

Abstract
Human epidermal growth factor receptors (HER, also known as ErbB) drive cellular proliferation, pro-survival and stress responses by activating several downstream kinases, in particular ERK, p38, JNK (SAPK), the PI3K/AKT, as well as various transcriptional regulators such as STAT3. When co-expressed, the first three members of HER family (HER1-3) can form homo- and hetero-dimers, and there is considerable evidence suggesting that the receptor dimers differentially activate intracellular signaling pathways. To better understand the interactions in this system, we pursued multi-factorial experiments where HER dimerization patterns and signaling pathways were rationally perturbed. We measured the activation of HER1-3 receptors and of the sentinel signaling proteins ERK, AKT, p38, JNK, STAT3 as a function of time in a panel of human mammary epithelial (HME) cells expressing different levels of HER1-3 stimulated with various ligand combinations. We hypothesized that the HER dimerization pattern is a better predictor of downstream signaling than the total receptor activation levels. We validated this hypothesis using a combination of model-based analysis to quantify the HER dimerization patterns, and by clustering the activation data in multiple ways to confirm that the HER receptor dimer is a better predictor of the signaling through p38, ERK and AKT pathways than the total HER receptor expression and activation levels. We then pursued combinatorial inhibition studies to identify the causal regulatory interactions between sentinel signaling proteins. Quantitative analysis of the collected data using the Modular Response Analysis (MRA) and its Bayesian Variable Selection Algorithm (BVSA) version allowed us to obtain a consensus regulatory interaction model, which revealed that STAT3 occupies a central role in the crosstalk between the studied pathways in HME cells. Results of the BVSA/MRA and cluster analysis were in agreement with each other.
Keywords: ErbB/HER/RTK signaling, signaling pathway, crosstalk, proliferation, pro-survival, STAT3, MAPK, mathematical modeling, cluster analysis, modular response analysis
INTRODUCTION
Because of their fundamental regulatory role in physiological processes such as cell proliferation, survival, and migration,1 Human Epidermal growth factor Receptor (HER, also known as ErbB) signaling has been extensively studied over the years. HER receptors belong to the receptor tyrosine kinase superfamily, and the first three members of this receptor family (EGFR/HER1, HER2 and HER3) are often co-expressed, and they play important roles in carcinogenesis.2, 3 HER receptors are highly homologous and their activation occurs through similar biochemical steps, although the individual receptors possess certain distinct properties. For example, HER2 receptors have no known ligand, and HER3 lacks intrinsic tyrosine kinase activity.4, 5 Upon ligand binding, receptors undergo conformation changes that favor the formation or stabilization of receptor hetero- and homo-dimers,6, 7 and almost every possible pairwise combination of HER dimers has been reported.8 Dimer formation patterns are affected by ligand availability,9 receptor expression levels,10 and kinetic dimerization hierarchy.11–13 Each HER receptor appears to have a characteristic repertoire of adaptor proteins depending on its dimerization partner.14 Which receptor-adaptor protein complexes are formed determines differential downstream signal transduction, and can trigger diverse biological responses.15, 16 Thus, the types of receptor homo- and hetero-dimers that are formed, and the signal transduction pathways that are activated depend upon the specifics of the system and treatment conditions. This severely complicates investigation of the HER signaling pathways and the HER-initiated cellular responses.17–20
Biomedical and biomolecular studies have shown that inhibition of HER dimerization and HER-mediated signaling can be an extremely effective therapeutic strategy.21 The role of HER receptors seems to be different in different cells lines and under different conditions. In HER2 positive breast cancers, heterodimer interactions between HER2 and its partners are often constitutively activated and their disruption has proven to be an effective means for inhibiting HER2-mediated aberrant responses.1, 22 HER3 overexpression has also been shown to correlate with poor prognosis in epithelial cancers.23, 24 Activation of pro-survival responses through the HER3 pathway can also lead to drug resistance in cancer treatment.25, 26 Further, interactions between members of the HER family and signaling redundancies can also contribute to drug resistance when specific inhibitors are used against a single HER receptor and the importance of HER3-HER1 dimers in drug resistance is increasingly recognized.21, 25, 27 Thus, the use of combination therapeutics that target multiple members of the HER family of receptors is advocated as an effective treatment strategy for a variety of tumors.25, 26, 28, 29
Receptor tyrosine kinases drive cellular responses via the activation of several distinct intracellular signaling pathways.30, 31 HER receptors activate pathways of several downstream kinases, in particular ERK, p38, JNK (SAPK), the PI3K/AKT signaling pathways, as well as various transcriptional regulators such as STAT3.1, 15, 32–42 These are the prominent sentinel proteins of the cellular processes associated with cell proliferation, pro-survival and stress-response pathways of cells. These inter-related signaling pathways can be stimulated to varying degrees by different receptor-adaptor protein complexes.14 The abundance of these complexes in turn is a function of the relative abundances of the various HER dimer types in the cell.
Many stimuli, including growth factors, cytokines, transforming agents, and carcinogens activate the ERK (extracellular signal-regulated kinase) pathway. Sustained ERK activation can have significant functional consequences.43, 44 For example, constitutive or prolonged ERK activation through HER receptors contributes to speedy cell migration45 and enhances cell transformation and resistance to apoptosis 46. The serine/threonine kinase AKT promotes cellular survival by blocking apoptosis, which it achieves by binding and regulating many downstream effectors; e.g., NF-κB, Bcl-2 family proteins, and MDM2.47 AKT also induces protein synthesis and therefore plays a role in tissue growth. AKT has been implicated in many types of cancers because of its possible role in angiogenesis and tumor development.48, 49 PI3K is a well-studied regulator of cellular growth 50 and is known to be activated by HER3, especially in HER2 over-expressing cancer cells 51, 52. HER3 in particular has a unique ability to efficiently engage the pro-survival PI3K/AKT pathway.53–55
ERK, p38 and JNK are the three major subgroups of the MAPK family.56 In contrast to the ERKs, which are mainly mitogen activated proliferation/differentiation factors, JNK and p38 are stress-activated proteins involved in apoptosis/survival. The p38 kinase regulates EGFR down-regulation, survival, and cellular migration,38, 57, 58 and it can suppress cell proliferation by antagonizing the JNK/c-JUN pathway.39 Although all MAPKs can be activated by HER receptors in different cellular systems, p38 and JNK phosphorylation may also occur in an EGFR-independent manner.59 STAT signaling promotes RAS/RAF/ERK signaling indirectly.60 Activation of MAPKs leads to the activation of STAT-family transcription factors, which promotes their translocation to the nucleus32, 61 where they elevate the transcription of cell proliferation related genes 32, 60.
Upon HER receptor stimulated activation of cell proliferation, pro-survival and stress-response signaling pathways, cellular responses are regulated collectively by these prominent sentinel proteins.30, 31 Identification of how the regulatory interactions between sentinel proteins are affected by the unavoidable simultaneous activation of multiple signaling pathways, i.e., pathway crosstalk, has been very challenging and requires multi-factorial experiments where HER dimerization patterns and crosstalk between signaling pathways is altered. Towards this goal, we have measured HER1-3 activation, and the activation of key signaling proteins ERK, AKT, p38, JNK, STAT3 as a function of time in a panel of human mammary epithelial (HME) cells expressing different levels of HER1-3 stimulated with various ligand combinations, e.g., when the signaling pathways were activated differentially in cells with same cellular background but with varying HER expression levels. Conditions covered in these studies resulted in large differences in receptor dimerization,13 and in the activation levels of investigated signaling proteins. These time-series measurements were supplemented with combinatorial measurements where we determined how the inhibition of one of the sentinel proteins affected the activation levels of the other sentinels. This data was used to identify the regulatory interactions between the studied sentinel proteins using the modular response analysis method.
EXPERIMENTAL MATERIALS AND METHODS
General considerations
There is considerable evidence that the HER dimerization pattern is an important determinant of the consequences of HER signaling,62, 63 which suggests that activation of the downstream signaling pathways would be functions of receptor dimer formation and activation.18 In this regard, we hypothesized that the HER dimerization pattern is a better predictor of downstream signaling than total receptor activation levels. Validation of this hypothesis and quantification of the relative potency of the HER dimer types for activating distinct downstream pathways would enable rational perturbation of the signaling patterns within a cell.
Due to the redundancy in downstream pathway activation, and the extensive crosstalk between signaling pathways, establishing the relationship between HER dimers and signaling requires: 1) the ability to quantify the HER dimerization patterns, and 2) multi-factorial experiments where HER dimerization patterns and signaling crosstalk is altered. Direct isolation and measurement of the receptor dimers is challenging, and one is largely restricted to the measurement of the total receptor activation levels. However, as we have recently shown that the dimer activation levels (point #1 above) can be derived from receptor activation data using an integrated experimental-computational approach.13 To satisfy the second requirement, we measured HER1-3 activation, and the activation of key signaling proteins ERK, AKT, p38, JNK, STAT3 as a function of time in a panel of HME cells expressing different levels of HER1-3 stimulated with various ligand combinations. Conditions in these studies result in large differences in receptor dimerization,13 and in the activation levels of investigated signaling proteins (cf., Results section below). Ability to obtain internally consistent and complementary data sets in a panel of HME cells with the same cellular background is a novel aspect of our study and it was critical for deriving statistically significant regulatory interactions from the experimental data.
Cell lines
The HME cell line 184A1L5 used in this study was originally provided by Martha Stampfer (Lawrence Berkeley Laboratory, Berkeley, CA). It expresses approximately 200,000 molecules of EGFR/ErbB1/HER1.64, 65 Cells were maintained at 37°C in 5% CO2 air in DFCI-1 medium supplemented with 12.5 ng/ml EGF (PeproTech, Rocky Hill, NJ) as described previously.66 At about 70–80% cell confluency, DFCI-1 medium was replaced with bicarbonate-free DFHB minimal medium lacking EGF, plus 0.1% bovine serum albumin. Cells were then brought to quiescence for 12–18 hours before treatment. We note that, unlike many model cell lines with limited physiological responses to growth factors, HME cells are an excellent model system for studying the properties of the HER system because, like many epithelium derived cell types, HME cells require EGFR activation for proper proliferation and migration responses.67 The 184A1 strain of HME cells that was used to develop our clone library retains the EGFR-dependent regulatory machinery of the primary cell type from which it was derived.68 This parental cell line expresses high levels of EGFR and relatively low copy numbers of HER2 and HER3. It is labeled the HER2−/3− cell line, and it was used to create the HER2 and HER3 overexpressing cell lines by retroviral transduction.65
Briefly, the retroviral MFG vector containing HER2 was constructed and transfected into the ψCRIP packaging cell line.69–71 The transfected clones with highest level of HER2 expression were expanded to produce viral supernatant. This supernatant was then used to infect the HME cells. Individual clones of retrovirus transduced HME cells were isolated using cloning rings. The degree of HER2 overexpression was determined by immunofluorescence, flow cytometry, and equilibrium-binding studies using labeled Fab fragments. HER3 expressing cells were derived in a similar retrovirus-based strategy. Retroviral pBM-IRESpuro vector containing HER3 and the puromycin resistance gene was transfected into the Phoenix packaging cells.72 Viral supernatant was collected from transiently transfected Phoenix cells. The supernatant was then used to infect the parental HME cells and the HER2 expressing cells (discussed in the previous paragraph) to create the HER3 expressing and HER2-HER3 co-expressing cell lines, respectively (Table 1). Growth medium for the HER2 and/or HER3 expressing cells was the same as that of the parental HME cells except for addition of the antibiotic G418 (250 μg/ml; Invitrogen, Carlsbad, CA) and/or puromycin (2 μg/ml; Sigma, St. Louis, MO) to ensure selection for HER2 and HER3, respectively. Utilized cell lines are labeled and referred to as R2a3b where a or b=+/− indicates that the cell line expresses/do not express (i.e., very low endogenous expression) the type 2 and type 3 HER receptors, respectively. We note that these cell lines were labeled with their clone names in our earlier studies13, 65 as R2− 3− (parental, or Par for short), R2+3− (24H), R2− 3+ (B5), and R2+3+ (D20) and these clone names are used interchangeably in this report as well.
TABLE 1.
Human Mammary Epithelial Cell Library
| Cell Lines * | Receptor Abundance (1,000 receptors/cell) | ||||
|---|---|---|---|---|---|
| Label | Clone tag | Description (HER1/2/3) | EGFR | HER2 | HER3 |
| HER2−3− | Par | Parental, very low HER2/3 (+/−/−) | 200 | 30 | 2 |
| HER2+3− or HER2+ | 24H | HER2 expressing (+/++/−) | 137 | 600 | 2 |
| HER2−3+ or HER3+ | B5 | HER3 expressing (+/−/+) | 189 | 30 | 28 |
| HER2+3+ | D20 | HER2/3 expressing (+/++/+) | 86 | 644 | 29 |
Cell labels and clone tags are used interchangeably in the text.
Signal initiation
To activate the cells, ligands were added to the culture medium, and cells were incubated at 37°C for fixed amounts of time. Stimulation of EGFR/HER1 with 12 ng/ml EGF, of HER3 with 40 ng/ml HRG, as well as the co-stimulation of both receptors via simultaneous addition of both ligands was studied. For simplicity, in the remainder of the paper, these ligand treatment conditions will be labeled and referred to with +E, +H, and +EH where E and H stand for EGF and HRG, respectively. No-ligand measurements provided the negative control as 0 min time point. We note that same ligand doses were used in the ELISA, Western blot and inhibition experiments described below to obtain consistent data sets.
Receptor activation measurements using ELISA
Phosphorylation levels of the receptors were measured at 5, 10, 20, 30, 60, 90 and 120 min after ligand stimulation. Data for the phosphorylation levels of HER1-3 receptors were collected by us earlier.13, 65 These measurements used the R&D DuoSet IC ELISA kits (R&D Systems Inc., Minneapolis, MN), and detailed descriptions can be found in our earlier studies.13, 17, 65 Briefly, cells were washed 3X with ice cold PBS and incubated at room temperature for one minute to allow surface receptor dephosphorylation. After another round of cold PBS washing, cells were solubilized with ice cold lysis buffer (1% NP-40, 20 mM pH 8.0 Tris buffer, 137 mM NaCl, 10% glycerol, 2 mM EDTA, supplemented with 1 mM heat activated sodium orthovanadate and 1% protease inhibitor cocktail III; Calbiochem, La Jolla, CA) for 20 min. Lysates were centrifuged at 13,000 rpm for 10 min at 4°C, and the supernatants were transferred into fresh microtubes. Obtained cell lysates were either analyzed immediately or stored at a −80°C freezer until needed.
ERK and AKT activation measurements using ELISA
Data for the phosphorylation levels of the kinases ERK and AKT were quantified in ELISA experiments by us earlier and has already been communicated.18 Briefly, R&D DuoSet IC ELISA kits were used and the manufacture’s protocol was followed in these measurements. Before each ELISA assay, protein concentration of cell lysate was measured using the Bicinchoninic Acid protein quantitation kit (Sigma, St. Louis, MO). ERK and AKT phosphorylation levels were measured at 5, 10, 20, 30, 45, 60, 90 and 120 min after ligand stimulation. Further details of these experiments can be found in Zhang et al.18
Western Blot analysis of p38, JNK and STAT3
Cells were grown to near confluency and were starved by replacing regular DFCI-1 culture medium with DFCI-1 medium lacking all supplements but 0.1% bovine serum albumin for 12–18 hours. Cells were stimulated at 37°C by adding the appropriate concentration of ligands for the desired durations. Afterwards, cells were washed twice with cold PBS and cell lysates were collected with a scraper. Cells were lysed using RIPA buffer containing protein protease and phosphatase inhibitors on ice for 10 to 30 min. Lysates were centrifuged at 12,000 rpm for 10 min at 4°C, and only the supernatants were kept in new microtubes. Cell lysates were either analyzed immediately or stored at −80°C until needed. Total protein concentration of cell lysate was determined by the Bicinchoninic Acid protein quantitation kit (Sigma, St. Louis, MO), and same total protein amount (25ug) was loaded onto the SDS-PAGE gel. NuPAGE Bis-Tris precast 4%–12% gradient gels with 15 wells were used. Proteins were transferred from SDS-PAGE gel to PVDF membrane using XCell SureLock™ Mini-Cell Electrophoresis System (Invitrogen). PVDF membrane were developed using ECL reagents and Western blot images were captured by Lumi-Imager F1 device and analyzed using the LumiAnalyst 3.1 software.
JNK, STAT3, and p38 phosphorylation levels were measured at 0, 15, 30, 60, 120, 240 and 480 min after ligand stimulation. For each experiment, at least two independent measurements were performed for every treatment condition. To be able to scale the data collected in different gels, we allocated two of the lanes to “standard” measurements, which allowed us to normalize the measurements across gels. As having common positive controls is necessary for reliable quantification, results for the stimulation with both EGF and HRG ligands at 15 min time point for the parental (R2−3−) and B5 (R2−3+) cells were measured in each gel and then used as the standard data for the gel-to-gel normalization. Additionally, to further improve the comparison of the results for the four cell-lines, each gel was designed to measure the activation of two cell lines. This allowed us to do quality check across experimental time series datasets.
Inhibition studies
To further investigate the crosstalk between the signaling pathways, we selectively inhibited the studied proteins one at a time and measured how the particular inhibitions affect the activities of the studied sentinels. Cell lysates for inhibitor-treated cell assays were collected as described above for the no-inhibition studies except that during cell starvation inhibitors were added to the medium and incubated for certain time. U0126 monoethanolate (ERK1/2 inhibitor), SB 203580 (p38 MAPK inhibitor), and AKT inhibitor VIII were added to starvation medium 2 hours before ligand stimulation all at 20 uM concentration. LY294002 (hydrochloride; PI3K inhibitor) was added to starvation medium 1 hour before ligand stimulation at a concentration of 10 uM. JNK inhibitor SP600125 and STAT3 inhibitor VIII were added to starvation medium 18 hours before ligand stimulation at 40 uM concentration. Phosphorylation levels of the studied proteins were measured 15 min after ligand stimulation in these inhibition studies using Western blots for p38, JNK and STAT3, and ELISAs for ERK and AKT as described in the previous sections.
Reagents
Epidermal growth factor (EGF, human recombinant) and heregulin-β1 (HRG, human recombinant) were purchased from Peprotech (Rocky Hill, NJ). Primary Western blot antibodies against total p38 MAPK (#9212), SAPK/JNK (#9258), and STAT3 (#9139), and against phosphorylated p38 MAPK (Thr180, Tyr182, #9215), SAPK/JNK (Thr183/Tyr185, #9255), and STAT3 (Tyr705, #9145) were purchased from Cell Signaling Technology (Danvers, MA). Primary antibody for β-actin (A2228) was purchased from Sigma. Anti-mouse IgG-peroxidase secondary antibody (A4416) was purchased from Sigma (St. Louis, MO), and anti-rabbit IgG HRP-linked secondary antibody (7074) was purchased from Cell Signaling Technology (Danvers, MA). U0126 monoethanolate (ERK1/2 inhibitor), SB 203850 (p38 MAPK inhibitor), AKT inhibitor VIII, and STAT3 inhibitor VIII were purchased from Calbiochem (EMD Millipore, Billerica, MA). PI3K inhibitor LY294002 (L9908) hydrochloride was purchased from Sigma (St. Louis, MO). JNK inhibitor SP600125 was purchased from LC Laboratories (Woburn, MA). ELISA assay kits were purchased from R&D Systems Inc. (Minneapolis, MN). G418 was purchased from Invitrogen (Carlsbad, CA), and puromycin was purchased from Sigma (St. Louis, MO). Cell culture supplements, SDS-PAGE gels, and PVDF membranes were purchased from Invitrogen (Carlsbad, CA). The high sensitivity peroxidase-based Electrochemiluminescence (ECL) immunoassay substrate for Western blot was purchased from Thermo Scientific (Pittsburg, PA). All the other reagents were purchased from Sigma (St. Louis, MO) unless otherwise indicated.
COMPUTATIONAL METHODS
HER1-3 dimerization
One of the aims of this study is to show that cellular signaling initiated by the HER receptors can be better characterized by the dimerization patterns between the HER family members. In other words, not the receptor phosphorylation per se but which receptor dimers are formed and activated is the determinant of the information flow through HER signaling pathways. This requires the quantification of the HER receptor dimer activation profiles. Although this information can in principle be obtained in co-IP type experiments, such experimental studies are technically challenging and do not provide the level of quantitation required for our analysis. We therefore measured the receptor phosphorylation instead, and used a model-based analysis to extract the HER receptor dimerization profiles from the collected experimental data. Our modeling approach is described in detail elsewhere.13, 73 Therefore, it will only be briefly summarized here.
Model used to analyze the receptor activation considers ligand binding to receptors, receptor dimerization and trafficking through endocytosis and degradation. All possible dimer combinations are included except HER3-HER3 pair, which is excluded because this dimer has very low kinase activity and is not formed at significant levels.5, 26, 74–78 Although HER3 has been shown to have some kinase activity, as a kinase it is ~1000 times less efficient than EGFR/HER1.79 Therefore, when other HER types are present, HER3 can be considered as kinase deficient for practical reasons. Additionally, even at its highest expression, HER3 levels in the cells is about 30,000 receptors, which is about an order of magnitude less than HER1 and HER2 in our HME cells (Table 1) as well as in many other epithelial cell types.13 Thus, even if they are formed, the number of HER3-HER3 receptor pairs will be low due to the availability of large number of HER1 or HER2 receptors for partnering and the contribution of HER3-HER3 pair to HER3 phosphorylation would be minute because of its low kinase activity. For these reasons, HER3-HER3 pair was not included in our model to avoid addition of extra parameters unnecessarily, i.e., to avoid parameter overestimation.
Model parameters were fitted to the experimental data to obtain its kinetic parameters. Parameter sets that explain the data the best were then used to compute the receptor dimerization and activation profiles. One key feature of our model is that total phosphorylation of an HER receptor type is expressed as a sum of the contributions from all possible dimer types. For example, phosphorylation of EGFR/HER1, pR1, can be expressed as the sum of contribution of phosphorylated HER1 to cellular signaling when it is in dimer complexes with HER1 (pR1r1), with HER2 (pR1r2), and with HER3 (pR1r3): pR1=pR1r1+pR1r2+pR1r3. We note that our analysis assumes that the phosphorylated forms of the proteins are active. Therefore, in the remainder of the paper, we use the terms “active” and “phosphorylated” receptors interchangeably.
Correlation Analysis
Above described experimental studies provided us the phosphorylation data as time series. Our analysis combined our earlier data (for HER1-3, ERK and AKT) with the new data (for p38, JNK, STAT3) that were collected as part of this study. Because of the limited number of lanes in SDS-PAGE gels, we were not able to measure the protein activation levels at all the time points that were used in our earlier ELISA experiments (cf. Experimental Methods section). We therefore used the 15 min time point in the Western Blot experiments and interpolated the ERK and AKT ELISA results for this time point by averaging their measured activation levels at 10 and 20 minutes.
A set of 12 time series data were collected for every protein; 3 ligand treatment conditions (+E, +H, and +EH) in 4 cell lines (R2−3−, R2−3+, R2+3−, and R2+3+). Obtained time series data was used to identify the correlations between the activation patterns of the proteins at different levels, and the cluster analysis was used to group the proteins. The analysis was performed using MATLAB’s clustering software. In the cluster analysis, 1−|Corr(X,Y)| values were used as the distance measure where Corr(X,Y) is the correlation between protein X and protein Y over the included conditions. We preferred to use the absolute value of the correlation in the distance measure to allow for the possibility that the protein activation patterns may be negatively correlated, which too imply functional relationships. However, significant correlations in the investigated data sets were all positive so reached conclusions would have been the same if a 1−Corr(X,Y) distance criteria was used. Hierarchical cluster analysis utilized the average distance criteria in assigning the clusters and the connection values. Corr computation only included the common time points, and correlations were calculated using the protein activation values at the same time points.
Availability of data for different cell lines and different ligand treatment conditions allowed us to pursue the cluster analysis at multiple levels: First, we combined the data for four cell lines and three ligand treatment cases and created an “all-data” set for the proteins. This set made it possible to investigate the overall correlations between the investigated proteins at a higher level. We then divided up the data to perform the analysis on a “cell-type” and “treatment-type” manner. In the former set, data about a protein for all ligand treatment cases were combined for each cell line to form cell-line based datasets. Similarly, in the latter set, data for the four cell lines for a particular ligand treatment condition was combined.
Additionally, the analysis was pursued by investigating the correlation between the downstream sentinels and receptors by 1) using the activation data for the receptors directly (i.e., without considering dimers), and 2) using the dimer contributions to the receptor phosphorylations. Comparison of these two approaches allowed us to validate our hypothesis. We note that, as it consists of closely related cell types and overlapping time spans, data collected and analyzed in this study was internally cohesive, and this has allowed for a systematic investigation.
Modular Response Analysis
Correlation analysis described above does not provide information about the causal regulatory interactions between studied proteins. Modular Response Analysis 80–82 (MRA) is a powerful method to reverse engineer a system to infer the existing regulatory interactions and their causality in a quantitative manner. In MRA, one starts with perturbing the system to measure how varying a module impacts the response of the other modules in the system. Observed variations are then used to derive the local response matrix elements rij, which quantifies the sensitivity of module i (i.e., activation of sentinel protein i in our case) to changes in module j of the network system, rij=∂xi/∂xj provided that activity of all other nodes are kept constant where xi denotes the activity of module i.81, 82 When multiple interactions may be involved, changes in the activities can be characterized with the global response matrix with elements Rij, which corresponds to the change in component i, xi, in response to a perturbation in component j, Pj, as Rij=∂lnxi/∂lnPj. For small enough variations Rij may be approximated as Rij≅2(xi(j) − xi(0))/(xi(j) + xi(0)) where xi(j) and xi(0) are the activity levels of module i after and before the perturbation to module j.81
In its original formulation, MRA was developed to estimate the local matrix elements that represent the strength of the involved interactions between modules. The local response matrix rij is calculated from the measured Rij by solving Σk rikRkj = 0, where the sum is over the nodes k (k ≠ i) that were perturbed in the experiments and rii=−1.81, 82 Typically the total least squares estimation is used in Monte Carlo based simulations to estimate the coefficients.83 Utilizing this approach, we have computed the estimated probability distributions of the response coefficients by forming random realizations of the node activities that were drawn from a normal distribution with a mean equal to those of the measured values. Standard deviation in the distribution was assumed to be 30% of the mean for each of the measured Rij values. This deviation was larger than the deviations observed in our Western blot experiments but we preferred to use a conservative standard deviation value in our analysis. Response matrices rij were calculated for 107 realizations of the randomly sampled data sets, and statistical distribution data obtained for the resulting local response coefficients were used to calculate the mean (μij=<rij>), standard deviation (σij), and coefficient of variation (CV=σij/μij) of the computed response coefficients. These MRA simulations were performed using the in-house written MATLAB codes.
Recently, Santra et al. have developed a variant of MRA, the Bayesian Variable Selection Algorithm (BVSA) that integrates Bayesian selection into MRA to infer regulatory interactions in biochemical networks.84 In BVSA, the constitutive MRA equation is changed to ΣkAikrikRkj=0, where the added variable Aij represents if direct interaction between modules i and j are present (Aij=1) or absent (Aij=0). For noisy datasets, which is the usual case in biological experiments, the global responses Rij contain uncertainties and the above equality usually does not hold exactly. The BVSA algorithm estimates the probability of module i being directly influenced by module j without having to estimate the connection coefficients as in the MRA algorithm. As a statistical inference algorithm, BVSA uses Bayesian statistics: Starting with a prior distribution, distributions are updated based on the experimental data using Bayes’ theorem to obtain the posterior distributions for the probabilities of direct connections between network modules. Sandra et al. utilized Markov Chain Monte Carlo sampling to obtain the posterior distributions for Aij, and decided on the inferred network topology by defining a threshold equal to the average posterior edge probability.84 We have used the Gibbs sampler MATLAB program provided as Supplementary Material to their publication in our simulations. Simulations were run for 1000 Gibbs scans (parameter: noit) and 5000 iterations (parameter: times). 50% of the early samples were assumed to be burn-ins and were not considered in the calculation of the posterior edge probabilities. Prior and posterior distributions were computed using the default values for the other parameters of the program.
In this study, we have combined these two algorithms in a two-stage approach to first determine which regulatory interaction links are likely to exist and then to determine the strength and type (activation/repression) of interactions. First, we have used BVSA to identify which interaction links may be present in the investigated biological network, i.e., find which Aij are not zero. In the second MRA stage, we have set the local response matrix rij elements corresponding to the interactions that were found to be non-existing in the first round to zero and computed the values of rij for the interactions that were found to exist in our network. The second stage allowed us to rank order the strengths of the interactions and to characterize if an interaction is activation or repression relation.
Since we have obtained consistent datasets in four cell lines with same cellular background, we also investigated if the detected interactions depended on the expression of either HER2 or HER3 receptors. This was achieved by grouping the perturbation response data in three different ways. First, we defined an “all dataset” by considering all four cell lines and all three ligand-treatment cases. For this set Rij data consisted of 12 separate conditions in which each sentinel protein was inhibited as perturbation for each condition and the activity (phosphorylation) changes of all proteins, i.e., xi(j), were measured to determine the Rij values. We also defined “HER2 and HER3 specific datasets” by comparing the responses in our cell lines. Cell lines in our library (Table 1) can be contrasted to each other to signify a particular HER receptor: For example, the principal difference between the R2−3− (Par) and R2+3− (24H) and between the R2−3+ (B5) and R2+3+ (D20) cell lines is the HER2 expression when HER1/EGFR and HER3 levels are roughly the same. Therefore, to form a “HER2 specific perturbation response dataset” from our measurements, we computed the differences between the measured responses in pairs of cell lines. I.e., the response matrix for HER2 specific data set was obtained by computing the Rij(R2+3−) − Rij (R2−3−) and Rij(R2+3+) − Rij(R2−3+) difference for every ligand combination. This resulted in a dataset for 6 conditions (2 cellular x 3 ligand treatments) where the main difference was the HER2 expression. “HER3 specific perturbation response dataset” for 6 different conditions was formed in a similar manner by computing Rij(R2−3+) − Rij(R2−3−) and Rij(R2+3+) − Rij(R2+3−) differences for every ligand combination where the main difference was the HER3 expression.
RESULTS
Correlations of JNK and STAT3 with other proteins
Our cluster analysis indicated that JNK and STAT3 activation patterns only weakly correlated to HER, p38, ERK, and AKT activations (Figs. 1A and 2A). Both STAT3 and JNK were more associated with the p38/ERK/HER1 cluster than the AKT/HER3 cluster (Figs. 1A and 2A). However, the linkage distances were large indicating that auxiliary JNK and STAT3 pathways have rather weak correlative associations with the p38, AKT, and ERK pathways. Correlation between JNK and STAT3 and other sentinels were also weak when the “treatment-type” and “cell-type” based datasets (cf., Computational Methods section) were used in the cluster analysis (data not shown). Modular response analysis results discussed below further indicated that the activation patterns of STAT3 and JNK were complex and they may be differentially regulated by both ERK and AKT pathways. We therefore exclude JNK and STAT3 from the detailed discussion of the activation pattern correlations, and discuss the involvement of JNK and STAT3 in the regulations of other proteins later below.
Figure 1.
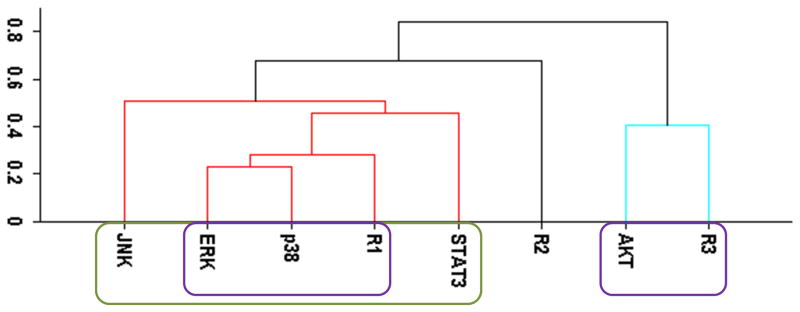
Cluster analysis of the time-dependent activation patterns of the investigated sentinel proteins and the HER1-3 receptors: (A) Analysis using the all data set, i.e., data combined for 4 cell lines and 3 ligand-treatments; (B) Ligand-treatment based analysis using the data grouped for 4 cell lines for EGF (E), HRG (H), and both ligands (EH) treatment conditions; and (C) Cell-type based analysis using the data grouped for the 3 ligand treatments for each cell line, i.e., for the cells R2−3− (− −), R2+3− (+−), R2−3+ (−+), and R2+3+ (++). The value of y-axis presents the score of 1−|Corr| where Corr is the correlation between the activities of the entities as discussed in the text.
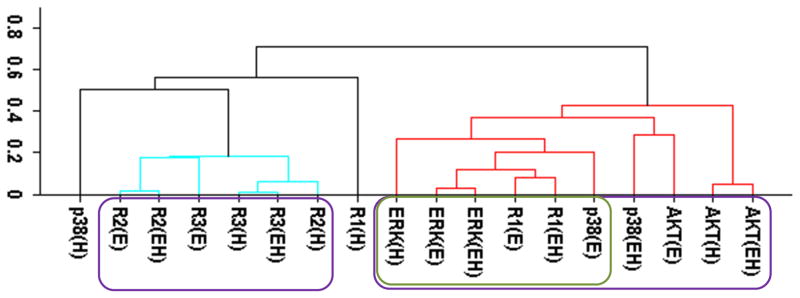
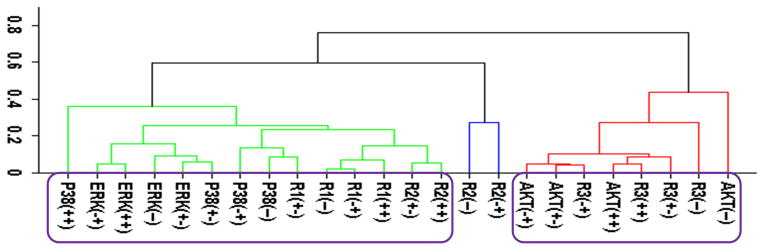
Figure 2.

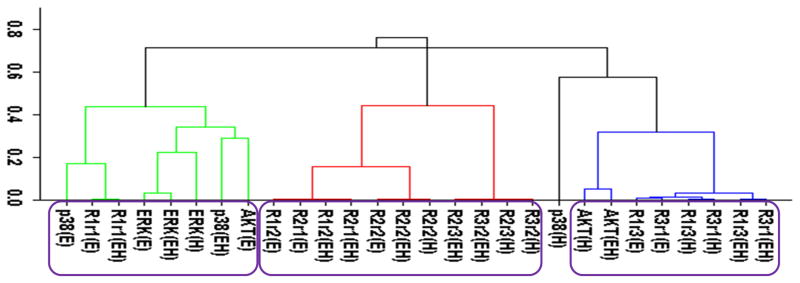
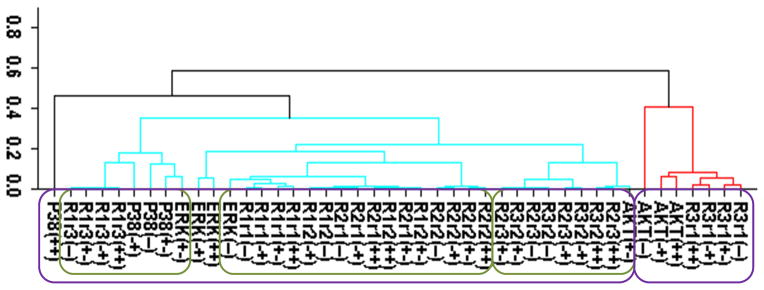
Cluster analysis of the time-dependent activation patterns of the investigated sentinel proteins and the HER receptor dimers. (A) Analysis using the all data set, i.e., data combined for 4 cell lines and 3 ligand-treatments; (B) Ligand-treatment based analysis using the data grouped for 4 cell lines for EGF (E), HRG (H), and both ligands (EH) treatment conditions; and (C) Cell-type based analysis using the data grouped for the 3 ligand treatments for each cell line, i.e., for the cells R2−3− (− −), R2+3− (+−), R2−3+ (−+), and R2+3+ (++). The value of y-axis presents the score of 1−|Corr| where Corr is the correlation between the activities of the entities as discussed in the text. In the plots, Rarb indicates the activation of HERa receptors in HERa:HERb dimer complexes.
Correlation analysis without consideration of receptor dimers
We first analyzed the data using the receptor phosphorylation information without considering receptor dimers. In this “receptor based analysis”, we address the question of whether ERK, AKT and p38 activation can be explained with the total HER receptor phosphorylation levels. This is the typical approach used in a large majority of studies in the literature.
Figure 1 reports the clusters when analysis was pursued using the all, treatment-type, and cell-type based datasets. All data set results (Fig. 1A) showed nicely clustered correlations between i) AKT and HER3, and ii) p38, ERK and HER1/EGFR. Even though HER2 is believed to be the mediator of HER3 signaling and its tumorigenic potency,1, 10 interestingly, its activation did not correlate well with the other receptors and sentinel proteins. Ability to partition the data to cell-type and treatment-type subsets allowed us to investigate the dependence of these high level correlations on receptor expression and treatment conditions. In the analysis based on ligand-treatment (Fig. 1B), AKT correlated more with HER1, ERK and p38 instead, while HER2 and HER3 clustered together. Whereas, the correlation between AKT and HER3 still held on a cell-type basis (Fig. 1C) but the tight grouping between p38, ERK and HER1 seemed to disintegrate at the cell-type level and HER1 correlated more with HER2.
Even though it may capture and reveal the correlations between EGFR/HER receptor activation and their signaling pathways, these results clearly illustrate that the interpretation of the data can depend on the way the receptor based analysis is performed. Most alarmingly, even though the analyzed data set was internally consistent, different analyses were found to lead to conflicting conclusions.
Correlation analysis using receptor dimers
We have repeated the cluster analysis using HER dimer activation levels instead of the experimentally measured receptor phosphorylation data. Contributions of dimer types to the overall receptor phosphorylations were inferred from the experimental data in a model-centric analysis (cf., Computational Methods section).
Figure 2 reports the clustering results for the receptor dimer-based approach. All data set results (Fig. 2A) paralleled the results in Fig. 1A: AKT clustered with HER3, and p38, ERK and HER1 clustered together. Figure 2A provides further details about which dimers participate in the correlations: AKT was most correlated with the HER3 signaling from the HER3:HER1 (R3r1) dimers. In contrast, p38 and ERK were correlated the most with HER1 signaling from its homo-dimers. As in the receptor based analysis discussed above, obtained clusters showed that activation patterns of HER2 correlated poorly with the activation patterns of the sentinel proteins for the signaling pathways. Figures 2B and 2C report the results when the cluster analysis was pursued using the ligand treatment-type and cell-type data subsets, respectively. Unlike the receptor-based analysis reported in the previous subsection, where different analyses led to conflicting correlations, ligand treatment type (Fig. 2B) and cell-type (Fig. 2C) dependent clustering agreed to a large degree with the results of the analysis with all data set. Most notably: i) AKT activation correlated the most with HER3 signaling from HER3:HER1 dimers in both the ligand treatment- and cell-type dependent analyses (Fig. 2B,C). Ligand treatment-based analysis indicated additional correlation between AKT and HER1 signaling from HER1:HER3 dimers. Interestingly, AKT activation when the receptors were stimulated with EGF better correlates with p38/ERK/HER1 activation (AKT(E) in Fig. 2B; cluster on the left) while HRG stimulation by itself, AKT(H), or together with EGF, AKT(EH), leads to a better correlation with signaling from the HER3:HER1 dimer (Fig. 2B; cluster on the right). This could be a reflection of the known multi-functionality of the AKT pathway where its contribution to cell signaling depends on the received cues.85 ii) In agreement with the all-data set results, analyses using data subsets did not indicate any strong correlation between HER2 signaling and sentinel proteins. iii) In the cell-line dependent analysis (Fig. 2C), p38 showed a stronger correlation with HER1 signaling from HER1:HER3 dimer and ERK was better correlated with HER1 signaling from HER1:HER1 homo- and HER1:HER2 hetero-dimers. However, p38 and ERK clusters merged at a score ~0.35, i.e., a correlation of 0.65, which was still a significant correlation. Thus, these results were in good agreement with the higher level analysis results (Fig. 2A). A notable difference in the analyses results for the p38/ERK/HER1 cluster was that HER2 containing dimers became part of this cluster in cell-line based analysis (Fig. 2C).
Modular response analysis of the inhibition studies
Investigation of the correlations between protein activation levels can reveal the associations between signaling pathways but they do not provide direct information regarding causal effects. We have therefore also pursued a systematic study where we have inhibited one of the proteins using specific inhibitors and measured the activation levels of the other studied proteins. This data (Supplementary Figures 1–6) would indicate the impact of the inhibition of an “effector”-protein on its “target”-protein. We have analyzed the obtained data using modular response analysis to infer a model for the interactions between ERK, AKT, PI3K, p38, JNK and STAT3 in our HME cells.
Our quantitative systems approach does not focus on a particular response condition but uses the general trends observed across all of the studied conditions to determine robust crosstalk interactions to obtain a consensus interaction model (Figure 3). A unique aspect of our study is that ability to obtain consistent datasets in four cell lines with same cellular background but different HER2/3 expression and ability to differentially activate particular HER receptor types using different ligand regimens allowed us to infer the interaction model in a receptor type specific manner. This was achieved by grouping the perturbation response data in three different ways, as described in the Computational Methods section.
Figure 3.

Consensus regulatory interaction diagram between the studied sentinel proteins as derived from the inhibition experiments described in the text. Arrows with a pointed end indicate activation interaction and with a blunt end indicate repression interaction. Tags next to the arrows indicate if the interaction was observed regardless of HER2 or HER3 expression (all); or particularly in HER2 (R2) or HER3 (R3) expressing cells. Further details are explained in the main text.
The probabilities of the regulatory reaction interactions as determined in the BVSA analysis are tabulated in Supplementary Table 1. To decide on the regulatory interactions, Sandra et al. used the mean of the computed response matrix element values as the threshold. Mean value of the three datasets μr was 0.34 in our case with a standard deviation σr of 0.27, which is quite lower than a mean of 0.5 that would be for random interactions. We have therefore classified the response matrix elements into 3 classes (Supplementary Table 1): Interactions with r values that are i) r < μr, ii) μr < r < μr+σr, and iii) r > μr+σr. Interactions in the first category are unlikely to be present and the ones in the last category are most likely to be present in our system. Interactions with r probabilities in the intermediate second category are probable but they are hard to estimate with our data and their prediction is uncertain.
We have then pursued MRA in the second stage of our analysis (Supplementary Table 2). We have first computed the response matrix element values by including all of the possible links in the network, i.e., all 5*4=20 binary possibilities were included. Second, MRA was pursued by including the interactions detected in the BVSA (Supplementary Table 2). Further details of this analysis are described in the Methods section above.
The consensus model shown in Figure 3 was obtained by combining the BVSA and MRA results, and it revealed that STAT3 occupies a central role in the crosstalk between studied proteins. Some of the interactions were consistently observed across different analysis. Most interestingly, as we have expected, our analysis has established that some of the regulatory interactions were more apparent in HER2 or HER3 expressing cell lines. We next discuss the combined analysis results for the identified interactions (Figure 3):
Interaction between AKT and STAT3
BVSA analysis (Supplementary Table 1) indicated that AKT is regulated by STAT3 and this regulation was enhanced with HER3 expression. MRA computations (Supplementary Table 2) supported this result and further revealed that AKT is activated by STAT3 and this activation was more pertinent in HER3 expressing cells. AKT in turn was found to activate STAT3 in all cases strongly (Supplementary Tables 1 and 2).
Interactions between ERK, STAT3 and JNK are bi-directional and HER2 dependent
BVSA analysis has indicated that ERK, STAT3 and JNK interact with each other in a bi-directional manner (Supplementary Table 1). MRA computations (Supplementary Table 2) supported this result and further revealed that ERK-JNK and STAT3-JNK are activating interactions while ERK and STAT3 repress each other. All of these interactions were predicted to be more dominant when HER2 is expressed with the exceptions that STAT3 was found to be activated by JNK in all cases and repressed by ERK when either HER2 or HER3 is expressed. Based on the derived interaction strengths, mutually repressive interaction between STAT3 and ERK is the most questionable of all the binary interactions between sentinels. This was reflected as large standard deviations in the interaction values derived for the regulation of ERK (Supplementary Table 2).
Interactions between p38, STAT3 and JNK are triangular and HER receptor expression dependent
Similar to the ERK-STAT3-JNK case, p38 was found to interact with STAT3 and JNK in a triangular manner. BVSA analysis indicated that these interactions may be HER expression dependent (Supplementary Table 1) and its findings were supported by the MRA analysis (Supplementary Table 2). STAT3-JNK interaction has been discussed in the previous paragraph. STAT3 and p38 were found to mutually repress each other in all cells (Supplementary Tables 1 and 2). JNK and p38 interaction was mutually activating in HER3 expressing cells (Supplementary Tables 1 and 2).
Comparison of the consensus model with cluster analysis results
The obtained consensus model can be compared and contrasted to the cluster analysis results to retrospectively understand the sources of the observed signaling correlations:
ERK and p38 correlation is indirect
Cluster analysis of the phosphorylation patterns discussed above indicated that ERK and p38 activations were closely correlated. Interestingly, modular response analysis predicted that there was no apparent direct impact of inhibiting ERK or p38 on the activation level of the companion protein. Derived model however illustrated that p38 and ERK activities are coordinated through STAT3 and JNK. Cluster analysis indicated that p38/ERK/HER1 activation correlated better with the HER2 containing dimers (Figure 2C). Obtained consensus model has validated this finding by showing that the triangulated interactions between ERK/JNK/STAT3 and JNK/STAT3/p38 are HER2 expression dependent. According to the model, ERK and p38 activity may be correlated through STAT3 (ERK is high, STAT3 is low, p38 is high; or ERK is low, STAT3 is high, p38 is low) or through JNK.
STAT3 is activated by AKT and suppressed by ERK
Our analysis has shown that AKT is an activator of STAT3 and ERK is a repressor of STAT3 in either HER2 or HER3 expressing cells (Figure 3, Supplementary Table 2). These results indicate contrasting roles for ERK and AKT in the regulation of STAT3 in HME cells when responding to stimulation with different growth factors. Therefore, STAT3’s correlative association with either ERK or AKT pathways is only partial, which gets reflected as low correlations in the cluster analysis.
JNK is mainly stimulated through ERK pathway and it is HER2 dependent
Obtained model indicated an interaction between ERK and JNK, particularly in the HER2 expressing cells (Fig. 3). In contrast, AKT stimulation was found to have low impact on JNK activation (Supplementary Table 1). These results supported the correlation analysis results (Figs. 1 and 2) that signaling through JNK pathway is more associated with the ERK pathway and likely to involve the EGFR/HER1 and HER2 receptors and not the HER3/AKT pathway.
STAT3 and JNK regulate p38 in a context dependent manner
We have found that STAT3 repressed p38 activation (Figure 3). JNK in general tends to contribute to this suppression by activating STAT3. However, in HER3 expressing cells JNK relieves p38’s suppression by activating it directly. Thus, the p38/JNK/STAT3 interactions are complex and context dependent in HME cells. When cell signaling is initiated through HER3/AKT pathway, activity of p38 would be repressed by STAT3, and enhanced through JNK. For the signaling via HER1/HER2/ERK pathway, p38 is suppressed by the concerted effort of STAT3 and JNK.
AKT activation
We have also investigated how AKT phosphorylation was affected by PI3K inhibition, and found that inhibition of PI3K almost diminishes the AKT activation (Supplementary Figure 6). Activation of AKT by PI3K is well established in the literature,86–88 and our results confirmed it for our cell lines. Our results also established that AKT and ERK activations were not impacted by the inhibition of each other (Supplementary Figures 1B and 2A) and there were not any detected interactions (Supplementary Tables 1 and 2). These results establish that the crosstalk between the two signaling pathways33 is not direct in our HME cells.
DISCUSSION
Having access to a closely related cell library in which multiple signaling pathways could be activated differentially via different HER receptors allowed us to pursue a systematic study of crosstalk between proliferation, pro-survival and stress response pathways in HME cells under growth factor stimulated signaling conditions. Our first aim was to establish the possible relationships between the HER family member receptors and several key proteins that are believed to be sentinels for the investigated signaling pathways. Towards this aim, we have collected the experimental data for four HME cell lines and for three ligand treatment conditions. Analysis of the collected data using correlation based cluster analysis has allowed us to establish that: i) AKT activation correlates the most with HER3 phosphorylation, in particular with the HER3 signaling from the HER3:HER1 dimer. ii) ERK and p38 activations correlate the most with HER1 phosphorylation, in particular HER1 signaling from its homo-dimers, and with the HER1:HER2 hetero-dimer signaling. iii) Even though HER2 is believed to be the mediator of HER3 signaling and its increased potency,3, 89–95 the HER2:HER3 dimers correlated only weakly with the activation of signaling proteins. iv) Activations of JNK and STAT3 did not correlate strongly with the activation patterns of the other studied proteins in HME cells under the investigated conditions. STAT3 and JNK were more associated with p38/ERK/HER1, albeit weakly, than with AKT/HER3. Modular response analysis results implied that the observed weak correlation was most likely due to the complex crosslink relationship between AKT and ERK pathways through STAT3.
Most importantly, our analysis led to the conclusion that a consistent analysis of the HER signaling pathways has to take dimer-based receptor signaling into account. There is considerable evidence that the HER dimerization pattern is an important determinant of the consequences of HER signaling,62, 63 which suggests that activation of the downstream signaling pathways would be functions of receptor dimer formation and activation.18 In this regard, we have hypothesized that the HER dimerization pattern is a better predictor of downstream signaling than total receptor activation levels. Correlation analysis using subsets of the experimental data led to cohesive interpretations only when the protein activation patterns were investigated in relationship to receptor signaling from dimer complexes. Analysis based on receptor measurements that did not consider dimer-dependent signaling resulted in conflicting interpretations. This finding can also be seen as a cause of alarm for the interpretation of experimental results when the dynamics of cells are stimulated through HER. Studies in the literature often investigate the activation profiles of the signaling proteins for a particular condition that is relevant for the addressed problem. Our results however clearly illustrated that the interpretation can depend on the way the analysis is performed.
Our correlation analysis did not take into account that there may be some time delay between the receptor activation and activation of the downstream elements. As we have shown earlier,18 the delay between the receptor and ERK/AKT activations is on the order of 3 minutes in the HME cells. So use of equal-time correlation in the reported analysis was reasonable.
To extend our analysis to identify possible causal regulatory relationships, we have collected the experimental data for how inhibition of a protein affects the activation levels of the other investigated proteins. Our ability to generate such data for multiple cell lines and ligand stimulation conditions made it possible to derive causal relationships that are consensus based instead of being applicable to particular conditions. This analysis led us to establish the regulatory interaction model between the studied proteins ERK, AKT, p38, STAT3, and JNK in our HME cells.
Derived consensus model, which is shown in Figure 3 and quantitatively described in Supplementary Tables 1 and 2, illustrates that the proliferation and pro-survival cell signaling pathways are tightly cross-linked in HME cells. MRA of the collected inhibition data led us to identify a central switch-like regulatory role for STAT3 in HME cells with contrasting mechanisms for ERK and AKT in the regulation of STAT3 when responding to stimulation with different growth factors where STAT3 is suppressed by the ERK pathway and activated by the AKT pathway. MRA also identified triangular regulatory patterns among sentinels, which can be considered as evidence of the possible multi-functionality of the studied pathways and how central signaling pathways may be utilized by cells for differentially regulating their responses as a function of their physiological state.
Our analysis indicated HER2 or HER3 receptor driven interactions as well. Triangular interactions between ERK, STAT3 and JNK are mainly operational in HER2 expressing cells while the interactions between p38, STAT3 and JNK have a more complex HER expression dependence and can be context dependent in HME cells. For the signaling via HER1/HER2/ERK pathway, p38 is suppressed by both STAT3 and JNK. When cell signaling is initiated through HER3/AKT pathway, activity of p38 would be repressed by STAT3 and enhanced through JNK. As JNK is regulated by ERK in HER2 expressing cells, ERK also contributes to STAT3’s regulation indirectly through JNK. Thus, STAT3 can couple the ERK and AKT pathways in either direction and its regulatory role depends on pathway stimulation through activation of HER2. Overall, our analysis has established that the crosstalk between the canonical ERK and AKT pathways can occur at multiple levels and it can be dependent on the involvement of particular HER receptors.
As seen in the consensus model (Figure 3), almost all of the identified interactions are bidirectional where the interactions are either both activating or repressing interactions in both directions. A bi-directional interaction that is activating in both directions is a positive feedback loop where an activating stimulation leads to the high activity of both proteins (i.e., both on or both off). A bi-directional interaction that is repressing in both directions is a feedback loop that leads to bi-stable dynamics where the activity of one of the proteins is high and of the companion protein is low (i.e., one is on and one is off as in a genetic toggle switch96). Dynamic response of bi-stable circuits depends on the initial conditions of the system when the kinetic rates, i.e., interaction strength, are comparable in forward and reverse directions. Protein with higher activity level initially would continue to stay active while the companion protein’s activity would be suppressed until a signal flips the states of the proteins. With these properties in mind, obtained likelihood of interaction to exist (Supplementary Table 1) and interaction strength and type (Supplementary Table 2) data may also be used to interpret the hierarchy of regulatory order in HME cells. For example, our analysis results indicated that STAT3 regulation by AKT to be more dominant than its regulation by p38 or JNK (Supplementary Table 2).
The strong correlation between ERK and p38 observed in the cluster analysis, and the lack of direct interaction between them as found in modular response analysis may appear as contradictory. However, based on the model shown diagrammatically in Fig. 3, ERK activation represses STAT3 and suppressed STAT3 activities lift the repression on p38. Thus, ERK activation can lead to p38 activation through STAT3 in a positively correlated manner. Such a positive correlation is also possible for the activation of p38 via JNK upon signaling through ERK pathway. Therefore, the results of the cluster analysis and the modular response analysis are not inconsistent with each other.
Involvement of p38, JNK, and STAT3 in the interplay between ERK and AKT signaling has been studied in the literature, where the conclusions seem to depend on the cell type and treatment conditions. To avoid such complications and possible contradictions, our study was designed to collect and analyze data that is internally consistent and abundant enough to allow more definitive conclusions. Model that we have constructed using the results of our analysis indicated that STAT3 is the focal point of the crosstalk between ERK and AKT pathways in the HME cells. STAT3 is aided by JNK and p38 in modulating the cellular responses to stimulation with growth factors. Our results indicated a less prominent role for JNK in coupling p38 activation to ERK. Overall structure of the signaling network connecting ERK and AKT to downstream elements consists of several positive and negative feedback loops, thus illustrating the complexity of the cellular signaling networks and that pathways do not function in isolation and wider profiling of pathways is needed to decipher the cellular responses reliably. We are in the process of extending these studies to (in)validate the derived network in other cell lines and to include the role of other related sentinel proteins.
These figures show the results of the inhibition studies to investigate the regulatory interactions between sentinel proteins. They show how the phosphorylation of a sentinel is affected by the inhibition of the other studied sentinel proteins, i.e., how the inhibition of a protein (regulator) impacts the phosphorylation of another protein (target). The x-axis in the figures shows the phosphorylation level of the target protein when the cells were stimulated with growth factors and no inhibitor was added. The y-axis shows the phosphorylation level of the target protein when the cells were first treated with an inhibitor and then stimulated with the ligands. Hollow points show the control results when the target was itself inhibited, i.e., these points should have low y-values. Solid points show the results when the regulator protein was inhibited. If the regulator and target protein is positively correlated (i.e., if regulator is activating its target), inhibition of the regulator protein would lower the phosphorylation of its target protein as well. Thus, those points would lie below the diagonal line. When the correlation is negative, the points would lie above the diagonal line because inhibition of the regulator protein would remove the suppression of its target and the phosphorylation of the target protein would go up. When there is no effect, the points would line along the diagonal. Results of all cell lines and ligand treatment cases are shown all together. Data points are marked with circles for R2−3− cells, with triangles for R2+3− cells, with diamonds for R2−3+ cells, and with squares for R2+3+ cells.
Supplementary Material
Supplementary Figure 1. Inhibition studies to investigate the interaction between sentinel proteins. This figure shows how the phosphorylation of ERK was affected by the inhibition of studied sentinel proteins. The x-axis shows the phosphorylation level of ERK when the cells were stimulated with growth factors and no inhibition was added. The y-axis shows the phosphorylation level of ERK when the cells were first treated with an inhibitor and then stimulated with the ligands. Hollow points show the control results when ERK was inhibited itself, i.e., these points should have low y-values. Solid points show the results when the regulator protein was inhibited. Data points are marked with circles for R2−3− cells, with triangles for R2+3− cells, with diamonds for R2−3+ cells, and with squares for R2+3+ cells. Regulator protein is: (A) p38, (B) AKT, (C) JNK, and (D) STAT3.
Supplementary Figure 2. Regulation of AKT by the studied sentinel proteins, Regulator → AKT (target). Details are as in Supplementary Figure 1. Regulator protein is: (A) ERK, (B) p38, (C) JNK, and (D) STAT3.
Supplementary Figure 3. Regulation of JNK by the studied sentinel proteins, Regulator → JNK (target). Details are as in Supplementary Figure 1. Regulator protein is: (A) ERK, (B) AKT, (C) p38, and (D) STAT3.
Supplementary Figure 4. Regulation of p38 by the studied sentinel proteins, Regulator → p38 (target). Details are as in Supplementary Figure 1. Regulator protein is: (A) ERK, (B) AKT, (C) JNK, and (D) STAT3.
Supplementary Figure 5. Regulation of STAT3 by the studied sentinel proteins, Regulator → STAT3 (target). Details are as in Supplementary Figure 1. Regulator protein is: (A) ERK, (B) AKT, (C) JNK, and (D) p38.
Supplementary Figure 6. Regulation of AKT by PI3K. Details are as in Supplementary Figure 1.
Acknowledgments
The research described in this paper was funded by the National Institutes of Health Grant 5R01GM072821-07 to H.R. Pacific Northwest National Laboratory is a multiprogram national laboratory operated by Battelle for the U.S. Department of Energy under Contract DE-AC06-76RL01830.
References
- 1.Yarden Y, Sliwkowski MX. Nat Rev Mol Cell Biol. 2001;2:127–137. doi: 10.1038/35052073. [DOI] [PubMed] [Google Scholar]
- 2.Normanno N, De Luca A, Bianco C, Strizzi L, Mancino M, Maiello MR, Carotenuto A, De Feo G, Caponigro F, Salomon DS. Gene. 2006;366:2–16. doi: 10.1016/j.gene.2005.10.018. [DOI] [PubMed] [Google Scholar]
- 3.Holbro T, Civenni G, Hynes NE. Exp Cell Res. 2003;284:99–110. doi: 10.1016/s0014-4827(02)00099-x. [DOI] [PubMed] [Google Scholar]
- 4.Harris RC, Chung E, Coffey RJ. Exp Cell Res. 2003;284:2–13. doi: 10.1016/s0014-4827(02)00105-2. [DOI] [PubMed] [Google Scholar]
- 5.Guy PM, Platko JV, Cantley LC, Cerione RA, Carraway KL., 3rd Proc Natl Acad Sci U S A. 1994;91:8132–8136. doi: 10.1073/pnas.91.17.8132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schlessinger J. Cell. 2002;110:669–672. doi: 10.1016/s0092-8674(02)00966-2. [DOI] [PubMed] [Google Scholar]
- 7.Dawson JP, Berger MB, Lin CC, Schlessinger J, Lemmon MA, Ferguson KM. Mol Cell Biol. 2005;25:7734–7742. doi: 10.1128/MCB.25.17.7734-7742.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sliwkowski MX, Schaefer G, Akita RW, Lofgren JA, Fitzpatrick VD, Nuijens A, Fendly BM, Cerione RA, Vandlen RL, Carraway KL., 3rd J Biol Chem. 1994;269:14661–14665. [PubMed] [Google Scholar]
- 9.Beerli RR, Hynes NE. J Biol Chem. 1996;271:6071–6076. doi: 10.1074/jbc.271.11.6071. [DOI] [PubMed] [Google Scholar]
- 10.Graus-Porta D, Beerli RR, Daly JM, Hynes NE. Embo J. 1997;16:1647–1655. doi: 10.1093/emboj/16.7.1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tzahar E, Waterman H, Chen X, Levkowitz G, Karunagaran D, Lavi S, Ratzkin BJ, Yarden Y. Mol Cell Biol. 1996;16:5276–5287. doi: 10.1128/mcb.16.10.5276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Duneau JP, Vegh AP, Sturgis JN. Biochemistry. 2007;46:2010–2019. doi: 10.1021/bi061436f. [DOI] [PubMed] [Google Scholar]
- 13.Shankaran H, Zhang Y, Tan YB, Resat H. Plos Computational Biology. 2013;9 doi: 10.1371/journal.pcbi.1003201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schulze WX, Deng L, Mann M. Mol Syst Biol. 2005;1:2005 0008. doi: 10.1038/msb4100012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jorissen RN, Walker F, Pouliot N, Garrett TP, Ward CW, Burgess AW. Exp Cell Res. 2003;284:31–53. doi: 10.1016/s0014-4827(02)00098-8. [DOI] [PubMed] [Google Scholar]
- 16.Yarden Y, Shilo BZ. Cell. 2007;131:1018. doi: 10.1016/j.cell.2007.11.013. [DOI] [PubMed] [Google Scholar]
- 17.Shankaran H, Zhang Y, Opresko L, Resat H. Biochemical and Biophysical Research Communications. 2008;371:220–224. doi: 10.1016/j.bbrc.2008.04.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang Y, Shankaran H, Opresko L, Resat H. IET Systems Biology. 2008;2:273–284. doi: 10.1049/iet-syb:20080116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Birtwistle MR, Hatakeyama M, Yumoto N, Ogunnaike BA, Hoek JB, Kholodenko BN. Mol Syst Biol. 2007;3:144. doi: 10.1038/msb4100188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen WW, Schoeberl B, Jasper PJ, Niepel M, Nielsen UB, Lauffenburger DA, Sorger PK. Mol Syst Biol. 2009;5:239. doi: 10.1038/msb.2008.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Britten CD. Mol Cancer Ther. 2004;3:1335–1342. [PubMed] [Google Scholar]
- 22.Sakai K, Yokote H, Murakami-Murofushi K, Tamura T, Saijo N, Nishio K. Cancer Sci. 2007;98:1498–1503. doi: 10.1111/j.1349-7006.2007.00553.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tanner B, Hasenclever D, Stern K, Schormann W, Bezler M, Hermes M, Brulport M, Bauer A, Schiffer IB, Gebhard S, Schmidt M, Steiner E, Sehouli J, Edelmann J, Lauter J, Lessig R, Krishnamurthi K, Ullrich A, Hengstler JG. J Clin Oncol. 2006;24:4317–4323. doi: 10.1200/JCO.2005.04.8397. [DOI] [PubMed] [Google Scholar]
- 24.Witton CJ, Reeves JR, Going JJ, Cooke TG, Bartlett JMS. Journal of Pathology. 2003;200:290–297. doi: 10.1002/path.1370. [DOI] [PubMed] [Google Scholar]
- 25.Engelman JA, Zejnullahu K, Mitsudomi T, Song Y, Hyland C, Park JO, Lindeman N, Gale CM, Zhao X, Christensen J, Kosaka T, Holmes AJ, Rogers AM, Cappuzzo F, Mok T, Lee C, Johnson BE, Cantley LC, Janne PA. Science. 2007;316:1039–1043. doi: 10.1126/science.1141478. [DOI] [PubMed] [Google Scholar]
- 26.Sergina NV, Rausch M, Wang D, Blair J, Hann B, Shokat KM, Moasser MM. Nature. 2007;445:437–441. doi: 10.1038/nature05474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rowinsky EK. Annu Rev Med. 2004;55:433–457. doi: 10.1146/annurev.med.55.091902.104433. [DOI] [PubMed] [Google Scholar]
- 28.Arteaga CL. Experimental Cell Research. 2003;284:122–130. doi: 10.1016/s0014-4827(02)00104-0. [DOI] [PubMed] [Google Scholar]
- 29.Normanno N, Campiglio M, De LA, Somenzi G, Maiello M, Ciardiello F, Gianni L, Salomon DS, Menard S. Ann Oncol. 2002;13:65–72. doi: 10.1093/annonc/mdf020. [DOI] [PubMed] [Google Scholar]
- 30.Kreeger PK, Lauffenburger DA. Carcinogenesis. 2010;31:2–8. doi: 10.1093/carcin/bgp261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kumar N, Afeyan R, Kim HD, Lauffenburger DA. Mol Pharmacol. 2008;73:1668–1678. doi: 10.1124/mol.107.043794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Katz M, Amit I, Yarden Y. Biochimica Et Biophysica Acta-Molecular Cell Research. 2007;1773:1161–1176. doi: 10.1016/j.bbamcr.2007.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Aksamitiene E, Kiyatkin A, Kholodenko BN. Biochemical Society Transactions. 2012;40:139–146. doi: 10.1042/BST20110609. [DOI] [PubMed] [Google Scholar]
- 34.Sheng G, Guo J, Warner BW. American Journal of Physiology-Gastrointestinal and Liver Physiology. 2007;293:G599–G606. doi: 10.1152/ajpgi.00182.2007. [DOI] [PubMed] [Google Scholar]
- 35.Barrantes ID, Nebreda AR. Biochemical Society Transactions. 2012;40:79–84. doi: 10.1042/BST20110676. [DOI] [PubMed] [Google Scholar]
- 36.Pawson T, Warner N. Oncogene. 2007;26:1268–1275. doi: 10.1038/sj.onc.1210255. [DOI] [PubMed] [Google Scholar]
- 37.Chiacchiera F, Grossi V, Cappellari M, Peserico A, Simonatto M, Germani A, Russo S, Moyer MP, Resta N, Murzilli S, Simone C. Cancer Letters. 2012;324:98–108. doi: 10.1016/j.canlet.2012.05.006. [DOI] [PubMed] [Google Scholar]
- 38.Frey MR, Dise RS, Edelblum KL, Polk DB. EMBO Journal. 2006;25:5683–5692. doi: 10.1038/sj.emboj.7601457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hui LJ, Bakiri L, Stepniak E, Wagner EF. Cell Cycle. 2007;6:2429–2433. doi: 10.4161/cc.6.20.4774. [DOI] [PubMed] [Google Scholar]
- 40.Nelson JM, Fry DW. Journal of Biological Chemistry. 2001;276:14842–14847. doi: 10.1074/jbc.M008786200. [DOI] [PubMed] [Google Scholar]
- 41.Neve RM, Holbro T, Hynes NE. Oncogene. 2002;21:4567–4576. doi: 10.1038/sj.onc.1205555. [DOI] [PubMed] [Google Scholar]
- 42.Wagner EF, Nebreda AR. Nature Reviews Cancer. 2009;9:537–549. doi: 10.1038/nrc2694. [DOI] [PubMed] [Google Scholar]
- 43.Raman M, Chen W, Cobb MH. Oncogene. 2007;26:3100–3112. doi: 10.1038/sj.onc.1210392. [DOI] [PubMed] [Google Scholar]
- 44.Marshall CJ. Cell. 1995;80:179–185. doi: 10.1016/0092-8674(95)90401-8. [DOI] [PubMed] [Google Scholar]
- 45.Joslin EJ, Opresko LK, Wells A, Wiley HS, Lauffenburger DA. J Cell Sci. 2007;120:3688–3699. doi: 10.1242/jcs.010488. [DOI] [PubMed] [Google Scholar]
- 46.Wu CJ, Qian X, O’Rourke DM. DNA Cell Biol. 1999;18:731–741. doi: 10.1089/104454999314872. [DOI] [PubMed] [Google Scholar]
- 47.Song G, Ouyang G, Bao S. J Cell Mol Med. 2005;9:59–71. doi: 10.1111/j.1582-4934.2005.tb00337.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang Y, Banerjee S, Wang Z, Xu H, Zhang L, Mohammad R, Aboukameel A, Adsay NV, Che M, Abbruzzese JL, Majumdar AP, Sarkar FH. Cancer Res. 2006;66:1025–1032. doi: 10.1158/0008-5472.CAN-05-2968. [DOI] [PubMed] [Google Scholar]
- 49.Chen J, Somanath PR, Razorenova O, Chen WS, Hay N, Bornstein P, Byzova TV. Nat Med. 2005;11:1188–1196. doi: 10.1038/nm1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Engelman JA, Luo J, Cantley LC. Nat Rev Genet. 2006;7:606–619. doi: 10.1038/nrg1879. [DOI] [PubMed] [Google Scholar]
- 51.Hsieh AC, Moasser MM. British Journal of Cancer. 2007;97:453–457. doi: 10.1038/sj.bjc.6603910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schoeberl B, Faber AC, Li D, Liang MC, Crosby K, Onsum M, Burenkova O, Pace E, Walton Z, Nie L, Fulgham A, Song Y, Nielsen UB, Engelman JA, Wong KK. Cancer Res. 2010;70:2485–2494. doi: 10.1158/0008-5472.CAN-09-3145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kim HH, Sierke SL, Koland JG. J Biol Chem. 1994;269:24747–24755. [PubMed] [Google Scholar]
- 54.Soltoff SP, Carraway KL, 3rd, Prigent SA, Gullick WG, Cantley LC. Mol Cell Biol. 1994;14:3550–3558. doi: 10.1128/mcb.14.6.3550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Schoeberl B, Pace EA, Fitzgerald JB, Harms BD, Xu L, Nie L, Linggi B, Kalra A, Paragas V, Bukhalid R, Grantcharova V, Kohli N, West KA, Leszczyniecka M, Feldhaus MJ, Kudla AJ, Nielsen UB. Sci Signal. 2009;2:ra31. doi: 10.1126/scisignal.2000352. [DOI] [PubMed] [Google Scholar]
- 56.Cobb MH. Prog Biophys Mol Biol. 1999;71:479–500. doi: 10.1016/s0079-6107(98)00056-x. [DOI] [PubMed] [Google Scholar]
- 57.Junttila MR, Li SP, Westermarck J. FASEB J. 2008;22:954–965. doi: 10.1096/fj.06-7859rev. [DOI] [PubMed] [Google Scholar]
- 58.Mueller KL, Powell K, Madden JM, Eblen ST, Boerner JL. Transl Oncol. 2012;5:327–334. doi: 10.1593/tlo.12163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Dong J, Ramachandiran S, Tikoo K, Jia Z, Lau SS, Monks TJ. American Journal of Physiology-Renal Physiology. 2004;287:F1049–F1058. doi: 10.1152/ajprenal.00132.2004. [DOI] [PubMed] [Google Scholar]
- 60.Rawlings JS, Rosler KM, Harrison DA. Journal of Cell Science. 2004;117:1281–1283. doi: 10.1242/jcs.00963. [DOI] [PubMed] [Google Scholar]
- 61.Sen M, Joyce S, Panahandeh M, Li CY, Thomas SM, Maxwell J, Wang L, Gooding WE, Johnson DE, Grandis JR. Clinical Cancer Research. 2012;18:4986–4996. doi: 10.1158/1078-0432.CCR-12-0792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Olayioye MA, Graus-Porta D, Beerli RR, Rohrer J, Gay B, Hynes NE. Mol Cell Biol. 1998;18:5042–5051. doi: 10.1128/mcb.18.9.5042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yen L, Benlimame N, Nie ZR, Xiao D, Wang T, Al Moustafa AE, Esumi H, Milanini J, Hynes NE, Pages G, Alaoui-Jamali MA. Mol Biol Cell. 2002;13:4029–4044. doi: 10.1091/mbc.E02-02-0084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hendriks BS, Opresko LK, Wiley HS, Lauffenburger D. Cancer Research. 2003;63:1130–1137. [PubMed] [Google Scholar]
- 65.Zhang Y, Opresko L, Shankaran H, Chrisler WB, Wiley HS, Resat H. BMC Cell Biol. 2009;10:78. doi: 10.1186/1471-2121-10-78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Band V, Sager R. Proc Natl Acad Sci U S A. 1989;86:1249–1253. doi: 10.1073/pnas.86.4.1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Dong J, Opresko LK, Dempsey PJ, Lauffenburger DA, Coffey RJ, Wiley HS. Proc Natl Acad Sci U S A. 1999;96:6235–6240. doi: 10.1073/pnas.96.11.6235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Stampfer MR, Pan CH, Hosoda J, Bartholomew J, Mendelsohn J, Yaswen P. Experimental Cell Research. 1993;208:175–188. doi: 10.1006/excr.1993.1236. [DOI] [PubMed] [Google Scholar]
- 69.Eming SA, Lee J, Snow RG, Tompkins RG, Yarmush ML, Morgan JR. J Invest Dermatol. 1995;105:756–763. doi: 10.1111/1523-1747.ep12325550. [DOI] [PubMed] [Google Scholar]
- 70.Di Fiore PP, Pierce JH, Kraus MH, Segatto O, King CR, Aaronson SA. Science. 1987;237:178–182. doi: 10.1126/science.2885917. [DOI] [PubMed] [Google Scholar]
- 71.Danos O, Mulligan RC. Proc Natl Acad Sci U S A. 1988;85:6460–6464. doi: 10.1073/pnas.85.17.6460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Garton KJ, Ferri N, Raines EW. Biotechniques. 2002;32:830–832. doi: 10.2144/02324rr01. 834 passim. [DOI] [PubMed] [Google Scholar]
- 73.Shankaran H, Zhang Y, Chrisler WB, Ewald JA, Wiley HS, Resat H. Molecular Biosystems. 2012;8:2868–2882. doi: 10.1039/c2mb25190f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Citri A, Skaria KB, Yarden Y. Experimental Cell Research. 2003;284:54–65. doi: 10.1016/s0014-4827(02)00101-5. [DOI] [PubMed] [Google Scholar]
- 75.Berger MB, Mendrola JM, Lemmon MA. FEBS Lett. 2004;569:332–336. doi: 10.1016/j.febslet.2004.06.014. [DOI] [PubMed] [Google Scholar]
- 76.Jura N, Shan YB, Cao XX, Shaw DE, Kuriyan J. Proc Natl Acad Sci U S A. 2009;106:21608–21613. doi: 10.1073/pnas.0912101106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Bublil EM, Pines G, Patel G, Fruhwirth G, Ng T, Yarden Y. Faseb Journal. 2010;24:4744–4755. doi: 10.1096/fj.10-166199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Telesco SE, Shih AJ, Jia F, Radhakrishnan R. Molecular Biosystems. 2011;7:2066–2080. doi: 10.1039/c0mb00345j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Shi FM, Telesco SE, Liu YT, Radhakrishnan R, Lemmon MA. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:7692–7697. doi: 10.1073/pnas.1002753107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Bruggeman FJ, Westerhoff HV, Hoek JB, Kholodenko BN. Journal of Theoretical Biology. 2002;218:507–520. [PubMed] [Google Scholar]
- 81.Kholodenko BN, Kiyatkin A, Bruggeman FJ, Sontag E, Westerhoff HV, Hoek JB. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:15245–15245. doi: 10.1073/pnas.192442699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Andrec M, Kholodenko BN, Levy RM, Sontag E. Journal of Theoretical Biology. 2005;232:427–441. doi: 10.1016/j.jtbi.2004.08.022. [DOI] [PubMed] [Google Scholar]
- 83.Santos SDM, Verveer PJ, Bastiaens PIH. Nature Cell Biology. 2007;9:324–U139. doi: 10.1038/ncb1543. [DOI] [PubMed] [Google Scholar]
- 84.Santra T, Kolch W, Kholodenko BN. Bmc Systems Biology. 2013;7 doi: 10.1186/1752-0509-7-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Grabinski N, Bartkowiak K, Grupp K, Brandt B, Pantel K, Jucker M. Cell Signal. 2011;23:1952–1960. doi: 10.1016/j.cellsig.2011.07.003. [DOI] [PubMed] [Google Scholar]
- 86.Rodland KD, Bollinger N, Ippolito D, Opresko LK, Coffey RJ, Zangar R, Wiley HS. J Biol Chem. 2008;283:31477–31487. doi: 10.1074/jbc.M800456200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Li H, Schmid-Bindert G, Wang D, Zhao Y, Yang X, Su B, Zhou C. Adv Med Sci. 2011;56:275–284. doi: 10.2478/v10039-011-0043-x. [DOI] [PubMed] [Google Scholar]
- 88.Krystal GW, Sulanke G, Litz J. Mol Cancer Ther. 2002;1:913–922. [PubMed] [Google Scholar]
- 89.Holbro T, Beerli RR, Maurer F, Koziczak M, Barbas CF, 3rd, Hynes NE. Proc Natl Acad Sci U S A. 2003;100:8933–8938. doi: 10.1073/pnas.1537685100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Zhang K, Sun J, Liu N, Wen D, Chang D, Thomason A, Yoshinaga SK. J Biol Chem. 1996;271:3884–3890. [PubMed] [Google Scholar]
- 91.Carraway KL, 3rd, Soltoff SP, Diamonti AJ, Cantley LC. J Biol Chem. 1995;270:7111–7116. doi: 10.1074/jbc.270.13.7111. [DOI] [PubMed] [Google Scholar]
- 92.Pinkas-Kramarski R, Soussan L, Waterman H, Levkowitz G, Alroy I, Klapper L, Lavi S, Seger R, Ratzkin BJ, Sela M, Yarden Y. Embo J. 1996;15:2452–2467. [PMC free article] [PubMed] [Google Scholar]
- 93.Alimandi M, Romano A, Curia MC, Muraro R, Fedi P, Aaronson SA, Di Fiore PP, Kraus MH. Oncogene. 1995;10:1813–1821. [PubMed] [Google Scholar]
- 94.Maurer CA, Friess H, Kretschmann B, Zimmermann A, Stauffer A, Baer HU, Korc M, Buchler MW. Hum Pathol. 1998;29:771–777. doi: 10.1016/s0046-8177(98)90444-0. [DOI] [PubMed] [Google Scholar]
- 95.Siegel PM, Ryan ED, Cardiff RD, Muller WJ. Embo J. 1999;18:2149–2164. doi: 10.1093/emboj/18.8.2149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Gardner TS, Cantor CR, Collins JJ. Nature. 2000;403:339–342. doi: 10.1038/35002131. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. Inhibition studies to investigate the interaction between sentinel proteins. This figure shows how the phosphorylation of ERK was affected by the inhibition of studied sentinel proteins. The x-axis shows the phosphorylation level of ERK when the cells were stimulated with growth factors and no inhibition was added. The y-axis shows the phosphorylation level of ERK when the cells were first treated with an inhibitor and then stimulated with the ligands. Hollow points show the control results when ERK was inhibited itself, i.e., these points should have low y-values. Solid points show the results when the regulator protein was inhibited. Data points are marked with circles for R2−3− cells, with triangles for R2+3− cells, with diamonds for R2−3+ cells, and with squares for R2+3+ cells. Regulator protein is: (A) p38, (B) AKT, (C) JNK, and (D) STAT3.
Supplementary Figure 2. Regulation of AKT by the studied sentinel proteins, Regulator → AKT (target). Details are as in Supplementary Figure 1. Regulator protein is: (A) ERK, (B) p38, (C) JNK, and (D) STAT3.
Supplementary Figure 3. Regulation of JNK by the studied sentinel proteins, Regulator → JNK (target). Details are as in Supplementary Figure 1. Regulator protein is: (A) ERK, (B) AKT, (C) p38, and (D) STAT3.
Supplementary Figure 4. Regulation of p38 by the studied sentinel proteins, Regulator → p38 (target). Details are as in Supplementary Figure 1. Regulator protein is: (A) ERK, (B) AKT, (C) JNK, and (D) STAT3.
Supplementary Figure 5. Regulation of STAT3 by the studied sentinel proteins, Regulator → STAT3 (target). Details are as in Supplementary Figure 1. Regulator protein is: (A) ERK, (B) AKT, (C) JNK, and (D) p38.
Supplementary Figure 6. Regulation of AKT by PI3K. Details are as in Supplementary Figure 1.