

Abstract
To maintain euglycemia in healthy organisms, hepatic glucose production is increased during fasting and decreased during the postprandial period. This whole process is supported by insulin levels. These responses are associated with the insulin signaling pathway and the reduction in the activity of key gluconeogenic enzymes, resulting in a decrease of hepatic glucose production. On the other hand, defects in the liver insulin signaling pathway might promote inadequate suppression of gluconeogenesis, leading to hyperglycemia during fasting and after meals. The hepatocyte nuclear factor 4, the transcription cofactor PGC1-α, and the transcription factor Foxo1 have fundamental roles in regulating gluconeogenesis. The loss of insulin action is associated with the production of pro-inflammatory biomolecules in obesity conditions. Among the molecular mechanisms involved, we emphasize in this review the participation of TRB3 protein (a mammalian homolog of Drosophila tribbles), which is able to inhibit Akt activity and, thereby, maintain Foxo1 activity in the nucleus of hepatocytes, inducing hyperglycemia. In contrast, physical exercise has been shown as an important tool to reduce insulin resistance in the liver by reducing the inflammatory process, including the inhibition of TRB3 and, therefore, suppressing gluconeogenesis. The understanding of these new mechanisms by which physical exercise regulates glucose homeostasis has critical importance for the understanding and prevention of diabetes.
Keywords: Physical exercise, TRB3, Insulin resistance, Liver, Hepatic glucose production
Background
Diabetes mellitus type 2 (DM2) is highly prevalent in modern society and is closely related to obesity. Its genesis is linked to insulin resistance (pre-diabetes condition) and there seems to be a triad of recurrent and intricate phenomena capable of contributing to the patient’s hyperglycemic condition with DM2. These are the following: (1) increased hepatic glucose production (via marked increase of glycogenolysis and gluconeogenesis molecules); (2) decreased insulin secretion and activity; and (3) reduced usage and storage of glucose in the body [1, 2].
The normal activity of glucagon and insulin signaling pathway in the liver is important for the control of hepatic glucose production. On one hand, glucagon promotes gluconeogenesis through two pathways: The first is via activating PGC-1α that will interact with FoxO1 and then increase the synthesis of phosphoenolpyruvate carboxykinase (PEPCK) and G6Pase, consequently increasing the gluconeogenesis in the liver [3]. The second way is via increasing Inositol 1,4,5-triphosphate (IP3), which acts on the calcium-releasing channels of the endoplasmic reticulum (ER) by releasing calcium (Ca) to the cytosol. In the cytosol, the Ca will act as a second messenger activating the cytoplasmic calcium-sensitive kinase and the calcium/calmodulin dependent-protein kinase II (CaMKII). In turn, CaMKII will activate MAPK p38α that will phosphorylate FoxO1 at Ser295 and 475, as well as Ser246; the phosphorylated FoxO1 will then enter the cell nucleus and consequently bind to PGC-1α to induce target genes that are responsible for the transcription of genes responsible for glycogenolysis and gluconeogenesis such as G6Pase and Phosphoenolpyruvate carboxykinase 1 (PCK1) in the liver [4–6]. On the other hand, insulin activation promotes a reduction in the expression and activity of the gluconeogenic enzymes, such as PEPCK and G6Pase, which results in a decreased hepatic glucose production [7–9]. In contrast, when hepatic insulin signaling is compromised, an inadequate suppression of gluconeogenic pathways will ensue, which will lead to high levels of glucose and insulin in postprandial and fasting states [7, 10].
The regular practice of physical exercise is a non-pharmacological strategy recognized and indicated for the prevention and treatment of diabetes. It was demonstrated in a previous study that lifestyle changes, including the practice of regular physical exercise and nutritional education, had higher protective effect against the development of diabetes when compared to the sole use of metformin [11]. This scientific evidence indicates that exercise is indeed an important strategy in preventing insulin resistance and should be used to circumvent the development of diabetes.
However, it is still not clear how physical exercise promotes a reduction in hyperglycemia. Furthermore, it is observed in the literature that most studies used skeletal muscle changes as their primary outcome [12–15]. In fact, many of the beneficial effects of physical exercise occur in other tissues which might not be directly involved in muscle contraction. This review was conducted to show how physical exercise could influence hepatic glucose production (with an emphasis on gluconeogenesis) and contribute to the maintenance of glucose homeostasis in a state of insulin resistance. In particular, this review elucidated the effects of physical exercise on TRB3 levels by minimizing its adverse effects on the insulin signaling pathway and the subsequent increase in gluconeogenesis in the liver.
Obesity and insulin resistance: the role of TRB3 protein
The relationship between obesity and insulin resistance has been intensively investigated and it is known that many biomolecules from the adipocytes are capable of inducing insulin resistance through different mechanisms. A recent study has shown that some types of fatty acids originating from the diet, especially saturated fatty acids, are able to bind to -and thus activate- membrane proteins referred to as toll like receptor 4 (TLR4) [16]. TLR4 are mediators of the immunological innate response and their activation promotes negative consequences on the insulin pathway, such as the activation of serine kinases [16, 17]. More specifically, the activation of TLR4 induces the activation of IKKβ (Ikappakinase beta) and JNK (c-jun N-terminal kinase), resulting in the phosphorylation of the insulin receptor (IR) and the insulin receptor substrates 1 and 2 (IRS-1/2) in a serin residue (instead of treonin), which leads to a decreased insulin signal in many tissues, including the liver [16].
TLR4 can also activate the complex called IKK/IKB/NFKB. When active, IKKβ promotes the phosphorylation of IKB, culminating in the dissociation of the IKB/NFKB complex. Once free in the cell, NFKB is able to translocate to the nucleus and promote the transcription of new inflammatory cytokines, such as TNF-α. This pro-inflammatory protein leads to an increased inflammatory level -also called a chronic low-grade inflammation- which seems to be strictly associated with obesity and insulin signaling impairment [16–18].
Notably, TLR4 is associated with TRB3 synthesis through the development of the ER stress [19]. The ER dysfunction arises as a consequence of TLR4 activation and can be modulated by anti-inflammatory cytokines [20]. Yao et al. observed that the activation of TLR4 by modified low-density lipoproteins is essential to induce ER stress in the macrophages [21]. Pierre et al. [22] demonstrated a strong relationship between TLR4 and ER stress; the authors showed that knockout mice for TLR-4 in the liver, fat, and muscle tissues and that were fed a high fat diet did not develop ER stress. TLR4/2 activation in the macrophages activates IRE1/XBP1s and contributes to proinflammatory cytokine production. Moreover, Koh and colleagues [23] reported that high fat diet, obesity, and T2DM induce stress reticulum and in response to this condition, TRB3 levels increase in muscle cells. Based on these findings, the authors hypothesized that impairments in insulin signaling could result from the increase in TRB3 levels. To confirm this hypothesis, the authors first overexpressed TRB3 and observed impairments in insulin signaling; on the other hand, when the transcription of TRB3 was silenced, the reverse phenomenon was observed in insulin signaling such as improved insulin sensitivity and glucose uptake. Further, when the authors induced animals to the ER stress, they observed an increase in TRB3 transcription levels concomitant with the losses in insulin signaling [23].
ER stress has been linked to insulin resistance in multiple tissues and is the major site in the cell for lipid and protein synthesis, folding, assembly, and trafficking, as well as cellular Ca2+ storage [24]. Another way how ER stress can induce the increase of TRB3 is through the increase of calcium in the cytosol. Obese people have higher calcium concentration in the cell due to the lipid-induced de-activation of the calcium pump [25] and to the opening of the IP3R ER calcium-channel. This is due to the activation of the glucagon receptor that is triggered by 2 processes: 1- The formation of IP3 by phospholipase C [26] and 2- The direct activation of the channel by PKA-mediated phosphorylation of IP3R [27]. This elevated calcium concentration will activate CaMKII, which will decrease p58IPK, induce ER stress, and activate PERK and ATF-4 pathways; this activation will in turn increase the synthesis of TRB3 [4].
In this regard, the TRB3 (a mammalian homolog of Drosophila tribbles) emerges as a negative regulator of the insulin signaling in several tissues, including the liver. Du et al. [28] found that in obese mice, there was an overexpression of the TRB3 protein that can be associated with a distal protein from the insulin pathway–the protein kinase B (Akt)– and can hence hinder Akt phosphorylation and its subsequent activation. Consequently, there was a reduction in insulin signaling on the insulin-sensitive tissues as well as other tissues such as the liver, triggering uncontrolled hepatic glucose production [28]. On the other hand, TRB3 inhibition reduces insulin resistance that is associated with obesity and helps control blood glucose levels [29].
Insulin resistance state: the role of TRB3 in suppressing Akt activity
TRB3 is part of a new family of proteins and consists of three members (TRB1, TRB2 and TRB3), which are associated with numerous proteins including transcription factors and protein kinases (as well as E3 ubiquitin ligase). By interacting with different factors, the TRBs regulate many biological processes, including cell growth, differentiation, and metabolism [28]. TRB1 interacts with the mitogen-activated protein kinase (MAPK) modulating its kinase activity, resulting in muscle cell proliferation [30, 31]. TRB2 interacts with transcription factors C/EBP and is implicated in adipocyte differentiation [32]. TRB3 isoform, which has been the focus of several studies, emerges as a key player in in the regulation of insulin signaling through inhibition of Akt activity [28, 33, 34]. The effects of TRB3 in the organism remain unclear; for instance, TRB3 can have positive effects in the adipocytes because it can increase fatty acid oxidation [35]; conversely, TRB3—induced by ER stress—can have negative effects in the liver because it can decrease insulin signaling by binding to Akt and inhibiting it [36].
The role of TRB3 on Akt activity has been studied in different animal tissues. The amounts of TRB3 RNA and protein were increased in livers of db/db diabetic mice as compared to those in wild-type mice [28]. In both the skeletal muscle and the liver, TRB3 has been proposed to be a suppressor of Akt activity, mainly during fasting period and/or diabetic state [28, 37, 38]. The main role of hepatic TRB3 on Akt activity is to bind to Akt and then inhibit its phosphorylation at Thr308 and Ser473motifs, which are the critical motifs for Akt activation induced by growth factors and insulin [28]. Interestingly, when TRB3 was knockdown in the liver, Akt became active and the serum glucose levels went back to normal [37]. Conversely, when TRB3 was overexpressed in the liver, it promoted hyperglycemia and glucose intolerance [28],
Further, when TRB3 is overexpressed in the liver it promoted hyperglycemia and glucose intolerance, the same frame was showed in db/db mice that have high concentration of TRB3 in the liver [28]. Some authors have suggested that, by interfering with Akt activation, TRB3 contributes to insulin resistance in individuals with susceptibility to type 2 diabetes. Recently, Yu and colleagues [39] suggested that liver transcription of TRB3 levels may also be regulated by phosphoserine aminotransferase 1 (PSAT1), an enzyme that acts on the synthesis of the protein serine. The authors showed that PSAT1 inhibited the transcription of TRB3 in the liver and these results were accompanied by an increased activity of Akt and consequently a higher sensitivity to insulin and glucose uptake. Moreover, human polymorphism of TRB3 (Q84R) leads to an important inhibition of Akt activity and is related to the development of insulin resistance and an increased risk of cardiovascular disease [40].
In the liver, the expression of TRB3 is induced during fasting period in order to reduce insulin signaling at a time when the nutrient supply is not abundant, preventing the organism to reach a state of hypoglycemia [28, 33, 34]. After a meal, when the circulating insulin levels are increased, there is a 50 % reduction in the expression of TRB3 [38]. Elevated levels of TRB3 protein and the increased association of TRB3 with Akt are related to insulin resistance and, consequently, obesity and diabetes development [28, 33, 38].
In the liver of patients with DM2, TRB3 expression is increased independent of nutrients’ excess or hyperinsulinemia, leading to defects in insulin signaling and triggering a reduction in glycogen synthesis that is mediated by insulin as well as an increase in gluconeogenesis, which leads to hyperglycemia [28, 33, 34]. The negative effects of TRB3 block the activities stimulated by insulin, such as the inhibitory effect on hepatic glucose production and the inactivation of transcription factors for gluconeogenic enzymes (such as Foxo1 and PGC-1α) [28, 38].
Studies in obesity-animal models (either induced by a high fat diet or genetically modified) showed an increase in TRB3 expression and in the TRB3/Akt association in the skeletal muscle tissue and the liver, with a consequent increase in blood glucose concentration, which later on led to the development of insulin resistance and DM2 [28, 29, 41].
On the other hand, the inhibition of TRB3 activity increases the phosphorylation of Akt in response to insulin in the liver [28, 38]. Previous studies have shown that the absence of TRB3 is related to increased insulin sensitivity [28, 33]. Studies developed by our group, seeking to investigate the acute effect of physical exercise, showed a reduction in the expression of TRB3 protein in the skeletal muscle and liver of exercised animals [29, 41]. This phenomenon was associated with increased insulin sensitivity in animals induced to obesity by a high fat diet [29, 41]. These findings shed light on the importance of investigating the effect of, not only an acute period of exercise, but also physical training (chronic exercise) on TRB3 protein and its role in restoring insulin sensitivity in the liver tissue. Despite the need to define many other effects of physical exercise on intracellular signaling pathways, all these findings open new perspectives for understanding the effect of physical exercise on glycemic control and glucose production in the liver.
The effect of exercise in the regulation of hepatic glucose production and the inhibitory effect of physical exercise on TRB3
Despite the beneficial effects of exercise on skeletal muscle and the increase in insulin sensitivity in animals and obese/diabetic patients, there is a lack of studies that demonstrate the role of physical exercise in regulating the insulin signaling pathway and the hepatic glucose production in the liver in these conditions.
In this regard, our research laboratory has sought to identify the effects of physical exercise on the liver tissue of obese and diabetic animals [29, 42–44]. The results have allowed a better understanding of the role of physical exercise in regulating some key proteins involved in glycogenolysis and gluconeogenesis in the liver.
A study that used a single session of aerobic exercise showed that exercise can decrease fasting hyperglycemia in obese mice induced by a high fat diet [42]. This effect on blood glucose was associated with a decrease in the association of Foxo1/PGC-1α inside the hepatocytes’ nucleus, consequently, it lowered the expression of the gluconeogenic enzymes PEPCK and G6Pase. There was also a reduction in hepatic glucose production, typically exaggerated under the condition of obesity and insulin resistance [42].
In the same direction, De Souza et al. [43] observed that a single session of physical exercise was able to decrease the expression of the nuclear factors Foxo1 and HNF-4α, both in genetically modified mice (ob/ob) and in Swiss obese mice induced by a high fat diet [43]. This cellular behavior was followed by a lower transcription of the gluconeogenic enzymes PEPCK and G6Pase and, consequently, a decrease in fasting glycemia in these two types of mice. Further, the obese animals subjected to physical exercise protocol had higher preservation and synthesis of hepatic glycogen [43]. Another study by Hoene et al. showed that after a single session of treadmill running, there was a rapid and pronounced increase of enzymes and transcription factors that regulated the hepatic metabolism such as G6Pase, pyruvate dehydrogenase kinase 4 (PDK4), IRS-2, PGC1- α, AMPK and Akt, which reinforces the idea that physical exercise might play a major role in TRB-3 activity [45].
To better understand the positive effect of physical exercise on insulin signaling in the liver of obese animals, Lima et al. investigated the effect of a single session of physical exercise on TRB3 protein levels and TRB3 association with Akt protein [29]. It was verified that the physical exercise reduced the expression of TRB3 and TRB3/Akt association. Consequently, such phenomenon was accompanied by an increase in Akt activity and its phosphorylation, leading to the inactivation of Foxo1. Associated with these adaptations, there was a reduction in the expression of the gluconeogenic enzyme PEPCK, both in ob/ob mice and in obese Swiss mice induced by a high fat diet [29]. Although these studies have demonstrated a significant effect of physical exercise on the TRB3 protein, they did not explain how this phenomenon happened.
To answer these questions, a study conducted by our research group described a new mechanism by which physical exercise could increase insulin signaling in the liver tissue while preserving Akt activity [44]. Aerobic physical training was able to increase the expression of endosomal adaptor protein expression (APPL1). This protein had the ability to inhibit the interaction between Akt and TRB3 in mice hepatocytes, which was accompanied by an increase in Akt activation in response to insulin stimulation [44].
Previous studies performed by Cheng et al. and Schenck et al. demonstrated that APPL1 regulated the activity of Akt [9, 46]. Both experiments with isolated cells, such as tissue analysis, showed that APPL1 competed directly with TRB3 in order to bind to Akt. Furthermore, overexpression of APPL1 could neutralize the inhibitory effect of TRB3 in the activation of Akt when stimulated by insulin and suppress the gluconeogenic program in the hepatocytes of obese mice [9].
Marinho et al. observed that aerobic training was able to increase the expression of APPl1 and Akt and the association between APPL1 and Akt protein in obese mice induced by a high fat diet. These results were confirmed by immunohistochemistry experiment, in which the obese mice submitted to physical training protocol showed higher levels of APPL1 in hepatic cells when compared to their sedentary pairs. Therefore, the increased expression of APPL1 resulted in an increased activation of Akt [44]. Moreover, it was also observed that physical training increased Foxo1 phosphorylation and decreased PGC-1α expression, reducing the association of these 2 proteins and consequently, the gluconeogenic program, by reducing the expression of PEPCK and G6Pase .
In addition to this phenomenon, physical training increased the hepatic glycogen content due to increased phosphorylation of glycogen synthase enzyme kinase 3 (GSK3), leading to dephosphorylation and activation of glycogen syntheses (GS) [44]. In the skeletal muscle and the liver, insulin can stimulate the accumulation of glycogen by increasing its synthesis. This effect is obtained by dephosphorylation of the glycogen synthase enzyme (GS), where such dephosphorylation is only possible from the inhibition of GSK3. Under insulin stimulation, Akt phosphorylates GSK3 and inactivates it, which reduces the rate of GS phosphorylation, thus, increasing its activity. Therefore, in the presence of insulin, glucose may be stored as glycogen [44]. The potential mechanism of inhibition of gluconeogenesis by physical training through increasing APPL1 and inhibiting TRB3 is summarized in Figs. 1, 2.
Fig. 1.
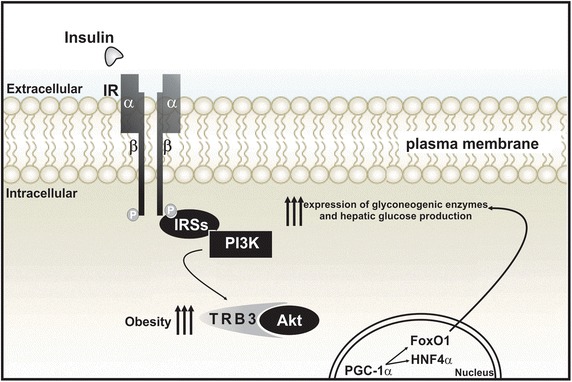
The increased expression of TRB3, induced by obesity or by the condition of insulin resistance, promotes a decrease in the phosphorylation of Akt and Foxo1, which enables the translocation of Foxo1 from the cytoplasm to the nucleus. Activated in the nucleus, the Foxo1 increases PGC-1α transcription and its association with this protein, starting the gluconeogenic program. As a consequence, there is an increase in the transcription of gluconeogenic enzymes, such as PEPCK and G6Pase in the liver resulting in hepatic glucose production
Fig. 2.
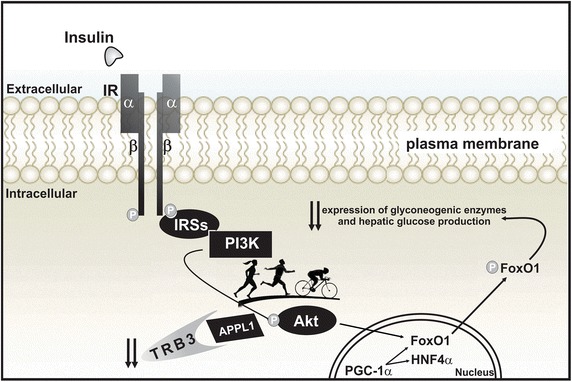
On the other hand, exercise markedly increases the expression of APPL1 and reduces the expression of TRB3, increasing hepatic insulin sensitivity and leading to the reduction of hyperglycemia due to a higher Akt phosphorylation. The activation of Akt allows the propagation of the insulin signal and the phosphorylation of Foxo1, which will exit the nucleus to the cytoplasm to decrease hepatic glucose production. These new findings allow us to better understand the molecular effects of physical exercise on the liver and its favorable effect on glycemic control and diabetes
Conclusion
In summary, it can be concluded that physical exercise is an effective treatment to improve insulin sensitivity and contribute to glycemic control. Moreover, its effects go beyond the skeletal muscle, with specific adaptations on molecular pathways involved in hepatic gluconeogenesis regulation. Although future studies are needed to elucidate the effects of physical exercise on the liver, it seems clear that, when performed regularly, it can play an important role in preventing the development of insulin resistance and diabetes through the APPL1/TRB3/Akt pathway. Among the pronounced effects achieved by physical training, the inhibition of TRB3 in the liver could be highlighted, which leads to a decrease in gluconeogenesis in obese and diabetic animals. It is noteworthy that many of the observed effects were obtained independent of changes in body composition; this reinforces the importance of maintaining a physically active lifestyle to prevent insulin resistance and DM2.
Authors’ contributions
All authors contributed equally to this work. All authors have read and approved the final manuscript.
Acknowledgements
The authors would like to thank the National Council of Scientific and Technological Development—CNPq, the Foundation for Research Support of São Paulo—FAPESP (#2012/01750-0; #2013/00554-6; #2013/21491-2), and FAEPEX.
Compliance with ethical guidelines
Competing interests The authors declare they have no competing interests.
Abbreviations
- TRB3
Mammalian homolog of Drosophila tribbles
- DM2
Diabetes mellitus type 2
- PEPCK
Phosphoenolpyruvate carboxykinase
- G6Pase
Glucose-6-phosphatase
- TLR4
Toll like receptor 4
- IKKβ
Ikappakinase beta
- JNK
c-Jun N-terminal kinase
- MAPK
Mitogen-activated protein kinase
- APPL1
Endosomal adaptor protein expression
- GSK-3
Glycogen synthase enzyme kinase 3
- GS
Glycogen synthase
Contributor Information
Rodolfo Marinho, Email: rodolfo-marinho@hotmail.com.
Rania A. Mekary, Email: rmekary@gmail.com
Vitor Rosetto Muñoz, Email: vitor.munoz93@gmail.com.
Ricardo José Gomes, Email: ricardojosegomes@yahoo.com.br.
José Rodrigo Pauli, Email: rodrigopaulifca@gmail.com.
Leandro Pereira de Moura, Phone: +55 (19) 3701-6680, Email: mouralp1@gmail.com.
References
- 1.American Diabetes Association. Diagnosis and classification of diabetes mellitus. Diabetes Care. 2006;29(Suppl 1):S43–8. [PubMed]
- 2.Matthaei S, Stumvoll M, Kellerer M, Haring HU. Pathophysiology and pharmacological treatment of insulin resistance. Endocr Rev. 2000;21:585–618. doi: 10.1210/edrv.21.6.0413. [DOI] [PubMed] [Google Scholar]
- 3.Puigserver P, Wu Z, Park CW, Graves R, Wright M, Spiegelman BM. A cold-inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell. 1998;92:829–839. doi: 10.1016/S0092-8674(00)81410-5. [DOI] [PubMed] [Google Scholar]
- 4.Ozcan L, Cristina de Souza J, Harari AA, Backs J, Olson EN, Tabas I. Activation of calcium/calmodulin-dependent protein kinase II in obesity mediates suppression of hepatic insulin signaling. Cell Metab. 2013;18:803–815. doi: 10.1016/j.cmet.2013.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ozcan L, Wong CCL, Li G, Xu T, Pajvani U, Park SKR, Wronska A, Chen B-X, Marks AR, Fukamizu A, Backs J, Singer HA, Yates JR, Accili D, Tabas I. Calcium signaling through CaMKII regulates hepatic glucose production in fasting and obesity. Cell Metab. 2012;15:739–751. doi: 10.1016/j.cmet.2012.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Asada S, Daitoku H, Matsuzaki H, Saito T, Sudo T, Mukai H, Iwashita S, Kako K, Kishi T, Kasuya Y, Fukamizu A. Mitogen-activated protein kinases, Erk and p38, phosphorylate and regulate Foxo1. Cell Signal. 2007;19:519–527. doi: 10.1016/j.cellsig.2006.08.015. [DOI] [PubMed] [Google Scholar]
- 7.De Souza CT, Araujo EP, Bordin S, Ashimine R, Zollner RL, Boschero AC, Saad MJA, Velloso LA. Consumption of a fat-rich diet activates a proinflammatory response and induces insulin resistance in the hypothalamus. Endocrinology. 2005;146:4192–4199. doi: 10.1210/en.2004-1520. [DOI] [PubMed] [Google Scholar]
- 8.Dentin R, Liu Y, Koo SH, Hedrick S, Vargas T, Heredia J, Yates J, 3rd, Montminy M. Insulin modulates gluconeogenesis by inhibition of the coactivator TORC2. Nature. 2007;449:366–369. doi: 10.1038/nature06128. [DOI] [PubMed] [Google Scholar]
- 9.Cheng KK, Iglesias MA, Lam KS, Wang Y, Sweeney G, Zhu W, Vanhoutte PM, Kraegen EW, Xu A. APPL1 potentiates insulin-mediated inhibition of hepatic glucose production and alleviates diabetes via Akt activation in mice. Cell Metab. 2009;9:417–427. doi: 10.1016/j.cmet.2009.03.013. [DOI] [PubMed] [Google Scholar]
- 10.Cintra DE, Pauli JR, Araujo EP, Moraes JC, de Souza CT, Milanski M, Morari J, Gambero A, Saad MJ, Velloso LA. Interleukin-10 is a protective factor against diet-induced insulin resistance in liver. J Hepatol. 2008;48:628–637. doi: 10.1016/j.jhep.2007.12.017. [DOI] [PubMed] [Google Scholar]
- 11.Knowler WC, Barrett-Connor E, Fowler SE, Hamman RF, Lachin JM, Walker EA, Nathan DM. Reduction in the incidence of type 2 diabetes with lifestyle intervention or metformin. N Engl J Med. 2002;346:393–403. doi: 10.1056/NEJMoa012512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ropelle ER, Pauli JR, Prada PO, de Souza CT, Picardi PK, Faria MC, Cintra DE, Fernandes MF, Flores MB, Velloso LA, Saad MJ, Carvalheira JB. Reversal of diet-induced insulin resistance with a single bout of exercise in the rat: the role of PTP1B and IRS-1 serine phosphorylation. J Physiol. 2006;577(Pt 3):997–1007. doi: 10.1113/jphysiol.2006.120006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Da Silva ASR, Pauli JR, Ropelle ER, Oliveira AG, Cintra DE, De Souza CT, Velloso LA, Carvalheira JBC, Saad MJA. Exercise intensity, inflammatory signaling, and insulin resistance in obese rats. Med Sci Sports Exerc. 2010;42:2180–2188. doi: 10.1249/MSS.0b013e3181e45d08. [DOI] [PubMed] [Google Scholar]
- 14.Hawley JA, Lessard SJ. Exercise training-induced improvements in insulin action. Acta Physiol. 2008;192:127–135. doi: 10.1111/j.1748-1716.2007.01783.x. [DOI] [PubMed] [Google Scholar]
- 15.Pauli JR, Ropelle ER, Cintra DE, Carvalho-Filho MA, Moraes JC, De Souza CT, Velloso LA, Carvalheira JBC, Saad MJA. Acute physical exercise reverses S-nitrosation of the insulin receptor, insulin receptor substrate 1 and protein kinase B/Akt in diet-induced obese Wistar rats. J Physiol. 2008;586:659–671. doi: 10.1113/jphysiol.2007.142414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Oliveira AG, Carvalho BM, Tobar N, Ropelle ER, Pauli JR, Bagarolli RA, Guadagnini D, Carvalheira JB, Saad MJ. Physical exercise reduces circulating lipopolysaccharide and TLR4 activation and improves insulin signaling in tissues of DIO rats. Diabetes. 2011;60:784–796. doi: 10.2337/db09-1907. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 17.Pauli JR, Cintra DE, De Souza CT, Ropelle ER. Novos mecanismos pelos quais o exercício físico melhora a resistência à insulina no músculo esquelético. Arq Bras Endocrinol Metabol. 2009;53(4):399–408. doi: 10.1590/S0004-27302009000400003. [DOI] [PubMed] [Google Scholar]
- 18.Da Luz G, Frederico MJ, da Silva S, Vitto MF, Cesconetto PA, de Pinho RA, Pauli JR, Silva AS, Cintra DE, Ropelle ER, De Souza CT. Endurance exercise training ameliorates insulin resistance and reticulum stress in adipose and hepatic tissue in obese rats. Eur J Appl Physiol. 2011;111:2015–2023. doi: 10.1007/s00421-010-1802-2. [DOI] [PubMed] [Google Scholar]
- 19.Masson GS, Nair AR, Dange RB, Silva-Soares PP, Michelini LC, Francis J. Toll-like receptor 4 promotes autonomic dysfunction, inflammation and microglia activation in the hypothalamic paraventricular nucleus: role of endoplasmic reticulum stress. PLoS One. 2015;10:e0122850. doi: 10.1371/journal.pone.0122850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cnop M, Foufelle F, Velloso LA. Endoplasmic reticulum stress, obesity and diabetes. Trends Mol Med. 2012;18:59–68. doi: 10.1016/j.molmed.2011.07.010. [DOI] [PubMed] [Google Scholar]
- 21.Yao L, Kan EM, Lu J, Hao A, Dheen ST, Kaur C, Ling E-A. Toll-like receptor 4 mediates microglial activation and production of inflammatory mediators in neonatal rat brain following hypoxia: role of TLR4 in hypoxic microglia. J Neuroinflammation. 2013;10:23. doi: 10.1186/1742-2094-10-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pierre N, Deldicque L, Barbé C, Naslain D, Cani PD, Francaux M. Toll-like receptor 4 knockout mice are protected against endoplasmic reticulum stress induced by a high-fat diet. PLoS One. 2013;8:e65061. doi: 10.1371/journal.pone.0065061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Koh H-J, Toyoda T, Didesch MM, Lee M-Y, Sleeman MW, Kulkarni RN, Musi N, Hirshman MF, Goodyear LJ. Tribbles 3 mediates endoplasmic reticulum stress-induced insulin resistance in skeletal muscle. Nat Commun. 1871;2013:4. doi: 10.1038/ncomms2851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang S, Kaufman RJ. The impact of the unfolded protein response on human disease. J Cell Biol. 2012;197:857–867. doi: 10.1083/jcb.201110131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fu S, Yang L, Li P, Hofmann O, Dicker L, Hide W, Lin X, Watkins SM, Ivanov AR, Hotamisligil GS. Aberrant lipid metabolism disrupts calcium homeostasis causing liver endoplasmic reticulum stress in obesity. Nature. 2011;473:528–531. doi: 10.1038/nature09968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hansen LH, Gromada J, Bouchelouche P, Whitmore T, Jelinek L, Kindsvogel W, Nishimura E. Glucagon-mediated Ca2+ signaling in BHK cells expressing cloned human glucagon receptors. Am J Physiol Cell Physiol. 1998;274:C1552–C1562. doi: 10.1152/ajpcell.1998.274.6.C1552. [DOI] [PubMed] [Google Scholar]
- 27.Wang Y, Li G, Goode J, Paz JC, Ouyang K, Screaton R, Fischer WH, Chen J, Tabas I, Montminy M. Inositol-1,4,5-trisphosphate receptor regulates hepatic gluconeogenesis in fasting and diabetes. Nature. 2012;485:128–132. doi: 10.1038/nature10988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Du K, Herzig S, Kulkarni RN, Montminy M. TRB3: a tribbles homolog that inhibits Akt/PKB activation by insulin in liver. Science (80). 2003;300:1574–7. [DOI] [PubMed]
- 29.Lima AF, Ropelle ER, Pauli JR, Cintra DE, Frederico MJ, Pinho RA, Velloso LA, De Souza CT. Acute exercise reduces insulin resistance-induced TRB3 expression and amelioration of the hepatic production of glucose in the liver of diabetic mice. J Cell Physiol. 2009;221:92–97. doi: 10.1002/jcp.21833. [DOI] [PubMed] [Google Scholar]
- 30.Sung HY, Guan H, Czibula A, King AR, Eder K, Heath E, Suvarna SK, Dower SK, Wilson AG, Francis SE, Crossman DC, Kiss-Toth E. Human tribbles-1 controls proliferation and chemotaxis of smooth muscle cells via MAPK signaling pathways. J Biol Chem. 2007;282:18379–18387. doi: 10.1074/jbc.M610792200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tang M, Zhong M, Shang Y, Lin H, Deng J, Jiang H, Lu H, Zhang Y, Zhang W. Differential regulation of collagen types I and III expression in cardiac fibroblasts by AGEs through TRB3/MAPK signaling pathway. Cell Mol Life Sci. 2008;65:2924–2932. doi: 10.1007/s00018-008-8255-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Naiki T, Saijou E, Miyaoka Y, Sekine K, Miyajima A. TRB2, a mouse Tribbles ortholog, suppresses adipocyte differentiation by inhibiting AKT and C/EBPbeta. J Biol Chem. 2007;282:24075–24082. doi: 10.1074/jbc.M701409200. [DOI] [PubMed] [Google Scholar]
- 33.Iynedjian PB. Lack of evidence for a role of TRB3/NIPK as an inhibitor of PKB-mediated insulin signalling in primary hepatocytes. Biochem J. 2005;386(Pt 1):113–118. doi: 10.1042/BJ20041425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Matsumoto M, Han S, Kitamura T, Accili D. Dual role of transcription factor FoxO1 in controlling hepatic insulin sensitivity and lipid metabolism. J Clin Invest. 2006;116:2464–2472. doi: 10.1172/JCI27047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Koo S-H, Satoh H, Herzig S, Lee C-H, Hedrick S, Kulkarni R, Evans RM, Olefsky J, Montminy M. PGC-1 promotes insulin resistance in liver through PPAR-alpha-dependent induction of TRB-3. Nat Med. 2004;10:530–534. doi: 10.1038/nm1044. [DOI] [PubMed] [Google Scholar]
- 36.Ohoka N, Yoshii S, Hattori T, Onozaki K, Hayashi H. TRB3, a novel ER stress-inducible gene, is induced via ATF4-CHOP pathway and is involved in cell death. EMBO J. 2005;24:1243–1255. doi: 10.1038/sj.emboj.7600596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Koh H-J, Arnolds DE, Fujii N, Tran TT, Rogers MJ, Jessen N, Li Y, Liew CW, Ho RC, Hirshman MF, Kulkarni RN, Kahn CR, Goodyear LJ. Skeletal muscle-selective knockout of LKB1 increases insulin sensitivity, improves glucose homeostasis, and decreases TRB3. Mol Cell Biol. 2006;26:8217–8227. doi: 10.1128/MCB.00979-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Matsushima R, Harada N, Webster NJ, Tsutsumi YM, Nakaya Y. Effect of TRB3 on insulin and nutrient-stimulated hepatic p70 S6 kinase activity. J Biol Chem. 2006;281:29719–29729. doi: 10.1074/jbc.M511636200. [DOI] [PubMed] [Google Scholar]
- 39.Yu J, Xiao F, Guo Y, Deng J, Liu B, Zhang Q, Li K, Wang C, Chen S, Guo F. Hepatic phosphoserine aminotransferase 1 regulates insulin sensitivity in mice via tribbles homolog 3. Diabetes. 2015;64:1591–1602. doi: 10.2337/db14-1368. [DOI] [PubMed] [Google Scholar]
- 40.Prudente S, Hribal ML, Flex E, Turchi F, Morini E, De Cosmo S, Bacci S, Tassi V, Cardellini M, Lauro R, Sesti G, Dallapiccola B, Trischitta V. The functional Q84R polymorphism of mammalian tribbles homolog TRB3 is associated with insulin resistance and related cardiovascular risk in caucasians from Italy. Diabetes. 2005;54:2807–2811. doi: 10.2337/diabetes.54.9.2807. [DOI] [PubMed] [Google Scholar]
- 41.Matos A, Ropelle ER, Pauli JR, Frederico MJ, de Pinho RA, Velloso LA, De Souza CT. Acute exercise reverses TRB3 expression in the skeletal muscle and ameliorates whole body insulin sensitivity in diabetic mice. Acta Physiol. 2010;198:61–69. doi: 10.1111/j.1748-1716.2009.02031.x. [DOI] [PubMed] [Google Scholar]
- 42.Ropelle ER, Pauli JR, Cintra DE, Frederico MJ, de Pinho RA, Velloso LA, De Souza CT. Acute exercise modulates the Foxo1/PGC-1alpha pathway in the liver of diet-induced obesity rats. J Physiol. 2009;587(Pt 9):2069–2076. doi: 10.1113/jphysiol.2008.164202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.De Souza CT, Frederico MJ, da Luz G, Cintra DE, Ropelle ER, Pauli JR, Velloso LA. Acute exercise reduces hepatic glucose production through inhibition of the Foxo1/HNF-4alpha pathway in insulin resistant mice. J Physiol. 2010;588(Pt 12):2239–2253. doi: 10.1113/jphysiol.2009.183996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Marinho R, Ropelle ER, Cintra DE, De Souza CT, Da Silva ASR, Bertoli FC, Colantonio E, D’Almeida V, Pauli JR. Endurance exercise training increases APPL1 expression and improves insulin signaling in the hepatic tissue of diet-induced obese mice, independently of weight loss. J Cell Physiol. 2012;227:2917–2926. doi: 10.1002/jcp.23037. [DOI] [PubMed] [Google Scholar]
- 45.Hoene M, Lehmann R, Hennige AM, Pohl AK, Haring HU, Schleicher ED, Weigert C. Acute regulation of metabolic genes and insulin receptor substrates in the liver of mice by one single bout of treadmill exercise. J Physiol. 2009;587(Pt 1):241–252. doi: 10.1113/jphysiol.2008.160275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schenck A, Goto-Silva L, Collinet C, Rhinn M, Giner A, Habermann B, Brand M, Zerial M. The endosomal protein Appl1 mediates Akt substrate specificity and cell survival in vertebrate development. Cell. 2008;133:486–497. doi: 10.1016/j.cell.2008.02.044. [DOI] [PubMed] [Google Scholar]