for positron emission tomography. Angew Chem, Int Ed. 2008;47(47):8998–9033. doi: 10.1002/anie.200800222. [DOI] [PubMed] [Google Scholar]
- 34.Zhang MR, Maeda J, Ito T, Okauchi T, Ogawa M, Noguchi J, Suhara T, Halldin C, Suzuki K. Synthesis and evaluation of N-(5-fluoro-2-phenoxyphenyl)-N-(2-[18F]fluoromethoxy-d2-5-methoxybenzyl)acetamide: a deuterium-substituted radioligand for peripheral benzodiazepine receptor. Bioorg Med Chem. 2005;13(5):1811–1818. doi: 10.1016/j.bmc.2004.11.058. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.