

Abstract
Background & Aims
Defects in intestinal innate defense systems predispose patients to inflammatory bowel disease (IBD). Reactive oxygen species (ROS) generated by nicotinamide-adenine dinucleotide phosphate (NADPH) oxidases in the mucosal barrier maintain gut homeostasis and defend against pathogenic attack. We hypothesized that molecular genetic defects in intestinal NADPH oxidases might be present in children with IBD.
Methods
After targeted exome sequencing of epithelial NADPH oxidases NOX1 and DUOX2 on 59 children with very early onset inflammatory bowel disease (VEOIBD), the identified mutations were validated using Sanger Sequencing. A structural analysis of NOX1 and DUOX2 variants was performed by homology in silico modeling. The functional characterization included ROS generation in model cell lines and in in vivo transduced murine crypts, protein expression, intracellular localization, and cell-based infection studies with the enteric pathogens Campylobacter jejuni and enteropathogenic Escherichia coli.
Results
We identified missense mutations in NOX1 (c.988G>A, p.Pro330Ser; c.967G>A, p.Asp360Asn) and DUOX2 (c.4474G>A, p.Arg1211Cys; c.3631C>T, p.Arg1492Cys) in 5 of 209 VEOIBD patients. The NOX1 p.Asp360Asn variant was replicated in a male Ashkenazi Jewish ulcerative colitis cohort. Patients with both NOX1 and DUOX2 variants showed abnormal Paneth cell metaplasia. All NOX1 and DUOX2 variants showed reduced ROS production compared with wild-type enzymes. Despite appropriate cellular localization and comparable pathogen-stimulated translocation of altered oxidases, cells harboring NOX1 or DUOX2 variants had defective host resistance to infection with C. jejuni.
Conclusions
This study identifies the first inactivating missense variants in NOX1 and DUOX2 associated with VEOIBD. Defective ROS production from intestinal epithelial cells constitutes a risk factor for developing VEOIBD.
Keywords: Inflammatory Bowel Disease, NADPH Oxidase, NOX1, DUOX2, Reactive Oxygen Species, VEOIBD
Abbreviations used in this paper: AJ, Ashkenazi Jewish; CGD, chronic granulomatous disease; DUOX2, dual oxidase 2; HA, human influenza hemagglutinin; IBD, inflammatory bowel disease; FAD, flavin adenine nucleotide; MAF, minor allele frequency; NADPH, nicotinamide-adenine dinucleotide phosphate; NOX1, NADPH oxidase 1; PAS, periodic acid–Schiff; PBS, phosphate-buffered saline; PMA, phorbol 12-myristate 13-acetate; ROS, reactive oxygen species; SNP, single-nucleotide polymorphism; UC, ulcerative colitis; VEOIBD, very early onset inflammatory bowel disease; WT, wild type
Summary.
NOX1 and DUOX2 are the predominant source of intestinal epithelial ROS. Here we identify novel NOX1 and DUOX2 variants associated with VEOIBD that result in reduced ROS production, Paneth cell metaplasia and defective host resistance to C. jejuni.
Inflammatory bowel disease (IBD), a complex disease associated with genetic predisposition and environmental factors, is characterized by recurrent intestinal inflammation and microbial dysbiosis. Genomewide association studies link adult IBD to alterations in genes involved in host-microbe interactions.1, 2 Nicotinamide adenine dinucleotide phosphate (NADPH) oxidase-generated reactive oxygen species (ROS) are intrinsic to the antimicrobial host defense system of professional phagocytes. Defective ROS production in patients with chronic granulomatous disease (CGD), a rare genetic disorder caused by inactivating alterations of genes required for formation of the penultimate phagocyte oxidase complex (CYBB, CYBA, NCF1, NCF2, NCF4), confers susceptibility to life-threatening bacterial and fungal infections.3 Up to 40% of CGD patients develop inflammatory colitis that mimics Crohn’s disease.4 Genetic variants in NCF4 and NCF2 that lead to partial attenuation in phagocyte oxidase (NADPH oxidase 2, NOX2) function without causing CGD have been associated with adult and very early onset IBD (VEOIBD).5, 6 We have recently shown that single-nucleotide polymorphisms (SNPs) and rare hypomorphic variants in all components of the NOX2 NADPH oxidase complex are associated with VEOIBD.7
A role for ROS production by intestinal epithelial cells in mucosal barrier function and intestinal homeostasis is just emerging.8 The predominant sources of ROS in the lining of the gastrointestinal tract are the NADPH oxidases NOX1 (NADPH oxidase 1) and DUOX2 (dual oxidase 2), with NOX1 expression restricted mainly to colon, caecum, and ileum, whereas DUOX2 can be found in all segments of the gut.9 NOX1 and DUOX2 are the catalytic subunits of multimeric, membrane-bound enzymes that generate upon stimulation superoxide and hydrogen peroxide by transfer of electrons from NADPH to molecular oxygen. We10 and others11, 12, 13 have reported NOX1/DUOX2-mediated ROS production in the intestine and its effect on bacterial pathogenicity and barrier integrity. Here, we describe the identification and characterization of missense mutations in NOX1 (NM_007052.4, location Xq22) and in DUOX2 (NG_016992, location 15q15.3) in patients diagnosed with VEOIBD.
Materials and Methods
Study Design
All results are presented according to the STrengthening the REporting of Genetic Association Studies (STREGA) guidelines.14 Fifty-nine IBD patients diagnosed under the age of 6 years were sequenced for NOX1 and DUOX2 by targeted exome sequencing using Agilent SureSelect target enrichment and sequencing (Agilent Technologies, Santa Clara, CA) on the Illumina HiSeq 2000/2500 (Illumina, San Diego, CA) with exon primer and sequencing protocols designed by the Beckman Coulter Genomics (beckmangenomics.com; Beckman Coulter, Brea, CA) as described previously elsewhere.15 Sanger sequencing was used to verify all genetic defects identified using targeted sequencing of the NOX1 and DUOX2 genes at the Centre for Applied Genomics (TCAG; http://www.tcag.ca; Hospital for Sick Children, Toronto, ON, Canada).
Single-nucleotide and insertion/deletion (indel) variants identified by targeted exome sequencing and validated by Sanger sequencing were automatically scanned and manually verified. Furthermore, all variants were also validated using Taqman performed by the Centre for Applied Genomics, Hospital for Sick Children.15, 16 Function and minor allele frequency (MAF) were searched for using the National Heart, Lung, and Blood Institute Exome Sequencing Project (ESP) Exome Variant Server (http://evs.gs.washington.edu/EVS/), the National Center for Biotechnology Information dbSNP (http://www.ncbi.nlm.nih.gov/projects/SNP/), the National Institute of Environmental Health Sciences FuncPred (http://snpinfo.niehs.nih.gov/snpinfo/snpfunc.htm), Polyphen2 (http://genetics.bwh.harvard.edu/pph2/),17 SIFT (http://sift.jcvi.org/),18 FastSNP (http://fastsnp.ibms.sinica.edu.tw/),19 Human Splicing Finder (http://www.umd.be/HSF/),20 and pfSNP (http://pfs.nus.edu.sg/).21
Setting
Patients included in the study were recruited from the Inflammatory Bowel Disease Clinic at the Hospital from Sick Children, University of Toronto. They were diagnosed with VEOIBD between the years 1994 and 2012 and had a confirmed diagnosis of IBD before the age of 6 years. Although there is no consensus on the definition of VEOIBD, we have used the stricter definition based on our recent modification (diagnosis <6 years of age)5, 22, 23 of the Paris classification.24 Our definition, which is more stringent and includes more severe cases that are more likely to cause monogenic forms of the disease, has been used to identify risk variants in this age group. There were no exclusion criteria for patients diagnosed with VEOIBD; however, patients with a known immunodeficiency or a clinical diagnosis of CGD were excluded because these patients were not defined as VEOIBD. The five identified patients were screened and were found negative for pathogenic mutations in IL10RA, IL10RB, IL10, XIAP, TTC7A, as well as genes involved in CGD (RAC1/2, NCF1/2/4, and CYBB, CYBA)23, 25 and NOD2 and ATG16L1 variants associated with IBD.
Participants
This was a cohort study that examined the genetics of VEOIBD patients. Fifty-five VEOIBD patients were recruited from the Hospital for Sick Children, Toronto, Canada. A second cohort of VEOIBD patients was recruited through NEOPICS (www.NEOPICS.org). The replication cohort comprised 1477 Crohn’s disease cases, 559 ulcerative colitis cases, and 2614 healthy controls, all with genetically verified Ashkenazi Jewish ancestry by principal components analysis.
Standard quality control procedures were applied, and we performed association testing using Fisher’s exact method, stratified by gender in 297 male ulcerative colitis (UC) cases, 262 female UC cases, 1708 male controls, and 906 female controls. Phenotypic information and DNA samples were obtained from the study participants with approval of the institutional review ethics board for IBD genetic studies at the Hospital for Sick Children and Mount Sinai Hospital Toronto.
Later onset UC cases were recruited through the National Institute of Diabetes and Digestive and Kidney Diseases Inflammatory Bowel Disease Genetics Consortium, the Cedars-Sinai Medical Center IBD Center in California and Mount Sinai Hospital in New York. Replication cohorts had ethics board approval for genetic and phenotypic studies at the individual institutions. Written informed consent was obtained from all participants/parents.
H&E and Periodic Acid–Schiff Staining in Patient Biopsy Samples
Colonic biopsy samples were fixed in 10% formaldehyde without methanol and afterward embedded in paraffin. For H&E staining, embedded paraffin tissues on slides were deparaffinized with xylene and afterward rehydrated with different percentages of ethanol. The slides were stained for 5 minutes with Meyer’s hematoxylin (Fisher Scientific, Fair Lawn, NJ) for nuclei and counterstained with eosin-Y (Fisher Scientific) for cytoplasm. Slides were mounted in Entellan (EMD Millipore, Billerica, MA). Photographs were taken using an epifluorescence light microscope (Leica Microsystems, Buffalo Grove, IL) and adjusted for brightness, contrast, and pixel size in Adobe Photoshop CS5 version 12.0 (Adobe System, San Jose, CA).
Modeling and Docking Procedure
Three-dimensional (3D) models of C-terminal domains of NOX1 and DUOX2 were generated using the homology modeling program Modeller 9v11 (http://www.salilab.org/modeller/).26 Blast of PDB was performed with the NOX1 FAD-binding domain, and a combination of several homologous structures served together with the 3D X-ray structure the NOX2 NADPH binding domain (PDB ID: 3A1F) as initial template. The modeling was performed with default parameters using the “allHmodel” protocol to include hydrogen atoms and the “HETATM” protocol to include FAD and NADPH. To compare the FAD and NADPH binding interaction between wild-type (WT) and sequence altered oxidases, the docking runs were performed with HADDOCK.27, 28 Docking was performed with most of the parameters set to default using the Web server version of HADDOCK with a Guru interface. To gain the Van der Waals, electrostatic, and desolvation energy for each enzyme - FAD or -NADPH model, HADDOCK automatically performed the molecular dynamics before and after each docking trial by including water into the calculation (detailed modeling procedure, publication in preparation).
Cell Culture and Transfection
Model cell lines were employed as intestinal epithelial cell lines, and primary colon cells express endogenous NOX1 and DUOX2. Cos7 cells are a suitable model system for NOX1-based oxidase reconstitution as they lack any functional NADPH oxidases, and NCI-H661 cells serve as a physiologically relevant model for DUOX oxidases.29 Cos7 cells stably expressing p22phox 30 were maintained in Dulbecco’s modified Eagle’s medium with 10% fetal bovine serum; for NCI-H661 cells stably expressing DUOXA2,29 RPMI 1640 medium with 10% fetal bovine serum was used. NOX1 was cloned into pcDNA3.1 with and without the N-terminal Myc epitope tag including a linker sequence. Influenza hemagglutinin (HA)-tagged DUOX2 in pcDNA3.1 was prepared by cloning the HA tag between amino acids D27 and A28. Mutations were introduced using site-directed mutagenesis and were verified by sequencing. NOX1 WT and missense variants were transiently transfected with NOXA1 and Myc-NOXO1 into Cos-p22phox cells (24 hours). HA-tagged DUOX2 WT and missense variants were transiently transfected into H661-DUOXA2 cells or together with DUOXA2 into Cos7 cells using X-tremeGENE (Roche Applied Science, Indianapolis, IN) (48 hours). For analysis of DUOX2 localization upon bacterial challenge, HT29 colon epithelial cells expressing endogenous NOX1 and NOD2 were stably transduced with lentivirus encoding for HA-tagged DUOX2 WT, DUOX2 R1211C, and DUOX2 R1492C in combination with WT DUOXA2.
Protein Isolation and Western Blotting
Cells were lysed in radioimmunoprecipitation assay buffer and after gel electrophoresis and blotting, membranes were probed with anti-HA (Covance Laboratories, Princeton, NJ), anti-DUOX2,31 anti-Myc (9E10), α-NOXA1,31 anti-NOX1,32 anti-p22phox FL-195 (Santa Cruz Biotechnology, Dallas, TX), anti-calnexin (BD Biosciences, San Jose, CA), and horseradish peroxidase-conjugated anti-rabbit or anti-mouse antibody (SouthernBiotech, Birmingham, AL). Proteins were visualized using enhanced chemiluminescence reagent (Pierce Biotechnology, Rockford, IL). Immunoblotting of p22phox or calnexin served as control.
ROS Assays
Superoxide production (NOX1) was measured using luminol enhanced chemiluminescence and stimulation with 1 mg/mL phorbol 12-myristate 13-acetate (PMA) for 30 minutes.33 Luminescence was measured on a Berthold Centro 960 LB in white 96-well plates. The chemiluminescence (relative light units, ΔRLU) readings were standardized against cellular protein (BCA assay).
H2O2 production (DUOX2) was measured using the homovanillic acid assay and addition of 1 μM thapsigargin.34 H2O2 production was standardized to H2O2 standard curves and cell lysate protein concentration. Empty vector transfection served as the control. For crypt ROS assays, Nox1−/− mice (Jackson Laboratory, Bar Harbor, ME) were transduced with lentivirus encoding empty vector, NOX1, NOX1 D330N, and NOX1 P360S. Briefly, the lentiviral titer was determined relative to p24 particles (QuickTiter Lentivirus Titer Kit; Cell Biolabs, San Diego, CA), and equal amounts of each lentivirus were intrarectally administered to Nox1−/− mice. Crypts were isolated from the intestine of euthanized mice 24 hours after lentiviral administration.
PMA-stimulated superoxide production was measured using L-012 enhanced chemiluminescence, and standardization was performed against total crypt protein concentration, as measured by BCA assay. ROS generation by transduced crypts was performed in two independent experiments (n = 2–3). Animal experiments were performed with ethics approval and authorization by the regulatory authority.
Flow Cytometry
H661-DUOXA2 cells expressing DUOX2 WT or variants were incubated with anti-HA antibody (Covance Laboratories) in fluorescence-activated cell sorting buffer on ice for 30 minutes without cell permeabilization. After incubation with anti-mouse Alexa Fluor 647, the cells were fixed in 1.5% paraformaldehyde and analyzed on an Accuri C6 flow cytometer (BD Biosciences).
Immunofluorescence
Cos cells expressing Myc-NOX1 WT or variants were treated with TAMRA-labeled Campylobacter jejuni for 15 minutes to visualize localization of NOX1 as described elsewhere10 while DUOX2-DUOXA2-expressing H661 cells were not stimulated. Cells were fixed in 3% paraformaldehyde, permeabilized in 0.5% Triton X-100, and stained with anti-DUOX2 or anti-Myc antibody, followed by goat anti-rabbit or anti-mouse Alexa Fluor 488 (Invitrogen/Life Technologies, Carlsbad, CA). HT29 cells expressing DUOXA2 and DUOX2 WT or missense variants were seeded on glass coverslips and treated with 300 μL of a clinical isolate of enteropathogenic Escherichia coli (EPEC) at optical density OD600 = 1 for 5 hours. Slides were washed, fixed, and permeabilized with 0.1% Triton X-100 and probed with antibodies against HA tag (Covance) and NOD2 (sc-30199, kind gift by P. Moynagh, National University of Ireland Maynooth), and 4′,6-diamidino-2-phenylindole (DAPI, blue). Images were acquired using a Zeiss LSM 700 microscope (Carl Zeiss, Thornwood, NY) and magnification 63× (oil) objective.
Colonic biopsies from control, disease control, and patients were fixed in 10% formaldehyde without methanol, embedded in paraffin, and processed for staining. Antigen retrieval was performed using high pressure-cooking with 1 mM EDTA at a pH 9.0 containing 0.05% Tween 20. Afterward, slides were blocked for 1 hour at room temperature with 5% bovine serum albumin in 1x phosphate-buffered saline (PBS) without calcium and magnesium containing 15% goat serum. Primary antibody incubation was performed overnight at 4°C. On the following day, the stained slides were washed three times for 10 minutes with 1x PBS without calcium and magnesium.
Secondary antibody incubation was performed at room temperature and in darkness for 1 hour. Slides were washed afterward three times for 10 minutes in darkness. Next, nuclear counterstaining with Hoechst 33342 Fluorescence Stain (Thermo Fisher Scientific, Waltham, MA) was performed at a dilution of 1:15,000. Finally, sections were mounted overnight with Vectorshield fluorescence mounting medium (Vector Laboratories, Burlingame, CA). Antibodies anti-beta catenin (BD Transduction Laboratories, BD Biosciences), anti-lysozyme (Abcam, Cambridge, MA), anti-CD24 (Abcam), and anti-EpCAM (Sigma-Aldrich, St. Louis, MO) were used at 1:100 dilution. Secondary antibodies were Alexa fluor 568 goat anti-rabbit and Alexa fluor 488 goat-anti mouse (both Invitrogen/Life Technologies). Images were acquired with an Olympus IX81 inverted fluorescence microscope (Olympus America, Center Valley, PA) equipped with a Hamamatsu C9100-13 back-thinned EM-CCD camera (Hamamatsu Photonics KK, Hamamatsu City, Japan) and Yokogawa CSU X1 spinning disk confocal scan head (Yokogawa Electric Corporation, Tokyo, Japan). Images were adjusted for contrast and brightness using the Volocity version 6.1.1 software (PerkinElmer Life and Analytical Sciences, Waltham, MA).
Virulence Assay
Adherence and invasion of C. jejuni 81-176 were assessed in NOX1 complex or DUOX2-DUOXA2 expressing Cos7 cells using the gentamicin protection assay.35 Plate grown C. jejuni 81-176 was washed and resuspended in tissue culture medium at OD600= 0.4 and added at multiplicity of infection 1000 to cells, followed by centrifugation at 250g for 5 minutes. After incubation for 3 hours at 37°C, the nonadherent and cell-associated bacteria were collected. For invasion, the infected and washed monolayers were incubated further with and without gentamicin (400 μg/mL) and incubated for an additional 2 hours at 37°C. The cells were lysed by the addition of 0.1% Triton X-100 in PBS for 10 minutes at 37°C. Bacterial counts for each assay were enumerated by serial dilution plating. All parameters were calculated as the average of the total number of colony-forming units/total initial inoculum.
Statistical Analysis
All functional experiments were conducted in triplicate with three repeats (n = 3), followed by an unpaired Student’s t test.
Results
Identification of NOX1 and DUOX2 Variants in VEOIBD
NOX1 and DUOX2 missense mutations were identified in five of 59 VEOIBD patients (age ≤6 years). All five patients presented with pancolitis without small bowel or perianal disease at diagnosis. None of the patients had systemic disease including thyroid disease or chronic infections, suggesting that defects were confined to the intestinal epithelium. SNPs and insertion/deletion variants were confirmed using Sanger sequencing and analyzed for potential function. Exon sequencing (Table 1, Table 2) identified a novel NOX1 variant (c.988G>A; p.P330S) in one male patient. Another rare variant (c.967G>A; rs34688635; p.D360N) was found in one male and one female patient. The missense variant NOX1 p.P330S is potentially damaging (Polyphen2 score: 0.995) and unique according to the Washington Exome Variant Server, while NOX1 p.D360N was predicted to be “probably damaging” by PolyPhen2 and was given a maximum evolutionary conservation score of 1 by the PhastCons program using 46 mammalian species. Variants in DUOX2 were also identified in VEOIBD patients (Table 1, Table 2). One of the patients was heterozygous for DUOX2 p.R1211C (c.4474G>A) and developed severe disease that necessitated colonic resection. The disease subsequently recurred at the resection site, a finding consistent with Crohn’s disease. The second variant was detected in a very early onset UC patient heterozygous for DUOX2 p.R1492C (c.3631C>T; rs374410986), who presented with pancolitis.
Table 1.
List of Variants Found in NOX1 and DUOX2 in Very Early Onset Inflammatory Bowel Disease Patients
| Gene | Variant | rs # | MAF/Minor Allele Count∗ | CADD Rank Score | Age at Diagnosis (y) | Gender | Diagnosis | Patient Summary |
|---|---|---|---|---|---|---|---|---|
| NOX1 | c.988G>A p.P330S |
Novel | Novel—no data available | 0.40694 | 1.8 | Male | IBD-U | Severe pancolitis Granuloma |
| NOX1 | c.967G>A p.D360N |
rs34688635 | T = 0.010/16 | 0.5415 | 5.3 | Female | UC | Pancolitis |
| 4.7 | Male | IBD-U | Pancolitis | |||||
| DUOX2 | c.4474G>A p.R1211C |
Novel | Novel—no data available | 0.90955 | 4.7 | Male | IBD-U | Severe pancolitis Colectomy, perforation Recurrence of disease |
| DUOX2 | c.3631C>Tpp.R1492C | rs374410986, Novel | Novel—no data available | 0.9002 | 4.3 | Male | UC | Pancolitis |
Note: CADD, Combined Annotation Dependent Depletion; DUOX2, dual oxidase 2; IBD-U, inflammatory bowel disease unclassified; MAF, minor allele frequency; NOX1, NADPH oxidase 1; UC, ulcerative colitis.
The minor allele frequencies are taken from 1000 Genomes of dbSNP.
Table 2.
Computational Analysis of NOX1 and DUOX2 Variants
| Gene | Variant | Chromosome | Position | rs# | Chromosomal position | Minor Allele Frequency (dbSNP) | Minor Allele Frequency (1000G) | Minor Allele Frequency (NHLBI exome variant frequencies) | ExAC v0.3 MAF | SIFT | SIFT Prediction | PolyPhen2 | PolyPhen2 Prediction | MutationTaster | MutationTaster Prediction | MutationAssessor | MutationAssessor Prediction | FATHMM | FATHMM Prediction | LRT Score | LRT Prediction | GERP++ | PhyloP | CADD Rank Score |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| NOX1 | P330S | X | 100105285 | Novel | c.988G>A | No MAF data | No MAF data | No MAF data | No MAF data | 0.051 | Tolerated | 1 | Probably Damaging | 1 | Disease Causing | 3.185 | Predicted Functional (Medium) | 2.44 | Tolerated | 0 | Deleterious | 3.87 | 1.767 | 0.40694 |
| NOX1 | D360N | X | 100105195 | rs34688635 | c.967G>A | T=0.010/16 (1%) | T=0.00529801 | 0.019881 | 0.018 | 0.042 | Tolerated | 0.085 | Possibly Damaging | 0 | Polymorphism Automatic | 2.225 | Predicted Functional (Medium) | -3.09 | Damaging | 0.000445 | Deleterious | 3.87 | 1.767 | 0.54147 |
| DUOX2 | R1211C | 15 | 45389874 | rs374410986 | c.3631C>T | No MAF data | No MAF data | A=7.7e-05 | A=0.00004118 | 0 | Damaging | 1 | Probably Damaging | 1 | Disease Causing | 3.37 | Predicted Functional (Medium) | 0 | Deleterious | 5.69 | 2.679 | 0.90955 | ||
| DUOX2 | R1492C | 15 | 45386811 | Novel | c.4474G>A | No MAF data | No MAF data | No MAF data | A=0.00004118 | 0 | Damaging | 1 | Probably Damaging | 1 | Disease Causing | 3.97 | Predicted Functional (High) | 0 | Deleterious | 5.68 | 2.838 | 0.9002 |
ExAC, Exome Aggregation Consortium; CADD, Combined Annotation Dependent Depletion; FATHMM, Functional Analysis through Hidden Markov Models; GERP, Genomic Evolutionary Rate Profiling; LRT Score, likelihood ratio test; MAF: Minor allele frequenct; PolyPhen2, Polymorphism Phenotyping v2; SIFT, Scale-invariant feature transform.
In an independent replication cohort of 150 VEOIBD patients, none of the NOX1 and DUOX2 missense variants were identified. Similarly, in the publicly available International IBD Genetics Consortium (http://www.ibdgenetics.org) database none of the NOX1 and DUOX2 missense variants were identified as this data set does not examine rare variants, only common polymorphisms, and the p.Asp360Asn variant is not analyzed by the immunochip.
Therefore, we took an alternate approach employing an array-based genotyping using the Illumina HumanExome v1.0 platform of 1477 Crohn’s disease (CD) cases, 559 UC cases, and 2614 healthy controls, all with genetically verified Ashkenazi Jewish (AJ) ancestry by principal components analysis. Using this approach we detected an association between D360N NOX1 and male AJ UC (MAFcase . 3.37%, MAFcontrol . 0.82%; odds ratio 4.22; P = 1.25 × 10−3). The association was not detected in either of the female AJ UC cases (MAFcase = 1.53%, MAFcontrol = 0.99%; odds ratio 1.55; P = .343), although the trend was in the same direction as observed in the AJ males cases. However, this trend was not observed in Crohn’s disease cases (MAFCD = 0.97%). The finding in an adult UC cohort suggests that pathways/processes involved in VEOIBD will have implications for adult IBD patients.
Histologic Analysis of NOX1/DUOX2 Variants
Histopathology analysis using HE and PAS staining (Figure 1A) was performed in biopsies from patients with the identified DUOX2 p.R1211C variant as well as a patient with the NOX1 p.D360N variant and compared with the healthy control and an IBD control biopsy. The disease control showed features of chronic and regenerative IBD, demonstrated by metaplastic Paneth cells within colonic crypts. The patient with the NOX1 p.D360N variant showed focal inflammation, increased cellularity of inflammatory cells adjacent to normal areas of unaffected colonic mucosa. The patient with the DUOX2 p.R1211C variant demonstrated more severe morphologic changes, with severe inflammation and crypt damage in the colonic mucosa when compared with the NOX1 variant.
Figure 1.

Characterization of selected patient biopsies. (A) HE and periodic acid–Schiff staining of colonic biopsy samples from an inflammatory bowel disease (IBD) control (Paneth cell metaplasia), a patient with the NOX1 D360N variant, and a patient with the DUOX2 R1211C variant. The patient with the NOX1 D360N variant shows focal inflammation, increased cellularity of inflammatory cells adjacent to normal area. The patient with the DUOX2 R1211C variant shows severe colitis with architectural distortion (crypt damage). Scale bar: 20 μm. (B) Immunofluorescence analysis with Paneth cell markers lysozyme and CD24 in colonic biopsy samples: lysozyme and EpCAM (red), β-catenin and CD24 (green), and nuclei (blue). Lysozyme was expressed in the crypts of the patients as well as the IBD control, but not in the healthy control. CD24 is expressed in colonic crypts in the IBD control and the patient with the DUOX2 R1211C variant, but neither in the healthy control nor the patient with the NOX1 variant. Scale bar: 10 μm.
Immunofluorescence staining was performed on colonic biopsy samples to determine whether Paneth cell metaplasia, a feature of chronic and regenerative change as a consequence of continuous inflammation within the colon, has occurred. Both markers, lysozyme and CD24, were highly positive in metaplastic Paneth cells of colonic crypt enterocytes in the disease control (see Figure 1B). Altered NOX1 appears not to progress cells into full metaplasia as seen by the absence of CD24 within crypt cells of the patient harboring NOX1 p.D360N. In colonic crypts of the patient with the DUOX2 p.R1211C variant, both lysozyme and CD24 were expressed, albeit not as prominent as observed within metaplastic Paneth cells in the IBD control.
Topologic Models of NOX1/DUOX2 Variants
The NOX1 NADPH oxidase is formed by heterodimerization of NOX1 with p22phox, followed by assembly with the regulatory proteins NOXO1, NOXA1, and Rac1-GTP.8 The cytosolic carboxyl terminus of NADPH oxidases harbors NADPH and FAD-binding regions, which are required for electron transport across the membrane via hemes where molecular oxygen is reduced to form superoxide. The identified NOX1 variants are located either just in front of FAD1 (p.P330S) or inside FAD2 (p.D360N) (Figure 2A). Pro330 and Asp360 are conserved in NOX1–4 proteins identified in vertebrates and lower organisms.36 CYBB missense variants (X-CGD) leading to loss or diminished ROS generation in neutrophils are located in close vicinity to the identified NOX1 variants (http://bioinf.uta.fi/CYBBbase).37 Modeling of NOX1 WT, NOX1 (p.P330S), or NOX1 (p.D360N) dehydrogenase domains was performed by combining the crystal structures of FAD-binding domains homologous to the NOX FAD with the partial structure of the dehydrogenase domain of NOX2 in the correct orientation (see Figure 2B).
Figure 2.

Modeling and functional characterization of NOX1 variants. (A) Topologic model depicting NOX1 very early onset inflammatory bowel disease (VEOIBD) variants (red), selected X-CGD CYBB (NOX2) variants (green), conserved residues boxed (blue). (B) Three-dimensional model of NOX1 wild-type (WT) (grey), NOX1 P330S (green), or NOX1 D360N (pink) dehydrogenase domains. NADPH, FAD, residue H339, and variant positions are marked. (C) ROS production by NOX1 WT and variants. (D) Protein expression of NOX1 and variants, Myc-NOXO1, NOXA1, and p22phox as loading control. (E) ROS production in murine Nox1−/− crypts transduced with NOX1 WT or variants. Phorbol 12-myristate 13-acetate (PMA) stimulation was at 200 seconds. (F) Localization of Myc-NOX1 WT or D360N (green) in C. jejuni (red) infected Cos-p22 cells. Scale bar: 10 μm; arrow indicates membrane localization. (G) Adhesion and invasion of Campylobacter jejuni in cells expressing NOX1 WT, P330S, or D360N. Error bars ± standard deviation n = 3; *P ≤ .05; **P ≤ .01; ****P ≤ .0001; comparing NOX1 WT to variants.
FAD and NADPH were docked to each NOX/DUOX model by using HADDOCK. FAD binds to NOX1 WT mainly with electrostatic interaction to His339 in the FAD1 domain and Asp360 in the FAD2 domain. Based on the model, Pro330 will be important for stabilization of the antiparallel β-structure that creates the FAD1 domain. Although Pro330 is not directly involved in FAD binding, the change Pro330Ser in NOX1 alters the position of His339 in the FAD1 domain, which decreases binding affinity of this variant for FAD.
The second NOX1 residue altered in VEOIBD, Asp360, is directly involved in FAD binding, and therefore a change to asparagine (D360N) weakens the interaction between FAD and NOX1. FAD binds to NOX1 with binding affinity in μM range; therefore, we predict that small structural changes in both FAD domains will compromise catalytic activity of the NOX1 enzyme. Debeurme et al38 reported disrupted FAD binding and diminished catalytic activity of NOX2 in selected CYBB variants.
Functional Characterization of NOX1 Variants
As structural analysis predicts that the catalytic activity of NOX1 variants will be compromised, we reconstituted WT and altered NOX1 complexes in an epithelial model cell system (Cos7) deficient in all NOX/DUOX isoforms. Both NOX1 p.P330S and NOX1 p.D360N variants displayed diminished catalytic activity (see Figure 2C). Basal and phorbol ester-stimulated ROS generation was significantly reduced for NOX1 missense variants (50%–60%), while the overall protein expression was comparable to WT NOX1 (see Figure 2D).
As patients could not be recalled for colon tissue evaluation, catalytic activity of NOX1 variants was also measured in a murine in vivo expression setting. Nox1 knockout mice were transduced with lentivirus encoding NOX1 WT and variants intrarectally, and ROS generation of isolated crypts was recorded 24 hours later. Similar to the results obtained in cell lines, ROS production in the crypts was reduced in the NOX1 variants when compared with NOX1 WT (see Figure 2E).
A reduction in epithelial ROS production will attenuate host protection from intestinal pathogens. Defective processing of responses to mucosal bacteria is recognized to play a central role in the development and perpetuation of intestinal inflammation in IBD. C. jejuni in particular has been associated with the initiation of IBD.39 C. jejuni uptake was used to visualize infection-associated translocation of NOX1 to membrane ruffles and to assess the antibacterial response.10 Stimulated membrane localization of NOX1 WT and NOX1 variants (NOX1 p.D360N shown) were comparable (see Figure 2F), but reduced ROS generation caused a 10-fold increase in bacterial invasion when cells harbored the NOX1 p.P330S or NOX1 p.D360N variants with reduced catalytic activity (see Figure 2G).
Functional Characterization of DUOX2 Variants
Inactivating mutations in DUOX2 or DUOXA2 have been linked to inherited permanent or transient congenital hypothyroidism,40 and to date over 23 DUOX2 mutations have been described in this context (HGMD, www.hgmd.cf.ac.uk/ac/gene) (Figure 3A). The two VEOIBD-associated DUOX2 variants are novel; in contrast to most of the reported DUOX2 variants, they are not located in the peroxidase homology domain or the EF hand regions. DUOX2 p.R1211C is placed in a polybasic region within an intracellular loop, and Arg1492 in DUOX2 is an integral part of the highly conserved GRP sequence in the NADPH3 domain (see Figure 3A).
Figure 3.

Modeling and functional characterization of DUOX2 variants. (A) Topologic model depicting DUOX2 very early onset inflammatory bowel disease (VEOIBD) variants (red), selected DUOX2 hypothyroidism variants (black), and conserved residues boxed (blue). (B) Three-dimensional model of DUOX2 wild-type (WT) (grey) and DUOX2 R1492C (pink) dehydrogenase domain. NADPH, FAD, and variant position are marked. (C) H2O2 release by DUOX2 WT and variants. (D) Protein expression of HA-DUOX2 WT and variants; calnexin served as loading control. (E) HA-DUOX2 WT and variant surface expression by flow cytometry (left) and localization by immunofluorescence (right) (green, arrow for membrane). Scale bar: 10 μm. (F) Adhesion and invasion of C. jejuni in cells expressing HA-DUOX2 WT, R1211C, or R1492C. Error bars ± standard deviation, n = 3; *P ≤ .05; **P ≤ .01; ***P ≤ .001; ****P ≤ .0001; comparing DUOX2 WT with variants.
As described for NOX1, the dehydrogenase domains of DUOX2 WT and DUOX2 p.R1492C were modeled onto the extended NOX2 structure; by use of HADDOCK, NADPH and FAD were docked to the structure (see Figure 3B). Structural analysis revealed that Arg1492 is part of the NADPH-binding pocket. NADPH binds to DUOX2 WT with strong electrostatic interactions to the residues Arg1421 and Arg1492 with a sum of −181.7 ± 76.4 kcal/mol and with weak Van der Waals interactions to Gly1385, Thr1463, Pro1520, Gly1521, and Met1520 with a sum of −30.9 ± 7.8 kcal/mol. Replacing Arg1492 with cysteine as in the DUOX2 p.R1492C variant does not change the DUOX2 structure or the position of other NADPH-interacting residues. However, the change is predicted to weaken the interaction between NADPH and DUOX2 by a factor of 2. How replacement of Arg1221 with cysteine will directly affect DUOX2 catalytic activity cannot be predicted because suitable structures for modeling do not exist, but in both NOX2 and NOX4 the analogous D loop participates in ROS production.41, 42
Functional analysis of DUOX2 variants was performed in the H661 cellular model system that represents a physiologic context for DUOX-DUOXA expression and is devoid of NOX1–5 activity.29 Both DUOX2 variants, when coexpressed with their dimerization partner DUOXA2, produced significantly less H2O2 than WT DUOX2 (see Figure 3C), although protein expression and cellular localization were not altered (see Figure 3D and E). DUOX2 has been functionally associated with NOD2 in transient overexpression conditions.43 HT29 colonic cells express endogenously a functional NOX1 complex and NOD2, and thus provide an appropriate context for analysis of putative DUOX2-NOD2 interactions.
DUOX2 or DUOX2 variants together with DUOXA2 were stably incorporated into HT29 cells, followed by exposure to enteropathogenic E. coli. DUOX2 WT or variants, localized on internal membrane structures before the challenge, translocated to the plasma membrane and cell-cell junctions. NOD2, on the other hand, remained in the intracellular compartment, albeit NOD2 protein expression was up-regulated (Figure 4). Thus, DUOX2 and NOD2 were not recruited simultaneously upon E. coli challenge.
Figure 4.
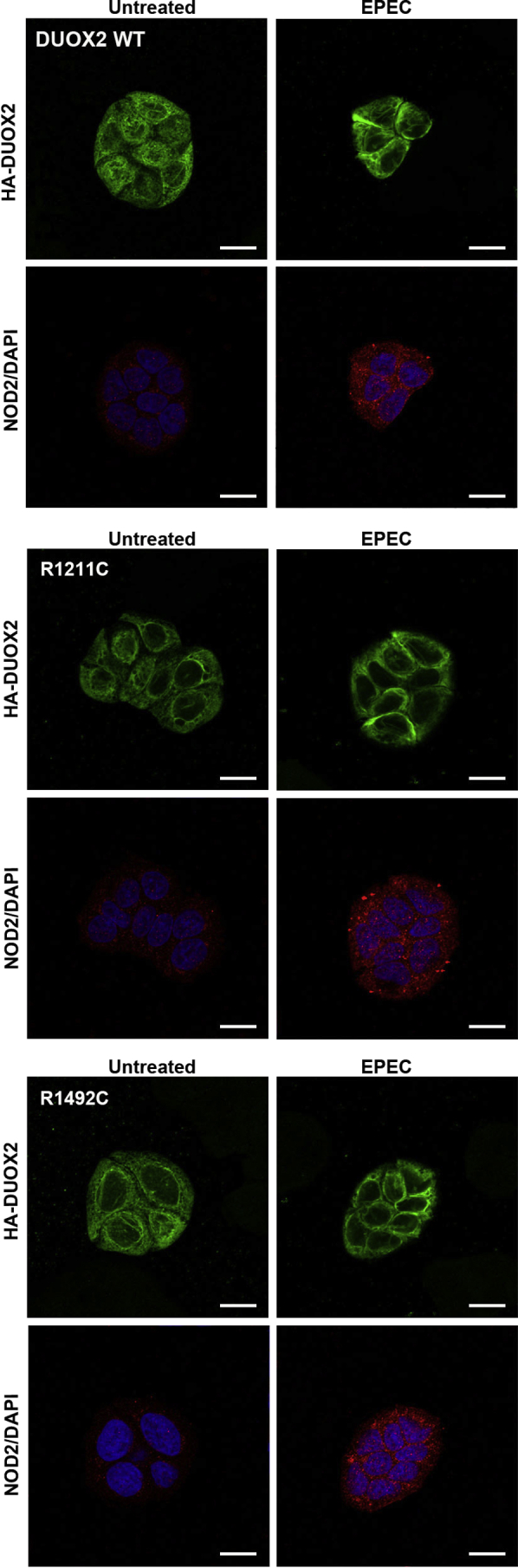
Bacteria-induced translocation of DUOX2 and variants does not involve NOD2 in colonic cells. HT29 cells stably expressing DUOX2 WT, DUOX2 R1211C, and DUOX2 R1492C were exposed to enteropathogenic Escherichia coli (EPEC) for 5 hours. Immunofluorescence images of DUOX2 (green), NOD2 (red), and nuclei (blue). Scale bar: 15 μm.
Stimulated H2O2 release in DUOX2 WT or variant-expressing HT29 cells mirrored the results obtained with H661 cells (data not shown). DUOX2-mediated H2O2 release at apical membranes has been linked to antimicrobial host defense and decreased C. jejuni virulence.10 Comparison of C. jejuni invasion in DUOX2 WT or DUOX2 variant-expressing (DUOX2 p.R1211C, DUOX2 p.R1492C) epithelial cells showed increased invasion when ROS generation was diminished (see Figure 3F).
Discussion
We have identified inactivating missense variants in each of the epithelial NADPH oxidases NOX1 (p.P330S, p.D360N) and DUOX2 (p.R1211C, p.R1492C) in five VEOIBD patients. Variants in X-linked NOX1 were found in two male VEOIBD patients, and NOX1 p.D360N was associated with male UC in an AJ ancestry case-control cohort, likely leading to increased or sustained disease severity.
The identification of rare functional variants contributing to the pathogenesis of VEOIBD has been observed with other genes, including the NOX2 NADPH oxidase complex,7 NOS2,44 IL10R,15 and XIAP.45, 46 The variants we identified in both NOX1 and DUOX2 are rare and not found in a replication VEOIBD cohort or data sets of common variants. However, all variants showed both pathologic and functional defects, indicating that these variants may contribute to disease susceptibility or pathogenesis. Further large-scale sequencing of pediatric- and adult-onset IBD may indicate a broader role of both NOX1 and DUOX2 in IBD pathogenesis, as observed in our AJ population.
Recently, altered DUOX2 expression was identified in ileum biopsies from pediatric Crohn’s disease patients.47 Further, ROS derived from NADPH oxidases is critical to control mucin granule accumulation in colonic goblet cells,12 and NOX1 has been shown to control the balance between goblet and absorptive cell types in murine colon.48 Interestingly, colonic biopsies from patients carrying either NOX1 p.D360N or DUOX2 p.R1211C variants showed abnormal CD24 and lysozyme expression (see Figure 1B), suggesting a role for these proteins in Paneth cell metaplasia.
The thyroid function of the two male VEOIBD patients harboring DUOX2 mutations was normal, although inactivating monoallelic and biallelic DUOX2 and DUOXA2 variants have been linked to hypothyroidism.49 In contrast to adult onset IBD, VEOIBD frequently encompasses a unique clinical presentation, with severe disease limited to the colon and with poor response to standard therapies.24 VEOIBD variants (NCF2,50 NOS2,44 IL10RA/B,15 TTC7A51) have usually been rare, suggesting that these patients may have a unique genetic susceptibility. Furthermore, we have recently shown that SNPs and rare variants in all components of the NOX2 NADPH oxidase complex are associated with VEOIBD.7 Similar to our recent observations with NOX2 NADPH oxidase complex variants leading to decreased ROS production in neutrophils,7 reduced mucosal ROS levels originating from NOX1 and DUOX2 variants play also a role in susceptibility to VEOIBD and perhaps other severe IBD phenotypes.
Intestinal NADPH oxidases connect to antibacterial autophagy and endosomal pathways important for mucus secretion and may modulate the interplay between commensal bacteria and pathogens.12, 13 Recent microbiome studies on a large pediatric cohort with new-onset Crohn’s disease assigned a unique role to changes in the rectal mucosal microbiota for disease classification.52 Changes in ROS generation at the mucosal surface will most likely result in dysbiosis, intestinal inflammation, and pathobiont development. Our functional studies provide strong support both for the pathogenic nature of the mutations identified in these VEOIBD patients and the role of epithelial ROS in protecting cells from bacterial attack.
Further phenotypic exploration of NOX/DUOX variants will be aided by studies in humans and improved animal models, as current IBD animal models seem often not to reflect human disease triggered by reduced ROS. For example, murine Cybb (Nox2) deficiency does not lead to spontaneous Crohn’s disease-like intestinal disease and gut inflammation, both observed in many CGD patients. Although Cybb knockout mice exhibit several hallmarks of CGD upon fungal or bacterial challenge, they were slightly protected in the dextran sodium sulfate–induced colitis mouse model.53 Similarly, Nox1 deficiency in the murine mucosa did not alter dextran sodium sulfate–colitis pathology,54 although combined Nox1 and Il10 deficiency caused spontaneous colitis in mice.55 Mice harboring an inactivating Duox2 variant or Duoxa deficiency showed severe hypothyroidism and increased colonization with Helicobacter felis.11, 56
In conclusion, our findings demonstrate that novel NOX1 and DUOX2 NADPH oxidase variants resulting in attenuated ROS production and impaired mucosal defense occur in children with VEOIBD. This may influence IBD pathogenesis beyond childhood.
Acknowledgments
The authors thank N. Corcionivoschi for technical assistance.
Footnotes
Conflicts of interest These authors disclose the following: Scott B. Snapper has received personal fees from Pfizer, Ironwood Pharmaceuticals, AbbVie, and Cubist, outside of the submitted work. Holm H. Uhlig is participating in ongoing project collaborations or reagent supply unrelated to the manuscript, including UCB Pharma, Eli Lilly, GSK, Tetralogics, Vertex, and MSD. The remaining authors disclose no conflicts.
Funding This study was funded by the Science Foundation Ireland (PI award, Stokes award to U.G.K.); the Children’s Medical and Research Foundation Ireland (to B.B., U.G.K.); National Institutes of Health grants DK062413, AI067068, DK046763, HS021747, the European Union, the CCFA, and the Lisa Z and Joshua L Greer Endowed Chair in IBD Genetics (to D.P.B.M.); the New York Crohn’s Foundation grant (to I.P.); National Institutes of Health grants U01 DK062429, DK062422, R01 DK092235, and the Ward-Coleman Translational Genetics Chair (to J.C.); the Medical Research Fund, Oxford (to H.H.U.); National Institutes of Health grant DK62431, the Atran Foundation, and the Morton Hyatt Family (to S.R.B.); Crohn’s and Colitis Canada (CCC), Canadian Association of Gastroenterology (CAG), and Canadian Institute of Health Research (CIHR) Fellowship (to A.E.); the RESTRACOMP fellowship from the Research Institute of the Hospital for Sick Children, Toronto, Canada (to C.T.); a CIHR–Operating Grant MOP119457 and MOP97756 (to A.M.M. and J.H.B.); and in part by Leona M. and Harry B. Helmsley Charitable Trust (to A.M.M., H.H.U., D.P.B.M., S.B.S., J.C.).
References
- 1.Jostins L., Ripke S., Weersma R.K. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature. 2012;491:119–124. doi: 10.1038/nature11582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Maloy K.J., Powrie F. Intestinal homeostasis and its breakdown in inflammatory bowel disease. Nature. 2011;474:298–306. doi: 10.1038/nature10208. [DOI] [PubMed] [Google Scholar]
- 3.van den Berg J.M., van Koppen E., Ahlin A. Chronic granulomatous disease: the European experience. PLoS One. 2009;4:e5234. doi: 10.1371/journal.pone.0005234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Marks D.J., Miyagi K., Rahman F.Z. Inflammatory bowel disease in CGD reproduces the clinicopathological features of Crohn’s disease. Am J Gastroenterol. 2009;104:117–124. doi: 10.1038/ajg.2008.72. [DOI] [PubMed] [Google Scholar]
- 5.Muise A.M., Snapper S.B., Kugathasan S. The age of gene discovery in very early onset inflammatory bowel disease. Gastroenterology. 2012;143:285–288. doi: 10.1053/j.gastro.2012.06.025. [DOI] [PubMed] [Google Scholar]
- 6.Somasundaram R., Deuring J.J., van der Woude C.J. Linking risk conferring mutations in NCF4 to functional consequences in Crohn’s disease. Gut. 2012;61:1097–1098. doi: 10.1136/gutjnl-2011-301344. [DOI] [PubMed] [Google Scholar]
- 7.Dhillon S.S., Fattouh R., Elkadri A. Variants in nicotinamide adenine dinucleotide phosphate oxidase complex components determine susceptibility to very early onset inflammatory bowel disease. Gastroenterology. 2014;147:680–689.e2. doi: 10.1053/j.gastro.2014.06.005. [DOI] [PubMed] [Google Scholar]
- 8.Lambeth J.D., Neish A.S. Nox enzymes and new thinking on reactive oxygen: a double-edged sword revisited. Annu Rev Pathol. 2014;9:119–145. doi: 10.1146/annurev-pathol-012513-104651. [DOI] [PubMed] [Google Scholar]
- 9.Rada B., Leto T.L. Oxidative innate immune defenses by Nox/Duox family NADPH oxidases. Contrib Microbiol. 2008;15:164–187. doi: 10.1159/000136357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Corcionivoschi N., Alvarez L.A., Sharp T.H. Mucosal reactive oxygen species decrease virulence by disrupting Campylobacter jejuni phosphotyrosine signaling. Cell Host Microbe. 2012;12:47–59. doi: 10.1016/j.chom.2012.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Grasberger H., El-Zaatari M., Dang D.T. Dual oxidases control release of hydrogen peroxide by the gastric epithelium to prevent Helicobacter felis infection and inflammation in mice. Gastroenterology. 2013;145:1045–1054. doi: 10.1053/j.gastro.2013.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Patel K.K., Miyoshi H., Beatty W.L. Autophagy proteins control goblet cell function by potentiating reactive oxygen species production. EMBO J. 2013;32:3130–3144. doi: 10.1038/emboj.2013.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jones R.M., Luo L., Ardita C.S. Symbiotic lactobacilli stimulate gut epithelial proliferation via Nox-mediated generation of reactive oxygen species. EMBO J. 2013;32:3017–3028. doi: 10.1038/emboj.2013.224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Little J.L., Higgins P.T., Ioannidis J.P.A. STREGA: Strengthening the Reporting of Genetic Associations: an extension of the STROBE Statement. Ann Intern Med. 2009;150:206–215. doi: 10.7326/0003-4819-150-3-200902030-00011. http://www.medicine.uottawa.ca/public-health-genomics/web/eng/strega.html Also available at: [DOI] [PubMed] [Google Scholar]
- 15.Moran C.J., Walters T.D., Guo C.H. IL-10R polymorphisms are associated with very-early-onset ulcerative colitis. Inflamm Bowel Dis. 2013;19:115–123. doi: 10.1002/ibd.22974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Muise A.M., Walters T., Xu W. Single nucleotide polymorphisms that increase expression of the guanosine triphosphatase RAC1 are associated with ulcerative colitis. Gastroenterology. 2011;141:633–641. doi: 10.1053/j.gastro.2011.04.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Adzhubei I.A., Schmidt S., Peshkin L. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kumar P., Henikoff S., Ng P.C. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009;4:1073–1081. doi: 10.1038/nprot.2009.86. [DOI] [PubMed] [Google Scholar]
- 19.Yuan H.Y., Chiou J.J., Tseng W.H. FASTSNP: an always up-to-date and extendable service for SNP function analysis and prioritization. Nucleic Acids Res. 2006;34:W635–W641. doi: 10.1093/nar/gkl236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Desmet F.O., Hamroun D., Lalande M. Human Splicing Finder: an online bioinformatics tool to predict splicing signals. Nucleic Acids Res. 2009;37:e67. doi: 10.1093/nar/gkp215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang J., Ronaghi M., Chong S.S. pfSNP: An integrated potentially functional SNP resource that facilitates hypotheses generation through knowledge syntheses. Hum Mutat. 2011;32:19–24. doi: 10.1002/humu.21331. [DOI] [PubMed] [Google Scholar]
- 22.Benchimol E.I., Mack D.R., Nguyen G.C. Incidence, outcomes, and health services burden of very early onset inflammatory bowel disease. Gastroenterology. 2014;147:803–813.e7. doi: 10.1053/j.gastro.2014.06.023. [DOI] [PubMed] [Google Scholar]
- 23.Uhlig H.H., Schwerd T., Koletzko S. The diagnostic approach to monogenic very early onset inflammatory bowel disease. Gastroenterology. 2014;147:990–1007.e3. doi: 10.1053/j.gastro.2014.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Levine A., Kugathasan S., Annese V. Pediatric onset Crohn’s colitis is characterized by genotype-dependent age-related susceptibility. Inflamm Bowel Dis. 2007;13:1509–1515. doi: 10.1002/ibd.20244. [DOI] [PubMed] [Google Scholar]
- 25.Uhlig H.H. Monogenic diseases associated with intestinal inflammation: implications for the understanding of inflammatory bowel disease. Gut. 2013;62:1795–1805. doi: 10.1136/gutjnl-2012-303956. [DOI] [PubMed] [Google Scholar]
- 26.Sali A., Blundell T.L. Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol. 1993;234:779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- 27.De Vries S.J., van Dijk M., Bonvin A.M. The HADDOCK web server for data-driven biomolecular docking. Nat Protoc. 2010;5:883–897. doi: 10.1038/nprot.2010.32. [DOI] [PubMed] [Google Scholar]
- 28.Van Dijk A.D., Bonvin A.M. Solvated docking: introducing water into the modelling of biomolecular complexes. Bioinformatics. 2006;22:2340–2347. doi: 10.1093/bioinformatics/btl395. [DOI] [PubMed] [Google Scholar]
- 29.Luxen S., Noack D., Frausto M. Heterodimerization controls localization of Duox-DuoxA NADPH oxidases in airway cells. J Cell Sci. 2009;122:1238–1247. doi: 10.1242/jcs.044123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yu L., Zhen L., Dinauer M.C. Biosynthesis of the phagocyte NADPH oxidase cytochrome b558. Role of heme incorporation and heterodimer formation in maturation and stability of gp91phox and p22phox subunits. J Biol Chem. 1997;272:27288–27294. doi: 10.1074/jbc.272.43.27288. [DOI] [PubMed] [Google Scholar]
- 31.Pacquelet S., Lehmann M., Luxen S. Inhibitory action of NoxA1 on dual oxidase activity in airway cells. J Biol Chem. 2008;283:24649–24658. doi: 10.1074/jbc.M709108200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Antony S., Wu Y., Hewitt S.M. Characterization of NADPH oxidase 5 expression in human tumors and tumor cell lines with a novel mouse monoclonal antibody. Free Radic Biol Med. 2013;65:497–508. doi: 10.1016/j.freeradbiomed.2013.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.von Lohneysen K., Noack D., Jesaitis A.J. Mutational analysis reveals distinct features of the Nox4-p22 phox complex. J Biol Chem. 2008;283:35273–35282. doi: 10.1074/jbc.M804200200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Martyn K.D., Frederick L.M., von Loehneysen K. Functional analysis of Nox4 reveals unique characteristics compared to other NADPH oxidases. Cell Signal. 2006;18:69–82. doi: 10.1016/j.cellsig.2005.03.023. [DOI] [PubMed] [Google Scholar]
- 35.Elsinghorst E.A. Measurement of invasion by gentamicin resistance. Methods Enzymol. 1994;236:405–420. doi: 10.1016/0076-6879(94)36030-8. [DOI] [PubMed] [Google Scholar]
- 36.Kawahara T., Quinn M.T., Lambeth J.D. Molecular evolution of the reactive oxygen-generating NADPH oxidase (Nox/Duox) family of enzymes. BMC Evol Biol. 2007;7:109. doi: 10.1186/1471-2148-7-109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kuhns D.B., Alvord W.G., Heller T. Residual NADPH oxidase and survival in chronic granulomatous disease. N Engl J Med. 2010;363:2600–2610. doi: 10.1056/NEJMoa1007097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Debeurme F., Picciocchi A., Dagher M.C. Regulation of NADPH oxidase activity in phagocytes: relationship between FAD/NADPH binding and oxidase complex assembly. J Biol Chem. 2010;285:33197–33208. doi: 10.1074/jbc.M110.151555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gradel K.O., Nielsen H.L., Schonheyder H.C. Increased short- and long-term risk of inflammatory bowel disease after salmonella or campylobacter gastroenteritis. Gastroenterology. 2009;137:495–501. doi: 10.1053/j.gastro.2009.04.001. [DOI] [PubMed] [Google Scholar]
- 40.Moreno J.C., Bikker H., Kempers M.J. Inactivating mutations in the gene for thyroid oxidase 2 (THOX2) and congenital hypothyroidism. N Engl J Med. 2002;347:95–102. doi: 10.1056/NEJMoa012752. [DOI] [PubMed] [Google Scholar]
- 41.Carrichon L., Picciocchi A., Debeurme F. Characterization of superoxide overproduction by the D-Loop(Nox4)-Nox2 cytochrome b(558) in phagocytes—differential sensitivity to calcium and phosphorylation events. Biochim Biophys Acta. 2011;1808:78–90. doi: 10.1016/j.bbamem.2010.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Von Lohneysen K., Noack D., Wood M.R. Structural insights into Nox4 and Nox2: motifs involved in function and cellular localization. Mol Cell Biol. 2010;30:961–975. doi: 10.1128/MCB.01393-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lipinski S., Till A., Sina C. DUOX2-derived reactive oxygen species are effectors of NOD2-mediated antibacterial responses. J Cell Sci. 2009;122:3522–3530. doi: 10.1242/jcs.050690. [DOI] [PubMed] [Google Scholar]
- 44.Dhillon S.S., Mastropaolo L.A., Murchie R. Higher activity of the inducible nitric oxide synthase contributes to very early onset inflammatory bowel disease. Clin Transl Gastroenterol. 2014;5:e46. doi: 10.1038/ctg.2013.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Latour S., Aguilar C. XIAP deficiency syndrome in humans. Semin Cell Dev Biol. 2015;39:115–123. doi: 10.1016/j.semcdb.2015.01.015. [DOI] [PubMed] [Google Scholar]
- 46.Speckmann C., Ehl S. XIAP deficiency is a mendelian cause of late-onset IBD. Gut. 2014;63:1031–1032. doi: 10.1136/gutjnl-2013-306474. [DOI] [PubMed] [Google Scholar]
- 47.Haberman Y., Tickle T.L., Dexheimer P.J. Pediatric Crohn disease patients exhibit specific ileal transcriptome and microbiome signature. J Clin Invest. 2014;124:3617–3633. doi: 10.1172/JCI75436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Coant N, Ben Mkaddem S, Pedruzzi E, et al. NADPH oxidase 1 modulates WNT and NOTCH1 signaling to control the fate of proliferative progenitor cells in the colon. Mol Cell Biol;30:2636–2650. [DOI] [PMC free article] [PubMed]
- 49.Muzza M., Rabbiosi S., Vigone M. The clinical and molecular characterization of patients with dyshormonogenic congenital hypothyroidism reveals specific diagnostic clues for DUOX2 defects. J Clin Endocrinol Metab. 2014;99:E544–E553. doi: 10.1210/jc.2013-3618. [DOI] [PubMed] [Google Scholar]
- 50.Muise A.M., Xu W., Guo C.H. NADPH oxidase complex and IBD candidate gene studies: identification of a rare variant in NCF2 that results in reduced binding to RAC2. Gut. 2012;61:1028–1035. doi: 10.1136/gutjnl-2011-300078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Avitzur Y., Guo C., Mastropaolo L.A. Mutations in tetratricopeptide repeat domain 7A result in a severe form of very early onset inflammatory bowel disease. Gastroenterology. 2014;146:1028–1039. doi: 10.1053/j.gastro.2014.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gevers D., Kugathasan S., Denson L.A. The treatment-naive microbiome in new-onset Crohn’s disease. Cell Host Microbe. 2014;15:382–392. doi: 10.1016/j.chom.2014.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bao S., Carr E.D., Xu Y.H. Gp91(phox) contributes to the development of experimental inflammatory bowel disease. Immunol Cell Biol. 2011;89:853–860. doi: 10.1038/icb.2011.4. [DOI] [PubMed] [Google Scholar]
- 54.Leoni G., Alam A., Neumann P.A. Annexin A1, formyl peptide receptor, and NOX1 orchestrate epithelial repair. J Clin Invest. 2013;123:443–454. doi: 10.1172/JCI65831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Treton X., Pedruzzi E., Guichard C. Combined NADPH oxidase 1 and interleukin 10 deficiency induces chronic endoplasmic reticulum stress and causes ulcerative colitis-like disease in mice. PLoS ONE. 2014;9:e101669. doi: 10.1371/journal.pone.0101669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Johnson K.R., Marden C.C., Ward-Bailey P. Congenital hypothyroidism, dwarfism, and hearing impairment caused by a missense mutation in the mouse dual oxidase 2 gene, Duox2. Mol Endocrinol. 2007;21:1593–1602. doi: 10.1210/me.2007-0085. [DOI] [PubMed] [Google Scholar]