

Abstract
Nanomedicine has advanced to clinical trials for adult cancer therapy. However, the field is still in its infancy for treatment of childhood malignancies such as acute lymphoblastic leukemia (ALL). Nanotherapy offers multiple advantages over conventional therapy. It facilitates targeted delivery and enables controlled release of drugs to reduce treatment-related side effects. Here, we demonstrate, that doxorubicin (DOX) encapsulated in polymeric nanoparticles (NPs) modified with targeting ligands against CD19 (CD19-DOX-NPs) can be delivered in a CD19-specific manner to leukemic cells. The CD19-DOX-NPs were internalized via receptor-mediated endocytosis and imparted cytotoxicity in a CD19-dependent manner in CD19 positive ALL cells. Leukemic mice treated with CD19-DOX-NPs survived significantly longer and manifested a higher degree of agility indicating reduced apparent systemic toxicity during treatment compared to mice treated with free DOX. We suggest that targeted delivery of drugs used in childhood cancer treatment should improve therapeutic efficacy and reduce treatment-related side effects in children.
Keywords: Nanoparticles, Doxorubicin, Acute Leukemia, CD19, Endocytosis, Xenograft Model
Graphical abstract
Introduction
Acute Lymphoblastic Leukemia or ALL, is the most prominent form of all pediatric cancers 1. Based on Leukemia and Lymphoma Society's Facts 2013, the disease accounted for one-third of cancer deaths in children and adolescents affected by blood cancers. ALL is characterized by massive proliferation of immature white blood cells (WBCs) or lymphoblastic cells in the blood and bone-marrow, that subsequently infiltrate and cause enlargement of various sites such as the liver and spleen (hepatosplenomegaly), lymph nodes (lymphadenopathy) and the central nervous system (causing headaches, sixth-nerve palsy, edema and vomiting). The uncontrolled immature blast proliferation restricts the development red blood cells (RBCs, critical to transport oxygen and nutrients), platelets (essential for blood clotting) and WBCs (vital to fight infections).
Existing treatment protocols in the clinic employ combinations of conventional chemotherapy, molecularly targeted therapies and immunotherapies, often in conjunction with radiation and hematopoietic stem cell transplantation. This achieves an overall five-year relative survival rates that exceed 90% in children treated for ALL 1. Although effective in controlling the growth of rapidly dividing malignant cells, conventional chemotherapeutic drugs do not differentiate between a normal dividing healthy cell and a rapidly dividing cancerous cell. As a consequence, treatment related side effects in more than 60% of pediatric cancer survivors has undermined the very significance of the 90% five-year relative survival rate achieved by combination therapies in pediatric leukemia 2.
To date, several nanoparticle-based drug delivery systems have been formulated and evaluated preclinically for cancer therapy 3-5. Some of these systems have advanced into clinical trials for adult cancer therapy. However, efforts to direct research, development and application of nanotherapeutic platforms in pediatric oncology is lagging 5. Liposomal vincristine sulfate (Marqibo®) is the first system to have entered into Phase I clinical trials for the treatment of pediatric ALL following its approval by the FDA to treat Philadelphia chromosome positive (Ph+) ALL in adults 6.
The CD19 antigen is a single chain, type 1 transmembrane glycoprotein with an extracellular domain composed of 280 amino acids. CD19 is expressed in nearly all stages of B-cell development and also on the majority of acute B-ALL cells 7-9. It is generally absent in normal T-lymphocytes, NK cells, monocytes, and granulocytes and its expression is seldom lost during malignant transformation 10. Since it is not shed from the surface of malignant cells and is internalized after antibody (Ab) binding, CD19 has been used as a target antigen for development of antibody-based therapeutics for B-lymphoid malignancies 11-16.
We recently reported that NPs derived from an amphiphilic block copolymer consisting of hydrophilic poly(ethylene glycol) (PEG) and hydrophobic poly(ε-caprolactone) (PCL) bearing pendent cyclic ketals were biocompatible in vitro and in vivo. Encapsulation of dexamethasone (a steroidal drug used for treating ALL) in these polymeric nanocarriers significantly extended survival and reduced disease symptoms in human xenograft models of ALL 17. Encouraged by these results, we engineered and tested our second generation polymeric NPs to deliver doxorubicin (DOX) specifically to leukemic cells. DOX belongs to the anthracycline class of drugs and is widely used for treating pediatric leukemia. The drug is primarily known to cause significant cardiomyopathy and congestive heart failure apart from other side effects including myelosuppression, nausea and vomiting, hair loss, diarrhea, mucositis, encephalopathy and hemorrhagic cystitis in children 18, 19. To target the ALL cells, the NP surface was modified to display antibodies (Abs) directed against CD19 (a B-lymphoblast cell surface antigen that shows enhanced expression on ALL cells with reduced expression on pluripotent stem cells).
In this study, we demonstrate that anti-CD19Ab-conjugated DOX-encapsulated NPs (CD19-DOX-NPs) specifically targets DOX into CD19-positive (CD19 (+)) B-ALL cells and induce apoptosis in a target specific manner in vitro. We also show that the CD19-DOX-NPs are rapidly internalized via clathrin-mediated endocytosis and impart target specific cytotoxicity at a lower concentration of the encapsulated drug compared to free DOX. We further show that CD19-DOX-NPs treated leukemic mice survive longer and manifested higher degree of agility compared to mice treated with free DOX. We suggest that targeted delivery of clincally used chemotherapeutics should improve or sustain current survival rates with reduced side effects in children treated for leukemia.
Experimental Section
Reagents, cell lines and mouse models
All chemicals necessary for the synthesis of the amphiphilic block copolymers were purchased from Sigma-Aldrich (St. Louis, MO) and were used as received unless otherwise indicated. Palmitic acid N-hydroxysuccinimide ester and avidin from egg white were also obtained from Sigma-Aldrich (St. Louis, MO). Biotinylated mouse anti-human CD19Ab and biotinylated mouse IgG isotype control was obtained from eBioscience (San Diego, CA). Solvents were purchased as anhydrous grade and used without further purification. Doxorubicin Hydrochloride (DOX-HCL) for in vitro and in vivo studies was purchased from Tocris Biosciences (Minneapolis, MN). Nile red (NR) used as a fluorescent probe for cellular tracking of NPs and sucrose was purchased from Sigma-Aldrich. Nystatin was obtained from Thermo-Fisher Scientific (Waltham, MA) and amiloride-hydrochloride was purchased from MP Biomedicals (Santa Ana, CA). DilC18 (7) tricarbocyanine probe (DiR) for in vivo biodistribution studies was acquired from Life Technologies (Grand Island, NY). Human acute leukemia cell lines RS4;11 (ATCC® CRL-1873™, established from a patient with B-ALL at first relapse) and REH (ATCC® CRL-8286™, also established from a patient with B-ALL at first relapse) were purchased from the American Type Culture Collection (ATCC, Manassas, VA). Both RS4;11 and REH cells were maintained in Roswell Park Memorial Institute (RPMI) media (Life Technologies) supplemented with 10% fetal bovine serum (FBS), 2 mmol/L l-glutamine, 25 U/mL penicillin, and 25 μg/mL streptomycin. The cell lines were maintained at 37°C under a humidified atmosphere of 95% air and 5% CO2. BALB/c mice used for in vivo pharmacokinetic and organ biodistribution analysis, and immune-compromised NSG-B2m mice used to develop preclinical B-ALL mouse models for therapeutic efficacy studies were all purchased from The Jackson Laboratory, Bar Harbor, Maine. Animal studies were approved by the Institutional Animal Care and Use Committee at the University of Delaware.
Preparation of DOX-loaded NPs with or without the targeting Ab
(A) Polymer synthesis
The amphiphilic block copolymer was synthesized via a ring-opening copolymerization of ε-caprolactone (CL) and 1,4,8-trioxaspiro-[4,6]-9-undecanone (TSU) using α-hydroxy, ω-methoxy PEG as the initiator, following previously reported procedures 20. The resultant copolymer had a composition of EG113CL152TSU25, a number-average molecular weight (Mn) of 40.6 kg/mol and a polydispersity index (PDI) of 1.57.
(B) Synthesis of avidin-palmitic acid conjugates (avidin-PA)
Avidin at a concentration of 0.25 mg/ml was reacted with palmitic acid N-hydroxysuccinimide ester (NHS-PA, 0.54 mg/ml) in a solvent mixture of DI H2O and dimethylformamide (DMF) (1:39, v/v). The reaction was conducted at 37°C for 4h. To remove excess fatty acid and hydrolyzed ester, the reactants were extensively dialyzed against DMF, followed by DI water using hydrated regenerated cellulose dialysis tubing with a molecular weight cutoff (MWCO) of 10KDa. Dry product was obtained after lyophilization.
(C) Preparation of drug/dye-loaded NPs
Prior to drug encapsulation, DOX-HCl was desalted to generate DOX following reported procedures 21. NPs were then formulated following a nanoprecipitation method 22. Briefly, an acetone/DMSO (1:1, v/v) solution of the block copolymer (10 mg/ml, 1ml) was added dropwise to a stirred aqueous phase (5 ml DI water). The mixture was stirred on a magnetic stir plate at 900 rpm for 2h at ambient temperature to obtain blank NPs. DOX, NR or DiR dye-loaded NPs were similarly prepared using an acetone/DMSO (1:1, v/v) solution of the block copolymer (10 mg/ml, 1ml) containing 2 mg/ml DOX, 0.1 mg/ml NR or 0.036 mg/ml DiR, respectively. The NP suspensions were subsequently centrifuged (2,880 g for 10min) to remove the large aggregates formed from polymers. The supernatant containing the NPs was then transferred to Amicon regenerated cellulose centrifuge filter units (MWCO=30KDa, EMD Millipore) and centrifuged (4500 g for 15min) to remove the free drug or dye and organic solvent. Subsequently, the drug or dye-loaded NPs were collected after thorough washing (three times) with PBS (pH 7.4) using centrifuge filters and immediately used for characterization and biological studies.
(D) Preparation of drug-loaded Ab-conjugated NPs
Drug or dye-loaded NPs with immobilized avidin-PA were prepared following the procedure described previously, with the addition of avidin-PA (0.125 mg/ml) in the stock polymer solution. Following centrifugation to remove the large polymer aggregates, un immobilized avidin-PA, free drug or dye, and organic solvent was removed by transferring the supernatent containing the NPs to Amicon regenerated cellulose centrifuge filter units (MWCO=100KDa, EMD Millipore). The purified avidin-PA-NPs were then re-suspended in PBS at a concentration of 10 mg/ml. Avidin-PA incoporation in the NPs was quantified by a bicinchoninic acid (BCA) assay following the manufacturer's protocols. The concentration of immobilized avidin-PA was derived from a series of standards at an avidin concentration of 6.25-100 μg/ml. NPs without avidin-PA was used as the control. Finally, biotinylated anti-CD19Abs (20 ul, 0.5 mg/ml) were added to avidin-PA-NP suspension (0.5ml, 10 mg/ml) in PBS, and the mixture was incubated at 37°C for 2h. Free Abs were removed by ultracentrifugation (162,635 g for 30min) and pure NPs were obtained by repeated washing with PBS (3 times). The resultant CD19-targeting NPs (CD19-NPs) was resuspended in PBS at desired concentrations for in vitro and in vivo testing.
NP characterization
(A) Particle size and surface charge
Various NP fomulations in PBS were analyzed by dynamic light scattering (DLS) analysis using the Zetasizer nanoZS (Malvern Instruments, Westborough, MA). The mean diameter was computed from the scattered light intensity using the Malvern software package based on the theory of Brownian motion and Stokes-Einstein equation. Zeta potential of NPs was measured by electrophoretic light scattering.
(B) Drug/dye loading and in vitro release
Freshly prepared NPs were lyophilized and the dry weight was noted. The powder was then dissolved in DMSO (1 ml) and DOX/NR concentration was determined by fluorescence using a plate reader (DTX880 Multimode Detector, Beckman Coulter, Fullerton, CA) at 485nm/595nm and at 535nm/595nm for DOX and NR, respectively. The concentration of DiR was quantified using a UV-Vis spectrophotometer (Agilent Technologies, Santa Clara, CA) at 750 nm. The loading content (LC) was defined as the amount of drug/dye (μg) loaded per milligram of NPs and the encapsulation efficiency (EE) was calculated as a percentage based on the amount of drug/dye encapsulated over that in feed.
For studying the in vitro release rates, freshly formulated NPs were incubated at 37°C in PBS under sink conditions for up to 7 days. The release media was collected at predetermined time points after removing NPs by centrifugation (4500 g for 15min) using regenerated cellulose membrane filters with MWCO of 30KDa for non-targeted and 300KDa for targeted NPs. The unfiltered NP suspension was then dispersed in fresh PBS to continue the release study. The filtrate containing the released drug or dye was then lyophilized and the resultant solid is dissolved in DMSO to estimate its concentration. The cumulative release was calculated by dividing the amount of drug or dye released in one specific measurement with the total mass initially loaded. All measurements were carried out in triplicate, and the results were indicated as the mean ± SD.
(C) Quantifying anti-CD19Abs on NPs
Serial dilutions of free biotinylated anti-CD19Abs were prepared and resolved on 10% SDS-PAGE gels and transferred overnight onto nitrocellulose membranes (Bio-Rad) in 20% methanol, 25mM Tris, and 192mM glycine. After transfer, the membranes were blotted with mouse HRP-conjugated streptavidin to bind to biotin and visualized by incubating the membrane with Enhanced Chemiluminescence Plus reagent (GE Healthcare, Piscataway, NJ) followed by exposure to X-ray film (Amersham Biosciences, Piscataway, NJ). A calibration curve obtained from using free anti-CD19Abs was used to determine the the level of anti-CD19Abs incorporated on the NPs.
Analyzing CD19 cellular levels
(A) Immunoblot
To examine and compare the total levels of CD19, B-Cell ALL (B-ALL) cells (REH or RS4; 11) at a density of 3 × 106 cells/dish were collected, washed with cold PBS twice and resuspended in a lysis buffer (20mM Tris-HCl, pH 7.5; 150mM NaCl; 1mM EDTA; 1mM EGTA; 1mM β-Glycerol Phosphate; 1mM Sodium Vanadate; 1.25mM Sodium Pyrophosphate; 1% (w/v) Triton X-100) with a 1% protease inhibitor cocktail (100mM Phenylmethylsulfonyl Fluoride, 1:100; 15 mg/ml mixture of Antipain, Leupeptin, Pepstatin, 1:1000; Sigma-Aldrich. St Louis, MO) at 4°C for 30min. Cell lysates were then prepared by sonication and removal of insoluble material by centrifugation (15,294 g) at 4°C for 15min. The protein concentration in the lysates was determined using a protein assay kit (DC protein assay reagent, Bio-Rad, Hercules, CA, USA). Equal amounts of protein (100 μg) were resolved on 10% SDS-PAGE gels and transferred overnight onto nitrocellulose membranes in 20% methanol, 25mM Tris, and 192mM glycine. After transfer, the membranes were blotted with a mouse monoclonal antibody against CD19 (1:1000, Cell Signaling, Danvers, MA) and visualized by HRP-conjugated secondary antibodies and Enhanced Chemiluminescence Plus reagent (GE Healthcare, Piscataway, NJ) followed by exposure to X-ray film (Amersham Biosciences, Piscataway, NJ). Subsequently, densitometric analysis of the blots was performed using Tina image analysis software to quantify CD19 levels.
(B) Flow cytometry
To quantify the cell surface levels of CD19, B-ALL cells (1.0 × 106) were stained with fluorescein isothiocyanate (FITC)-conjugated mouse anti-human CD19Ab (eBioscience) for 30min at 4°C. Cells were then washed three times with cold PBS and analyzed using a BD Acuri C6 Flow Cytometer® System. For each sample, FITC fluorescence emitted by approximately 10,000 cells was measured using the FL-1 detector (wavelength 530 nm). Data was analyzed using BD Accuri CFlow® software.
Cell uptake of non-targeted and targeted NPs
(A) Treatment with NPs, CD19-NPs and IgG-NPs
B-ALL cells (REH or RS4; 11) were seeded at a density of 300, 000 cells/ml and incubated at 37°C for 1h in the presence or absence of Nile red (543Ex/650Em) encapsulated non-targeted NPs (NR-NPs) or anti-CD19Ab-conjugated NPs (CD19-NR-NPs) or an irrelevant isotype control IgG-conjugated NPs (IgG-NR-NPs) at a final concentration equivalent to 1μM of encapsulated DOX. When indicated, 100 μg of free rabbit anti-human CD19Abs (Cell Signaling) or free irrelevant isotype control IgG (EMD Millipore) was added to the cells at 4°C, 30min prior to incubating with the NPs. To visualize cellular uptake of NPs by confocal laser scanning microscopy (CLSM) analysis, the treated cells were washed three times with cold PBS and mounted on Poly Prep Slides™ (Sigma Aldrich, St. Louis, USA), fixed with 2% paraformaldehyde solution and embedded in ProLong Antifade Kit® mounting medium (Life Technologies, Grand Island, NY) for fluorescent immunocytochemistry. Image acquisition was performed by sequential scanning using a Leica TCS SP5 laser-scanning confocal microscope and subsequently processed by merging of the fluorescence channels using the software LSM (Leica Microsystems, Mannhein, Germany). The cell uptake levels of NR-NPs and CD19-NR-NPs in REH and RS4;11 B-ALL cells were quantified using the ImageJ analysis software (NIH, Bethesda, MD). Statistical significance of the values obtained were then determined by performing multiple t-tests using the Holm-Sidak method, with alpha=5.0%.
(B) Treatment with endocytic inhibitors
To examine the endocytic mode of uptake for CD19-NPs, RS4; 11 cells were seeded at a density of 300,000 cells/ml and pre-treated at 37°C for 30min with one of the following inhibitors: 0.1M hypertonic sucrose (an inhibitor of clathrin-mediated endocytosis) 23; 20 μg/ml Nystatin (an inhibitor of caveolae-mediated endocytosis) 24; and 8 μg/ml amiloride-hydrochloride (an inhibitor of macropinocytosis) 24. Subsequently, the cells were incubated with CD19-NR-NPs at a final concentration equivalent to 1μM of encapsulated DOX for 30min in cell culture media at 37°C, washed and processed for CLSM imaging to visualize uptake of NPs as explained previously. When indicated, RS4; 11 cells were co-treated with fluorescein isothiocyanate-conjugated transferrin (FITC-Tf, Life Technologies) and CD19-NR-NPs at 37°C and processed for further analysis by CLSM imaging.
Cytotoxicity evaluation of DOX formulations
(A) Evaluation of toxicity
The toxicity of free DOX or CD19-DOX-NPs was tested and compared between B-ALL cell lines REH and RS4; 11. When indicated, 100μg of free anti-CD19Ab or free irrelevant isotype control IgG was added to the cells at 4°C, 30min prior to incubating with the DOX nanoformulations. The assays were performed in 96-well cell culture plates which were maintained at 37°C in 5% CO2 atmosphere. The cells were seeded at an initial density of 30,000 cells per well and treated with free DOX or CD19-DOX-NPs at an equivalent dose of 100nM DOX for 1h. Following treatment, the cells were washed with PBS at least two times and left for further incubation up to 48h and 72h. At the end of each incubation period, cell viability was measured by Cell Titer-Blue® Viability Assay. The measurements were then expressed as the percentage of viable cells compared to the survival of untreated cells defined as the maximum cell viability.
(B) Evaluation of apoptosis
B-ALL RS4; 11 cells were plated in 10 cm dishes at a density of 3 × 106 cells/dish and incubated with CD19-DOX-NPs at doses ranging from 1pM to 10μM of encapsulated DOX for 48h. Leukemia cells untreated or treated with 10μM DOX in free form were considered as negative and positive experimental controls respectively. The cell lysates were prepared and protein concentrations were estimated as described earlier. Equal amounts of protein (90 μg) were resolved on 10% SDS-PAGE gels and transferred overnight onto nitrocellulose membranes and blotted with a rabbit polyclonal against cleaved PARP(1:1000, Cell Signaling, Danvers, MA) and a mouse monoclonal antibody against actin (1:10000, Cell Signaling) as explained previously.
HPLC/MS/MS analysis of cell associated DOX (intracellular and cell surface bound) levels in targeted and non-targeted cells
To determine the concentration levels of cell associated DOX (caDOX), B-ALL cells (REH or RS4;11) were seeded in 12 well plates at a density of 300,000 cells/ml and incubated in the presence of either free DOX or CD19-DOX-NPs at concentrations corresponding to 100nM, 1μM or 10μM of DOX equivalents at 37°C for 1h. Subsequently, the cells were lysed and centrifuged at 20,817 g. The supernatant was then subjected to acetonitrile extraction and quantitation of DOX was determined by LC/MS/MS using an established procedure 25. DOX was initially purified by chromatography on a Poroshell 120 EC-C18 column (2.7 μm, 3.0 × 50 mm) at 50°C and eluted with a gradient from 0.1% formic acid to 100% methanol in 0.1% formic acid (1.0 – 3.0min) using an Agilent 1260 Infinity high-performance liquid chromatography (HPLC) system. Solvent flow rate was 0.4 mL/min and total run time per sample was 8min, including column equilibration. DOX was detected using an Agilent 6460 Triple Quad MS/MS equipped with an ESI source. MS conditions were: gas temperature 350°C and flow rate 10 L/min; sheath gas temperature 400°C, flow rate 12 L/min; nebulizer pressure 45 psi; capillary 3500 V and detector in positive ion mode. DOX primary ion was 544 and fragment ion 361 with fragmentor set at 120 V and collision energy 30 eV. A standard curve was created using pure DOX for quantitation of the drug in the supernatant. The assay was linear up to 5μM with a limit of quantitation of 10nM and limit of detection of 3nM using an injection volume of 5 μl.
In vivo pharmacokinetics and biodistribution
To analyze plasma pharmacokinetics; female BALB/c mice (4-6 weeks of age; 3 per time point) were injected via the tail vein with a single dose of 100 μl of CD19-NPs (≈2.5 mg/kg DOX) loaded with a lipophilic “far-red” dye DiR (750Ex/830Em) resuspended in PBS (CD19-DiR-NPs). At 0.08, 0.25, 0.5, 1, 2, 6, 8 and 12h post NP injection, 100 μl of peripheral blood was collected from the mice by submandibular bleeding in heparinized tubes to prevent the blood from clotting. Blood was then centrifuged (455 g, 15min) and CD19-DiR-NP fluorescence levels in the plasma were analyzed by using a highly sensitive multi-label microplate reader (Plate Chameleon V, Hidex, Finland) to assess its plasma half-life. The NP levels were then estimated by comparing with standards prepared in plasma.
To investigate the organ biodistribution and clearance rates of CD19-NPs, female BALB/c mice (4-6 weeks of age; 3 per time point) received intravenous injections of 100 μl of CD19-DiR-NPs (≈2.5 mg/kg DOX). Subsequently, liver, spleen, lung, heart, stomach, kidney and brain were dissected from euthanized mice 6h, 12h, 24h, 48h, 96h, 1week, 2weeks and a month post administration. The organs were then homogenized in tissue lysis buffer (20mM Tris-HCL, pH 7.5; 150mM Sodium Chloride; 1mM EDTA; 1mM EGTA; 1mM β-Glycerol Phosphate; 1mM Sodium Vanadate; 2.5mM Sodium Pyrophosphate; 1% (w/v) Triton X-100; 1% (w/v) IGEPAL; 0.5 % (w/v) Deooxycholate; 1 % (w/v) SDS) and 1% protease inhibitor cocktail at 4°C for 1h. The lysates were used to quantify the CD19-DiR-NP fluorescence levels in various organs using the microplate reader and the NP levels were estimated by comparing with standards prepared in tissue lysis buffer. BALB/c mice treated with saline were included as controls for the experiment and to establish the measurement settings based on background fluorescence.
Measurement of agility and monitoring of survival
RS4; 11 cells (5×106) were injected via the tail vein into female NSG-B2m mice (6 - 8 weeks old; 8 per group). Three days later, the mice were given intraperitoneal injections of saline, free DOX, IgG-DOX-NPs or CD19-DOX-NPs at dose equivalents of 2.5 mg/kg DOX, once a week. The physical activity of mice was measured with a Low Profile Wireless Running Wheel (Med Associates Inc., St. Albans, VT, USA) every 7 days over the course of the treatment and data was analyzed with the activity software SOF-860 provided by Med Associates. The treatment efficacy was finally determined using Kaplan-Meier survival curves and the wheel running activity was recorded and plotted to determine agility. Animals were sacrificed when they depicted signs of morbidity, including hind-limb paralysis or excessive weight loss, according to IACUC guidelines.
Statistical Analysis
All experiments were carried out in triplicates, and results are indicated as the mean ± SE unless otherwise indicated. All graphs have been generated and analyzed using Prism nonlinear regression software (Graphpad Software, Version 6.0c). The survival data in efficacy studies are presented using Kaplan–Meier plots and the data was analyzed using log-rank (Mantel–Cox) test. A p < 0.05 was considered significant.
Results
(A) NP formulation and characterization
Particle size analysis by dynamic light scattering (DLS) (Table 1) showed that the DOX-NPs exhibited an intensity average size of 81 ± 4 nm and a volume average size of 62 ± 3 nm. Similarly, NR-NPs and DiR-NPs displayed the intensity average size of 80 ± 8 nm and 88 ± 1 nm, and the volume average size of 63 ± 2 nm and 73 ± 1 nm, respectively. The average sizes from intensity and volume were in agreement with each other and reflected the size distribution of major population of the NPs. The results in Table 1 also revealed that incorporation of anti-CD19Abs significantly increased the size of nanoparticles. In addition, measurement of zeta potential values revealed that IgG conjugation to NPs significantly decreased the surface charge (-5mV for non-targeted vs -42mV for anti-CD19Ab-conjugated and -50mV for IgG-conjugated NPs).
Table 1. Summary of size, loading content and encapsulation efficiency of different types of NPs.
| Samples | By Intensity (nm) | By Volume (nm) | PDI | Loading Efficiency (μg/mg) | Encapsulation Efficiency (%) |
|---|---|---|---|---|---|
| Blank NPs | 75 ± 1 | 65 ± 1 | 0.073 | - | - |
| DOX-NPs | 81 ± 4 | 62 ± 3 | 0.094 | 71.3 ± 4.7 | 42.8 ± 2.8 |
| NR-NPs | 80 ± 8 | 63 ± 2 | 0.106 | 34 ± 04 | 34.4 ± 4.0 |
| DiR-NPs | 83 ± 1 | 69 ± 1 | 0.133 | 3.1 ± 0.3 | 86.4 ± 8.4 |
| Anti-CD19 Blank NPs | 82 ± 1 * | 68 ± 1 * | 0.145 | - | - |
| Anti-CD19 DOX-NPs | 83 ± 1 | 69 ± 1 * | 0.139 | 72.1 ± 6.4 | 45.4 ± 2.1 |
| Anti-CD19 NR-NPs | 88 ± 1 | 75 ± 3 * | 0.068 | 3.5 ± 0.1 | 35.3 ± 1.0 |
| Anti-CD19 DiR-NPs | 88 ± 1 * | 73 ± 1 * | 0.137 | 3.2 ± 0.4 | 89.1 ± 11.1 |
: indicates that the size of the targeted NPs is significantly larger than that of the corresponding non-targeted NPs. For example, in terms of volume, the size of anti-CD19 DOX NPs is significantly larger than that of DOX-NPs. All quantitative measurements were performed on three replicate samples. Statistical significance was evaluated using a two-tailed Student's t-test. A P value of <0.05 was considered to be statistically different.
Protein assays showed that an average of 8.2 ± 1.2 μg of avidin was immobilized onto 1 mg of NPs. Quantification of the surface density of biotinylated anti-CD19Abs by immunoblot analysis revealed approximately 120 ng of Ab per mg of NPs (Supplementary Information, Figure S1).
(B) Drug/dye loading and release
DOX was efficiently entrapped in the polymeric NPs with a loading content and encapsulation efficiency of 71.3 ± 4.7 μg/mg and 42.8 ± 2.8 % for non-targeted-NPs, and of 72.1 ± 6.4 μg/mg and 45.4 ± 2.1% for targeted-NPs, respectively (Table 1). In vitro release was evaluated by incubating targeted or non-targeted DOX-NPs in PBS under sink conditions at 37°C for up to 7 days. For non-targeted NPs, an average of 18.5 ± 2.7 wt% of DOX was released per day from day 0 to day 3, and a slower DOX release rate of 8.4 ± 4.8 wt% per day was obtained from day 3 to day 7. By day 7, a total of 88.9 ± 4.8 wt% of the initially loaded DOX was released. The release rate of DOX from targeted-NPs was estimated as 15.8 ± 2.2 wt% and 12.2 ± 2.7 wt% from day 0 to day 3 and day 3 to day 7, respectively (Figure 1).
Figure 1. In vitro release profiles of.
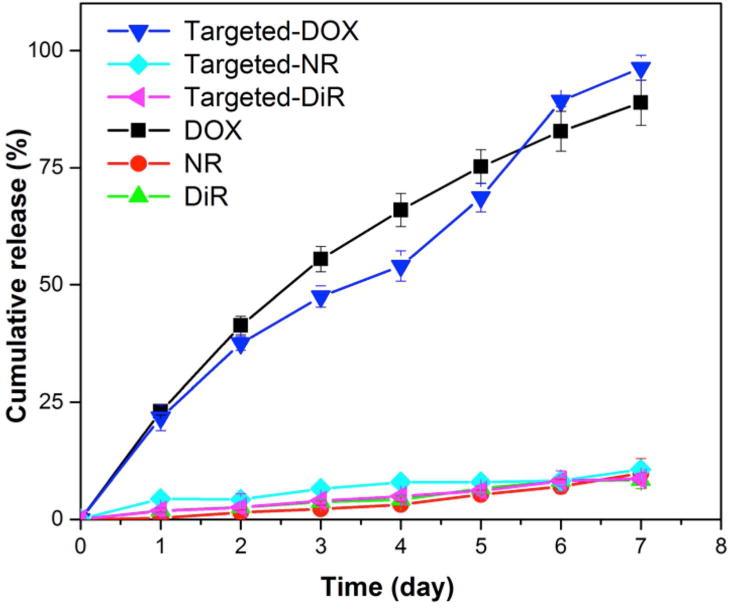
DOX (black - targeted; blue - non-targeted), NR (turquoise - targeted; red - non-targeted), and DiR (pink - targeted; bright green - non-targeted) from NPs in PBS (pH 7.4) at 37°C. Data shown are averages of three independent experiments.
Separately, the loading content and encapsulation efficiency for NR in non-targeted NPs were quantified as 3.4 ± 0.4 μg/mg and 34.4 ± 4.0 %, respectively; and for DiR loaded non-targeted NPs, those values were 3.1 ± 0.3 μg/mg and 86.4 ± 8.4 %, respectively. The loading content and encapuslation efficiency of the dyes in targeted-NPs were similar to that in the non-targeted ones (Table 1). The release profiles revealed limited dye release (equal or less than 10 wt%) by day 7 from both the non-targeted and targeted NPs (Figure 1).
(C) CD19-specific targeting and uptake of anti-CD19Ab-conjugated NPs
The ability of CD19-NPs to function as a targeted drug delivery system was examined by comparing uptake of particles by the cell lines REH and RS4;11, two B-ALL cell lines that express different levels of CD19. Immunoblot analysis revealed that RS4;11 cells express approximately 3 times as much CD19 protein than REH cells (P = 0.0214, Figures 2A and B). Flow cytometry confirmed enhanced cell surface expression of CD19 in RS4;11 cells (Figure 2C). These cell lines were then utilized to characterize the CD19-specific targeting of NPs.
Figure 2. Quantification of CD19 total and surface expression levels in REH and RS4;11 BALL cells.
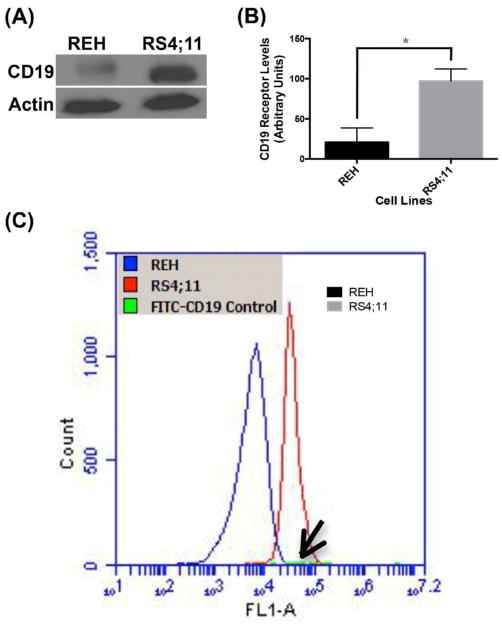
(A and B) Immunoblot and quantification of the CD19 levels. (C) Flow cytometry analysis of surface levels of CD19 (Note FITC-(anti-CD19Ab) control, arrow). Quantified data shown are averages of two independent experiments. Statistical significance was determined using a two-tailed Student's t-test where p value < 0.05 was considered significant
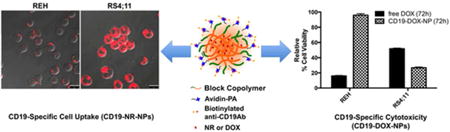
Cell binding and uptake studies with REH and RS4;11 leukemia cells were performed using NR encapsulated NPs that were either non-targeted (NR-NPs) or conjugated with an irrelevant isotype control IgG (IgG-NR-NPs) or anti-CD19Abs (CD19-NR-NPs). CLSM imaging revealed that uptake levels of CD19-NR-NPs were more than 5-times in RS4;11 cells than in REH cells (P=0.0076, Figure 3A upper panels, and 3B, left bars). Interestingly, the uptake of NR-NPs in REH cell line was significantly higher than RS4;11 cells (P=0.03) indicating that CD19 targeting reduces the non-specific uptake. (Figure 3A lower panels and 3B right bars). No uptake was observed for IgG-NR-NPs (Figure 3C). These data show that the anti-CD19Ab-conjugated NPs were taken up by a CD19-dependent uptake mechanism.
Figure 3. CD19 specific targeting and uptake of anti-CD19Ab conjugated NPs.
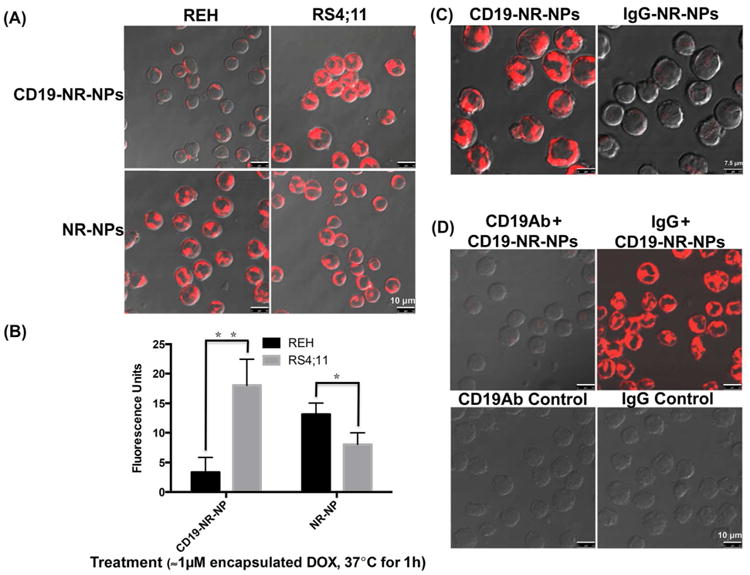
(A) CLSM analysis to compare uptake of CD19-NR-NPs and NR-NPs (final concentration ≈1μM of encapsulated DOX) in REH and RS4;11 cells for 1h at 37°C. The uptake pattern was similar at 5min. Note that even after 1h exposure, REH cells shows minimal uptake. Scale bar, 10 μm. (B) Quantification of cell uptake levels of CD19-NR-NPs and NR-NPs in REH and RS4;11 cells. (C) CLSM analysis to compare uptake of CD19-NR-NPs with isotype matched non-specific uptake of IgG-NR-NPs (final concentration ≈1μM of encapsulated DOX) in RS4;11 cells for 1h at 37°C. Scale bar, 7.5 μm. (D). CLSM analysis to confirm CD19 dependent uptake of CD19-NR-NPs (final concentration ≈1μM of encapsulated DOX) in RS4;11 cells for 1h at 37°C. Scale bar, 10 μm. Quantified data shown are averages of three independent experiments. Statistical significance was determined using the Holm-Sidak method, with alpha = 5.000%.
(D) Competition assays confirmed specific binding and uptake of anti-CD19Ab-conjugated NPs
A competition assay was conducted to confirm the CD19-specific uptake of targeted NPs. RS4;11 cells were pre-incubated with excess of free anti-CD19Abs or free irrelevant isotype matched IgG for 30min at 4°C prior to treatment with CD19-NR-NPs. CLSM imaging showed that pre-treatment with free anti-CD19Abs reduced uptake of CD19-NR-NPs in RS4;11 cells but that pre-incubation with excess control IgG does not (Figure 3D, upper panels), confirming CD19-specific uptake of the targeted NPs. There was no background fluorescence either in free anti-CD19Ab or irrelevant IgG treated cells (Figure 3D, lower panels).
(E) Analysis of endocytic mechanism for anti-CD19Ab-conjugated NPs
To investigate the mechanism of uptake of CD19-NPs in CD19(+) RS4;11 cells, the effect of various endocytic pathway inhibitors was analyzed. Compared to Nystatin (caveolae-mediated endocytic inhibitor 24 or A-Hcl (a macropinocytosis inhibitor 24, sucrose (clathrin-mediated endocytic inhibitor) 23, inhibited uptake of CD19-NR-NP (Figure 4A). In addtion, FITC-Tf (a well-established marker for clathrin-mediated endocytosis) 26, colocalized with CD19-NR-NPs (Figure 4B), demonstrating that anti-CD19Ab-conjugated NPs are internalized primarily into endosomes via clathrin-dependent endocytosis.
Figure 4. Analysis of endocytic mechanism for anti-CD19Ab conjugated NPs.
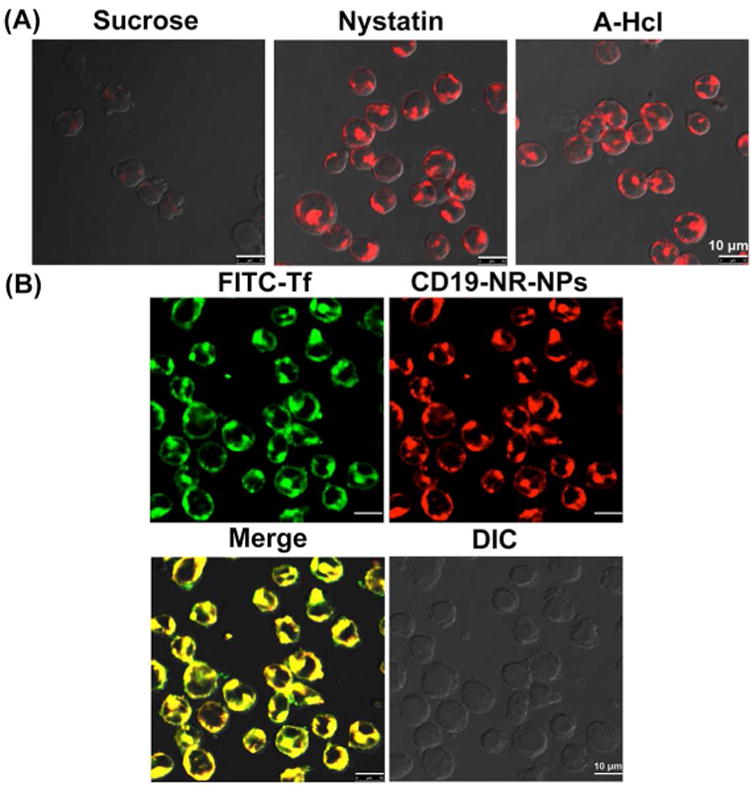
(A) Inhibiting clathrin-mediated uptake of CD19-NR-NPs (final concentration ≈1μM of encapsulated DOX) in RS4;11 cells for 30 min at 37°C. Scale bar, 10 μm. (B) Colocalization of FITC-transferrin and CD19-NR-NPs in RS4;11 cells. Scale bar, 10 μm.
(F) CD19-mediated induction of apoptosis in leukemia cells
After confirming CD19-mediated internalization of NPs, we investigated whether DOX loaded targeted NPs induce CD19-dependent cytotoxicity. For this purpose, cells were incubated with free DOX or CD19-DOX-NPs for 1h, washed to remove the excess drug present in the media and incubated for an additional 72h in drug free medium. REH cells were more sensitive to free DOX than RS4;11 cells. Further, free DOX significantly reduced the viability of REH cells than CD19-DOX-NPs (P=3.335067e-012). In contrast, the latter cells were even more sensitive to CD19-DOX-NPs while 95% REH cells remained viable when treated with the targeted NPs (Figure 5A).
Figure 5. CD19-mediated induction of apoptosis in leukemia cells.
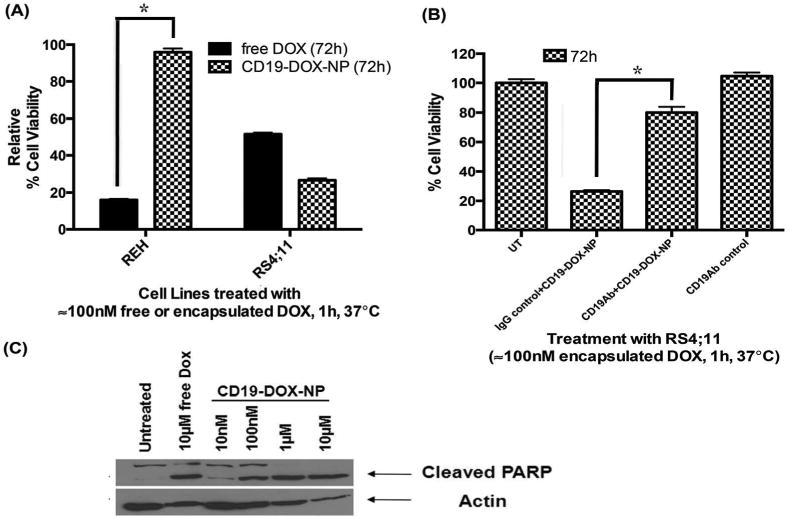
(A) Comparison of toxicity induced by free DOX, and CD19-DOX-NPs (≈100nM DOX) in REH and RS4;11 cells. (B) Competition assay to confirm CD19-receptor dependent induction of toxicity by CD19-DOX-NPs (≈100nM DOX) in RS4;11 cells. (C) Dose-dependent cleaved PARP levels due to treatment of CD19-DOX-NPs with RS4;11 cells (48h). Quantified data shown are averages of three independent experiments. Statistical significance was determined using the Holm-Sidak method, with alpha = 5.000%.
To confirm that selective toxicity is induced via the CD19-mediated uptake of CD19-DOX-NPs, competition experiments were performed by pre-incubating RS4;11 cells with excess of free irrelevant isotype matched IgG or free anti-CD19Abs for 30min at 4°C prior to treatment with CD19-DOX-NPs. Pretreatment with control IgG did not affect the toxicity of CD19-DOX-NPs. On the other hand, free anti-CD19Abs did affect the toxicity CD19-DOX-NPs (P=3.668123e-008, Figure 5B). Furthermore, CD19-DOX-NPs induced dose-dependent cleavage of PARP (Figure 5C), in RS4;11 cells indicating apoptotic cell death.
(G) CD19 targeting increases the cell-associated DOX (intracellular and cell surface bound) levels in RS4;11 cells
Treatment with DOX (1 and 10μM) reduced viability in REH and RS4;11 irrespective of the method of delivery (Supporting Information, Figures S2 and S3). However, CD19-DOX-NPs (≈100nM DOX) reduced the viability of RS4;11 but not REH cells (Figure 5A). Reduced uptake of CD19-targeted NPs in REH cells should reduce levels of cell associated DOX (caDOX) and as a consequence have a reduced effect on viability of these cells. To test this possibility, we treated REH and RS4;11 cell lines with increasing doses of free DOX and CD19-DOX-NPs (≈100nM, 1μM, 10μM DOX) and quantified caDOX levels using HPLC/MS/MS (Figure 6A). The caDOX levels in free DOX treated REH and RS4;11 cells were similar (Figure 6B). By contrast, in CD19-DOX-NP treated cells, the caDOX levels in RS4;11 cells was 4-10 times higher than in REH cells (P = 0.0119 for 100nM DOX, P = 0.0157 for 1μM DOX and P = 0.0166 for 1μM DOX, Figure 6C). These results indicate that specific targeting of NPs via CD19 results in elevation of caDOX to levels that can cause loss of viability in RS4;11 cells. It is also notewothy that at the same doses of DOX, caDOX levels in RS4;11 cells were not very different on treatment with targeted NPs compared to free DOX (Supporting Information, Figure S4). However, the caDOX levels in free DOX treated REH cells are an order of magnitude higher than the targeted NPs treated cells (Supporting Information, Figure S5).
Figure 6. CD19-targeting increases the cell associated DOX (intracellular and cell surface bound, caDOX) levels in RS4;11 cells.
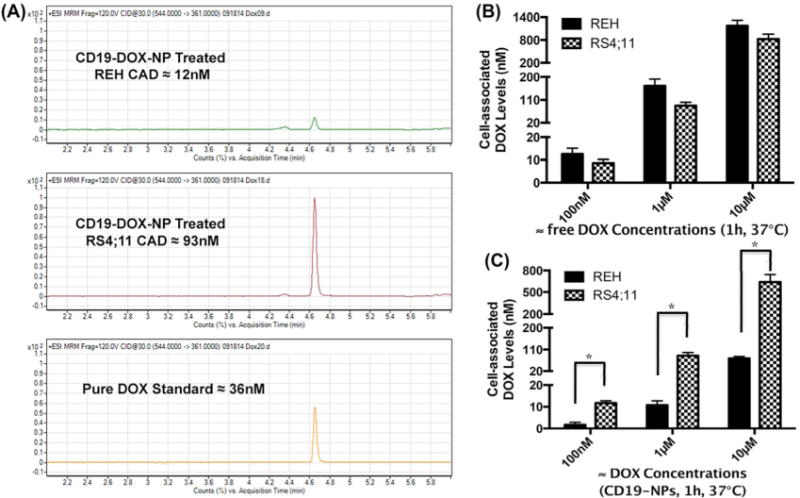
(A) HPLC/MS/MS representative quantitation of caDOX levels in REH and RS4;11 cells treated with CD19-DOX-NPs (≈1μM DOX, for 1h at 37°C). (B) REH and RS4;11 caDOX levels on treatment with free DOX for 1h at 37°C. (C) REH and RS4;11 caDOX levels on treatment with CD19-DOX-NPs for 1h at 37°C. Quantified data shown are averages of three independent experiments. Statistical significance was determined using the Holm-Sidak method, with alpha = 5.000%.
(H) Plasma levels and biodistribution of anti-CD19Ab-conjugated NPs in mice
In order to evaluate plasma levels of CD19-DiR-NPs, peripheral blood samples drawn from mice at various time points were assessed for “DiR” (750Ex/830Em) fluorescence levels. Soon after the tail vein injection of NPs, there was an initial spike in the plasma at 5min, which then dropped and sustained in circulation for approximately 8h only to further drop at 12h and 24h. (Figure 7A).
Figure 7. Plasma levels and biodistribution of anti-CD19Ab conjugated NPs in mice (3 per time point).
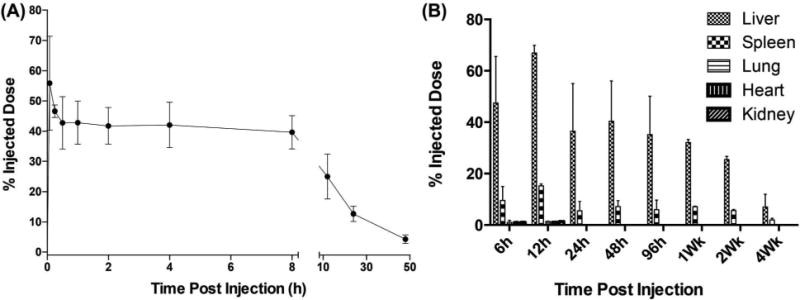
(A) Plasma distribution of CD19-DiR-NPs in BALB/c mice. (B) Biodistribution and clearance of CD19-DiR-NPs in mice.
To assess tissue distribution levels, BALB/c mice injected via the tail vein with CD19-DiR-NPs (2.5mg/kg DOX) were euthanized at different time points to harvest liver, spleen, heart, lung, kidneys and brain. Six hours after treatment, CD19-DiR-NP fluorescence was primarily detected in liver and spleen with lower levels in kidney, lung and heart. Levels increased at 12h and then slowly declined over the next 4 weeks (Figure 7B).
(I) CD19-DOX-NPs Enhance Therapeutic Efficacy in a Pre-clinical Mouse Model of ALL
To test the in vivo efficacy of CD19-DOX-NPs, an ALL human xenograft model, developed in NSG-B2m mice, received intraperitoneal injections at 2.5 mg/kg DOX once a week. The mice were randomized into four groups (8/group) to receive saline, free DOX, IgG-DOX-NPs, or CD19-DOX-NPs. Kaplan-Meier survival curves showed that mice that received CD19-DOX-NPs survived significantly longer than those treated with saline (P = 0.0021) or the group treated with free DOX (P = 0.0369) or IgG-DOX-NPs (P = 0.0163) (Figure 8).
Figure 8. CD19-DOX-NPs enhance therapeutic efficacy and prolongs survival in pre-clinical leukemia mouse models (8 per treatment group).
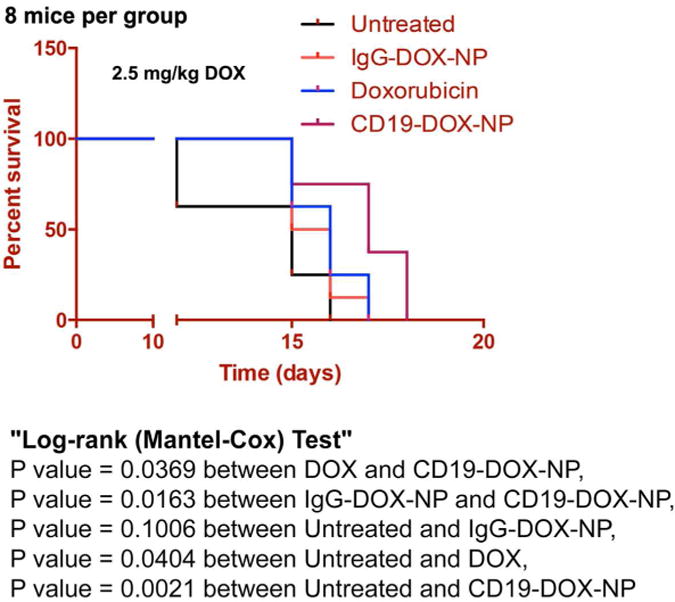
Efficacy of CD19-DOX-NPs in xenograft model of ALL. (Survival rate is presented in a Kaplan-Meier plot). CD19-DOX-NPs (≈2.5 mg/kg DOX) significantly prolonged survival in comparison with groups treated with saline, IgG-DOX-NPs and free DOX (≈2.5 mg/kg DOX).
During the survival study we also monitored the physical activiy of different groups using a computerized low profile wireless running wheel which monitors the agility of the treated mice. Interestingly, the group that received CD19-DOX-NPs manifested high agility in comparison with the other groups during the treatment (Supporting Information, Figure S6).
Discussion
Nanoparticle (NP) based targeted drug delivery has a promising future in medicine due to its multiple applications in cancer treatment: it can improve the half-life of drugs, reduce dosage, improve drug solubility, reduce immunogenicity and minimize non-specific exposure of toxic drugs. While many of these applications have advanced for solid tumors, targeted delivery approaches are minimally developed for blood cancers especially in childhood leukemia. In this study, we utilized amphilic block copolymer NPs with anti-CD19Abs (CD19-NPs) as a targeting moiety to deliver DOX specifically to ALL cells. We demonstrated that the targeting of DOX to ALL cells and its induction of cytotoxicity is mediated in a CD19-dependent manner. We also show that CD19-NPs were internalized rapidly in a clathrin-dependent manner resulting in elevated levels of DOX only in targeted cells. We further show that CD19-DOX-NPs treated leukemic mice survived longer and manifested higher degree of agility indicating reduced apparent systemic toxicity compared to mice treated with free DOX. This is the first time CD19-targeted polymeric NPs have been utilized in ALL mouse models to show advantages over conventional chemotherapy. We suggest that targeted drug delivery for liquid tumors such as ALL may be advantageous due to the ease of access to tumor cells, unlike solid tumors where targeting is complex on account of multiple phsyical barriers that NPs have to overcome 27.
Previously, we demonstrated that NPs formulated from amphiphilic block copolymers consisting of hydrophilic poly(ethylene glycol) (PEG) and hydrophobic polyester bearing pendent cyclic ketal groups efficiently encapsulated Dexamethasone which is comparable to values other platforms have achieved 28, 29. The incorporation of pendent cyclic ketal groups on the hydrophobic portion on the polymer backbone increased the chain flexibility while decreasing polymer crystallinity which improved the drug loading capacity and its release profile 20. To immobilize the targeting moiety, we utilized avidin which is chemically modified with a fatty acid (i.e. palmitic acid), and then incorporated onto the surface of NPs during the nanoprecipitation process. The palmitic acid preferentially partitions within the hydrophobic core of NPs, and the hydrophilic avidin head group is displayed on the particle surface along with the mPEG segments. Biotinylated Abs were then linked to the NP surface via the anchored avidin. A similar approach has been reported previously, incorporating avidin-fatty acid conjugates onto poly (lactic-co-glycolic acid) (PLGA) microspheres 30. We further demonstrated that the fatty acid modified avidin can be efficiently anchored onto the surface of NPs, exhibiting an average surface modification efficiency of 8.2 μg avidin per mg of NPs. This is comparable to values reported for other systems 30. Subsequently, biotinylated anti-CD19Abs were linked onto the surface of the avidin-PA-NPs. We achieved a DOX encapsulation efficiency of about 45%, irrespective of the presence or absence of avidin in NPs, indicating that addition of avidin does not compromise the encapsulation efficiency of DOX. In addition, the in vitro release profiles revealed that incorporation of avidin did not affect the release kinetics of DOX from the NPs. The limited release of dyes - NR and DiR – from the NPs could be attributed to its strong hydrophobic nature 17. The high retention of the hydrophobic fluorescent probes by NPs demonstrates the feasibility to use NR and DiR to track the location of NPs without undesirable dye leaking concerns 17.
One of the most important characterization studies of targeted NPs is validation of target specificity. We utilized multiple approaches to confirm the specific targeting of CD19-targeted NPs: 1. RS4;11 cells that have higher expression levels of CD19 than REH cells showed more rapid uptake of CD19-NPs. 2. Uptake of IgG-NPs by RS4;11 cells was much lower than uptake of CD19-NPs. This could be atributed to the highly negative zeta potential of IgG-conjugated NPs. 3. Pre-incubation of RS4;11 cells with free anti-CD19Abs almost completely blocked the uptake of CD19-NPs. 4. Viability of RS4;11 cells was significantly reduced when treated with CD19-NPs containing 100nM of encapsulated DOX, but REH cells were unaffected. 5. Free anti-CD19Abs significantly blocked the toxicity of CD19-DOX-NPs in RS4;11 cells. 6. caDOX levels in RS4; 11 cells treated with CD19-DOX-NPs at 100nM of encapsulated DOX was 4-fold higher than in REH cells. Taken togther, these results demonstrate that DOX is delivered and toxicity is induced in a CD19-dependent manner in CD19(+) leukemia cells.
Free hydrophobic drugs enter the cells via diffusion across the plasma membrane. By contrast, NP mediated drug delivery utilizes the cellular internalization machinery to deliver drugs into the cells. Internalization mechanisms include macropinocytosis, caveolae, and clathrin-dependent endocytosis 31. Our results show that CD19-NPs are internalized into endosomes of RS4;11 cells by a clathrin-dependent mechanism, demonstrated by inhibition of uptake by sucrose and co-localization with FITC-transferrin – a clathrin-dependent endocytic marker. Clathrin-dependent uptake results in rapid drug accumulation within the cells. We determined the caDOX levels by HPLC/MS/MS to determine how much DOX is needed to induce cytotoxicity. caDOX levels in free DOX and CD19-DOX-NPs treated RS4;11 cells were comparable (Supporting Information, Figure S4). However, caDOX levels in REH cells treated with free DOX were more than 6-fold higher than the CD19-DOX-NPs treated cells (Supporting Information, Figure S5). These results show that delivery of DOX into cells expressing high levels of CD19 by endocytosis of targeted NPs is as efficient as free DOX and that the targeted NPs enhance selectivity for targeting of DOX to such cells.
In a previous study we showed that plasma levels of 110 nm diameter ECT2-NPs were stable for up to 2h compared to the 8h stability seen in this study for smaller 75 nm diameter CD19-NPs. This is consistent with literature data showing better plasma profiles for smaller particles 32-34. In vivo studies demonstrated that survival of mice treated with CD19-DOX-NPs was significantly better than untreated mice (P = 0.0021), free DOX treated mice (P = 0.0369) and IgG-DOX-NP treated mice (P = 0.0163). In this severe model of ALL, therapeutic efficacy of all three treatments was limited, but the enhanced efficacy of the targeted NPs does provide proof-of-concept that such drug formulations can be effective against ALL. Our in vitro data indicated that drug delivery of the targeted NPs would be more specific than free DOX for leukemia cells than cells that do not express high levels of CD19 and consequently the targeted NPs would be expected to be less toxic to other cells in animals. A larger toxicity study is required to determine whether the targeted NPs are safer than free DOX.
ALL is an acute and an aggressive form of blood cancer. It progresses and disseminates rapidly throughout the body. Current therapies to combat ALL require the use of multiple drugs given in combination. In the study presented here, we used a single drug and were able to demonstrate efficacy with just one or two low-level doses of drug. It is also important to note that the limited amount of drug released by a controlled delivery system as described here is not sufficient to combat the rapidly progressing disease. However, low dose of available drug reduces the apparent systemic toxicity which is supported by the high level of agility observed in our CD19-DOX-NP treated mice.
In conclusion, results of this study demonstrate the potential for targeted NPs to selectively deliver drugs that can be effective against ALL. These encouraging results should lead to future studies that examine increased safety profiles of these drug formulations, particularly in relation to non-specific effects on other organs and tissues. Non-specific effects are of particular concern for pediatric ALL where non-targeted cytotoxic drugs can cause lifelong problems for treated patients.
Supplementary Material
Figure S1. A representative immunoblot used to quantify biotinylated anti-CD19Ab levels on NPs.
Figure S2. Treatment of REH cells with free DOX or CD19-DOX-NPs (≈100nM, 1μM or 10μM DOX) for 1h at 37°C. CD19-DOX-NPs did not affect the cell viability at 100nM DOX, but reduced the viability at higher concentrations of 1 and 10μM DOX.
Figure S3. Treatment of RS4;11 cells with free DOX or CD19-DOX-NPs (≈100nM, 1μM or 10μM DOX) for 1h at 37°C. CD19-DOX-NPs reduced the cell viability at all concentrations of DOX.
Figure S4. Treatment with free DOX and CD19-targeting achieves similar cell associated DOX (intracellular and cell surface bound, caDOX) levels in RS4;11 cells. RS4;11 caDOX levels on treatment with free DOX or CD19-DOX-NPs (≈100nM, 1μM or 10μM DOX) for 1h at 37°C.
Figure S5. CD19-targeting reduces the cell associated DOX (intracellular and cell surface bound, caDOX) levels in REH cells. REH caDOX levels on treatment with free DOX or CD19-DOX-NPs (≈100nM, 1μM or 10μM DOX) for 1h at 37°C.
Figure S6. CD19-DOX-NPs maintained high agility factor in leukemic mice. Agility Factor (normalized to saline) = no. of wheel rotations on day 15 ÷ no. of wheel rotations on day 1. CD19-DOX-NPs (≈2.5 mg/kg DOX) maintained a high agility factor in comparison with groups treated with saline, IgG-DOX-NPs and free DOX (≈2.5 mg/kg DOX). This indicates reduced apparent systemic toxicity in leukemic mice treated with CD19-DOX-NPs.
Acknowledgments
This work is supported by National Institutes of Health (P20GM103464, and P20 GM103446), the Sigma Xi Grants-in-Aid of Research Award (Vinu Krishnan), Alex's Lemonade Stand Foundation for Childhood Cancer Research's Pediatric Oncology Student Training Grant Award (Vinu Krishnan), Delaware Health Sciences Alliance, Andrew McDonough B + Foundation, Kids Runway for Research, Nemours Foundation, and funds from the University of Delaware. We thank Dr. Gwen Talham, Frank M. Warren and Ming A. Pham for help with the in vivo studies at the University of Delaware's Office of Laboratory Animal Medicine.
Footnotes
Supporting Information (SI): This material is available at free of charge via the Internet at http://pubs.acs.org.
References
- 1.Facts 2012 - The Leukemia & Lymphoma Society. The Leukemia & Lymphoma Society; [Google Scholar]
- 2.Stuart FA, Segal TY, Keady S. Adverse psychological effects of corticosteroids in children and adolescents. Arch Dis Child. 2005;90(5):500–6. doi: 10.1136/adc.2003.041541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Davis ME, Chen ZG, Shin DM. Nanoparticle therapeutics: an emerging treatment modality for cancer. Nat Rev Drug Discov. 2008;7(9):771–82. doi: 10.1038/nrd2614. [DOI] [PubMed] [Google Scholar]
- 4.Kamaly N, Xiao Z, Valencia PM, Radovic-Moreno AF, Farokhzad OC. Targeted polymeric therapeutic nanoparticles: design, development and clinical translation. Chem Soc Rev. 2012;41(7):2971–3010. doi: 10.1039/c2cs15344k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Krishnan V, Rajasekaran AK. Clinical Nanomedicine: a solution to the chemotherapy conundrum in pediatric leukemia therapy. Clin Pharmacol Ther. 2013 doi: 10.1038/clpt.2013.174. [DOI] [PubMed] [Google Scholar]
- 6.FDA approves Marqibo to treat rare type of leukemia. 2012 Aug 9; http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm315027.htm.
- 7.Uckun FM, Jaszcz W, Ambrus JL, Fauci AS, Gajl-Peczalska K, Song CW, Wick MR, Myers DE, Waddick K, Ledbetter JA. Detailed studies on expression and function of CD19 surface determinant by using B43 monoclonal antibody and the clinical potential of anti-CD19 immunotoxins. Blood. 1988;71(1):13–29. [PubMed] [Google Scholar]
- 8.Cooper LJ, Al-Kadhimi Z, DiGiusto D, Kalos M, Colcher D, Raubitschek A, Forman SJ, Jensen MC. Development and application of CD19-specific T cells for adoptive immunotherapy of B cell malignancies. Blood cells, molecules & diseases. 2004;33(1):83–9. doi: 10.1016/j.bcmd.2004.03.003. [DOI] [PubMed] [Google Scholar]
- 9.Tedder TF. CD19: a promising B cell target for rheumatoid arthritis. Nature reviews Rheumatology. 2009;5(10):572–7. doi: 10.1038/nrrheum.2009.184. [DOI] [PubMed] [Google Scholar]
- 10.Loken MR, Shah VO, Dattilio KL, Civin CI. Flow cytometric analysis of human bone marrow. II. Normal B lymphocyte development. Blood. 1987;70(5):1316–24. [PubMed] [Google Scholar]
- 11.Manzke O, Berthold F, Huebel K, Tesch H, Diehl V, Bohlen H. CD3xCD19 bispecific antibodies and CD28 bivalent antibodies enhance T-cell reactivity against autologous leukemic cells in pediatric B-ALL bone marrow. International journal of cancer Journal international du cancer. 1999;80(5):715–22. doi: 10.1002/(sici)1097-0215(19990301)80:5<715::aid-ijc15>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- 12.Szatrowski TP, Dodge RK, Reynolds C, Westbrook CA, Frankel SR, Sklar J, Stewart CC, Hurd DD, Kolitz JE, Velez-Garcia E, Stone RM, Bloomfield CD, Schiffer CA, Larson RA. Lineage specific treatment of adult patients with acute lymphoblastic leukemia in first remission with anti-B4-blocked ricin or high-dose cytarabine: Cancer and Leukemia Group B Study 9311. Cancer. 2003;97(6):1471–80. doi: 10.1002/cncr.11219. [DOI] [PubMed] [Google Scholar]
- 13.Mitchell P, Lee FT, Hall C, Rigopoulos A, Smyth FE, Hekman AM, van Schijndel GM, Powles R, Brechbiel MW, Scott AM. Targeting primary human Ph(+) B-cell precursor leukemia-engrafted SCID mice using radiolabeled anti-CD19 monoclonal antibodies. Journal of nuclear medicine : official publication, Society of Nuclear Medicine. 2003;44(7):1105–12. [PubMed] [Google Scholar]
- 14.Sapra P, Moase EH, Ma J, Allen TM. Improved therapeutic responses in a xenograft model of human B lymphoma (Namalwa) for liposomal vincristine versus liposomal doxorubicin targeted via anti-CD19 IgG2a or Fab' fragments. Clinical cancer research : an official journal of the American Association for Cancer Research. 2004;10(3):1100–11. doi: 10.1158/1078-0432.ccr-03-0041. [DOI] [PubMed] [Google Scholar]
- 15.Cheng WW, Allen TM. Targeted delivery of anti-CD19 liposomal doxorubicin in B-cell lymphoma: a comparison of whole monoclonal antibody, Fab' fragments and single chain Fv. Journal of controlled release : official journal of the Controlled Release Society. 2008;126(1):50–8. doi: 10.1016/j.jconrel.2007.11.005. [DOI] [PubMed] [Google Scholar]
- 16.FDA approves Blincyto to treat a rare form of acute lymphoblastic leukemia. 2014 Dec 3; http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm425549.htm.
- 17.Krishnan V, Xu X, Barwe SP, Yang X, Czymmek K, Waldman SA, Mason RW, Jia X, Rajasekaran AK. Dexamethasone-Loaded Block Copolymer Nanoparticles Induce Leukemia Cell Death and Enhances Therapeutic Efficacy: A Novel Application in Pediatric Nanomedicine. Mol Pharm. 2012 doi: 10.1021/mp300350e. doi:org/10.1021/mp300350e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lipshultz SE, Colan SD, Gelber RD, Perez-Atayde AR, Sallan SE, Sanders SP. Late cardiac effects of doxorubicin therapy for acute lymphoblastic leukemia in childhood. N Engl J Med. 1991;324(12):808–15. doi: 10.1056/NEJM199103213241205. [DOI] [PubMed] [Google Scholar]
- 19.Lipshultz SE, Lipsitz SR, Sallan SE, Dalton VM, Mone SM, Gelber RD, Colan SD. Chronic progressive cardiac dysfunction years after doxorubicin therapy for childhood acute lymphoblastic leukemia. J Clin Oncol. 2005;23(12):2629–36. doi: 10.1200/JCO.2005.12.121. [DOI] [PubMed] [Google Scholar]
- 20.Wang X, Gurski LA, Zhong S, Xu X, Pochan DJ, Farach-Carson MC, Jia X. Amphiphilic Block Co-polyesters Bearing Pendant Cyclic Ketal Groups as Nanocarriers for Controlled Release of Camptothecin. J Biomater Sci Polym Ed. 2010;22(10):1275–1298. doi: 10.1163/092050610X504260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Altreuter DH, Dordick JS, Clark DS. Nonaqueous biocatalytic synthesis of new cytotoxic doxorubicin derivatives: Exploiting unexpected differences in the regioselectivity of salt-activated and solubilized subtilisin. Journal of the American Chemical Society. 2002;124(9):1871–1876. doi: 10.1021/ja015977y. [DOI] [PubMed] [Google Scholar]
- 22.Xu X, Sabanayagam CR, Harrington DA, Farach-Carson MC, Jia X. A hydrogel-based tumor model for the evaluation of nanoparticle-based cancer therapeutics. Biomaterials. 2014;35(10):3319–30. doi: 10.1016/j.biomaterials.2013.12.080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Heuser JE, Anderson RG. Hypertonic media inhibit receptor-mediated endocytosis by blocking clathrin-coated pit formation. J Cell Biol. 1989;108(2):389–400. doi: 10.1083/jcb.108.2.389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Plummer EM, Manchester M. Endocytic uptake pathways utilized by CPMV nanoparticles. Molecular pharmaceutics. 2013;10(1):26–32. doi: 10.1021/mp300238w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Arnold RD, Slack JE, Straubinger RM. Quantification of Doxorubicin and metabolites in rat plasma and small volume tissue samples by liquid chromatography/electrospray tandem mass spectroscopy. Journal of chromatography B, Analytical technologies in the biomedical and life sciences. 2004;808(2):141–52. doi: 10.1016/j.jchromb.2004.04.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Benmerah A, Lamaze C. Clathrin-coated pits: vive la difference? Traffic. 2007;8(8):970–82. doi: 10.1111/j.1600-0854.2007.00585.x. [DOI] [PubMed] [Google Scholar]
- 27.Christiansen J, Rajasekaran AK. Biological impediments to monoclonal antibody-based cancer immunotherapy. Molecular cancer therapeutics. 2004;3(11):1493–501. [PubMed] [Google Scholar]
- 28.Yoo HS, Park TG. Biodegradable polymeric micelles composed of doxorubicin conjugated PLGA-PEG block copolymer. Journal of controlled release : official journal of the Controlled Release Society. 2001;70(1-2):63–70. doi: 10.1016/s0168-3659(00)00340-0. [DOI] [PubMed] [Google Scholar]
- 29.Shuai X, Merdan T, Schaper AK, Xi F, Kissel T. Core-cross-linked polymeric micelles as paclitaxel carriers. Bioconjugate chemistry. 2004;15(3):441–8. doi: 10.1021/bc034113u. [DOI] [PubMed] [Google Scholar]
- 30.Fahmy TM, Samstein RM, Harness CC, Mark Saltzman W. Surface modification of biodegradable polyesters with fatty acid conjugates for improved drug targeting. Biomaterials. 2005;26(28):5727–36. doi: 10.1016/j.biomaterials.2005.02.025. [DOI] [PubMed] [Google Scholar]
- 31.Xiang S, Tong H, Shi Q, Fernandes JC, Jin T, Dai K, Zhang X. Uptake mechanisms of non-viral gene delivery. Journal of controlled release : official journal of the Controlled Release Society. 2012;158(3):371–8. doi: 10.1016/j.jconrel.2011.09.093. [DOI] [PubMed] [Google Scholar]
- 32.Jiang W, Kim BY, Rutka JT, Chan WC. Nanoparticle-mediated cellular response is size-dependent. Nat Nanotechnol. 2008;3(3):145–50. doi: 10.1038/nnano.2008.30. [DOI] [PubMed] [Google Scholar]
- 33.Peer D, Karp JM, Hong S, Farokhzad OC, Margalit R, Langer R. Nanocarriers as an emerging platform for cancer therapy. Nat Nanotechnol. 2007;2(12):751–60. doi: 10.1038/nnano.2007.387. [DOI] [PubMed] [Google Scholar]
- 34.Petros RA, DeSimone JM. Strategies in the design of nanoparticles for therapeutic applications. Nature reviews Drug discovery. 2010;9(8):615–27. doi: 10.1038/nrd2591. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. A representative immunoblot used to quantify biotinylated anti-CD19Ab levels on NPs.
Figure S2. Treatment of REH cells with free DOX or CD19-DOX-NPs (≈100nM, 1μM or 10μM DOX) for 1h at 37°C. CD19-DOX-NPs did not affect the cell viability at 100nM DOX, but reduced the viability at higher concentrations of 1 and 10μM DOX.
Figure S3. Treatment of RS4;11 cells with free DOX or CD19-DOX-NPs (≈100nM, 1μM or 10μM DOX) for 1h at 37°C. CD19-DOX-NPs reduced the cell viability at all concentrations of DOX.
Figure S4. Treatment with free DOX and CD19-targeting achieves similar cell associated DOX (intracellular and cell surface bound, caDOX) levels in RS4;11 cells. RS4;11 caDOX levels on treatment with free DOX or CD19-DOX-NPs (≈100nM, 1μM or 10μM DOX) for 1h at 37°C.
Figure S5. CD19-targeting reduces the cell associated DOX (intracellular and cell surface bound, caDOX) levels in REH cells. REH caDOX levels on treatment with free DOX or CD19-DOX-NPs (≈100nM, 1μM or 10μM DOX) for 1h at 37°C.
Figure S6. CD19-DOX-NPs maintained high agility factor in leukemic mice. Agility Factor (normalized to saline) = no. of wheel rotations on day 15 ÷ no. of wheel rotations on day 1. CD19-DOX-NPs (≈2.5 mg/kg DOX) maintained a high agility factor in comparison with groups treated with saline, IgG-DOX-NPs and free DOX (≈2.5 mg/kg DOX). This indicates reduced apparent systemic toxicity in leukemic mice treated with CD19-DOX-NPs.