

Abstract
Interrogating fragment libraries by X-ray crystallography is a powerful strategy for discovering allosteric ligands for protein targets. Cryocooling of crystals should theoretically increase the fraction of occupied binding sites and decrease radiation damage. However, it might also perturb protein conformations that can be accessed at room temperature. Using data from crystals measured consecutively at room temperature and at cryogenic temperature, we found that transient binding sites could be abolished at the cryogenic temperatures employed by standard approaches. Changing the temperature at which the crystallographic data was collected could provide a deliberate perturbation to the equilibrium of protein conformations and help to visualize hidden sites with great potential to allosterically modulate protein function.
Keywords: allosterism, biophysics, ligand discovery, structural biology, thermodynamics, X-ray diffraction
Fragment-based ligand discovery (FBLD) uses small-molecule fragments (<250 Da) to increase the probability of finding weak hits.[1] Additionally, these fragments have less molecular complexity and can therefore sample chemical space more efficiently than the larger molecules found in conventional high-throughput screening libraries. Successful FBLD campaigns have rapidly improved the affinity of these hits[2] and advanced several lead molecules to modulate biology or disease.[3] Fragment-screening methods, including surface plasmon resonance and NMR spectroscopy, can measure binding affinities and provide initial structure–activity relationships (SARs). X-ray crystallography provides key insights for FBLD by identifying the binding sites of hits and structurally guiding medicinal chemistry efforts to optimize the fit (and ultimately the affinity) between the ligand and the binding site.[4]
Even weak affinity hits can be identified by most FBLD X-ray crystallography experiments, because highly concentrated solutions of ligands are soaked into the protein crystal. The crystal is then cryocooled to protect against radiation damage[5] and to halt any destructive effects of the solvent or ligands on the crystal lattice.[6] Cryocooling also offers tremendous benefits for the transportation of crystals and automated crystal screening by using robotics. An additional, but rarely considered, potential advantage of cryocooling is that thermodynamics favor ligand binding at lower temperatures. For example, given a driving force of 2.16 kcal mol−1 for the standard Gibbs free energy of ligand binding (corresponding to a 26 mm Kd at room temperature) and a concentration of 33 mm for the ligand soaked into the crystal, we would expect the percentage of ligand-bound receptors to be 56 % at 293 K (room temperature), 88 % at 200 K (the glass-transition temperature), and 99.9 % at 100 K (cryogenic temperature; see the Supporting Information). Therefore, at high, but non-saturating, ligand concentrations, greater ligand occupancy at cryocooled temperatures would increase the observable electron density for weak ligands at receptor binding sites.
In addition to active-site ligands, X-ray crystallographic fragment screens often identify hits to secondary binding sites that are distant from the active site and require protein conformational flexibility to become accessible to ligands. Initial hits can be subsequently refined to affect allosteric inhibition or activation, as has been demonstrated against targets such as HIV reverse transcriptase[7] and 3-phosphoinositide-dependent kinase 1.[8] These “cryptic” binding sites can be invisible to experimental techniques such as crystallography[9] because the energy gap between the pocket-forming (high-energy) state and the pocket-occluding (ground) state is too large. Therefore, identifying partially occupied ligands that show only weak electron density is especially important when searching for allosteric modulators, because the electron density will reflect the equilibrium between the pocket-occluding and ligand-bound states. As for active sites of enzymes, cryocooling should increase the fraction of ligands bound at the cryptic sites, which creates a potential advantage for identifying weak cryptic-site binders that can be developed to affect allosteric responses.
However, recent studies suggest that collection of X-ray diffraction data at cryogenic temperatures could mask alternate conformational states that are accessible to the protein at room temperature.[10] By altering the equilibrium of protein conformations, cryocooling may therefore stabilize the pocket-occluding states of cryptic binding sites and oppose the predicted enhanced ligand occupancy at lower temperatures. We have recently found that considering such high-energy/low-occupancy states, which are present only at room temperature, can be essential for discovering new ligands using flexible receptor docking.[11] This approach identified ligands that stabilize specific alternative loop conformations of the cavity site of cytochrome c peroxidase (CcP-ga).[11] Interestingly, in the apo structure (determined at cryogenic temperatures), the conformation that is preferred by the most potent compounds is not significantly populated. These results may also reflect non-equilibrium kinetic considerations of cryocooling,[12] because cryocooling may occur faster than some protein conformational changes, ligand binding/dissociation events (kon/koff), and ligand diffusion through vitrifying solvent channels. Herein, we have investigated the effect of cryocooling on fragment ligand-binding sites of CcP, one of which is a cryptic site that is only observed upon ligand binding and is not visible in the apo structures. Our studies demonstrate how distinct binding sites can be differentially affected by the tradeoffs between enhancing ligand occupancy upon cryocooling and altering the population of higher energy conformational states that can present new ligand-binding sites.
To systematically probe the impact of temperature on fragment binding, we obtained crystallographic data at both cryogenic temperature and room temperature (RT) from single crystals for ligand-free (apo) CcP-ga and five different ligand-bound CcP-ga complexes (Supporting Information). To minimize the difference between the datasets we 1) used 2-methyl-2,4-pentanediol (MPD) as a precipitant and cryoprotectant to allow for the re-collection of data on the same crystal at cryogenic temperature, 2) collected data on the same crystal volume, 3) matched the crystal size to the synchrotron beam size. This approach minimizes differences in the chemical composition of the crystal, local differences in crystal quality, and differences in the distribution of soaked compounds, respectively. For each crystal, we first measured the diffraction pattern at RT, and then, after flash-cooling the crystal in liquid nitrogen, we re-measured the diffraction pattern of the same crystal at cryogenic temperatures. Changes in the resolution and mosaicity were minimal and typical for well-cooled crystals (Supporting Information, Table S3). Using the same method, we also managed to collect eight complete RT datasets on the same crystal volume, three of which were before the data quality decreased to below 2.1 Å (see also the Supporting discussion and Supporting Information, Figure S8). To compare the structural impact of cryocooling and ligand binding, we calculated the distance of each protein residue from the protein center-of-mass as a function of temperature and as a function of ligand state (bound or apo). The distribution of differences between these distances (e.g., CcP-ligand1 at RT—CcP-ligand1 at cryogenic temperature) provides an estimate of the anisotropy of the structural perturbation of the protein.[13] Although conformational changes are required to accommodate different ligands,[11] we found that the protein structure is more perturbed by temperature than by ligand binding (Figure 1 A; Figure S1 and Table S8).
Figure 1.
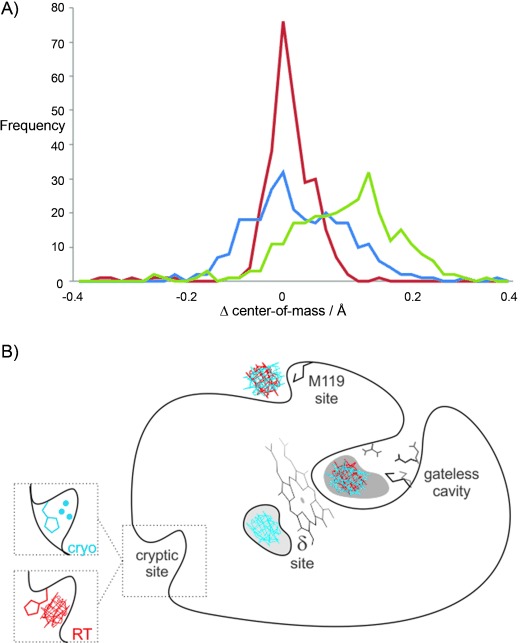
Protein structure is more perturbed by temperature and than by ligand binding. A) The distance of each protein residue from the protein center-of-mass is compared between two structures either at different temperatures (green line) or in different ligand states (apo versus with benzimidazole) at the same temperature (red=RT and blue=cryogenic temperature). All temperature pairs were collected consecutively on the same crystal. The amount of offset from zero reflects the expected thermal contraction of the protein upon freezing (green line). The broader distributions indicate structural heterogeneity upon ligand binding at cryogenic temperature (blue line) and structural heterogeneity of the same structure collected at different temperatures (green line). The narrow distribution of the different ligand states at RT (red line) suggests that, at cryogenic temperature, the protein structure is non-specifically perturbed by cryocooling rather than showing a response unique to ligand binding. B) Multiple protein sites display ligand electron density at different temperatures. Electron density was observed at both RT (red mesh) and cryogenic temperature (blue mesh) for the primary cavity site and the M119 surface site, whereas the heme proximal δ-site and the H96 cryptic binding site are temperature sensitive.
For example, the peaks of the distributions comparing datasets at two different temperatures (RT versus cryogenic) with the same ligand state are offset from zero, which shows that there is thermal contraction of the protein. Also, the distributions are broad, which indicates that there is heterogeneity in the structures (Figure 1 A). Comparing the apo to the ligand-bound protein structures at the same temperature reveals that peaks are centered at zero. However, the distributions are much narrower at RT than at cryogenic temperatures, which indicates a decrease in precision at cryogenic temperature, commonly defined as an increase in random errors. Note that the observed decrease in the atomic displacement distributions[12] (B factors) observed at cryogenic temperatures would normally be interpreted as evidence for increased precision (Tables S1 and S2).
However, the broad distributions of the cryocooled structures suggest much larger structural differences, which are likely due to nonspecific perturbations caused by the cryocooling process and not due to ligand binding. In contrast, the comparisons between the RT structures isolate those structural responses that are unique to ligand binding. This discrepancy between the RT and the cryogenic data is counterintuitive, because we would expect dissimilarities to be amplified between the ligand-stabilized and the ligand-free structures upon increasing the thermal motion at RT. These results indicate that cryocooling can have a large and inconsistent impact on the conformations of residues throughout the protein (Figure S2), which may misinform structure-based drug and probe design.
Next, we examined the binding site and other protein regions for large temperature-dependent changes in electron-density distributions. For the ligand benzimidazole, as expected, we observed consistent electron density for the ligand at both temperatures for the primary binding site.[11] We were surprised to observe ligand density at three additional distal sites (Figure 1 B). As with the primary binding site, ligand electron density appears at both temperatures in a second, surface-exposed site near Met119 (Figures 1 B; Figure S3 B). In a third site, near the δ-heme edge, a known CcP substrate site,[14] we observed ligand density only at cryogenic temperatures and not at RT (Figures 1 B; Figure S3 A). Benzimidazole also occupied a fourth site, near His96, but only at RT (Figures 1 B and 2). This fourth site can be classified as a cryptic site because the binding pocket is not apparent in the apo structure at either room- or cryogenic temperature. Access of the ligand to this cryptic site is controlled by an alternative conformation of His96, which is correlated with the presence of the benzimidazole (Figure 2) and can be identified by a secondary electron-density peak using Ringer[15] (Figure 2 A). Although the cryogenic data were collected on the same crystal volume as the RT data, the cryogenic temperature electron-density maps are consistent only with the “closed” His96 rotamer and three water molecules occluding the cryptic site (Figure 2 B). This result illustrates how the population of alternative conformations can be perturbed by temperature, thus altering the potential for observing a ligand in a binding pocket (Figure 1 B).
Figure 2.
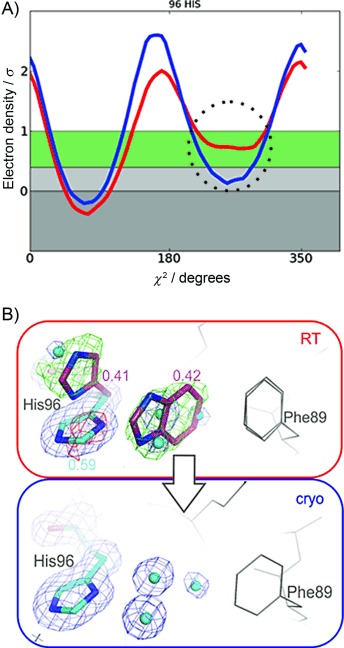
Benzimidazole binding to the transient cryptic site is only observed at RT. A) Electron density sampling around His96 Chi-2 using Ringer reveals an electron density peak (dotted circle) for the alternative conformation at room temperature (red line), but not cryogenic temperature (blue line). This minor “open” state would have been missed if the conventional 1σ cutoff to distinguish electron density signal from noise was used or only cryogenic data were available. b) Difference electron density contoured at 3σ (green and red mesh) confirms the presence of the alternative His rotamer and ligand presence at RT (red box). Both the ligand and the alternative His96 conformer were excluded from the crystallographic refinement but are shown in magenta as refined automatically to their final occupancies. At cryogenic temperature (blue box) we observed no ligand electron density but instead only the pocket-filling His96 rotamer and water molecules (blue spheres) that occluded the cryptic site. Note that observing ligand binding to the cryptic site only at RT is unexpected, because thermodynamics should favor binding at cryogenic temperatures.
To probe the biological relevance of the cryptic binding site, we moved from the CcP-ga model system, which has a cavity engineered to bind small molecules,[11, 16] to CcP-wt, which contains the radical-forming Trp191 residue that is involved in long-range electron transfer at the active site.[17] We first confirmed that benzimidazole occupied the cryptic binding site in CcP-wt. Co-crystals with benzimidazole diffracted to a resolution of 2.6 Å and showed electron density for the ligand in the cryptic binding site for three of the four protein copies within the crystallographic asymmetric unit.
Refinement of the data unambiguously revealed His96 to be in an open state (Figure S4), but no ligand density at the Met119 residue or δ-site could be detected at this resolution. To determine the binding affinity of benzimidazole to the cryptic binding site, we monitored ligand binding through the saturable perturbation of the CcP heme Soret band.[16b] In the CcP-ga cavity site, benzimidazole can occupy multiple sites and has low micromolar affinity for the primary binding site (Figure S5). However, in the wild-type protein, Trp191 blocks this high-affinity binding site, which allowed us to isolate the binding affinity of benzimidazole to the lower affinity site. We determined an affinity of benzimidazole for CcP-wt of 26 mm (Figure S6), which corresponds to a ligand efficiency (LE)[18] of 0.24 kcal mol−1 per ligand heavy atom (HA; Figure 2 A). Given that the cryptic site is too far away (25 Å) from the heme to elicit a direct Soret shift, we were initially surprised to observe an allosteric Soret band shift for benzimidazole. Although we detected no steric coupling networks connecting the cryptic binding site to the heme by using CONTACT[19] in our CcP-ga crystal structures at RT (Figure S7), a recent EPR study proposes the cryptic binding site to be a biologically relevant site for substrate oxidation that is remote from the heme.[20] This alternative electron transfer pathway includes the nearby Tyr71 residue as a reactive intermediate[20] and contrasts with a model where small-molecule binding to sites other than the δ-heme edge was suggested to be nonspecific.[21]
The discrepancy between a binding site being occupied by benzimidazole at RT and unoccupied in the same crystal at cryogenic temperature is counterintuitive and suggests that cryocooling can “overwhelm” the driving forces of ligand binding. To explain the reduction of benzimidazole occupancy from approximately 50 % at room temperature to below the detection limit of about 5 % at cryogenic temperature, cryocooling must counteract the temperature-dependent effects on the ligand–protein equilibrium. To estimate the penalty on occupancy from cryocooling, we assume both cryocooling and ligand soaking to be at equilibrium, although the heterogeneous structural perturbations we observed in response to cryocooling could suggest that the system has not equilibrated at the cryocooled temperature. However, on the typical timescale of freezing (100 ms),[12] we are neither in the fast-cooling regime where RT states are frozen-in nor in the slow-cooling regime where the crystal has had time to equilibrate. Therefore, intermediate states of slow and fast equilibrating domains are likely differentially trapped. We related the energy penalty (ΔG) as a function of ligand-soaking concentration [L], standard affinity at room temperature (Kd site), minimum detectable occupancy (occmin), temperature T, and the gas constant R:
| (1) |
See Supporting Information for the derivation of this equation.
Given our experimental conditions of soaking a ligand with a Kd of 26 mm at a concentration of 33 mm and crossing the glass-transition temperature of 200 K upon cooling, the allosteric cryocooling penalty must be at least 1.3 kcal mol−1 to render the ligand invisible below 5 % occupancy. The cryocooling penalty includes the solvent glass transition, unusual temperature dependencies of other enthalpic or entropic terms, and allosteric lattice changes to the protein ensemble. All of these mechanisms are likely to be important for increasing the magnitude of the cryocooling penalty of CcP residue His96.[12]
In contrast to the behavior of benzimidazole at the cryptic binding site, we identified two examples where the cryocooling penalty does not dominate over other contributions to binding. First, 2-amino-5-methylthiazole binds to the cryptic binding site at both temperatures (Figure 3 A). When the soaking concentration is 100 mm, 2-amino-5-methylthiazole (kd=68 mm; LE=0.22) shows higher crystallographic occupancy at cryogenic temperatures than at RT (75 % versus 54 %, respectively; Figure 3 A); this mirrors our expectation of 99.6 % occupancy at 100 K and 59 % occupancy at 298 K. Second, cryocooling can promote compounds to bind to other sites that do not require a conformational change to form a pocket. For the δ-site, for example, we only observed benzimidazole density at cryogenic temperatures (Figures 1 B and S3). Based on these results, which suggest that the cryocooling penalty would disfavor binding at the cryptic site, and the fact that the Protein Data Bank is dominated by data collected at cryogenic temperatures,[5] we expected to find more δ-site binders than cryptic site binders in the previously determined structures. We inspected 136 electron-density maps of previously determined structures of three CcP variants (CcP-wt, CcP-W191G, CcP-ga) from the PDB and struggled to find significant electron density in the δ-site, whereas we found several examples of ligand densities in the cryptic binding site (Figure 3 B). Counterintuitively, this suggests a lower driving force for ligands to bind to the δ-site. Because the δ-site is accessible without local side-chain conformational changes, the lower observed frequency might also hint at allosteric lattice effects that reduce binding at this site. Collectively, these results illustrate how cryocooling can have counteracting effects on ligand occupancy at fragment-binding sites.
Figure 3.
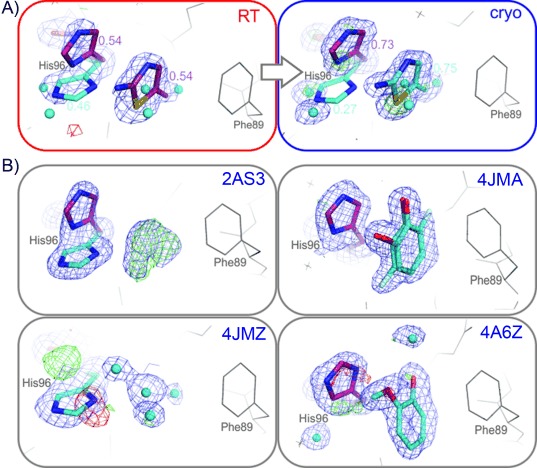
Cryptic site binders are prevalent in the PDB. A) 2-amino-5-methylthiazole binds to the cryptic site at both temperatures, RT (red box) and cryogenic (blue box). Both the ligand and the alternative His96 conformation (included in refinement) refined to higher ligand occupancy at cryogenic temperatures (0.75 versus 0.54 at RT). B) Electron density maps in other CcP structures, as deposited in the PDB, show evidence of ligand binding with an “open” His96 conformer, either unmodeled (phenol in PDB structure 2AS3, and N-methyl-1H-benzimidazol-2-amine in 4JMZ) or modeled (3-fluorocatechol in 4JMA, and guaiacol in 4A6Z). 2 mFo−DFc maps shown as blue mesh (rendered at 1σ), mFo−DFc maps in green and red (3σ).
Heterogeneous and non-equilibrium contributions to protein–ligand interactions upon cryocooling make it difficult to assign an exact value for this penalty beyond our previously mentioned estimate of 1–2 kcal mol−1. However, our observations illustrate that the cryocooling penalty plays a dominant role in determining the net binding of a ligand in a cryogenically frozen protein.
Our observation that the cryptic binding site was occupied at RT but not at cryogenic temperature contradicts the thermodynamic expectation that a higher fraction of sites should be bound at cryogenic temperatures. This suggests shifting temperatures as a general strategy to modulate the energy landscape of protein–ligand binding and overcome cryocooling penalties in favor of populating and revealing transient sites. We note that these cryocooling effects can be especially problematic at ligand concentrations around or below the Kd, which equals the ligand concentration at which half of the protein molecules are bound. Although the observation of differential binding depends on a lucky choice of concentration and compound soaking time, as for benzimidazole in CcP-ga, the chance of observing a secondary ligand-binding site increases with concentration—at both temperatures. Fragments will be particularly affected, because they intrinsically achieve low binding affinities even at high ligand efficiencies, and the penalties may therefore overwhelm binding upon cryocooling. To counteract this effect, very high soaking concentrations would have to be used to achieve a sufficient fraction of receptors bound to a ligand. However, preparing such high concentration stock solutions is often impractical, because it is limited by the solubility of compound and the sensitivity of the protein to organic solvents (like DMSO) or the compound itself.
Allosteric ligand-binding sites offer great potential for modulating protein function, but are often difficult to visualize. However, cryptic binding sites, with the potential to allosterically modulate protein function, can be discovered serendipitously, even for well-studied proteins, by using a fragment-based approach. Herein, we demonstrate that shifting the temperature at which the crystallographic data are collected can deliberately perturb the protein to help visualize such cryptic binding sites. To shift the population of conformational states towards the energetically less-accessible states and detect new binding sites for low-affinity, less-soluble fragments, we suggest a dual strategy: collect both datasets, if possible. This approach will complement mutagenesis efforts designed to stabilize specific protein conformations and may help identify cryptic binding sites, which could substantially extend the targets that can be probed to dissect biological mechanisms or enable therapeutic intervention.[9, 22] For fragments, rather than invalidating cryogenic data, RT data collection has potential as an orthogonal method that could unleash some of the unused potential within FBLD. However, even without RT data, we suspect that many existing electron-density maps may contain evidence of unmodeled ligands partially occupying such sites; especially when high concentrations of ligand were used to soak the crystals, as is typical of fragment-based methods. While we cannot link remote binding sites to function from structure alone, FBLD-investigators specifically looking to find or optimize allosteric binding ligands may find promise in exploring the full landscape of ligand binding. In those cases, our strategy of shifting conformational equilibria by shifting temperature may become illuminating, even crucial.
Experimental Section
Experimental details are given in the Supporting Information.
Acknowledgments
We thank Allison Doak for help with the protein preparation and Dr. Anja Fischer, Dr. Christoph Rademacher, and Chelsea Bidlow for critical reading of this manuscript. M.F. and B.K.S. are supported by GM59957. J.S.F. is a Searle Scholar, Pew Scholar, and Packard Fellow, and is supported by grants: NIH OD009180, NIH GM110580, and NSF STC-1231306. Data collection at BL831 at the Advanced Light Source is supported by the Director, Office of Science, Office of Basic Energy Sciences, of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231, and the Program Breakthrough Biomedical Research, which is partially funded by the Sandler Foundation.
Supporting Information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re-organized for online delivery, but are not copy-edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
miscellaneous_information
References
- 1.Fischer M, Hubbard RE. Mol. Interventions. 2009;9:22–30. doi: 10.1124/mi.9.1.7. [DOI] [PubMed] [Google Scholar]
- 2.Hajduk PJ, Meadows RP, Fesik SW. Science. 1997;278:497–499. doi: 10.1126/science.278.5337.497. [DOI] [PubMed] [Google Scholar]
- 3a.Murray CW, Verdonk ML, Rees DC. Trends Pharmacol. Sci. 2012;33:224–232. doi: 10.1016/j.tips.2012.02.006. [DOI] [PubMed] [Google Scholar]
- 3b.Lipinski C, Hopkins A. Nature. 2004;432:855–861. doi: 10.1038/nature03193. [DOI] [PubMed] [Google Scholar]
- 4.Patel D, Bauman JD, Arnold E. Prog. Biophys. Mol. Biol. 2014;116:92–100. doi: 10.1016/j.pbiomolbio.2014.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Garman E. Curr. Opin. Struct. Biol. 2003;13:545–551. doi: 10.1016/j.sbi.2003.09.013. [DOI] [PubMed] [Google Scholar]
- 6.Davies TG, Tickle IJ. Top. Curr. Chem. 2012;317:33–59. doi: 10.1007/128_2011_179. [DOI] [PubMed] [Google Scholar]
- 7.Bauman JD, Patel D, Dharia C, Fromer MW, Ahmed S, Frenkel Y, Vijayan RS, Eck JT, Ho WC, Das K, Shatkin AJ, Arnold E. J. Med. Chem. 2013;56:2738–2746. doi: 10.1021/jm301271j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sadowsky JD, Burlingame MA, Wolan DW, McClendon CL, Jacobson MP, Wells JA. Proc. Natl. Acad. Sci. USA. 2011;108:6056–6061. doi: 10.1073/pnas.1102376108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bowman GR, Geissler PL. Proc. Natl. Acad. Sci. USA. 2012;109:11681–11686. doi: 10.1073/pnas.1209309109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10a.Fraser JS, Clarkson MW, Degnan SC, Erion R, Kern D, Alber T. Nature. 2009;462:669–673. doi: 10.1038/nature08615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10b.Fraser JS, van den Bedem H, Samelson AJ, Lang PT, Holton JM, Echols N, Alber T. Proc. Natl. Acad. Sci. USA. 2011;108:16247–16252. doi: 10.1073/pnas.1111325108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fischer M, Coleman RG, Fraser JS, Shoichet BK. Nat. Chem. 2014;6:575–583. doi: 10.1038/nchem.1954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Halle B. Proc. Natl. Acad. Sci. USA. 2004;101:4793–4798. doi: 10.1073/pnas.0308315101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13a.Rader SD, Agard DA. Protein Sci. 1997;6:1375–1386. doi: 10.1002/pro.5560060701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13b.Tilton RF, Jr, Dewan JC, Petsko GA. Biochemistry. 1992;31:2469–2481. doi: 10.1021/bi00124a006. [DOI] [PubMed] [Google Scholar]
- 14.Gumiero A, Murphy EJ, Metcalfe CL, Moody PC, Raven EL. Arch. Biochem. Biophys. 2010;500:13–20. doi: 10.1016/j.abb.2010.02.015. [DOI] [PubMed] [Google Scholar]
- 15.Lang PT, Ng H-L, Fraser JS, Corn JE, Echols N, Sales M, Holton JM, Alber T. Protein Sci. 2010;19:1420–1431. doi: 10.1002/pro.423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16a.Barelier S, Boyce SE, Fish I, Fischer M, Goodin DB, Shoichet BK. PLoS One. 2013;8:e69153. doi: 10.1371/journal.pone.0069153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16b.Rocklin GJ, Boyce SE, Fischer M, Fish I, Mobley DL, Shoichet BK, Dill KA. J. Mol. Biol. 2013;425:4569–4583. doi: 10.1016/j.jmb.2013.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Volkov AN, Nicholls P, Worrall JA. Biochim. Biophys. Acta Bioenerg. 2011;1807:1482–1503. doi: 10.1016/j.bbabio.2011.07.010. [DOI] [PubMed] [Google Scholar]
- 18.Hopkins AL, Groom CR, Alex A. Drug Discovery Today. 2004;9:430–431. doi: 10.1016/S1359-6446(04)03069-7. [DOI] [PubMed] [Google Scholar]
- 19.van den Bedem H, Bhabha G, Yang K, Wright PE, Fraser JS. Nat. Methods. 2013;10:896–902. doi: 10.1038/nmeth.2592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Miner KD, Pfister TD, Hosseinzadeh P, Karaduman N, Donald LJ, Loewen PC, Lu Y, Ivancich A. Biochemistry. 2014;53:3781–3789. doi: 10.1021/bi500353p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Murphy EJ, Metcalfe CL, Nnamchi C, Moody PC, Raven EL. FEBS J. 2012;279:1632–1639. doi: 10.1111/j.1742-4658.2011.08425.x. [DOI] [PubMed] [Google Scholar]
- 22.Hopkins AL, Groom CR. Nat. Rev. Drug. Discov. 2002;1:727–730. doi: 10.1038/nrd892. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
miscellaneous_information