

Abstract
AIM
To identify the pathogenic mutations in a Chinese pedigree affected with Usher syndrome type II (USH2).
METHODS
The ophthalmic examinations and audiometric tests were performed to ascertain the phenotype of the family. To detect the genetic defect, exons of 103 known RDs -associated genes including 12 Usher syndrome (USH) genes of the proband were captured and sequencing analysis was performed to exclude known genetic defects and find potential pathogenic mutations. Subsequently, candidate mutations were validated in his pedigree and 100 normal controls using polymerase chain reaction (PCR) and Sanger sequencing.
RESULTS
The patient in the family occurred hearing loss (HL) and retinitis pigmentosa (RP) without vestibular dysfunction, which were consistent with standards of classification for USH2. He carried the compound heterozygous mutations, c.721 C>T and c.1969 C>T, in the MYO7A gene and the unaffected members carried only one of the two mutations. The mutations were not present in the 100 normal controls.
CONCLUSION
We suggested that the compound heterozygous mutations of the MYO7A could lead to USH2, which had revealed distinguished clinical phenotypes associated with MYO7A and expanded the spectrum of clinical phenotypes of the MYO7A mutations.
Keywords: Usher syndrome, mutation, MYO7A, next-generation sequencing
INTRODUCTION
Usher syndrome (USH) is the most common form of hereditary syndrome characterized by sensorineural hearing loss (HL) and retinitis pigmentosa (RP). The worldwide prevalence has been estimated to be ranged from 1/25 000 to 4/25 000[1]. According to the degree of HL and the presence or not of vestibular dysfunction, USH is divided into three clinical subtypes. USH type I (USH1) is the most severe form characterized by prepubertal onset of RP, profound HL, and vestibular dysfunction. USH type II (USH2) is characterized by postpubertal onset RP and moderate deafness without vestibular dysfunction. USH type III (USH3) is featured as postlingual deafness, teenage-onset RP and varying degree of vestibular dysfunction[2]. To date, at least 16 loci have been mapped for the three types of USH in human chromosomes and 12 genes have been identified responsible so far (RetNet; https://sph.uth.edu/Retnet/home.htm). Six causative genes have been reported for USH1: MYO7A, USH1C, CDH23, PCDH15, USH1G and CIB2[3]. Three genes have been described for USH2: USH2A, GPR98 and DFNB31. Three genes have been described for USH3: CLRN1, HARS and PDZD7[1]. It is well-known that the USH causative genes have usually been associated with several phenotypes. For example MYO7A has been identified to be associated with not only USH1 but also autosomal dominant deafness and autosomal recessive deafness. However it has rarely been correlated with USH2. The relationship of genotype and phenotype is remarkable and interesting for USH.
The molecular diagnosis of USH is made challenging by widely spread over the coding regions of these causative genes. Recently, next-generation sequencing (NGS) technology has provided a high-throughput and cost-effective method for identification of the causative genes for USH which could sequences the candidate genes rapidly and accurately[4]–[6]. Moreover, successful identification of USH causative mutation provides more probability for analysis the relationship between genotype and phenotype.
In the present study, we utilized targeted genes NGS approaches to identify the genetic defects in a Chinese family with USH2. We identified the compound heterozygous MYO7A mutations, c.721 C>T and c.1969 C>T, segregated with USH2 phenotype in the pedigree. MYO7A c.721 C>T had been reported to be associated with USH1 while c.1969 C>T with hereditary HL. To our knowledge, there has rarely been reported about the relationship between MYO7A mutation and USH2.
SUBJECTS AND METHODS
This study was approved by the Ethical Review Board of the Chinese PLA General Hospital and conformed to the provisions of the Declaration of Helsinki. Written informed consents were signed by all participants.
Pedigree
The pedigree described in this study was from Anhui Province in China, it was identified and followed up clinically at the Chinese PLA General Hospital. A detailed medical history was obtained by interviewing family members. The patients from the family underwent comprehensive ophthalmological examinations and audiologic assessments. Ophthalmological examinations included best-corrected visual acuities (BCVA), slit lamp biomicroscopy, fundus photography, Goldmann perimetry and electroretinogram (ERG). ERGs were also performed and recorded according to the standards of the International Society for Clinical Electrophysiology of Vision (ISCEV)[7]. The severity of hearing impairment was defined as mild (20-40 dB), moderate (41-70 dB), severe (71-90 dB) and profound (>91 dB)[8]. A series of vestibular tests, which included alternate binaural bithermal caloric irrigations with 30°C and 44°C water and videonystagmography (VNG), were further conducted on the patients as detailed previously[9].
Targeted Sequence Capture and Next-generation Sequencing
A custom-made capture array (NimbleGen, Madison, USA) from Beijing Genomics Institute-Shenzhen, was designed to capture the coded exons of the 103 RDS genes, in which contained 12 known USH genes. Genomic DNA of the patient and his unaffected parents were extracted from peripheral blood using a QIAmp DNA Blood Midi Kit (Qiagen, Hilden, Germany) following the manufacturer's instructions. The quantity of DNA to be tested was chosen on the basis of standard laboratory methods. DNA was fragmented to 200-300 bp using an ultrasonoscope (Covaris S2, Massachusetts, USA). The libraries were pooled and hybridized to the custom capture array for 72h at 42°C. Hybridization and washing procedure were performed according to Nimblegen's standard protocol. Sequencing was performed by the HiSeq2000 Analyzers (Illumina, San Diego, USA) to produce paired-end reads (90 bps at each end) according to the manufacturer's instructions[10],[11]. Image analysis and base calling were performed using the Illumina Pipeline.
Filtering Procedures of Detected Variants
To detect the potential pathogenic variants of the proband, the 90 bp clean reads were then subjected to alignment with the human genome reference from the NCBI database (HG19) by using the Burrows-Wheeler Aligner (BWA) Multi-Vision software package (http://sourceforge.net/projects/bio-bwa/). Single-nucleotide variants (SNVs) and indels were identified using Short Oligonucleotide AlignmentProgram (SOAP, http://soap.genomics.org.cn) and Samtools, respectively. All SNVs were determined using the NCBI dbSNP(http://hgdownload-test.cse.ucsc.edu/goldenPath/hg19/database/), HapMap Project (ftp://ftp.ncbi.nlm.nih.gov/hapmap), and 1000 Genome Project (ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp) [11],[12].
Mutations Validation
To ensure the accuracy of results of NGS, Sanger sequencing of mutations was subsequently performed in the pedigree. The targeted sites were amplified by polymerase chain reaction (PCR) and sequenced by ABI 3730 Genetic Analyzer (Applied Biosystems, CA, USA). The specific primers listed in Table 1 were designed using Primer3 (http://bioinfo.ut.ee/primer3-0.4.0/). The sequencing results were compared with gene reference sequences respectively to confirm the variants.
Table 1. Primers used for amplification of potential pathogenic mutations in MYO7A.
| Mutation | Exon | Forward primer (5′- 3′) | Reverse primer (5′- 3′) | Product size (bp) |
| c.721 C>T | 7 | TGGATTGAGCAGCAGGTCTT | GGAAGCAGGCAGCAATACG | 391 |
| c.1969 C>T | 17 | GCCTTTCTGAGCCTTTGTCTG | GTCTTGTTCCTTACACCACCAT | 478 |
RESULTS
Clinical Phenotype of the Chinese Usher Syndrome Pedigree
The family originated from Anhui Province in the Central China Region, the proband had an unaffected brother and his father and mother also had no symptoms and signs of USH (Figure 1). The proband was found hysteresis response to sounds without a delay in sitting independently and toddler late one year after birth. Up to now, he was 12 years old, his language function was well-developed and did not occurred vestibular dysfunction. He was diagnosed with moderate to severe bilateral sensorineural hearing impairments according to his pure tone audiometry result, nevertheless, his alternate binaural bithermal caloric testing and VNG were all within normal (Figure 2). The patient begun to complain about night blindness at 6 years old. When he was twelve, the central BCV of the patient was normal but the fundus photographs showed extensive atrophy of the retinal pigment epithelium changes and attenuated retinal blood vessels (Figure 3A, 3B). His vision field got concentric narrowing, and the residual central visual field were 10 degrees (Figure 3C, 3D). ERG showed undetectable wave in both rod and cone responses (Figure 4).
Figure 1. Pedigree of the family with USH2.
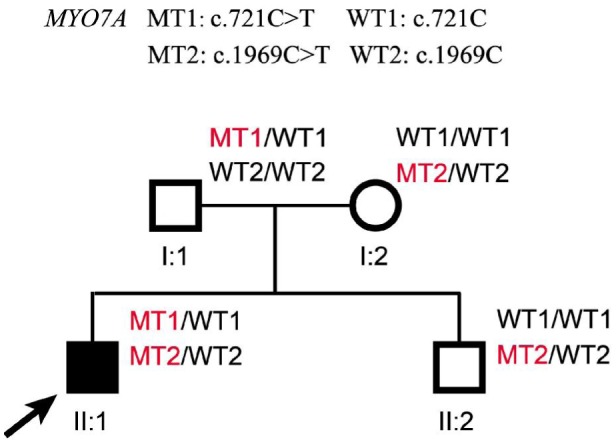
Normal males and female are shown with empty squares and circle, respectively. Affected patient is shown with filled symbols. The proband (II: 1) is indicated by arrow. MT: Mutant type; WT: Wild type.
Figure 2. Bilateral audiometry result of the proband.

A, B: Pure tone audiometry result of the proband reveals moderate to severe bilateral sensorineural hearing impairments.
Figure 3. Bilateral fundus photography and visual field of the proband.
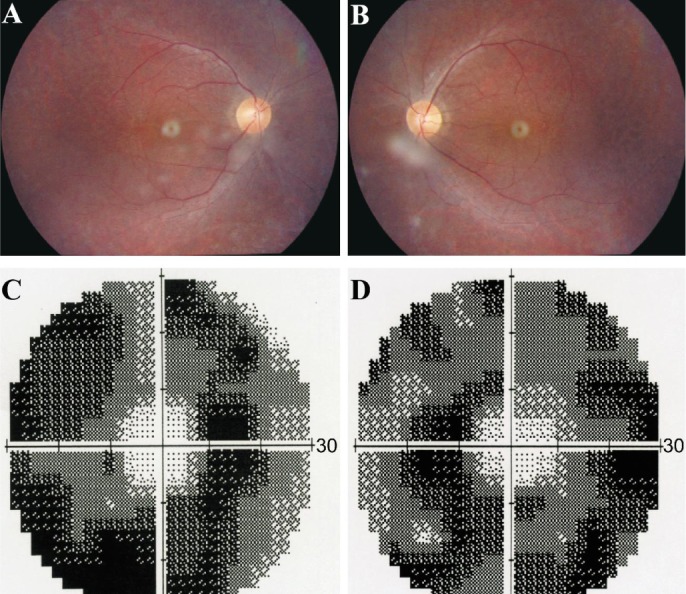
A, B: Bilateral fundus photography showing atrophy of the retinal pigment epithelium and attenuation of retinal arterioles, while the optic disc color were normal of the both eyes of the proband; C, D: Bilateral vision field got concentric narrowing, for the residual central visual field were 10 degrees.
Figure 4. Bilateral EGR result of the proband.

The proband demonstrated undetectable ERG in both dark-adapted and light-adapted responses.
Sequencing Data Analysis
The MYO7A compound heterozygous mutations, c.721C>T and c.1969C>T, were identified in the proband of the family (Figure 5). The mutation of c.721C>T in exon 7 caused a substitution of cysteine for arginine change at codon 241 (p.Arg241Cys, p.R241C), which corresponds to the missense change in the head region of the MYO7A protein[13]. The other missense mutation, c.1969C>T in exon 17 resulted in a substitution of tryptophan for arginine at codon 657 (p.Arg657Trp, p.R657W). The p.Arg657Trp mutation is hypothesized to injure the correspondence between the active binding site and the lever arm. Beacuse it is located at the junction between two connectors of the motor relay and the SH1 helix, which commands the rotation of the lever arm[14]. The compound heterozygous mutations were confirmed with direct Sanger sequencing (Figure 5) and cosegregated with the USH phenotypes. The proband's father only carried one mutation of c.721 C>T, his mother and unaffected brother only carried another one of c.1969 C>T (Figure 1). Both of the two mutations didn't occur in 100 normal controls database. The possible impact of the amino acid substitution was examined with SIFT and PolyPhen tools available online. The SIFT scores of both mutations (MYO7A c.721C>T and c.1969C>T) were 0 and PolyPhen scores were 1, which suggested these mutations were damaging.
Figure 5. Pathogenic mutations detected by Sanger sequencing.
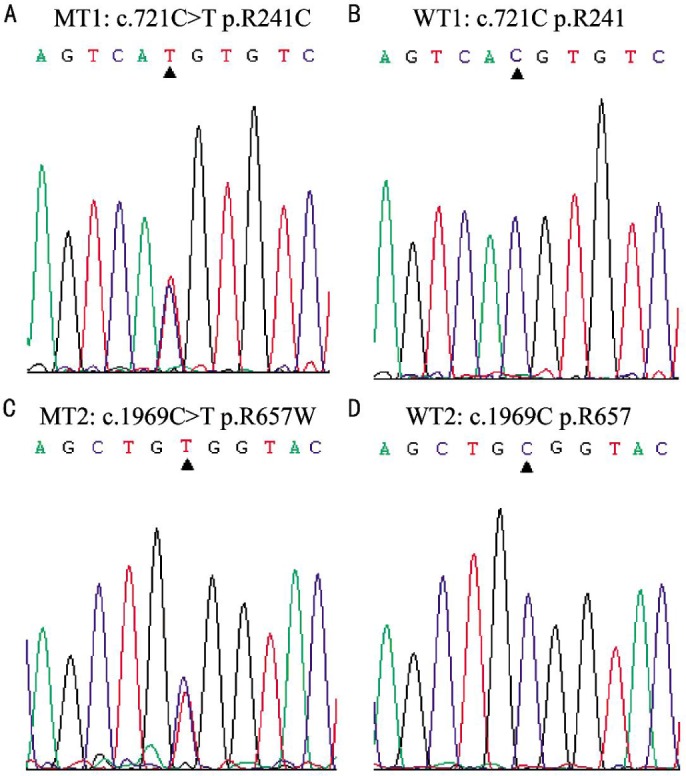
DNA sequence analysis for the proband II:1 showed the presence of compound heterozygous c.721 C>T (p.Arg241Cys) and c.1969 C>T (p.Arg657Trp) mutations. A and B showed mutant and wild type sequences of c.721C (p.Arg241) in MYO7A, respectively. C and D showed mutant and wild type sequences of c.1969 C (p.Arg657) in MYO7A, respectively. MT: Mutant type; WT: Wild type.
DISCUSSION
In the present study, we performed a NGS of targeted 103 known causative genes of inherited RD including 12 USH genes in a Chinese family with USH2. Two compound heterozygous mutations in the MYO7A gene (NM_000260), c.721 C>T and c.1969 C>T, were identified in the proband, which were validated by Sanger sequencing and verified to be cosegregated with the phenotypes of RP and congenital hearing impairment in the family.
The MYO7A gene, spanning 120 kb and containing 49 exons, encodes an unconventional myosin. It plays an important role in the retinal pigment epithelium and photoreceptor cells, as well as in cochlear and vestibular neuroepithelia as a kind of molecular motor. It was suggested that mutations in MYO7A could cause autosomal dominant deafness-11 (DFNA11; MIM#601317), autosomal recessive nonsyndromic deafness (DFNB2; MIM# 600060) and USH1 (MIM# 276900)[15]. Nearly 53.2% of USH1 cases were associated with MYO7A variants[16]. To the best of our knowledge, there have not been previous reports about that MYO7A mutations could cause USH2 clinical subtype in details except for c.1343+1G>A and c.2837T>G[17]. The MYO7A missense mutation c.721C>T was detected in USH1 patients from England and Scotland and c.1969C>T was found in Jewish-Turkey proband with hereditary HL[14],[18]. In hereditary HL family caused by MYO7A c.1969C>T, a medical history including vision problems and motor development basides HL was collected from each patient, however fundoscopy and vestibulo-ocular reflex were not examined. In present research, the patient from the family with the mutations of c.721 C>T and c.1969 C>T in MYO7A occurring moderate to severe HL and RP without vestibular dysfunction, was consistent with standards of classification for USH2. Why the known MYO7A mutations (c.721 C>T and c.1969 C>T) resulting in USH1 and hereditary HL were identified to be associated with USH2 in Chinese patients? We thought there might be some possible reasons: firstly, the difference in the ethnic background and/or environmental factors would influence the relationship of genotype and phonotype. Almost all the previous researches about these two MYO7A mutations were performed in the England, Scotland, Jewish-Turkey, and Pakistani families, nevertheless the present study focused attention on the Chinese family. It has been demonstrated that environmental and/or genetic factors could have an influence on the severity of the HL and USH[19],[20], therefore it was suggested that environmental and/or genetic factors also could affect the phenotypes. Secondly, the patients with MYO7A c.1969 C>T in the past study didn't performed the clinical symptoms of the RP at the time of diagnosis. For example, when the Pakistani DFNB2 family were reassessed 7y later after diagnosis, 5 of 12 adult patients (>25y of age) were found to have mild RP[21]. Thirdly, there are stochastic factors make influnce on the expression of the phenotype as well, for even a pair of monozygotic twins with disease due to USH2A show different phenotypes[22].
Compared with direct sequencing in a conservative estimation, the NGS technology has not only maintained the trait of high accuracy, but also greatly reduced the cost and greatly improved the speed of sequencing. It has been proved to be a fast and efficient screening approach to perform the Genetic diagnosis for USH, and make it possible to screen the USH causative genes and RD genes at the same time[2]–[5],[23],which helps us to detect the causative mutations and exclude the condition that phenotypes of HL and RP were associated with different genes, which made the phenotype looked very similar to USH2.
In conclusion, in this research we confirmed that the compound heterozygous mutations, c.721 C>T and c.1969 C>T, could result in USH2 in a Chinese family. Our finding expands the spectrum of clinical phenotypes of MYO7A mutations. It's also been proved that the NGS technology is high-efficiency strategy for molecular diagnosis, which can also help us better understand the relationship of genotype-phenotype in the disease.
Acknowledgments
Foundation: Supported by the Postdoctoral Science Foundation of China (No. 2014M562542).
Conflicts of Interest: Zhai W, None; Jin X, None; Gong Y, None; Qu LH, None; Zhao C, None; Li ZH, None.
REFERENCES
- 1.Mathur P, Yang J. Usher syndrome: Hearing loss, retinal degeneration and associated abnormalities. Biochim Biophys Acta. 2015;1852(3):406–420. doi: 10.1016/j.bbadis.2014.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Besnard T, Garcia-Garcia G, Baux D, Vache C, Faugere V, Larrieu L, Leonard S, Millan J M, Malcolm S, Claustres M, Roux AF. Experience of targeted Usher exome sequencing as a clinical test. Mol Genet Genomic Med. 2014;2(1):30–43. doi: 10.1002/mgg3.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Liu F, Li P, Liu Y, Li W, Wong F, Du R, Wang L, Li C, Jiang F, Tang Z, Liu M. Novel compound heterozygous mutations in MYO7A in a Chinese family with Usher syndrome type 1. Mol Vis. 2013;19:695–701. [PMC free article] [PubMed] [Google Scholar]
- 4.Liu T, Jin X, Zhang X, Yuan H, Cheng J, Lee J, Zhang B, Zhang M, Wu J, Wang L, Tian G, Wang W. A novel missense SNRNP200 mutation associated with autosomal dominant retinitis pigmentosa in a Chinese family. PloS One. 2012;7(9):e45464. doi: 10.1371/journal.pone.0045464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shanks ME, Downes SM, Copley RR, et al. Next-generation sequencing (NGS) as a diagnostic tool for retinal degeneration reveals a much higher detection rate in early-onset disease. Eur J Hum Genet. 2013;21(9):274–280. doi: 10.1038/ejhg.2012.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fu Q, Wang F, Wang H, et al. Next-generation sequencing-based molecular diagnosis of a chinese patient cohort with autosomal recessive retinitis pigmentosa. Invest Ophthalmol Vis Sci. 2013;54(6):4158–4166. doi: 10.1167/iovs.13-11672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bach M, Brigell MG, Hawlina M, Holder GE, Johnson MA, McCulloch DL, Meigen T, Viswanathan S. ISCEV standard for clinical pattern electroretinography (PERG): 2012 update. Doc ophthalmol. 2013;126(1):1–7. doi: 10.1007/s10633-012-9353-y. [DOI] [PubMed] [Google Scholar]
- 8.Yang T, Wei X, Chai Y, Li L, Wu H. Genetic etiology study of the non-syndromic deafness in Chinese Hans by targeted next-generation sequencing. Orphanet J Rare Dis. 2013;8:85. doi: 10.1186/1750-1172-8-85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pieke Dahl S, Kimberling WJ, Gorin MB, Weston MD, Furman JM, Pikus A, Möller C. Genetic heterogeneity of Usher syndrome type II. J Med Genet. 1993;30(10):843–848. doi: 10.1136/jmg.30.10.843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wei X, Ju X, Yi X, et al. Identification of sequence variants in genetic disease-causing genes using targeted next-generation sequencing. PloS one. 2011;6(12):e29500. doi: 10.1371/journal.pone.0029500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Qu LH, Jin X, Xu HW, Li SY, Yin ZQ. Detecting novel genetic mutations in Chinese Usher syndrome families using next-generation sequencing technology. Mol Genet Genomic. 2015;290(1):353–363. doi: 10.1007/s00438-014-0915-4. [DOI] [PubMed] [Google Scholar]
- 12.Jin X, Qu LH, Meng XH, Xu HW, Yin ZQ. Detecting genetic variations in hereditary retinal dystrophies with next-generation sequencing technology. Mol Vis. 2014;20:553–560. [PMC free article] [PubMed] [Google Scholar]
- 13.Shahzad M, Sivakumaran TA, Qaiser TA, et al. Genetic analysis through OtoSeq of Pakistani families segregating prelingual hearing loss. Otolaryngol Head Neck Surg. 2013;149(3):478–487. doi: 10.1177/0194599813493075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brownstein Z, Abu-Rayyan A, Karfunkel-Doron D, Sirigu S, Davidov B, Shohat M, Frydman M, Houdusse A, Kanaan M, Avraham KB. Novel myosin mutations for hereditary hearing loss revealed by targeted genomic capture and massively parallel sequencing. Eur J Hum Genet. 2014;22(6):768–775. doi: 10.1038/ejhg.2013.232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu XZ, Walsh J, Mburu P, Kendrick-Jones J, Cope MJ, Steel KP, Brown SD. Mutations in the myosin VIIA gene cause non-syndromic recessive deafness. Nat Genet. 1997;16(2):188–190. doi: 10.1038/ng0697-188. [DOI] [PubMed] [Google Scholar]
- 16.Le Quesne Stabej P, Saihan Z, Rangesh N, Steele-Stallard HB, Ambrose J, Coffey A, Emmerson J, Haralambous E, Hughes Y, Steel KP, Luxon LM, Webster AR, Bitner-Glindzicz M. Comprehensive sequence analysis of nine Usher syndrome genes in the UK National Collaborative Usher Study. J Med Genet. 2012;49(1):27–36. doi: 10.1136/jmedgenet-2011-100468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rong W, Chen X, Zhao K, Liu Y, Liu X, Ha S, Liu W, Kang X, Sheng X, Zhao C. Novel and recurrent MYO7A mutations in Usher syndrome type 1 and type 2. PloS One. 2014;9(5):e97808. doi: 10.1371/journal.pone.0097808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bharadwaj AK, Kasztejna JP, Huq S, Berson EL, Dryja TP. Evaluation of the myosin VIIA gene and visual function in patients with Usher syndrome type I. Exp Eye Res. 2000;71(2):173–181. doi: 10.1006/exer.2000.0863. [DOI] [PubMed] [Google Scholar]
- 19.Liu XZ, Hope C, Walsh J, Newton V, Ke XM, Liang CY, Xu LR, Zhou JM, Trump D, Steel KP, Bundey S, Brown SD. Mutations in the myosin VIIA gene cause a wide phenotypic spectrum, including atypical Usher syndrome. Am J Hum Genet. 1998;63(3):909–912. doi: 10.1086/302026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zina ZB, Masmoudi S, Ayadi H, Chaker F, Ghorbel AM, Drira M, Petit C. From DFNB2 to Usher syndrome: variable expressivity of the same disease. Am J med Genet. 2001;101(2):181–183. doi: 10.1002/ajmg.1335. [DOI] [PubMed] [Google Scholar]
- 21.Ben Rebeh I, Moriniere M, Ayadi L, Benzina Z, Charfedine I, Feki J, Ayadi H, Ghorbel A, Baklouti F, Masmoudi S. Reinforcement of a minor alternative splicing event in MYO7A due to a missense mutation results in a mild form of retinopathy and deafness. Mol Vis. 2010;16:1898–1906. [PMC free article] [PubMed] [Google Scholar]
- 22.Bernal S, Meda C, Solans T, Ayuso C, Garcia-Sandoval B, Valverde D, Del Rio E, Baiget M. Clinical and genetic studies in Spanish patients with Usher syndrome type II: description of new mutations and evidence for a lack of genotype-phenotype correlation. Clin Genet. 2005;68(3):204–214. doi: 10.1111/j.1399-0004.2005.00481.x. [DOI] [PubMed] [Google Scholar]
- 23.Huang XF, Xiang P, Chen J, Xing DJ, Huang N, Min Q, Gu F, Tong Y, Pang CP, Qu J, Jin ZB. Targeted exome sequencing identified novel USH2A mutations in Usher syndrome families. PloS One. 2013;8(5):e63832. doi: 10.1371/journal.pone.0063832. [DOI] [PMC free article] [PubMed] [Google Scholar]