

Abstract
Background. Previous studies have demonstrated an association between antibiotic use and the development of skin abscesses. We tested the hypothesis that alterations in the composition of the cutaneous microbiota may predispose individuals to skin abscesses.
Methods. We studied 25 patients with skin abscesses and 25 age-matched controls, who each completed a questionnaire. Skin swab samples were obtained for DNA analysis from 4 sites around the abscess site (hereafter, “peri-abscess specimens”) and from similar sites on the patient's contralateral side and on healthy control subjects. DNA was extracted and analyzed by quantitative polymerase chain reaction (qPCR) and high-throughput sequencing. The purulent abscess drainage was sent for culture.
Results. Fifteen patients with abscess were infected with Staphylococcus aureus. Use of nuc qPCR to quantitate S. aureus revealed a significantly greater frequency of positive results for peri-abscess and contralateral skin samples, compared with control skin specimens. Analysis of community structure showed greater heterogeneity in the control samples than in the peri-abscess and contralateral samples. Metagenomic analysis detected significantly more predicted genes related to metabolic activity in the peri-abscess specimens than in the control samples.
Conclusions. The peri-abscess microbiome was similar to the contralateral microbiome, but both microbiomes differed from that for control patients. Host characteristics affecting microbial populations might be important determinants of abscess risk.
Keywords: skin infection, MRSA, skin microbiome, abscess
Patients commonly present to emergency departments with skin abscesses, and the incidence appears to be increasing [1]. While many factors contribute to the development of infection with community-acquired methicillin-resistant Staphylococcus aureus (MRSA), such as participation in contact sports, incarceration, injection drug use, and, for men, having sex with men, the strongest risk factors are having a household contact with MRSA infection and having a history of recent antibiotic use [2]. Systemic antibiotics, as well as topical agents, including antibacterial soaps and cleansers, can suppress or eliminate normal skin bacteria [3]; antibiotic use may alter the indigenous microbiota for several weeks or longer [3]. Among patients presenting to the emergency department with skin abscesses due to MRSA, antibiotic use within the prior month has been associated with an increased risk [2].
The composition of the normal cutaneous microbiota is complex; analysis of skin bacteria indicates multiple species, with particular compositions depending on body site [4–10]. The skin microbiome can be divided into dry, moist, and sebaceous sites [6]. As at other sites, the residential bacteria may play a role in defenses against pathogenic bacteria [3]. Our hypothesis is that the perturbation of the normal protective bacterial population predisposes individuals to contracting MRSA and developing skin abscesses. We tested this hypothesis by comparing the microbiota in patients with skin abscesses to the microbiota in control patients without abscesses. We hypothesized that, compared with controls, individuals who developed skin abscesses would have an abnormal microbiota, either at sites adjacent to the abscess, indicating a local diathesis, or more distantly, consistent with a more generally perturbed microbiota.
METHODS
Patients Studied
We examined the cutaneous microbiota from 25 adult patients (age, ≥18 years) with skin abscesses and compared those results with the microbiota present in 25 age-matched controls. Our study population consisted of patients who presented with a skin abscess to the emergency department at Carolinas Medical Center between 9 July 2009 and 18 December 2011. Age-matched (±5 years) controls from the same population were recruited to provide samples from the same body site as for the patient with the abscess to whom they were matched. All patients provided written informed consent, and the protocol was approved by the Carolinas Medical Center institutional review board (protocol 06-09-04A). A questionnaire about risk factors for abscesses included recent prior use of antibiotics and antibacterial soaps. Patients were included in the study if the abscess was sufficiently large to be drained and for purulent material to be sent to the microbiology laboratory for culture.
Patients were excluded from the study if they had abscesses of the face near the eyes, nose, mouth, or ears where specimens could not be obtained, or if they had diabetes or renal failure, immunosuppressive illnesses, including human immunodeficiency virus infection (with or without AIDS), cancer, or congenital immunosuppressive illnesses; had preexisting dermatologic conditions, including eczema or chronic blistering; had used any immunosuppressive medications (eg, prednisone and chemotherapy) within the prior month; had used chlorhexidine (a skin cleanser) in the prior 14 days; or had had the abscess site cleaned with antiseptic solution, including Betadine, chlorhexidine, or alcohol, in the emergency department. Patients were also excluded if the abscess was drained before the peri-abscess skin samples (ie, samples obtained from the skin around the abscess site) could be obtained. Patients in the control group were subject to the same exclusion criteria but also were excluded if they had a history of MRSA skin abscess.
Specimen Collection
The study evaluated swab specimens collected from the peri-abscess site, from the identical but unaffected contralateral site, and from the same site in the unaffected matched control subject. Skin swab samples were obtained at each of the 4 cardinal points of the compass 3 cm from the abscess and from the equivalent control (ie, contralateral and matched control) sites. Skin samples were obtained for microbial analysis by stroking the skin for 60 seconds with a cotton-tipped swab soaked in a solution of normal saline and Tween 20, as described elsewhere [11]; 2 samples were obtained at each site. The head of the swab was cut from the handle, and swabs were centrifuged for 5 minutes at 6700 ×g. The supernatants were stored at −80°C. The purulent drainage from the abscess, which was lanced and drained after peri-abscess skin specimen collection in all cases, was cultured by standard methods in the Carolinas Medical Center clinical microbiology laboratory.
Analysis of S. aureus Isolates From the Abscesses
PCR was used to screen for Panton-Valentine leukocidin (PVL; lukF-PV and lukS-PV) and arginine catabolic mobile element (ACME) and to determine staphylococcal cassette chromosomal mec (SCCmec) types, as described previously [12–14]. All strains were spa typed [15]. Clonal complexes (CCs) were assigned on the basis of the spa-typing results, using the multilocus sequence typing mapping database (http://spa.ridom.de/mlst).
Swab Sample Processing
DNA was extracted from the swab suspensions, using the MoBio PowerLyzer Power Soil DNA Isolation Kit (MoBio Laboratories, Carlsbad, California), according to the manufacturer's protocol. Samples then were divided into aliquots for quantitative polymerase chain reaction (qPCR) analysis and Illumina sequencing with MiSeq.
qPCR Analysis
Sets of primers and TaqMan MGB probes were developed on the basis of the universal bacterial and Staphylococcus species 16S ribosomal RNA (rRNA) genes [11], S. aureus nuc [16], and mecA of MRSA (forward primer, 5′-CAATACAATCGCACATACATTA [designed for this study]; reverse primer, 5′-CATACATAAATGGATAGACGTC [designed for this study]; and probe, 5′-AACAGGTGAATTATTAGCAC). Standards for the qPCR assays were amplified from S. aureus USA300 strain SA11–41, using specific primers for each of the above genes. qPCRs were performed as follows: 0.5 µM forward and reverse primers, 0.1 µM hydrolysis probe, 10 µL of Light Cycler 480 Probes Master (Roche Applied Sciences), and 1 μL of extracted DNA in a final 20-μL volume. The assays were performed using the LightCycler 480 II PCR system (Roche Applied Sciences), each assay was run in duplicate, and the results were analyzed using the LightCycler480 II program.
High-Throughput Sequencing
For each extracted DNA sample, the V4 region of the bacterial 16S rRNA gene was amplified in triplicate reactions, using primer set 515F/806R, which nearly universally amplifies bacterial and archaeal 16S rRNA genes [17, 18]. PCR reactions contained 11 μL of molecular biology–grade water (Corning Cellgro), 10 μL of 5-PRIME Hot Master Mix (5-PRIME), 1.0 μL of each of the forward and reverse primers (5 μM final concentration for each), and 2.0 μL of genomic DNA. Reactions were held at 94°C for 3 minutes to denature the DNA; run for 35 amplification cycles at 94°C for 45 seconds, 50°C for 60 seconds, and then 72°C for 90 seconds; and completed with a final extension step of 10 minutes at 72°C. Amplicons from each sample were quantified using the Quant-iT PicoGreen dsDNA Assay Kit (ThermoFischer Scientific, Waltham, Massachusetts); equal amounts of DNA from each sample were then pooled, followed by PCR purification (Qiagen, Germantown, Maryland). DNA concentrations in these subpools were quantified with the Qubit high-sensitivity dsDNA Assay (ThermoFischer Scientific) and combined at equal concentration. The Illumina MiSeq platform was used for DNA sequencing, in the NYULMC Genome Technology Center, spiking with 40% PhiX DNA to increase sequence diversity. The sequencing was performed in 3 separate reads: (1) 151-base pair forward read, (2) 12-base pair barcode read, and (3) 151-base pair reverse read. Only reads that passed the Illumina quality filter were used for downstream analysis.
Data Analysis
Sequence data were processed with QIIME v1.7.0 [19], as described elsewhere [20]. Briefly, sequences were demultiplexed and quality filtered, using default QIIME parameters, and clustered into operational taxonomic units (OTUs) with a sequence similarity threshold of 97%, with UCLUST [21]. 16S rRNA OTUs were picked using an open-reference OTU protocol with the following parameters: –max_accepts 1 –max_rejects 8 –stepwords 8 –word_length 8. The sequence reads were clustered using the Greengenes 97% reference data set (http://greengenes.secondgenome.com; May 2013 release) [22, 23]. Of the 1.98 million Illumina reads from the V4 region of bacterial 16S rRNA genes that passed the QIIME quality filters, 92% matched a reference sequence at ≥97% nucleotide sequence identity, and those failing to match were discarded. Taxonomy was assigned to the retained clusters (OTUs) on the basis of the Greengenes reference sequence, and the Greengenes phylogeny was used for all downstream community comparisons. With pooling of the reads of the 4 samples from each site, the number of sequences per subject ranged from 6166 to 89 978 (mean, 24 292 sequences per sample). To avoid biases caused by differences in sample sequencing depth, analyses were conducted on data rarefied to 6000 sequences per sample.
Linear discriminant analysis (LDA) effect size [24] was used via the Galaxy Browser to detect significant changes in the relative abundance of microbial taxa among the different groups. To reduce the number of features, analysis was limited to taxa with a relative abundance ≥0.1% in any sample. Significance thresholds were performed at the default settings [24]. Metagenomic content of the microbiota samples was predicted from the 16S rRNA profiles, and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway functions were categorized at level 3, using the Phylogenetic Investigation of Communities by Reconstruction of Unobserved States (PICRUSt) tool [25].
Statistical Analysis
Significance was determined using the Student t test, Mann–Whitney U test, 1-way analysis of variance (ANOVA), Friedman test, and χ2 test, depending on the data type. Differences with a P value of <.05 were noted as statistically significant. Demographic data were analyzed by means of techniques designed for matched case/control studies, using χ2 analysis and the Fisher exact test.
RESULTS
Demographic Characteristics
The demographic characteristics of the 25 patients with skin abscesses were generally similar to those of the 25 age-matched controls (Table 1). Most significantly, 56% of the patients with abscess had a past history of abscess, compared with only 1 control (P < .001). The frequency of antibiotic use in the prior 3 months trended toward being significantly greater among patients with abscesses (P = .067). The frequency of antimicrobial soap use was high in both groups.
Table 1.
Demographic and Clinical Characteristics of 25 Patients With Abscesses and 25 Matched Control Subjects
| Characteristic | Patients | Controls | P Value |
|---|---|---|---|
| Age, y, mean | 34.8 | 35.3 | .91 |
| Female sex | 48 | 52 | >.99 |
| Ethnicity | |||
| Black | 72 | 68 | >.99 |
| White | 24 | 28 | |
| Hispanic | 4 | 4 | |
| Antibiotic use in past 3 mo | 32 | 12 | .067 |
| Antimicrobial soap use | 64 | 68 | .77 |
| Household contact with MRSA | 16 | 12 | >.99 |
| Incarceration past 3 y | 16 | 40 | .11 |
| History of abscess | 56 | 4 | .0001 |
| Hospitalization in past 3 mo | 4 | 4 | >.99 |
| Chief complaint | |||
| Abscess | 100 | … | |
| Trauma/laceration | 28 | ||
| Abdominal pain | 16 | ||
| Hypertension | 8 | ||
| Dyspnea/cough | 12 | ||
| Dental problem | 8 | ||
| Vaginitis/urinary tract infection | 16 | ||
| Other | 12 | ||
Data are % of subjects, unless otherwise indicated.
Abbreviation: MRSA, methicillin-resistant Staphylococcus aureus.
Role of S. aureus
Of the 25 patients with abscesses, 15 were positive for S. aureus, and 10 were positive for other pathogens (Figure 1). Of the patients with S. aureus infections, 11 had cultures positive for MRSA, and 2 had no cultures obtained but were positive for S. aureus by PCR in the nuc and mecA analyses. We combined these groups for MRSA analysis; thus, 13 subjects were MRSA positive. Two of the 15 patients had methicillin-susceptible S. aureus, rather than MRSA. Of the patients with other pathogens, 3 cultures grew coagulase-negative Staphylococcus species, 2 grew mixed flora, 1 had no growth, and 1 each grew Enterococcus species, Proteus species, Streptococcus agalactiae, and Corynebacterium species. Abscesses occurred in all 3 (dry, moist, and sebaceous) cutaneous zones (Supplementary Table 1). In the dry areas, S. aureus caused 83% of the abscesses, compared with 38% in the moist or sebaceous areas (P = .04, by the Fisher exact test).
Figure 1.

Study design and bacteriologic characteristics of the study subjects. Abbreviations: MRSA, methicillin-resistant Staphylococcus aureus; MSSA, methicillin-susceptible Staphylococcus aureus.
All 10 MRSA strains analyzed were pandemic sequence type 8/pulsed-field gel electrophoresis type USA300, as determined by conventional genotyping (spa type, SCCmec element subtype, presence of PVL, and ACME). Semiquantitative comparison of hemolytic toxin levels revealed that all 10 strains were indistinguishable from one another and from prototype USA300 clones, such as strain LAC. Thus, selection for mutants with altered virulence was not present in the strains identified in this study.
Results of nuc qPCR for quantitation of S. aureus were positive significantly more often among both the peri-abscess and contralateral skin samples, compared with the control samples (Figure 2A and Table 2). qPCR detected mecA-positive bacteria more commonly in both the peri-abscess and contralateral skin samples than in control samples, using a low (>0.2 log10 copies) threshold for detection (P = not significant; Table 3). However, samples with higher mecA gene levels (≥0.5 log10 copies) were more common in both the peri-abscess and contralateral sites than in control samples. Positivity for mecA in control patients in the absence of detecting nuc suggests the presence of methicillin-resistant staphylococcal species other than S. aureus (Figure 2A). The presence of S. aureus in abscesses was associated with significant differences between the peri-abscess and control samples in measures of both nuc and mecA status, confirming the usefulness of these assays (Figure 2B). Each PCR assay is a separate analysis, and PCR varies in efficiency when different primers are used and expected amplicon sizes vary. For the Staphylococcus species PCR, the amplicon size is about 270 bp, and for the total 16S PCR, the amplicon size is about 370 bp. Therefore, the absolute numbers of amplicons detected are not directly comparable.
Figure 2.
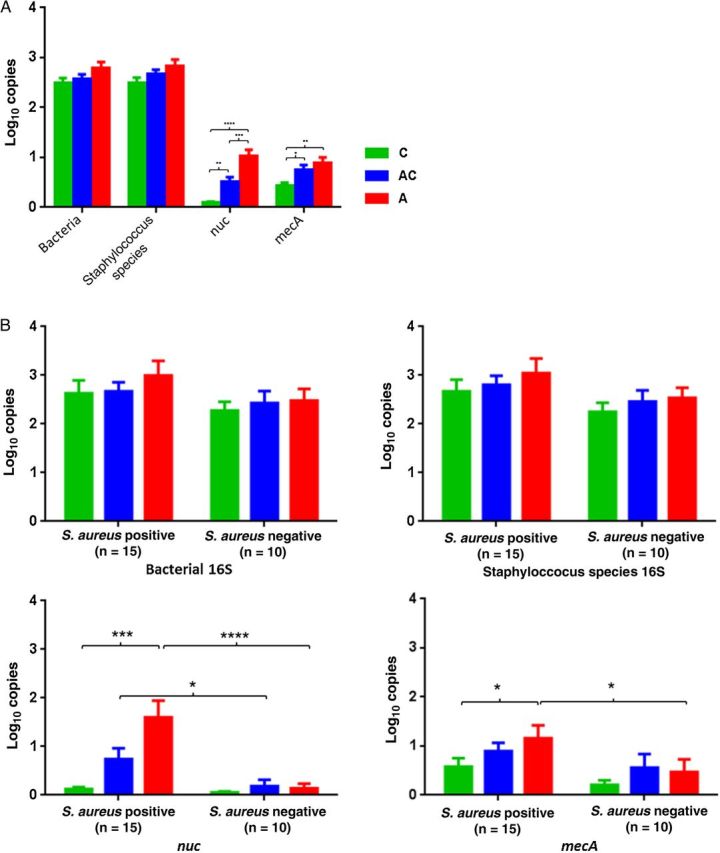
Comparison of quantitative polymerase chain reaction (qPCR) positivity in cutaneous samples from 50 study subjects. A, Mean log10 copies (±standard error of the mean [SEM]) for each gene obtained by qPCR assays for the 4 samples per site in 25 patients (2 sampled sites each) and 25 controls (1 sampled site each). Green denotes specimens from matched control subjects (C), blue denotes contralateral specimens from patients (AC), and red denotes peri-abscess specimens from patients (A). Significance was determined by 1-way analysis of variance with the Tukey analysis for multiple comparisons. Significant differences were found between all 3 comparisons for nuc and mecA, except for A vs AC for nuc. *P < .05, **P < .01, ***P < .001, and ****P < .0001. B, Enumeration of 4 qPCR comparisons in the samples from patients and controls, based on the S. aureus status of the abscess. Mean log10 copies (±SEM) for each indicator gene obtained by qPCR assays; total bacterial and Staphylococcus populations were determined by detection of 16S ribosomal RNA, the presence of S. aureus was determined on the basis of nuc detection, and methicillin-resistant Staphylococcus aureus was determined on the basis of mecA detection. The colors of the 3 groups of samples are defined as described in panel A. Significant differences were found between C and A for S. aureus (P = .0003) and for mecA (P = .041) in the samples from the S. aureus–positive groups, using the Friedman method with the Dunn multiple comparisons adjustment. Significant differences were also obtained according to S. aureus status for nuc (P < .0001) and mecA (P = .046) in the peri-abscess samples and for nuc (P = .025) in the contralateral samples, by the Mann–Whitney U test.
Table 2.
Frequency of Staphylococcus aureus nuc Detection by Quantitative Polymerase Chain Reaction in Skin Samples From 25 Patients With Abscesses and 25 Matched Controls, by nuc Load
| No. of Positive Sites | >Log10 0.2 Copies/µL |
≥Log10 0.5 Copies/µL |
≥Log10 1.0 Copies/µL |
||||||
|---|---|---|---|---|---|---|---|---|---|
| Controls | Patients |
Controls | Patients |
Controls | Patients |
||||
| AC | A | AC | A | AC | A | ||||
| 4 | 1 | 8 | 11 | 0 | 2 | 8 | 0 | 2 | 6 |
| 3 | 2 | 1 | 2 | 0 | 3 | 3 | 0 | 1 | 2 |
| 2 | 7 | 3 | 2 | 1 | 3 | 0 | 0 | 3 | 3 |
| 1 | 8 | 8 | 5 | 1 | 2 | 2 | 1 | 2 | 1 |
| Total | |||||||||
| Subjects | 18 | 20 | 20 | 2 | 10a | 13b | 1 | 8a | 12c |
| Sites | 32 | 49a | 59c | 3 | 25d | 43d | 1 | 19d | 37d |
Abbreviations: A, samples from peri-abscess sites; AC, samples from the identical but unaffected contralateral sites.
a P < .05 vs control, by χ2 analysis.
b P < .01 vs control, by χ2 analysis.
c P < .001 vs control, by χ2 analysis.
d P < .0001 vs control, by χ2 analysis.
Table 3.
Frequency of Methicillin-Resistant Staphylococcus aureus mecA Detection by Quantitative Polymerase Chain Reaction in Skin Samples From 25 Patients With Abscesses and 25 Matched Controls, by mecA Load
| No. of Positive Sites | >Log10 0.2 Copies/µL |
≥Log10 0.5 Copies/µL |
≥Log10 1.0 Copies/µL |
||||||
|---|---|---|---|---|---|---|---|---|---|
| Controls | Patients |
Controls | Patients |
Controls | Patients |
||||
| AC | A | AC | A | AC | A | ||||
| 4 | 5 | 8 | 10 | 3 | 8 | 8 | 2 | 5 | 5 |
| 3 | 5 | 4 | 0 | 2 | 4 | 2 | 1 | 4 | 4 |
| 2 | 3 | 2 | 5 | 3 | 2 | 4 | 3 | 4 | 3 |
| 1 | 3 | 3 | 2 | 6 | 1 | 2 | 2 | 2 | 0 |
| Total | |||||||||
| Subjects | 16 | 17 | 17 | 14 | 15 | 16 | 8 | 15a | 12 |
| Sites | 44 | 51 | 52 | 30 | 49b | 48b | 19 | 42c | 38b |
Abbreviations: A, samples from peri-abscess sites; AC, samples from the identical but unaffected contralateral sites.
a P < .05 vs control, by χ2 analysis.
b P < .01 vs control, by χ2 analysis.
c P < .001 vs control, by χ2 analysis.
Microbiota Characteristics and Relatedness of the Samples
As defined above, the minimal sequence depth used was 6166 (mean, 24 292), which permits a rich analysis of colonizing taxa; depth was not significantly different between the 3 groups of specimens studied (P = .32; data not shown). Analysis of community structure (β-diversity) showed no pattern of clustering according to specimen type; however, mean pairwise distances in the unweighted Unifrac analysis were significantly greater in the control samples than in the peri-abscess and contralateral samples (Figure 3). The findings indicate that the populations of cutaneous microbiota present in the control samples were more distinct from one another than those present in the peri-abscess and contralateral samples. The intergroup distances between the contralateral and peri-abscess samples were significantly smaller than those between either sample type and the control samples, indicating higher similarity among microbiota between samples from the affected subject. There was a decreasing trend in α-diversity from control to contralateral to peri-abscess samples, but differences were not significant between groups in any index, as determined by 1-way ANOVA with the Tukey correction for multiple comparisons (data not shown).
Figure 3.

β-diversity for 75 cutaneous samples, based on the sample site (for patients, specimens from the peri-abscess region [A] and contralateral region [AC]; for controls, specimens from the site complementary to the abscess location on the matched patient [C]). A, Clustering of study subjects by Staphylococcus aureus status, using principal coordinates analysis based on unweighted Unifrac distances. The 75 samples from 50 subjects were divided into 3 groups on the basis of sampled sited (A, AC, and C). Red denotes S. aureus positivity (n = 15 for each group), and green denotes S. aureus negativity (n = 10 for each group). By Adonis testing, P > .05 based on the S. aureus status, in all three groups. B, Intergroup and intragroup β-diversity. Mean pairwise unweighted Unifrac distances (±standard error of the mean [SEM]) are shown. Significance was determined by 1-way ANOVA with the Tukey method for correction for multiple comparisons. **P < .01, ***P < .001, and ****P < .0001 for differences in unweighted Unifrac distances. No significant differences were observed between the groups in the weighted pairwise comparisons.
By neighbor-joining analysis, 18 of 25 subjects (72%) had microbiota from the peri-abscess and contralateral samples on the same branch (Figure 4A); in that analysis, one cluster each was predominantly from moist skin, from dry skin, or from a mixture of the 3 skin types.
Figure 4.
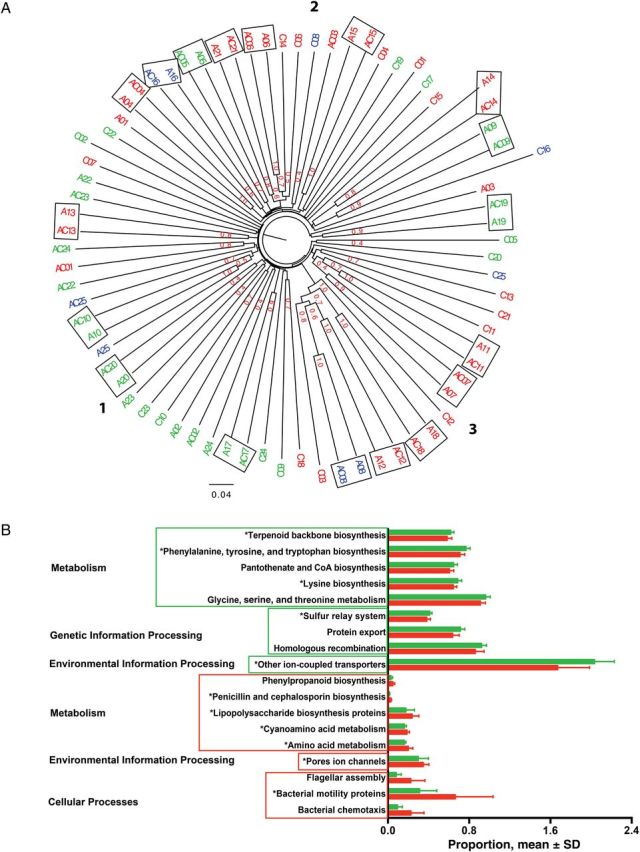
Clustered displays of taxonomic and functional data from 50 subjects. A, Unweighted pair group method with arithmetic mean cluster of the 75 samples from the 50 subjects, based on the unweighted Unifrac distance matrix with Jackknife analysis. The samples from the same microenvironment type are indicated by the same color, as follows: dry sites, red (n = 36); moist sites, green (n = 30); and sebaceous sites, blue (n = 9). The boxed samples are from the same subject. The 3 clusters are shown with bold numbers. In cluster 1, 20 of 27 samples (74%) are from moist skin. In cluster 3, 13 of 18 samples (72%) are from dry skin. Cluster 2 is a mixture of the 3 skin types. Jackknife support fractions of ≥0.5 are shown. B, Phylogenetic Investigation of Communities by Reconstruction of Unobserved States–predicted metagenome with significant differences in relative abundance. Samples are from patients with abscesses who did (n = 8; red) or did not (n = 17; green) receive antibiotics, and functional gene pathways categories with linear discriminant analysis scores of ≥2 are shown. Five of 9 functional gene pathways (56%) in both groups are constituents of the metabolism category. The red box indicates gene pathways with significant abundance in the patients with a history of relatively recent antibiotic use; the green box indicates significant abundance in the patients without recent antibiotic use history. *P < .05 for gene pathway categories that are significantly different after false-discovery rate correction. Abbreviation: SD, standard deviation.
Of 2800 OTUs represented in the samples, 1929 were present in all 3 groups (data not shown). Analysis of the major phyla showed no obvious patterns differentiating the samples according to specimen type (peri-abscess, contralateral, and control; Supplementary Figure 1A). However, within the S. aureus-positive group, a significantly greater percentage of sequences in the control samples was from the genus Brevibacillus, compared with the contralateral samples (P = .03), Within the S. aureus-negative group, the percentage of sequences from the genus Lactococcus was significantly higher in the control specimens, compared with the contralateral specimens (P = .02; Supplementary Figure 1B). Skin samples exposed to the outside world often have plant bacterial sequences present [26]. For 66 of 75 cutaneous samples in our study, sequences representative of the order Streptophyta were present (range, 3–2784 sequences/subject; mean± SD, 158 ± 446), generally at low levels, reflecting such exposure.
Using LEfSE to detect taxa with differential abundance comparing samples from S. aureus–positive patients and matched controls, we found 9 microbial taxa at different levels among the 3 groups of specimens; 4 of 5 differentially abundant taxa from controls belonged to the class Clostridiales (Supplementary Figure 2A). Similarly, differences were noted in 13 taxa between the S. aureus–positive and S. aureus–negative samples (Supplementary Figure 2B). Comparison of samples from patients who received antibiotics in the prior 3 months to those from patients who did not, 13 taxa also were significantly different (Supplementary Figure 2C); the patients who used antibiotics showed enrichment of Actinobacteria, as well as Erysipelotrichaceae (from order to family).
Metagenomic Analyses
To identify microbiome functions that might be characteristic of the samples from the subjects studied, PICRUSt was used with the Greengenes OTU database to derive information on the relative abundance of KEGG pathways, generating metagenomic data. Among the most abundant pathways (≥1%) present in the 3 types of specimens, only 4 KEGG gene pathways related to genetic information processing and metabolic activity were significantly more abundant in the peri-abscess specimens than in control specimens (data not shown). Analysis of paired peri-abscess and control specimens obtained from the same subjects (Supplementary Figure 2D) showed that 11 of 13 genes (85%) with increased abundance belonged to the genetic information processing functional category. In contrast, no functional gene pathways in control specimens were from this category. S. aureus positivity among patients was associated with significant differences, particularly in the metabolic functional category (data not shown). Similarly, recent antibiotic exposure was associated with significant differences in 18 genetic categories, mostly related to metabolism (Figure 4B).
DISCUSSION
Although the majority of skin abscesses were found to be due to S. aureus, 40% were due to other pathogens, a finding consistent with other studies of patients with skin abscesses presenting to emergency departments [1]. Research on skin and soft tissue infections is hindered by difficulties in obtaining reliable microbiology data, but we provided evidence that analysis of skin swab samples can identify S. aureus as a causative agent, even without lancing the abscess. Future studies should clarify whether this technique can be used to identify other pathogens that cause purulent skin infections. Differentiating purulent from nonpurulent skin infections also can be difficult [1]. While ultrasonography may be useful, future studies should clarify whether molecular analysis of adjacent skin for S. aureus or other pathogens can provide additional clinical guidance.
We demonstrated that S. aureus abscesses correlated with high concentrations of S. aureus not only in peri-abscess specimens but also in contralateral skin specimens. Finding high levels of S. aureus, including MRSA, on the contralateral side suggests a general diathesis for S. aureus. An alternative explanation is that the high contralateral concentrations represent a form of spillover from the abscess. However, since the samples were collected before the abscess was drained, iatrogenic contamination is unlikely.
The type of cutaneous microenvironment (dry, moist, or sebaceous) is an important factor in the composition of the local microbiota [6, 8]. Although we found a significant trend toward MRSA occurring more often in dry skin areas, further studies are needed. For wounds occurring in the moist and sebaceous areas, pathogens can differ from those in dry areas [27]. We found that the taxa present in the peri-abscess specimens and the contralateral (ie, unaffected) specimens are more similar to one another than to those in unaffected control specimens. This finding confirms the intrapersonal conservation of taxa present when controlling for site [4] and highlights the interpersonal differences within which microbial pathogenesis must be understood.
Although no large-scale patterns were observed between persons with and those without MRSA infection, there were differences in the skin microbiota identified by LEfSE analysis. For example, samples from subjects with recent antibiotic use showed enrichment of gram-negative bacteria (Comamonadaceae, a family of the Betaproteobacteria). PICRUSt analysis [25] revealed that the collective peri-abscess microbes and those on the contralateral side had gene representation signatures consistent with higher metabolic activity, particularly associated with genetic information processing, than signatures for microbes from control specimens. One explanation is that the abscess environment selects for neighboring organisms that are enriched in these pathways, but the increased representation in the unaffected (contralateral) side is evidence against that possibility. Alternatively, the altered metagenome per se is a diathesis for abscess formation or is a biomarker for such diathesis. Independent confirmation of these findings will be needed.
The study is limited by the relatively small sample size, by our a priori restriction of enrollment to patients without underlying diseases, and by a lack of analysis of the microbiome present at other sites, such as the nares. Thus, future analyses of patients with cutaneous abscesses should include sampling of the microbiota of the nares, inguinal region, and fingertips in affected patients (and controls), as well as in patients with diabetes, who have enhanced abscess risk, which will enable better testing of hypotheses relating to causal inferences.
In conclusion, we found that the peri-abscess and contralateral microbiota were similar in taxonomic composition and gene representation but that these characteristics differed from those of microbiota from control specimens. These findings suggest that general microbiota-host interactions might be important determinants of cutaneous abscess risk. We plan to extend and broaden these studies.
Supplementary Data
Supplementary materials are available at The Journal of Infectious Diseases online (http://jid.oxfordjournals.org). Supplementary materials consist of data provided by the author that are published to benefit the reader. The posted materials are not copyedited. The contents of all supplementary data are the sole responsibility of the authors. Questions or messages regarding errors should be addressed to the author.
Notes
Acknowledgments. We thank the NYULMC Genome Technology Core, for technical assistance (this shared resource is partially supported by National Cancer Institute grant P30CA016087 at the Laura and Isaac Perlmutter Cancer Center); and Moriah Rogers and Jessica Kearney-Bryan, for assistance with identifying appropriate patients and obtaining the specimens.
Financial support. This work was supported by the US Army (program W81XWH-11-1-0739), the National Institutes of Health (UH2 AR057506), the Diane Belfer Program in Human Microbial Ecology, the Cannon Foundation, and the Silverman Foundation.
Potential conflicts of interest. J. M. H. is a subinvestigator on a Gilead pharmaceutical trial unrelated to this work. All other authors report no potential conflicts of interest.
All authors have submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest. Conflicts that the editors consider relevant to the content of the manuscript have been disclosed.
References
- 1.Singer AJ, Talan DA. Management of skin abscesses in the era of methicillin-resistant Staphylococcus aureus. N Engl J Med 2014; 370:1031–47. [DOI] [PubMed] [Google Scholar]
- 2.Moran GJ, Krishnadasan A, Gorwitx RJ. Methicillin-resistant S. aureus infections among patients in the emergency department. N Engl J Med 2006; 355:666–74. [DOI] [PubMed] [Google Scholar]
- 3.Sullivan A, Edlund C, Nord CE. Effect of antimicrobial agents on the ecological balance of human microflora. Lancet Infect Dis 2001; 1:101–14. [DOI] [PubMed] [Google Scholar]
- 4.Gao Z, Tseng C, Pei Z, Blaser MJ. Molecular analysis of human forearm superficial skin bacterial biota. Proc Natl Acad Sci U S A 2007; 104:2927–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fierer N, Hamady M, Lauber CL, Knight R. The influence of sex, handadness and washing on the diversity of hand surface bacteria. Proc Natl Acad Sci U S A 2008; 105:17994–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Grice EA, Kong HH, Conlan S, et al. Topographical and temporal diversity of the human skin microbiome. Science 2009; 324:1190–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Grice EA, Segre JA. The skin microbiome. Nat Rev Microbiol 2011; 9:244–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Costello EK, Lauber CL, Hamady M, et al. Bacterial community variation in human body habitats across space and time. Science 2009; 326:1694–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Findley KF, Oh J, Yang J, et al. Topographic diversity of fungal and bacterial communities on human skin. Nature 2013; 498:367–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Huttenhower C, Sathirapongsasuti J, Segatam N, et al. Structure, function and diversity of the healthy human microbiome. Nature 2012; 486:207–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gao Z, Perez IP, Chen Y, et al. Quantitation of major of major human cutaneous bacterial and fungal populations. J Clin Microbiol 2010; 48:3575–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen L, Mediavilla JR, Oliveira DC, Willey BM, de Lencastre H, Kreiswirth BN. Multiplex real-time PCR for rapid Staphylococcal cassette chromosome mec typing. J Clin Microbiol 2009; 47:3692–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Diep BA, Palazzolo-Ballance AM, Tattevin P, et al. Contribution of Panton-Valentine leukocidin in community-associated methicillin-resistant Staphylococcus aureus pathogenesis. PLoS One 2008; 3:e3198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lina G, Piemont Y, Godail-Gamot F, et al. Involvement of Panton-Valentine leukocidin-producing Staphylococcus aureus in primary skin infections and pneumonia. Clin Infect Dis 1999; 29:1128–32. [DOI] [PubMed] [Google Scholar]
- 15.Shopsin B, Gomez M, Montgomery SO, et al. Evaluation of protein A gene polymorphic region DNA sequencing for typing of Staphylococcus aureus strains. J Clin Microbiol 1999; 37:3556–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Redel H, Gao Z, Li H, et al. Quantitation and composition of cutaneous microbiota in diabetic and nondiabetic men. J Infect Dis 2013; 207:1105–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Walters WA, Caporaso JG, Lauber CL, Berg-Lyons D, Fierer N, Knight R. Primer-Prospector: de novo design and taxonomic analysis of barcoded polymerase chain reaction primers. Bioinformatics 2011; 27:1159–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Caporaso JG, Lauber CL, Walters WA, et al. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J 2012; 6:1621–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Caporaso JG, Kuczynski J, Stombaugh J, et al. QIIME allows analysis of high-throughput community sequencing data. Nat Methods 2010; 7:335–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yatsunenko T, Rey FE, Manary MJ, et al. Human gut microbiome viewed across age and geography. Nature 2012; 486:222–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Edgar RC. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010; 26:2460–1. [DOI] [PubMed] [Google Scholar]
- 22.DeSantis TZ, Hugenholtz P, Larsen N, et al. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl Environ Microbiol 2006; 72:5069–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McDonald D, Price MN, Goodrich J, et al. An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J 2012; 6:610–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Segata N, Izard J, Waldron L, et al. Metagenomic biomarker discovery and explanation. Genome Biol 2012; 12:R60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Langille MG, Zaneveld J, Caporaso JG, et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat Biotechnol 2013; 31:814–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fiere N, Hamady M, Lauber CL, et al. The influence of sex, handedness, and washing on the diversity of hand surface bacteria. Proc Natl Acad Sci U S A 2008; 105:17994–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kanafani ZA, Sexton DJ, Pien BC, et al. Postoperative joint infections due to Proprionobacterium species. Clin Infect Dis 2009; 49:1083–5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.