

Abstract
The aryl hydrocarbon receptor (AhR) is a ligand-activated transcription factor that mediates the toxicity of dioxin and serves multiple developmental roles. In the adult brain, while we now localize AhR mRNA to nestin-expressing neural progenitor cells in the dentate gyrus (DG) of the hippocampus, its function is unknown. This study tested the hypothesis that AhR participates in hippocampal neurogenesis and associated functions. AhR deletion and activation by the potent environmental toxicant, 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD), adversely impacted neurogenesis and cognition. Adult AhR-deficient mice exhibited impaired hippocampal-dependent contextual fear memory while hippocampal-independent memory remained intact. AhR-deficient mice displayed reduced cell birth, decreased cell survival, and diminished neuronal differentiation in the DG. Following TCDD exposure, wild-type mice exhibited impaired hippocampal-dependent contextual memory, decreased cell birth, reduced neuronal differentiation, and fewer mature neurons in the DG. Glial differentiation and apoptosis were not altered in either TCDD-exposed or AhR-deficient mice. Finally, defects observed in TCDD-exposed mice were dependent on AhR, as TCDD had no negative effects in AhR-deficient mice. Our findings suggest that AhR should be further evaluated as a potential transcriptional regulator of hippocampal neurogenesis and function, though other sites of action may also warrant consideration. Moreover, TCDD exposure should be considered as an environmental risk factor that disrupts adult neurogenesis and potentially related memory processes.
Keywords: Neurogenesis, dioxin, dentate gyrus, fear conditioning, proliferation, differentiation
Introduction
2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD; Dioxin) is a ubiquitous environmental chemical that has been linked to immune dysfunction, reproductive defects, and cancer (White and Birnbaum, 2009). Developmental TCDD exposures have also been reported to produce cognitive deficits (Haijima et al., 2010). However, whether adult TCDD exposure affects brain structure and function is unknown. TCDD exerts its toxicity by binding to the aryl hydrocarbon receptor (AhR), a ligand-activated basic Helix-Loop-Helix/Per-Arnt-Sim (bHLH/PAS) protein that regulates the expression of growth regulatory genes (Bock and Kohle, 2006). While the normal physiological functions of AhR are unresolved, previous reports indicate AhR regulates neurogenesis in invertebrate systems (Qin and Powell-Coffman, 2004; Kim et al., 2006). We recently identified AhR expression in neural progenitor cells (NPCs) in the mouse cerebellum and prenatal forebrain and discovered that TCDD exposure interfered with developmental neurogenesis (Williamson et al., 2005; Collins et al., 2008; Latchney et al., 2011). Additionally, cerebellar neurogenesis was abnormal in AhR-deficient mice (Collins et al., 2008). Because the AhR is expressed in the adult hippocampus, including granule cells of the neurogenic dentate gyrus (Petersen et al., 2000), we hypothesize that the AhR regulates neurogenesis and associated functions, and that these processes will be disrupted by TCDD exposure.
Hippocampal NPCs proliferate in the subgranular zone (SGZ) and then differentiate into mature dentate granule neurons between 1-4 weeks after birth (van Praag et al., 2002). Newly generated cells subsequently integrate into hippocampal networks encoding spatial memory (Kee et al., 2007) and, after a period of maturation, become physiologically indistinguishable from existing granule neurons (van Praag et al., 2002). Although the functional role of adult neurogenesis is uncertain, it is proposed that new neurons contribute to learning and memory (Inokuchi, 2011). For example, associative learning has been connected to the survival of newborn hippocampal cells (Anderson et al., 2011). Conversely, learning and memory deficits result when adult neurogenesis is ablated (Jessberger et al., 2009). While most evidence for the relationship between adult neurogenesis and cognitive processing has been correlative, the continuous incorporation of newborn neurons into the hippocampal network appears to be optimized according to cognitive demands (Zhao et al., 2008). Although the intrinsic molecular signals that orchestrate SGZ neurogenesis and function are complex, there is evidence that bHLH/PAS transcription factors participate in regulating neurogenesis (Pleasure et al., 2000).
To examine the role of the AhR in adult hippocampal neurogenesis and function, we first demonstrated the presence of AhR mRNA in nestin-expressing NPCs in the SGZ of the adult dentate gyrus. We next evaluated NPC birth, death, and differentiation in AhR-deficient and age-matched wild-type mice. We also examined the effect of TCDD on NPC maturation. Learning and memory were assessed by a contextual and auditory fear-conditioning paradigm in both AhR-deficient and TCDD-exposed mice. We demonstrate that both AhR-deficient and TCDD exposed wild-type mice exhibited compromised hippocampal-dependent contextual memory, reduced NPC birth, and diminished neuronal differentiation. Moreover, AhR was required to mediate the adverse effects of TCDD. Although additional studies are necessary, our findings are consistent with the possibility that AhR participates in the molecular signaling cascade that controls adult hippocampal neurogenesis, revealing a novel target for TCDD neurotoxicity.
Methods
Experimental Animals
For studies in wild-type mice, male and female C57BL/6J mice were purchased from Taconic Farms at 10 weeks of age. Upon arrival, mice were allowed to acclimate to their home cage for 2 weeks prior to initiation of experiments. All mice used for our experiments were from the same generation. Breeding pairs of AhR-/- mice were provided by Dr. Paige Lawrence (University of Rochester). In order to maintain fertility, AhR-/- mice modified at exon 1 (Lahvis and Bradfield, 1998) were regularly backcrossed onto wild-type C57BL background for at least 10 generations and a knockout colony was established. All mice were genotyped prior to being used for experiments. Homozygous AhR knockout mice (AhR-/-) from the same generation were used for the AhR-/- study. AhR-/- mice were observed daily in the mornings and afternoons during routine colony maintenance. AhR-/- mice do not exhibit overt behavioral differences such as seizure activity or abnormal grooming, compared to wild-types. The body weights of AhR-/- mice were not significantly different from wild-type mice across all experiments including those related to proliferation (KO=22.9±1.1, WT=25.7±0.89, mean weight (g) ± SEM, n=10, p=0.073) and cell survival (KO=22.6±1.0, WT=24.3±1.3, n=5, p=0.36). Breeding pairs of nestin-CFPnuc mice were provided by Dr. Grigori Enikolopov (Cold Spring Harbor Laboratory) and a colony was established. Nestin-CFPnuc mice constitutively express cyan fluorescent protein in the nucleus (CFPnuc) of nestin-positive neural progenitor cells. Mice were housed five/cage in standard, ventilated mouse cages in a temperature- and humidity-controlled environment and maintained on a 12h light/dark cycle with food and water provided ad libitum. All housing conditions and procedures were kept in accordance with the guidelines set by the University of Rochester University Committee on Animal Resources and the American Association for Laboratory Animal Science. Investigators took all steps to reduce the number of mice used, and to minimize animal suffering.
Immunoblot Analysis
Protein lysates were harvested in ice cold phosphate buffered saline (PBS) containing 0.1% Triton X-100, 1% PMSF, 1% EDTA, and 1% antiprotease cocktail (all from Sigma, St. Louis, MO). Total protein concentrations were determined by the BCA protein assay kit (Pierce, Rockford, IL). Proteins (10 μg) were fractionated on 10% polyacrylamide gels and transferred to polyvinylidene difluoride (PVDF) membranes (BioRad, Hercules, CA). Membrane was blocked with 5% powdered milk containing 0.2% Tween-20 and probed for AhR (1:1000, Biomol, Plymouth Meeting, PA) and β-actin (1:5000, Sigma) overnight at 4°C. Membrane was then probed with a horseradish peroxidase-conjugated secondary antibody (Jackson Immunoresearch Laboratories, West Grove, PA) for 1 h at room temperature. Proteins were visualized with LumiGLO chemiluminescent substrate reagent (Kirkegaard & Perry Laboratories, Gaithersburg, MD).
Flow Cytometry
Hippocampii from 12-week old nestin-CFPnuc mice were regionally dissected and dissociated to a single-cell suspension by enzymatic degradation using a neural tissue dissociation kit from Miltenyi Biotec (Auburn, CA) according to the manufacturer's protocol (# 130-092-628). For each of the 3 independent cell sorting preparations, hippocampii were dissected from the brains of 2 nestin-CFPnuc mice, for a total of 6 mice. Cells were then immediately sorted for CFP fluorescence using the BD FACSAria II cell sorter (BD Biosciences). Approximately 2.0 × 105 events were gated from the original parent population. Approximately 2% of the gated population was sorted as CFP-positive. All of the CFP-positive cells that were recovered were used for subsequent mRNA analysis. Wild-type mice were used to establish a non-fluorescent baseline.
RNA Isolation, Reverse Transcription, and Real Time Polymerase Chain Reaction
RNA was isolated from nestin-CPFnuc sorted cells using the Qiagen RNeasy Mini Kit as per the manufacturer's instructions. Samples were quantified by spectroscopy and resuspended in DEPC (0.1% diethylpyrocarbonate)-treated water. First strand cDNA was synthesized from 1 μg RNA using the SuperScript III First Strand cDNA Synthesis Kit (Invitrogen). Standard PCR was accomplished with PCR primers and the SuperScript One-Step RT-PCR kit (Invitrogen) according to the manufacturer's protocol. Intron spanning primers with the following sequences were designed using Primer Express Software to reduce genomic amplification. AhR: (F) CGGCTGAACACAGAGTTAGACC; (R) CTGACGCTGAGCCTAAGAACAG. Resulting PCR products were revealed by agarose gel electrophoresis. Non-reverse transcribed mRNA served as a negative control. mRNA isolated from C17.2 neural progenitor cells, in which we previously demonstrated to express AhR (Latchney et al., 2011) served as a positive control.
In vivo TCDD exposure and BrdU administration
For cell proliferation studies, male and female C57BL/6J mice (12 weeks of age) were gavaged with a single dose of 0.5μg/kg TCDD (Cambridge Isotopes, Cambridge, MA) dissolved in olive oil (vehicle) or with vehicle alone at 8:00a.m. and maintained for 8h, 24h, 6 weeks, or 8 weeks. During the last 8h of the exposure period, mice received 4 (50 mg/kg body weight; Sigma) intraperitoneal (i.p.) injections of 5-bromo-2-deoxyuridine (BrdU, Sigma) at 2h intervals. Mice were sacrificed 2h after the last BrdU injection. For cell survival and differentiation studies, 12-week old male C57BL/6J mice received 4 i.p. injections of BrdU at 2h intervals, starting at 8:00a.m. Two hours after the last BrdU injection (4:00p.m.), mice were gavaged with 0.5μg/kg TCDD or with vehicle alone and maintained for 4 weeks. For studies involving AhR-/- mice, 12-week old male and female C57BL/6J wild-type and AhR-/- mice received 4 i.p. injections of BrdU at 2h intervals, starting at 8:00a.m., and sacrificed 2h or 4 weeks after the last BrdU injection.
Fear Conditioning
Mice underwent contextual and auditory fear conditioning to assess hippocampal-dependent and -independent memory processes as previously published (Hein et al., 2010; Matousek et al., 2010). The fear conditioning chamber is equipped with a fan and house light controller that is set at 24VDC (Volt Direct Current, Coulbourn FreezeFrame Fan/House light controller, model ACT-130). The house light provided modest lighting to allow experimenters to view the freezing behavior of the mice. For 3 days before fear conditioning, mice were transported from the colony room to the testing room, handled for 2 min each, and returned to the colony room to acclimate them to experimenter manipulation. At 9:00a.m. on conditioning day, mice were individually allowed to explore the conditioning context, which consisted of a Plexiglas chamber and metal floor grid (model H10-11 M; Coulbourn Instruments, Whitehall, PA). After 3 min, 15s of white noise (80 dB) was presented co-terminating with a 2 s 0.75 mA foot shock. This noise-shock pairing was repeated twice for a total of 3 shocks with an interval of 30s between shocks. 24h later, mice were re-exposed to the conditioning chamber for 5 min each to test contextual fear memory. Mice were then tested for freezing to a novel context and the auditory stimulus. Mice were placed in a novel context consisting of a 15cm open-topped plastic cylinder with bedding on the floor for 3 min followed by re-exposure to the white noise for 3 min. All data were video recorded using FreezeFrame Video-Based Conditioned Fear System and analyzed by Actimetrics Software (Coulbourn Instruments) in a blinded fashion.
Immunohistochemistry
All mice were anesthetized with sodium pentobarbital and perfused transcardially with 0.1M phosphate buffer (PB) containing 2 IU/mL heparin and 0.5% w/v sodium nitrite followed by 4% paraformaldehyde in 0.1M PB. Brains were removed, post-fixed in 4% paraformaldehyde overnight, and transferred to 30% sucrose until equilibrated. The entire hippocampus (-0.82 to -4.24 mm Bregma) was sectioned on a freezing, sliding microtome into 30μm coronal sections and stored in cryoprotectant at -20°C. Immunohistochemistry was performed on free-floating sections as previously described (Collins et al., 2008). Sections were washed in 0.1M PB to remove cryoprotectant, followed by permeabilization in phosphate buffered saline containing 0.3% triton X-100 (PBST). Heat-induced antigen retrieval was performed by microwave heating to 90°C in 0.1 M sodium citrate buffer (pH 9.0). Sections were then incubated with 2N HCl for 60 min to denature DNA, rinsed, incubated with 3% hydrogen peroxide for 30 min to quench endogenous peroxidases, and rinsed again. Tissue was then blocked in 10% normal goat serum in PBST for 1h, and incubated with rat monoclonal antibody against BrdU (1:800; Accurate Chemical, Westbury, NY) in 0.3% PBST with 1% normal goat serum overnight at 4°C. After rinsing, sections were incubated with biotinylated goat anti-rat IgG (1:350, Vector Laboratories, Burlingame, CA) antibody in 0.3% PBST and 1% normal goat serum for 2h at room temperature. After rinsing, sections were incubated in an avidin-biotin-horseradish peroxidase solution (Vector Laboratories) for 1h at room temperature, then incubated in a 3,3′-diaminobenzidine (DAB) fast-tab solution (Sigma). Sections were rinsed in PB and mounted onto Superfrost Plus slides (VWR, West Chester, PA), dried, and coverslipped with Clarion mounting media (Sigma). Positive staining was not seen in mice that did not receive BrdU or in sections in which the primary antibody was omitted.
For immunofluorescent staining, sections were processed for double labeling of BrdU with various neurogenesis markers simultaneously. Free-floating sections were pre-treated with 2N HCl as above and incubated overnight 4°C with rat monoclonal anti-BrdU (1:800; Accurate Chemical, Westbury, NY) along with rabbit polyclonal anti-cleaved caspase-3 (1:500, Cell Signaling Technology, Danvers, MA), guinea pig polyclonal anti-doublecortin (1:1000, Millipore, Temecula, CA), biotinylated mouse anti-NeuN (1:1000, Millipore), rabbit polyclonal anti-GFAP (1:1000, Millipore), or rabbit polyclonal anti-Ki67 (1:500, Vector Laboratories) in 0.3% PBST with 1% normal goat serum. After rinsing, sections were incubated with the appropriate Alexa Fluor secondary antibody (Molecular Probes, Eugene, OR) in 0.3% PBST and 1% normal goat serum for 2h at room temperature. Hippocampal sections from nestin-CFPnuc were stained with a rabbit anti-GFP antibody conjugated to Alexa Fluor 488 (1:400, Molecular Probes). DAPI was used as a fluorescent counterstain. Sections were rinsed of excess secondary antibody prior to mounting onto Superfrost Plus slides, dried, and coverslipped with Fluoromount aqueous mounting media (Sigma). Omission of the primary antibody resulted in a lack of specific staining, thus serving as a negative control.
Image Analysis
Following immunohistochemistry, brightfield or fluorescent staining of positive cells from every eighth coronal section containing the dentate gyrus were visualized and quantified on a Nikon Eclipse 80i microscope (Nikon Instruments, Tokyo, Japan). Staining was quantified throughout the entire hippocampus (-0.82 to -4.24 mm Bregma). In sections processed for BrdU alone using DAB, exhaustive counting of BrdU-positive cells from all sections consisting of the dentate gyrus (including the GCL, SGZ, and hilus) were quantified in a manner similar to the optical fractionator method (Mandyam et al., 2004; 2007). Cells were visualized and quantified at 400× (0.63 NA) with continual adjustment throughout the z-plane to capture all positive cells throughout the thickness of the section. The total number of BrdU-positive cells were summed for each mouse and multiplied by 8 to obtain an estimate of the total number of BrdU-positive cells per dentate gyrus as previously described (Mandyam et al., 2004; 2007). This quantification method was also used to obtain an estimate of the total number of Ki67- and DCX-positive cells per dentate gyrus. All image capturing and quantification was performed in a blinded fashion. Initial data analysis from the AhR-/- and TCDD-exposure studies did not reveal any significant gender differences. Thus, male and female mice were combined for final data analysis.
In sections processed for double fluorescence immunohistochemistry, BrdU-, cleaved caspase-3, NeuN-, and GFAP-positive cells were subjected to phenotypic analysis using either a Nikon Eclipse 80i microscope under 400× with continual adjustment throughout the z-plane or with an Olympus FV1000 laser scanning confocal microscope under 60× objective with a 2× virtual zoom. This analysis was used to calculate the ratio of single-positive (BrdU) or double-positive (BrdU/CC3, BrdU/NeuN, BrdU/GFAP) cells in relation to the total number of BrdU-positive cells from each mouse. For confocal images, images were taken with Olympus FV1000 software. Scanning and optical sectioning in the Z-plane was carried out using multitrack scanning with 0.5μm optical section thickness. Stacks of Z-images were reconstructed into a final 3D image. All fluorescent image capturing and quantification was performed in a blinded fashion.
Statistical Analyses
Data are expressed as means ± S.E.M. Sample sizes are indicated in the figure legends. Data was analyzed in Prism (GraphPad Software 5.0, San Diego, CA) using 2-way ANOVA or Student's t-test where appropriate. The type of statistical test performed is indicated in the figure legends. Analyses were considered significant when p < 0.05. Bonferroni post-hoc tests further analyzed significant ANOVAs.
Results
Nestin-positive neural progenitor cells in the adult dentate gyrus express AhR
With immunoblotting, we first verified the absence of AhR expression in the AhR knockout mice (Fig 1a). Although it has previously been shown that AhR is expressed in the adult dentate gyrus (Petersen et al., 2000), the specific cell types in which AhR is present remains unidentified. To determine if proliferating NPCs in the adult dentate gyrus express AhR, we took advantage of a transgenic mouse line in which nestin-positive progenitor cells constitutively express cyan fluorescent protein in the nucleus (CFPnuc). As expected, CFP expression was restricted to the SGZ of the dentate gyrus, where proliferating NPCs normally reside (Fig 1b). These CFP-positive cells were then isolated via flow cytometry and the sorted CFP cells were subsequently used for RT-PCR analysis. RT-PCR results from 3 independent preparations of CFP sorted cells confirm the presence of AhR in nestin-positive NPCs of the adult dentate gyrus (Fig 1c). Since NPCs are key participants for ongoing hippocampal neurogenesis and associated functions, it is plausible that abnormal AhR function, via deletion or activation by TCDD, could impair NPC maturation and potentially related cognitive functions.
Figure 1. Nestin-positive neural progenitor cells in the adult dentate gyrus express AhR.
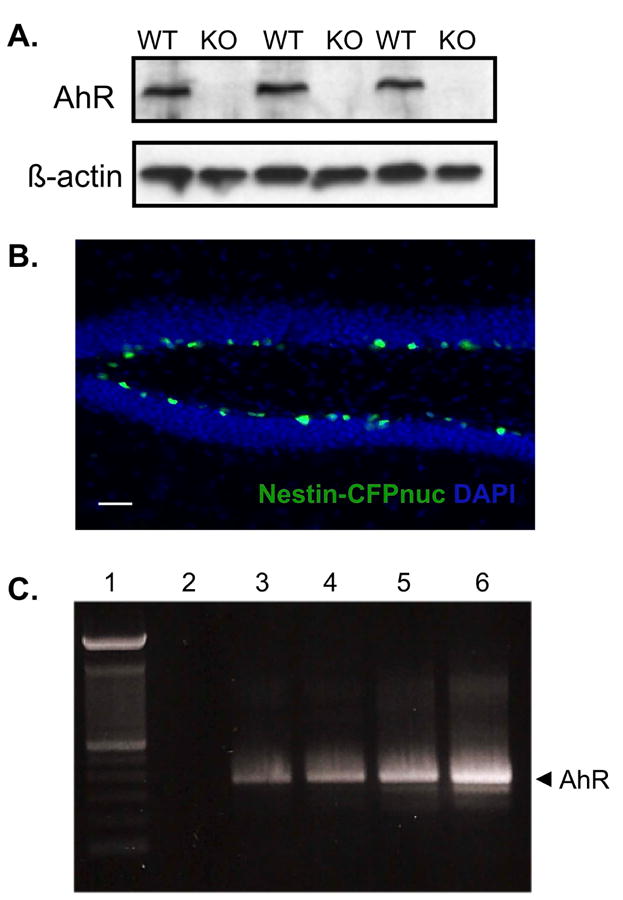
(A)Protein lysates were isolated from the hippocampus of 3 adult wild-type (WT) and 3 adult AhR knockout (KO) mice. AhR protein content (10μg) was analyzed by immunoblotting. The corresponding β-actin blot served as a loading control. (B) Representative image of the dentate gyrus from an adult nestin-CFPnuc mouse. CFP-positive cells (green) were restricted to the SGZ of the dentate gyrus. Scale bar=100μm. (C) Representative gel from RT-PCR analysis for AhR mRNA. Lane 1: 100bp DNA ladder, Lane 2: Negative control (non-reverse transcribed mRNA). Lanes 3-5: 3 independent preparations of CFP sorted cells from nestin-CFPnuc mice. Lane 6: Positive control (mRNA isolated from C17.2 cells).
AhR-deficient mice exhibit hippocampal-dependent memory impairment
To determine the influence of AhR on hippocampal-dependent memory processes, twelve-week old AhR-/- and wild-type mice underwent contextual fear conditioning to evaluate hippocampal-dependent and hippocampal-independent memory processes. No significant differences in freezing behavior between wild-type and AhR-/- mice were found during the 3 min acclimation period prior to shock presentation (Fig 2a). Following the acclimation period, mice were presented with a white noise co-terminating with a foot shock. This noise-shock pairing was repeated twice for a total of 3 shocks with an interval of 30s between shocks. Freezing during the intervals following shock presentation increased across trials (F(2, 36)=12.85, p<0.0001), but did not vary by genotype (F(1, 36)=0.6876, p=0.4178), suggesting that all mice processed the shock similarly. Likewise, freezing to the auditory stimulus increased across trials (F(2, 36)=15.84, p<0.0001), but did not vary by genotype (F(1, 36)=0.1860, p=0.1894), suggesting that the white noise stimulus was processed and the noise-shock pairing was formed by all mice (Fig 2b).
Figure 2. AhR deletion impairs hippocampal-dependent memory.
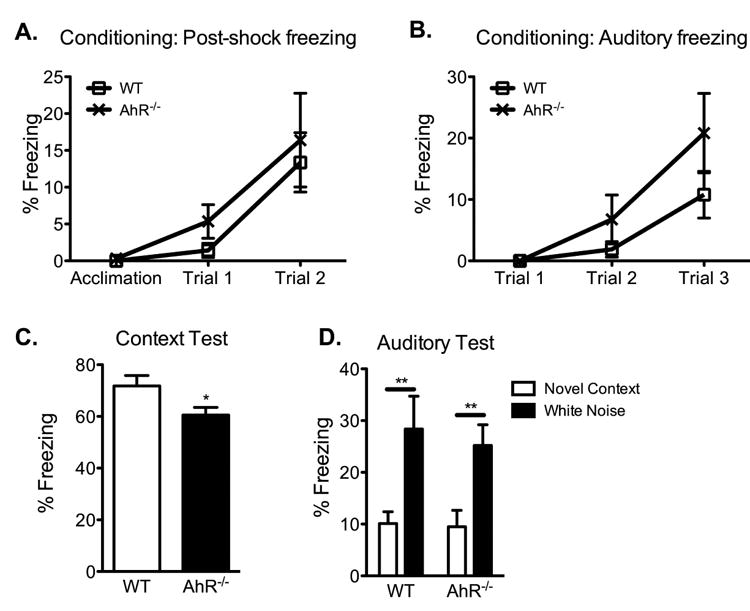
(A and B) Mean percent freezing during the acquisition, post-shock intervals, and auditory stimuli during conditioning increased with repeated pairing but did not differ between wild-type and AhR-/- mice (N=10 mice/genotype; 2-way ANOVA). (C) In the hippocampal-dependent context test, AhR-/- mice show reduced freezing to the conditioned context compared to age-matched wild-type animals (N=10 mice/genotype; Student's t-test, * p<0.05). (D) Both wild-type and AhR-/- mice showed low freezing to a novel context and elevated freezing to the conditioned white noise stimulus (N=10 mice/genotype; 2-way ANOVA with Bonferroni post-hoc test, ** p<0.01). Results are expressed as means ± S.E.M.
When tested the following day for contextual fear memory, AhR-/- mice froze significantly less to the conditioned context than age-matched wild-types (p<0.05, Fig 2c). In the auditory test, all mice demonstrated low levels of freezing to a novel context and elevated freezing to the conditioned white noise stimulus (F(1, 36)=16.22, p=0.0003), with no differences between the genotypes in either measure (F(1, 36)=0.2035, p=0.6546; Fig 2d). The intact memory for the auditory stimulus in AhR-/- mice demonstrate that AhR-/- mice do not have poor memory in general or decreased freezing ability and are capable of forming new memories that are independent of the hippocampus. Therefore, these results demonstrate that AhR deletion disrupts hippocampal-dependent but not hippocampal-independent memory processes, suggesting that AhR contributes to normal hippocampal memory function.
AhR-deficient mice exhibit reduced cell proliferation, cell survival, and neuronal differentiation in the adult dentate gyrus
To examine if AhR-deficient mice have abnormal neurogenesis, twelve-week old AhR-/- and age-matched wild-type mice were given 4 i.p. BrdU injections, one every 2h, to label newborn cells. Mice were sacrificed 2h after the last BrdU injection (Fig 3a). BrdU incorporation, a marker of S-phase entry and an index of cell birth, revealed a 31% decrease in labeled cells in AhR-/- mice compared to age-matched wild-type mice (p<0.01, Fig 3b). The number of Ki67-positive cells was also significantly decreased in AhR-/- mice (p<0.01, Fig 3c), confirming reduced NPC proliferation. To determine if the reduction in newly born cells was due to an increase in apoptosis, quantitative analysis of cells labeled with BrdU and the apoptotic marker cleaved caspase-3 (CC3) was conducted. This analysis revealed no differences in the number of CC3-single positive or in BrdU- and CC3-double positive cells in all groups (Fig 3d). The lack of increased cell death was subsequently confirmed by the absence of an increase in the number of pyknotic nuclei in AhR-/- mice (Veh=320±78, TCDD=333±91, mean ± SEM, n=10, p>0.05 student's t-test). Furthermore, the reduced proliferation is also not a result of a smaller dentate gyrus, as there was no difference in the estimated volume of this structure (KO=5.6×108±0.58×108, WT=5.8×108±0.56×108, mean volume (μm3) ± SEM, n=10, p>0.05 student's t-test). These results suggest AhR contributes to regulation of cell generation in the hippocampus and that the decline in new cell production in the AhR-/- mice is not due to increased cell death or a smaller dentate gyrus.
Figure 3. Reduced cell proliferation and neuronal differentiation in the dentate gyrus of AhR-deficient mice.
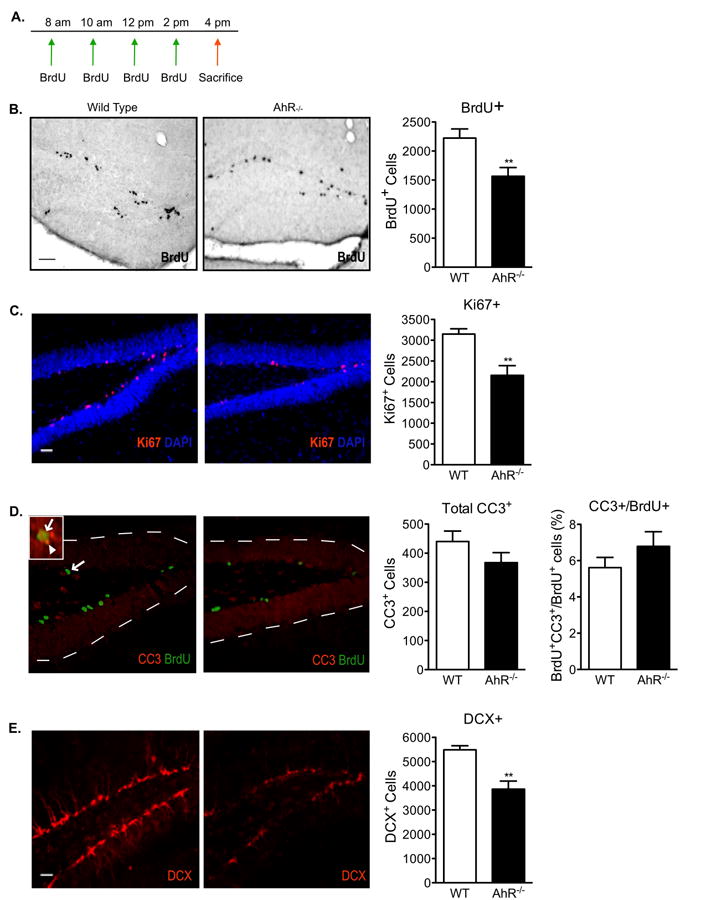
(A)Paradigm used to examine neural progenitor cell birth in wild-type and AhR-/- mice. Representative images and quantitative analysis of (B) BrdU, (C) Ki67, (D) Cleaved Caspase-3 (CC3) and CC3-BrdU double positive, and (E) doublecortin (DCX) positive cells in the dentate gyrus of wild-type and AhR-/- mice. Scale bar=100μm in (B) and 500μm in (C-E). The arrow in the low magnification image points to a double-positive BrdU+/CC3+ cell. The inset is a higher magnification image of the double-positive cell (arrow in inset points to the BrdU+ green signal and arrowhead points to the CC3+ red signal). Data represents means ± S.E.M. (N=10 mice/genotype); Student's t-test (* p<0.05, ** p < 0.01).
Following NPC birth, newborn hippocampal cells undergo cell fate decisions in which they commit to a specific cell lineage and undergo an extensive maturation process that takes 4 to 6 weeks to complete (van Praag et al., 2002). Initial analysis of the early neuronal differentiation marker, doublecortin (DCX), in AhR-/- mice demonstrated that AhR may also be involved in neuronal differentiation. AhR-/- mice exhibited a 39% reduction in DCX expressing cells compared to age-matched wild-type mice (p<0.01, Fig 3e).
To further evaluate NPC maturation, twelve-week old wild-type and AhR-/- mice were administered 4 BrdU injections, one every 2h, to label newly born cells and were sacrificed 4 weeks later (Fig 4a), giving sufficient time for the newborn BrdU labeled cells to develop into mature cells. The survival of the dividing NPCs, estimated as the total number of BrdU-positive cells at 4 weeks, was significantly reduced by 32% in the AhR-/- mice (p<0.05, Fig 4b, 4c). Unexpectedly, the reduced number of surviving BrdU cells in AhR-/- mice could not be explained by an increase in apoptosis, as CC3 counts did not differ compared to wild-type (Fig 4b, 4c). While it is difficult to reconcile decreased survival without an increase in apoptosis, this discrepancy is commonly reported (Cameron and Gould, 1996; Cameron and McKay, 1999; Biebl et al., 2000) and can partially be explained by the brief window of time in which cell death can be reliably detected (Harburg et al., 2007; Sierra et al., 2010). The precise phenotypic differentiation state of the BrdU-positive cells at 4 weeks was defined by double immunostaining for BrdU and the neuronal marker (NeuN) or astroctye marker (Glial Fibrillary Acidic Protein, GFAP). In addition, the expression of DCX, an early neuronal differentiation marker, was assessed. Similar to what was observed in our previous experiment, AhR-/- mice exhibited a 27% reduction in total DCX expressing cells (p<0.05), indicating reduced neuronal differentiation. Further analysis revealed a 28% reduction in BrdU- and NeuN-double positive cells (p<0.05, Fig 4b, 4c), perhaps indicating that newborn cells did not differentiate into mature neurons. Instead, it is possible that the newborn cells could be diverted from the neuronal lineage to become astrocytes. However, the percentage of BrdU-positive cells maturing into GFAP-positive astrocytes was not altered (Fig 4b, 4c). Taken together, these results suggest that the impairment of several stages of neurogenesis, including cell proliferation, survival, and neuronal differentiation, could potentially be associated with the memory impairment observed in AhR-/- mice.
Figure 4. Cell survival and neuronal differentiation is reduced in AhR-/- mice.
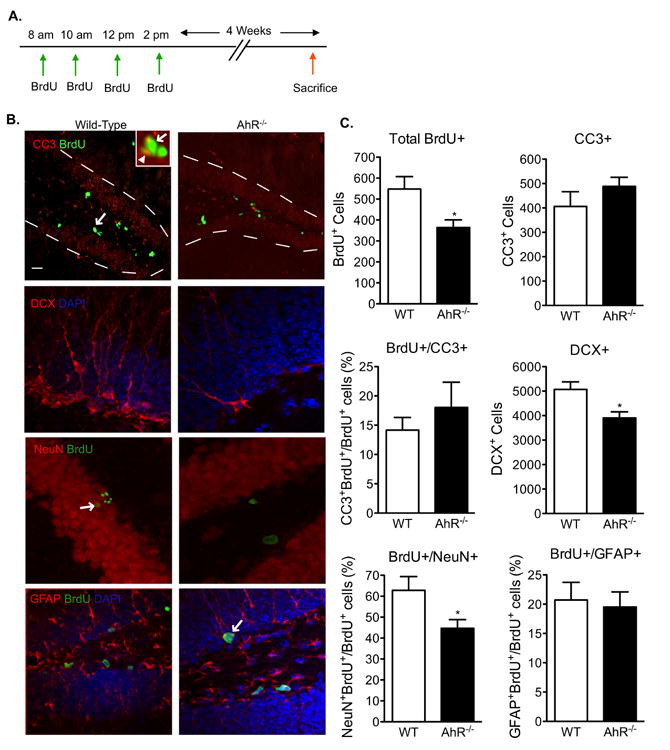
(A)Paradigm used to examine the survival and differentiation of newly born cells in AhR-/- and WT mice 4 weeks after BrdU labeling. (B) Representative images of the dentate gyrus stained for BrdU/CC-3, DCX, NeuN/BrdU, and GFAP/BrdU. Overlays are shown. Scale bar=100μm for BrdU/CC3 image and 10μm for all others. The arrow in the BrdU/CC3 low magnification image points to a to double-positive BrdU+/CC3+ cell. The inset is a higher magnification image of the double-positive cell (arrow in inset points to the BrdU+ green signal and arrowhead pionts to the CC3+ red signal). The arrow in the BrdU/NeuN and BrdU/GFAP images points to a double-positive cell. (C) Quantification revealed that cell survival (total BrdU+ cells) was reduced whereas apoptosis (CC-3) was not altered in AhR-/- mice. Early (DCX+ cells) and late (NeuN- and BrdU-double positive cells) neuronal differentiation was reduced in AhR-/- mice. Maturation of newborn cells into GFAP+ astrocytes was not affected. Results are expressed as means ± S.E.M. (N=5 mice/group); Student's t-test (* p<0.05).
Hippocampal-dependent memory is impaired following TCDD exposure
TCDD, a high affinity AhR ligand, is a ubiquitous and persistent environmental toxicant. Most of what is known about TCDD toxicity comes from acute, high-level accidental exposures (White and Birnbaum, 2009). Human exposure, however, largely comes from low levels of TCDD through dietary intake (Mato et al., 2007). Given the foregoing evidence that normal AhR expression may contribute to adult hippocampal neurogenesis and function, we determined whether activation of the AhR with TCDD also impacts these processes, as the neurological outcomes of low-dose exposures are undefined. Twelve-week old mice were gavaged with a single dose of vehicle or 0.5μg/kg TCDD and 6 weeks later they underwent the same contextual fear-conditioning paradigm used in the AhR-/- study (Fig 5a). The fear conditioning was performed 6 weeks post exposure to allow adequate time for the progenitor cells born at the time of exposure to develop into mature, functional cells capable of participating in the hippocampal circuitry. No significant differences in freezing behavior between vehicle and TCDD-exposed mice were found during the 3 min acclimation period prior to shock presentation (Fig 5b). Freezing during the intervals following shock presentation increased across trials (F(2, 36)=23.44, p<0.0001), but did not vary with TCDD exposure (F(1, 36)=0.0023, p=0.9623), suggesting that all mice processed the shock similarly. Likewise, freezing to the auditory stimulus increased across trials (F(2, 36)=27.60, p<0.0001), but did not vary with TCDD exposure (F(1, 36)=0.2268, p=0.6396), suggesting that the white noise stimulus was processed and the noise-shock pairing was formed by all mice (Fig 5c).
Figure 5. TCDD exposure leads to hippocampal-dependent memory impairment.
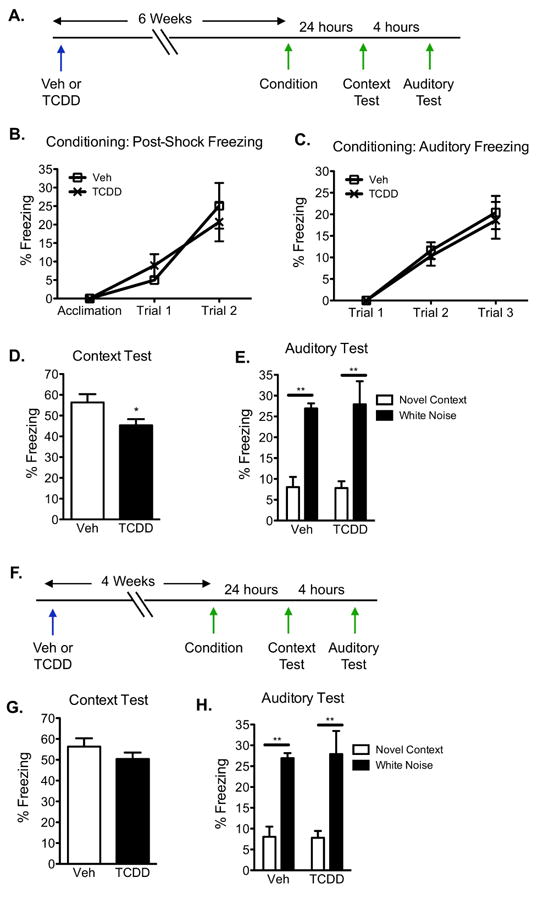
(A)Contextual fear conditioning paradigm to test for hippocampal-dependent and independent function. (B and C) Mean percent freezing during the acquisition, post-shock intervals, and auditory stimuli during conditioning increased with repeated pairing but did not differ between vehicle and TCDD exposed mice (N=10 mice/genotype; 2-way ANOVA). (C) In the hippocampal-dependent context test, TCDD exposed mice froze less to the conditioned context compared to vehicle exposed animals (N=10 mice/genotype; Student's t-test, * p<0.05). (D) All mice showed low freezing to a novel context and elevated freezing to the conditioned white noise stimulus (N=10 mice/genotype; 2-way ANOVA with Bonferroni post-hoc tests, ** p<0.01). Results are expressed as means ± S.E.M.
In the hippocampal-dependent context test, TCDD exposed mice exhibited a 19% reduction in freezing to the conditioned context compared to vehicle-exposed mice (p<0.05, Fig 5d). In the auditory test, all mice demonstrated low levels of freezing to a novel context and elevated freezing to the conditioned white noise stimulus (F(1, 36)=28.95, p<0.0001), with no differences with TCDD exposure in either measure (F(1, 36)=0.001828, p=0.9661; Fig 5e). These results demonstrate that AhR activation by a single, environmentally relevant low-dose of TCDD in adult mice compromises a hippocampal-dependent form of memory.
Because we postulate that the reduction in contextual fear memory is associated with the lack of new neuron development, we explored whether TCDD exposure had an effect at an earlier timepoint prior to the full functional incorporation of new neurons. Therefore, we repeated the same experiment 4 weeks following TCDD exposure (Fig 5f), a timepoint when new neurons have just completed their development into mature neurons but are not yet functionally incorporated into the hippocampal circuitry. At 4 weeks post-exposure, no significant differences in freezing behavior between vehicle and TCDD-exposed mice were found during the 3 min acclimation period prior to shock presentation. Freezing during the intervals following shock presentation increased across trials (2-way ANOVA, F(2, 36)=19.01, p<0.0001), but did not vary with TCDD exposure (2-way ANOVA, F(1, 36)=0.07253, p=0.7908; data not shown). Likewise, freezing to the auditory stimulus increased across trials (2-way ANOVA, F(2, 36)=29.26, p<0.0001), but did not vary with TCDD exposure (2-way ANOVA, F(1, 36)=1.284, p=0.2622; data not shown).
In the hippocampal-dependent context test, there was no difference in freezing behavior to the conditioned context between vehicle- and TCDD-exposed mice (p=0.254, Fig 5g). In the auditory test, all mice demonstrated low levels of freezing to a novel context and elevated freezing to the conditioned white noise stimulus (F(1, 36)=37.05, p<0.0001), with no differences with TCDD exposure in either measure (F(1, 36)=0.01401, p=0.906, Fig 5h). Considering there was a deficit in hippocampal-based memory at 6 weeks post exposure when new neurons become functionally incorporated, but not at an earlier timepoint, these observations support the notion that impairments in hippocampal-associated functions may be correlated to a reduction in neurogenesis.
TCDD exposure reduces cell proliferation in the dentate gyrus without inducing apoptosis
To examine the effects of TCDD exposure on cell birth and apoptosis, twelve-week old mice were gavaged with a single dose of 0.5μg/kg TCDD or vehicle and administered the same BrdU injection paradigm used in the AhR-/- study to label newborn cells. Proliferation was analyzed by BrdU and Ki67 immunohistochemistry 8 and 24h post-exposure (Fig 6a). TCDD exposure reduced S-phase entry by 15% after 8h (p<0.05), and by 34% at 24h (p<0.01) compared to vehicle exposed mice (Fig 6b). The number of Ki67-positive cells was also significantly reduced 24h after TCDD exposure (p<0.01), but not at 8h (Fig 6c). This could suggest that cells cycling in S-phase are initially more vulnerable to TCDD's inhibitory effects than cells cycling in other phases of the cell cycle. To determine if the reduction induced by TCDD was due to increased apoptosis, we examined CC3 and BrdU double-labeled cells. Quantitative analysis of CC3-single positive and BrdU- and CC3-double labeled cells revealed no differences between vehicle and TCDD exposed mice (Fig 6d). The absence of increased apoptosis in TCDD-exposed mice was confirmed by detecting similar numbers of pyknotic nuclei in vehicle control mice. (Veh=340±45, TCDD=364±54, mean ± SEM, n=10, p>0.05 student's t-test). The reduced proliferation in TCDD-exposed mice was also not a result of a smaller dentate gyrus because there were no differences in the estimated volume of this structure (Veh=5.4×108±0.57×108, TCDD=5.3×108±0.58×108, mean volume (μm3) ± SEM, n=10, p>0.05 student's t-test).
Figure 6. TCDD reduces cell proliferation in adult hippocampus without inducing cell death.
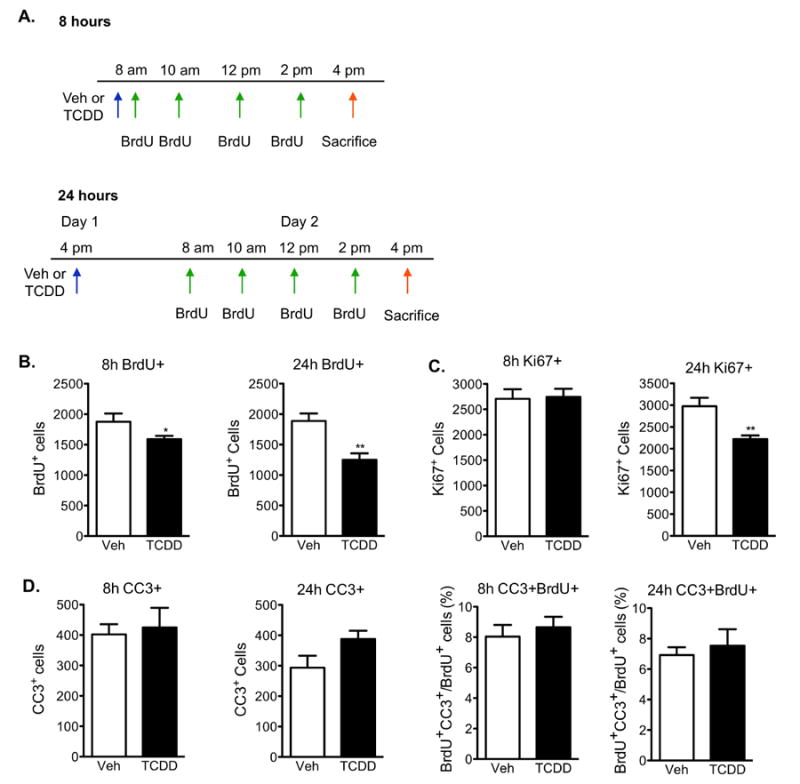
(A)Paradigm used to investigate neural cell proliferation in the dentate gyrus 8h and 24h following TCDD exposure. (B) Quantification of BrdU+ cells in the SGZ demonstrated a reduction in S-phase entry 8h after TCDD exposure compared to vehicle-treated animals, which became more pronounced at 24h. (C) Quantification of Ki67-positive cells revealed a significant reduction in newborn cells in all phases of the cell cycle following 24h exposure to TCDD, but not at 8h. (D) Quantification of CC3-single and CC3- and BrdU-double positive cells relative to total number of BrdU-positive cells in the SGZ demonstrated that TCDD does not induce apoptosis at either timepoint. All results are expressed as means ± S.E.M. (N=10 mice/group); Student's t-test (* p<0.05, ** p <0.01).
Neuronal differentiation, but not glial differentiation, is reduced following TCDD exposure
We next questioned whether TCDD exposure would ultimately lead to fewer mature neurons in the dentate gyrus. To investigate the effect of TCDD on the survival and differentiation of the newborn NPCs, twelve-week old mice were first administered BrdU to label newly born cells then gavaged with a vehicle or 0.5μg/kg TCDD 2h after the last BrdU injection. Mice were sacrificed 4 weeks later, giving sufficient time for the newborn BrdU labeled cells to develop into mature cells (Fig 7a).
Figure 7. Neuronal, but not glial, differentiation is reduced 4 weeks following TCDD exposure.
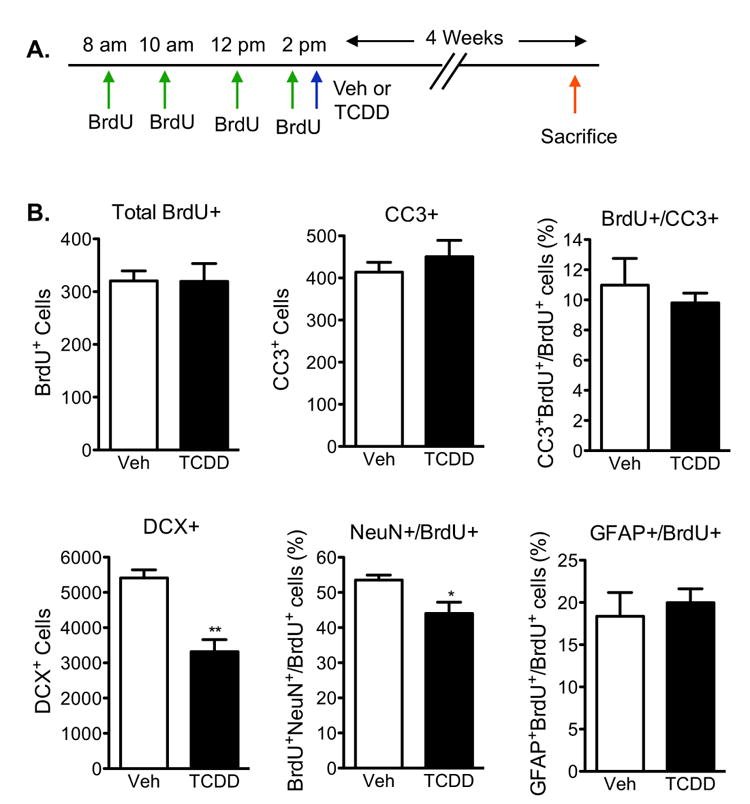
(A)Paradigm used to examine the survival and differentiation of newly born cells 4 weeks following TCDD exposure. (B) Quantification revealed that cell survival (total BrdU+ cells) and cell death (CC-3) were not affected with TCDD exposure. Early (DCX+ cells) and late (NeuN- and BrdU-double positive cells) neuronal differentiation was reduced with TCDD exposure. Maturation of newborn cells into GFAP+ astrocytes was not affected with TCDD exposure. Results are expressed as means ± S.E.M. (N=5 mice/group); Student's t-test (* p<0.05, ** p <0.01).
The survival of the dividing NPCs, estimated as the total number of BrdU-positive cells at 4 weeks, did not differ between vehicle and TCDD-exposed mice (Fig 7b). Likewise, TCDD did not induce apoptosis as indicated by no changes in CC3 counts (Fig 7b). Phenotypic differentiation of the BrdU-positive cells at 4 weeks was defined by performing double immunostaining for BrdU and NeuN or GFAP as well as analyzing DCX. TCDD exposure elicited a 40% reduction in total DCX expressing cells (p<0.01), indicating reduced neuronal differentiation. Further analysis revealed a 21% reduction in BrdU- and NeuN-double positive cells (p<0.05, Fig 7b), possibly indicating the inability of newborn cells to fully differentiate into mature neurons. Instead of differentiating into mature granule neurons, the newborn cells could be diverted from the neuronal lineage to become astrocytes or oligodendrocytes. However, the percentage of BrdU-positive cells maturing into GFAP-positive astrocytes was not altered with TCDD exposure (Fig 7b). Taken together, these results suggest that TCDD exposure interfered with neuronal differentiation, while cell survival and glial differentiation were unaffected.
TCDD exposure leads to a sustained reduction in neuronal differentiation, but not cell birth, up to 8 weeks post exposure
To address whether TCDD exposure leads to a long-term reduction in NPC birth, twelve-week old mice were gavaged with vehicle or 0.5μg/kg TCDD and newborn cells were labeled with BrdU 6 or 8 weeks post exposure (Fig 8a). Mice were sacrificed 2h after the last BrdU injection. Immunohistochemical analyses demonstrate that the reduction in cell birth seen 8 and 24h post TCDD exposure (Fig 6a) does not persist 6 and 8 weeks after exposure (Fig 8b). In contrast, there was a sustained reduction in neuronal differentiation, as evidenced by reduced DCX expression, 6 weeks (p<0.01) and 8 weeks (p<0.05) post exposure (Fig 8c). The reduction in DCX-positive cells in TCDD-exposed mice is not a result of a smaller dentate gyrus since the estimated volume of the dentate gyrus did not differ with TCDD (at 6 weeks: Veh=5.8×108±0.6×108, TCDD=5.7×108±0.59×108; at 8 weeks: Veh=5.6×108±0.55×108, TCDD=5.7×108±0.6×108, mean volume (μm3) ± SEM, n=5, p>0.05 student's t-test).
Figure 8. Sustained reduction in neuronal differentiation, but not cell birth, 6 and 8 weeks post TCDD exposure.
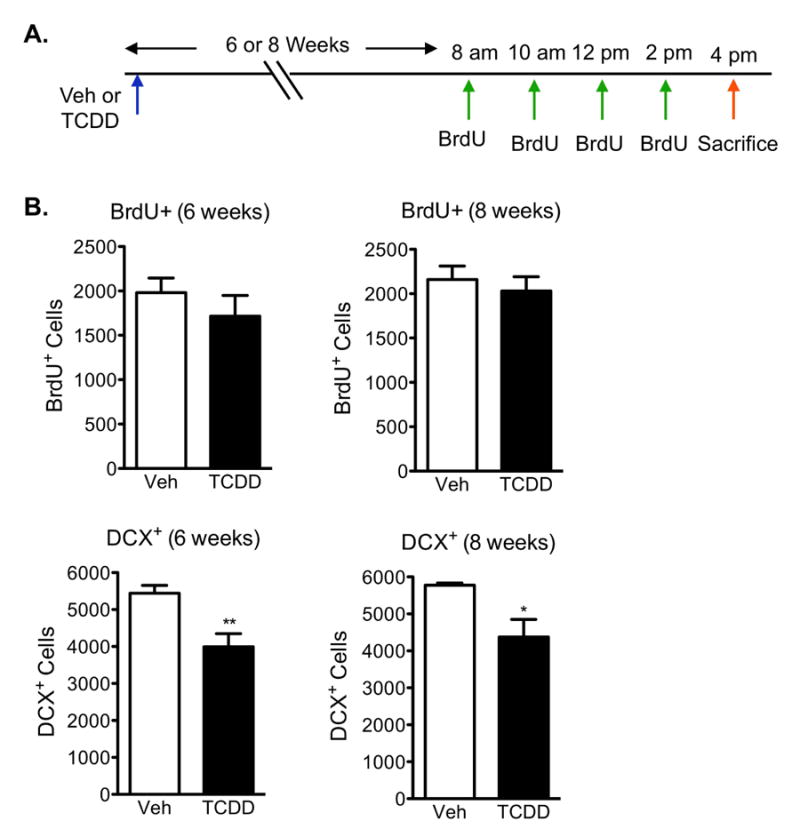
(A)Paradigm used to analyze long-term effects on neural cell birth and differentiation following TCDD exposure. (B) Quantification of BrdU+ cells in the SGZ demonstrated no significant changes in cell birth 6 and 8 weeks after TCDD exposure compared to vehicle-exposed mice. (C) Quantification of DCX+ cells in the SGZ demonstrated a persistent reduction in DCX expression 6 and 8 weeks after TCDD exposure. Results are expressed as means ± S.E.M. (N=5 mice/group); Student's t-test (* p<0.05, ** p <0.01).
TCDD exposure does not alter cell birth in AhR-deficient mice
As a high affinity ligand for the AhR, TCDD is known to require this receptor to exert its toxicity in other organ systems. Mice deficient for the AhR are resistant to the adverse effects of TCDD exposure (Bunger et al., 2003; 2008). To determine whether the neurotoxic effects observed in the TCDD-exposed mice require the AhR, twelve-week old wild-type and AhR-/- mice were exposed to a vehicle or 0.5μg/kg TCDD for 24h and labeled with BrdU to measure cell birth (see 24h exposure timeline in Fig 6a). As observed in our previous experiment, TCDD-exposed wild-type mice exhibited a 34% reduction in cell birth compared to vehicle-exposed wild-types (Fig 9). Similarly, AhR-/- mice, whether exposed to a vehicle or TCDD, both exhibited reductions in BrdU-positive cells compared to vehicle-exposed wild-types, specifically, 32% and 35% respectively (Fig 9). However, there were no significant differences in BrdU-positive cells between vehicle- and TCDD-exposed AhR-/- mice. These studies indicate that TCDD had no additive effect over that of receptor deletion, suggesting that TCDD toxicity requires the AhR, as has been previously reported in numerous studies in AhR-/- mice (Bunger et al., 2003; 2008).
Figure 9. AhR-deficient mice are resistant to TCDD-induced neurotoxicity.
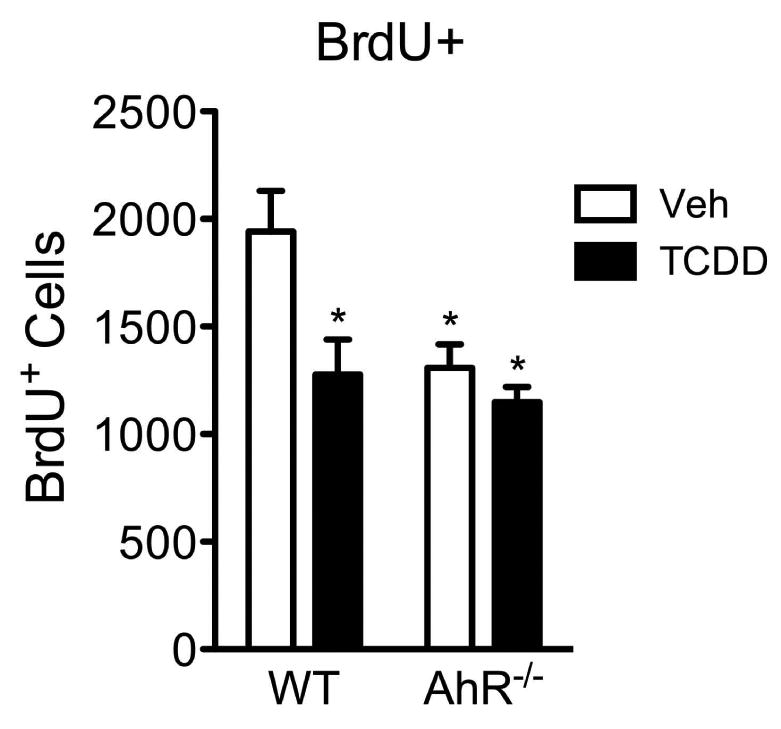
Quantification of BrdU+ cells in the SGZ revealed no difference in BrdU-positive cells between vehicle- and TCDD-exposed AhR-/- mice, indicating TCDD neurotoxicity requires the AhR. Results are expressed as means ± S.E.M. (N=3 mice/group); 2-way ANOVA with Bonferroni post-hoc tests (* p<0.05 versus vehicle-exposed wild-types).
Discussion
Our observations indicate that AhR deletion or activation negatively impact hippocampal NPC birth and differentiation associated with reduced hippocampal-dependent memory function. The presence of receptor expression in SGZ precursors is consistent with the hypothesis that AhR deletion or activation directly alters signaling pathways that orchestrate hippocampal neurogenesis and related functions. However, since our studies do not modulate AhR function selectively in hippocampal NPC, alternative sites of action must also be considered. For example, there could be subtle alterations in glucose or lipid metabolism (Sato et al., 2008; Takeda et al., 2011; Kachaylo et al., 2012; Tanos et al., 2012), circadian rhythms (Pendergast and Yamazaki, 2012), monoamine neurotransmitters (Byers et al., 2006; Tanida et al., 2009), thyroid hormones or adrenal cortical functions (Han et al., 2011; Spaulding, 2011; Leijs et al., 2012) among others, that could contribute to the observed impairments in SGZ NPC maturation and hippocampal function (Desouza et al., 2005; Kapoor et al., 2010; Snyder et al., 2011). Therefore, it remains to be determined whether the deficits in neurogenesis and behavior result directly from AhR signaling events in NPCs.
In addition to its well-characterized role in xenobiotic detoxification, AhR also functions as a modulator of cellular signaling pathways during development in the absence of an exogenous ligand (Puga et al., 2005). However, endogenous ligands for AhR remain elusive. Apart from the observed neurogenic deficits, we determined that hippocampal memory processing is abnormal in AhR-/- mice, suggesting an associated functional link for the receptor. We also demonstrated that TCDD produces similar hippocampal neurogenic and cognitive impairments as seen in AhR-/- mice. Moreover, these adverse responses were mediated by the AhR, since AhR-/- mice were resistant to TCDD-induced neurotoxicity. It is conceivable that environmental modulation of AhR activity by TCDD diverts the receptor from performing normal physiological activity, which results in impaired hippocampal function and neurogenesis. However, it remains a formal possibility that absence of a TCDD effect in AhR-/- mice represents a “floor effect” in that a constant minimum amount of neurogenesis remains despite the presence of inhibitory signals. Together our findings identify the AhR as a novel bHLH/PAS transcription factor that has the potential to contribute to signaling events that modulate adult hippocampal neurogenesis and related cognitive functions.
Although AhR expression in the adult dentate gyrus has previously been shown, the specific cell types expressing AhR remained unknown (Petersen et al., 2000). Using a transgenic mouse line, we built upon the previous study by demonstrating that nestin-positive NPCs express AhR in the SGZ of the adult neurogenic dentate gyrus. Since nestin-positive NPCs are critical participants in the hippocampal neurogenic cascade, it is plausible that AhR expression and activity plays a role in regulating NPC proliferation and differentiation. AhR expression in NPCs also renders these cells as targets for the potent neurotoxicant, TCDD. However, additional studies are necessary to fully delineate the potential direct roles of AhR deletion or activation on hippocampal NPC maturation and function.
In both AhR-/- and TCDD-exposed wild-type mice, there was a substantial reduction of newborn cells in the adult dentate gyrus, as shown by a decrease in BrdU- and Ki67-positive cells. We speculate that the decrease in newborn cells is not due to cell death, as there was no evidence of increased activated caspase-3 staining or numbers of pyknotic nuclei in hippocampal cells compared to the respective control mice in each study. However, it is possible that our studies missed the short timeframe when apoptotic cells are detectable in the SGZ (Harburg et al., 2007; Sierra et al., 2010). A more likely possibility is that the decrease in newborn cells represents a failure of NPCs to proliferate, ultimately depleting the precursor pool and leading to fewer mature neurons. Supporting this interpretation, AhR-/- and TCDD-exposed wild-type mice both exhibited deficits in neuronal differentiation and had fewer mature granule cells. Alternatively, the cell cycle length of newborn cells could either be shortened or prolonged, also leading to reduced NPC proliferation and delayed neuronal differentiation. We cannot conclude that the reduced proliferation contributed to the diminished neuronal differentiation, because these outcomes could be independent of each other. However, the precise stage during which AhR deletion or activation impedes differentiation of NPCs into neurons remains to be determined. Our data do not reveal that modulation of AhR activity altered fate determination of newborn SGZ cells. Moreover, our studies did not detect a reduction in dentate gyrus volume, but it is still possible that granule cell density could be altered. Further investigations involving a more comprehensive time course and stereological evaluations are necessary to resolve the observed differentiation defect and potential impact on granule cell density.
Although our studies do not provide evidence that AhR is directly involved in hippocampal neurogenesis or hippocampal-dependent behavior, we propose that a functional AhR in nestin-positive NPCs contributes to proper maintenance of the NPC pool and/or NPC cell cycle kinetics, which is important for balancing NPC proliferation with neuronal differentiation. This hypothesis is supported by the similarity between our findings related to NPC maturation and the observations described in inducible Notch knockout (iKO) mice, where decreased NSCs in these mice were interpreted as a failure to maintain the precursor reservoir in the adult brain, leading to fewer mature neurons (Ables et al., 2010). We propose that abnormal AhR function, via genetic deletion or the untimely activation of this receptor by TCDD, leads to abnormal neurogenesis and cognitive processing. This hypothesis raises the important issue of indirect and/or secondary effects that may occur in AhR-/- and TCDD-exposed mice and additional work is necessary to evaluate the numerous potential indirect mechanisms that might contribute to our observed deficits in neurogenesis and hippocampal dependent memory processing. Our findings in AhR-deficient mice should also be interpreted with some caution because as with all lifelong global knockout mice, the possibility that our observations are indirectly due to adaptive changes during development or from signaling events unrelated to NPCs must be also be considered, since AhR is widely expressed throughout the CNS and other organ systems. Future studies using inducible, cell-specific AhR knockout mice are required to draw a more direct link between AhR deletion and the associated neurological impairments in the adult hippocampus.
Whereas it was unexpected that AhR-/- and dioxin-exposed wild-type mice would exhibit phenotypic similarities, there are two reasons that might explain our experimental outcomes. One possible explanation is that TCDD has been shown to rapidly downregulate AhR, resulting in an expression deficiency analogous to the AhR-/- mice (Pollenz, 2002). However, this reason cannot fully explain our observed results because the AhR is not permanently downregulated following ligand exposure. Instead, AhR protein is eventually recycled and returned to basal levels (Pollenz and Buggy, 2006). A second possibility is that TCDD causes persistent activation of the AhR, thereby inducing a toxic response and preventing AhR from performing its functional roles. The toxic response from TCDD exposure may be due to the ligand displacing the AhR from promoter regions of gene clusters involved in cell growth to favor the regulation of xenobiotic metabolism genes. Consequently, cell growth related genes may become transcriptionally repressed, resulting in abnormal growth regulation (Sartor et al., 2009), which could interfere with neurogenesis and associated function.
The similar deficits in hippocampal mediated behavior and neurogenesis following AhR deletion or activation were initially surprising. However, there are other examples where either increases or decreases in transcription factors give similar functional and/or structural phenotypes in the brain, in both animal models and human disease. For instance, engrailed2 deficient and overexpression transgenic mouse mutants exhibited a similar autistic-related cerebellar phenotype (Millen et al., 1994; Kuemerle et al., 1997; Baader et al., 1998; 1999). The loss or gain of function in MECP2, a transcriptional repressor, also generated similar neurological aberrations in humans and experimental animals (Chahrour and Zoghbi, 2007). With regard to the AhR, two studies indicated that AhR deficient or TCDD-exposed wild-type mice modulate immune system T cells in a similar manner (Quintana et al., 2008; Veldhoen et al., 2008). The similarities observed between AhR-/- and TCDD-exposed wild-type mice in our studies suggest that the AhR may serve dual roles in mediating xenobiotic metabolism and hippocampal neurogenesis. Therefore, both the expression and activity levels of the AhR are potentially important in regulating adult hippocampal structural and functional outcomes.
Our findings that aberrant neurogenesis in both AhR-/- and TCDD-exposed wild-types produced parallel deficits in one type of hippocampal-based memory are consistent with the accumulating evidence that adult-born NPCs participate in hippocampal-dependent cognitive processing (Clelland et al., 2009; Stone et al., 2011). The observations that neuronal differentiation (DCX-positive cells) was persistently impaired several weeks following TCDD exposure are consistent with the idea that immature neurons undergoing maturation are important contributors to hippocampal function (Aasebo et al., 2011; Marin-Burgin et al., 2012). The absence of a sustained reduction in newborn cells, even though there was a functional deficit, further supports the idea that newborn NPCs less than one week old lack synaptic connections and are unable to make significant contributions to hippocampal function (Deng et al., 2009). However, contextual fear conditioning may not be a discrete measure of neurogenesis dependent memory acquisition. There is also controversy about the role of the hippocampus in contextual fear conditioning, since other brain regions such as the amygdala are thought to participate in this learning and memory paradigm (Goosens and Maren, 2001; Onishi and Xavier, 2010). Moreover, the hippocampus is involved in functions beyond contextual memory such as mood regulation (Lagace et al., 2010; Sahay et al., 2011). Future studies are required not only to more thoroughly evaluate hippocampal function but also to establish a connection with neurogenesis and behavioral outcomes related to AhR signaling activity.
Our data suggest the abnormal hippocampal neurogenesis resulting from AhR deletion or inappropriate activation by TCDD could lead to a gradual depletion of NPCs and compromised neuronal maturation, thereby potentially accounting for the impaired formation of hippocampal-based memories. Our current work adds to existing research related to the impact of TCDD exposure during developmental neurogenesis by demonstrating that adult hippocampal neurogenesis may also be vulnerable to environmentally-relevant doses of TCDD. It has been previously shown that in utero and lactational TCDD exposure also produces cognitive deficits in adulthood (Haijima et al., 2010). Additionally, whereas the functional implications of our data are conjectural, it is noteworthy to contemplate the relationship between defective AhR signaling and age-related cognitive impairments. Deficiencies in neurogenesis have previously been observed in mouse models of neurodegenerative diseases, including Alzheimer's disease (Winner et al., 2011). Given that TCDD body burdens increase with age (Schecter et al., 2006), it is possible that developmental or adult exposure may be a risk factor for age-related cognitive dysfunction. Therefore, further research is warranted to identify the precise mechanism by which developmental or adult TCDD exposure perturbs hippocampal neurogenesis in order to determine the potential contribution of this ubiquitous environmental chemical in developing age-related cognitive disorders.
Acknowledgments
This work was supported by NIH grants RO1ES016357, T32ES07026, P30ES01247, and T32NS051152. We acknowledge Bryan Thompson and Diana Navarro for technical assistance.
Footnotes
No conflicts of interests exist.
References
- Aasebo IE, Blankvoort S, Tashiro A. Critical maturational period of new neurons in adult dentate gyrus for their involvement in memory formation. Eur J Neurosci. 2011;33:1094–1100. doi: 10.1111/j.1460-9568.2011.07608.x. [DOI] [PubMed] [Google Scholar]
- Ables JL, Decarolis NA, Johnson MA, Rivera PD, Gao Z, Cooper DC, Radtke F, Hsieh J, Eisch AJ. Notch1 is required for maintenance of the reservoir of adult hippocampal stem cells. J Neurosci. 2010;30:10484–10492. doi: 10.1523/JNEUROSCI.4721-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson ML, Sisti HM, Curlik DM, 2nd, Shors TJ. Associative learning increases adult neurogenesis during a critical period. Eur J Neurosci. 2011;33:175–181. doi: 10.1111/j.1460-9568.2010.07486.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baader SL, Sanlioglu S, Berrebi AS, Parker-Thornburg J, Oberdick J. Ectopic overexpression of engrailed-2 in cerebellar Purkinje cells causes restricted cell loss and retarded external germinal layer development at lobule junctions. J Neurosci. 1998;18:1763–1773. doi: 10.1523/JNEUROSCI.18-05-01763.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baader SL, Vogel MW, Sanlioglu S, Zhang X, Oberdick J. Selective disruption of “late onset” sagittal banding patterns by ectopic expression of engrailed-2 in cerebellar Purkinje cells. J Neurosci. 1999;19:5370–5379. doi: 10.1523/JNEUROSCI.19-13-05370.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biebl M, Cooper CM, Winkler J, Kuhn HG. Analysis of neurogenesis and programmed cell death reveals a self-renewing capacity in the adult rat brain. Neurosci Lett. 2000;291:17–20. doi: 10.1016/s0304-3940(00)01368-9. [DOI] [PubMed] [Google Scholar]
- Bock KW, Kohle C. Ah receptor: dioxin-mediated toxic responses as hints to deregulated physiologic functions. Biochem Pharmacol. 2006;72:393–404. doi: 10.1016/j.bcp.2006.01.017. [DOI] [PubMed] [Google Scholar]
- Bunger MK, Glover E, Moran SM, Walisser JA, Lahvis GP, Hsu EL, Bradfield CA. Abnormal liver development and resistance to 2,3,7,8-tetrachlorodibenzo-p-dioxin toxicity in mice carrying a mutation in the DNA-binding domain of the aryl hydrocarbon receptor. Toxicol Sci. 2008;106:83–92. doi: 10.1093/toxsci/kfn149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunger MK, Moran SM, Glover E, Thomae TL, Lahvis GP, Lin BC, Bradfield CA. Resistance to 2,3,7,8-tetrachlorodibenzo-p-dioxin toxicity and abnormal liver development in mice carrying a mutation in the nuclear localization sequence of the aryl hydrocarbon receptor. J Biol Chem. 2003;278:17767–17774. doi: 10.1074/jbc.M209594200. [DOI] [PubMed] [Google Scholar]
- Byers JP, Masters K, Sarver JG, Hassoun EA. Association between the levels of biogenic amines and superoxide anion production in brain regions of rats after subchronic exposure to TCDD. Toxicology. 2006;228:291–298. doi: 10.1016/j.tox.2006.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cameron HA, Gould E. Distinct populations of cells in the adult dentate gyrus undergo mitosis or apoptosis in response to adrenalectomy. J Comp Neurol. 1996;369:56–63. doi: 10.1002/(SICI)1096-9861(19960520)369:1<56::AID-CNE4>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- Cameron HA, McKay RD. Restoring production of hippocampal neurons in old age. Nat Neurosci. 1999;2:894–897. doi: 10.1038/13197. [DOI] [PubMed] [Google Scholar]
- Chahrour M, Zoghbi HY. The story of Rett syndrome: from clinic to neurobiology. Neuron. 2007;56:422–437. doi: 10.1016/j.neuron.2007.10.001. [DOI] [PubMed] [Google Scholar]
- Clelland CD, Choi M, Romberg C, Clemenson GD, Jr, Fragniere A, Tyers P, Jessberger S, Saksida LM, Barker RA, Gage FH, Bussey TJ. A functional role for adult hippocampal neurogenesis in spatial pattern separation. Science. 2009;325:210–213. doi: 10.1126/science.1173215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins LL, Williamson MA, Thompson BD, Dever DP, Gasiewicz TA, Opanashuk LA. 2,3,7,8-Tetracholorodibenzo-p-dioxin exposure disrupts granule neuron precursor maturation in the developing mouse cerebellum. Toxicol Sci. 2008;103:125–136. doi: 10.1093/toxsci/kfn017. [DOI] [PubMed] [Google Scholar]
- Deng W, Saxe MD, Gallina IS, Gage FH. Adult-born hippocampal dentate granule cells undergoing maturation modulate learning and memory in the brain. J Neurosci. 2009;29:13532–13542. doi: 10.1523/JNEUROSCI.3362-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desouza LA, Ladiwala U, Daniel SM, Agashe S, Vaidya RA, Vaidya VA. Thyroid hormone regulates hippocampal neurogenesis in the adult rat brain. Mol Cell Neurosci. 2005;29:414–426. doi: 10.1016/j.mcn.2005.03.010. [DOI] [PubMed] [Google Scholar]
- Goosens KA, Maren S. Contextual and auditory fear conditioning are mediated by the lateral, basal, and central amygdaloid nuclei in rats. Learn Mem. 2001;8:148–155. doi: 10.1101/lm.37601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haijima A, Endo T, Zhang Y, Miyazaki W, Kakeyama M, Tohyama C. In utero and lactational exposure to low doses of chlorinated and brominated dioxins induces deficits in the fear memory of male mice. Neurotoxicology. 2010;31:385–390. doi: 10.1016/j.neuro.2010.04.004. [DOI] [PubMed] [Google Scholar]
- Han G, Ding G, Lou X, Wang X, Han J, Shen H, Zhou Y, Du L. Correlations of PCBs, DIOXIN, and PBDE with TSH in children's blood in areas of computer E-waste recycling. Biomed Environ Sci. 2011;24:112–116. doi: 10.3967/0895-3988.2011.02.004. [DOI] [PubMed] [Google Scholar]
- Harburg GC, Hall FS, Harrist AV, Sora I, Uhl GR, Eisch AJ. Knockout of the mu opioid receptor enhances the survival of adult-generated hippocampal granule cell neurons. Neuroscience. 2007;144:77–87. doi: 10.1016/j.neuroscience.2006.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hein AM, Stasko MR, Matousek SB, Scott-McKean JJ, Maier SF, Olschowka JA, Costa AC, O'Banion MK. Sustained hippocampal IL-1beta overexpression impairs contextual and spatial memory in transgenic mice. Brain Behav Immun. 2010;24:243–253. doi: 10.1016/j.bbi.2009.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inokuchi K. Adult neurogenesis and modulation of neural circuit function. Curr Opin Neurobiol. 2011;21:360–364. doi: 10.1016/j.conb.2011.02.006. [DOI] [PubMed] [Google Scholar]
- Jessberger S, Clark RE, Broadbent NJ, Clemenson GD, Jr, Consiglio A, Lie DC, Squire LR, Gage FH. Dentate gyrus-specific knockdown of adult neurogenesis impairs spatial and object recognition memory in adult rats. Learn Mem. 2009;16:147–154. doi: 10.1101/lm.1172609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kachaylo EM, Yarushkin AA, Pustylnyak VO. Constitutive androstane receptor activation by 2,4,6-triphenyldioxane-1,3 suppresses the expression of the gluconeogenic genes. Eur J Pharmacol. 2012;679:139–143. doi: 10.1016/j.ejphar.2012.01.007. [DOI] [PubMed] [Google Scholar]
- Kapoor R, van Hogerlinden M, Wallis K, Ghosh H, Nordstrom K, Vennstrom B, Vaidya VA. Unliganded thyroid hormone receptor alpha1 impairs adult hippocampal neurogenesis. FASEB J. 2010;24:4793–4805. doi: 10.1096/fj.10-161802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kee N, Teixeira CM, Wang AH, Frankland PW. Preferential incorporation of adult-generated granule cells into spatial memory networks in the dentate gyrus. Nat Neurosci. 2007;10:355–362. doi: 10.1038/nn1847. [DOI] [PubMed] [Google Scholar]
- Kim MD, Jan LY, Jan YN. The bHLH-PAS protein Spineless is necessary for the diversification of dendrite morphology of Drosophila dendritic arborization neurons. Genes Dev. 2006;20:2806–2819. doi: 10.1101/gad.1459706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuemerle B, Zanjani H, Joyner A, Herrup K. Pattern deformities and cell loss in Engrailed-2 mutant mice suggest two separate patterning events during cerebellar development. J Neurosci. 1997;17:7881–7889. doi: 10.1523/JNEUROSCI.17-20-07881.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagace DC, Donovan MH, DeCarolis NA, Farnbauch LA, Malhotra S, Berton O, Nestler EJ, Krishnan V, Eisch AJ. Adult hippocampal neurogenesis is functionally important for stress-induced social avoidance. Proc Natl Acad Sci U S A. 2010;107:4436–4441. doi: 10.1073/pnas.0910072107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lahvis GP, Bradfield CA. Ahr null alleles: distinctive or different? Biochem Pharmacol. 1998;56:781–787. doi: 10.1016/s0006-2952(98)00134-8. [DOI] [PubMed] [Google Scholar]
- Latchney SE, Lioy DT, Henry EC, Gasiewicz TA, Strathmann FG, Mayer-Proschel M, Opanashuk LA. Neural precursor cell proliferation is disrupted through activation of the aryl hydrocarbon receptor by 2,3,7,8-tetrachlorodibenzo-p-dioxin. Stem Cells Dev. 2011;20:313–326. doi: 10.1089/scd.2009.0529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leijs MM, ten Tusscher GW, Olie K, van Teunenbroek T, van Aalderen WM, de Voogt P, Vulsma T, Bartonova A, Krayer von Krauss M, Mosoiu C, Riojas-Rodriguez H, Calamandrei G, Koppe JG. Thyroid hormone metabolism and environmental chemical exposure. Environ Health. 2012;11(Suppl 1):S10. doi: 10.1186/1476-069X-11-S1-S10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandyam CD, Harburg GC, Eisch AJ. Determination of key aspects of precursor cell proliferation, cell cycle length and kinetics in the adult mouse subgranular zone. Neuroscience. 2007;146:108–122. doi: 10.1016/j.neuroscience.2006.12.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandyam CD, Norris RD, Eisch AJ. Chronic morphine induces premature mitosis of proliferating cells in the adult mouse subgranular zone. J Neurosci Res. 2004;76:783–794. doi: 10.1002/jnr.20090. [DOI] [PubMed] [Google Scholar]
- Marin-Burgin A, Mongiat LA, Pardi MB, Schinder AF. Unique processing during a period of high excitation/inhibition balance in adult-born neurons. Science. 2012;335:1238–1242. doi: 10.1126/science.1214956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mato Y, Suzuki N, Katatani N, Kadokami K, Nakano T, Nakayama S, Sekii H, Komoto S, Miyake S, Morita M. Human intake of PCDDs, PCDFs, and dioxin like PCBs in Japan, 2001 and 2002. Chemosphere. 2007;67:S247–255. doi: 10.1016/j.chemosphere.2006.05.105. [DOI] [PubMed] [Google Scholar]
- Matousek SB, Hein AM, Shaftel SS, Olschowka JA, Kyrkanides S, O'Banion MK. Cyclooxygenase-1 mediates prostaglandin E(2) elevation and contextual memory impairment in a model of sustained hippocampal interleukin-1beta expression. J Neurochem. 2010;114:247–258. doi: 10.1111/j.1471-4159.2010.06759.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millen KJ, Wurst W, Herrup K, Joyner AL. Abnormal embryonic cerebellar development and patterning of postnatal foliation in two mouse Engrailed-2 mutants. Development. 1994;120:695–706. doi: 10.1242/dev.120.3.695. [DOI] [PubMed] [Google Scholar]
- Onishi BK, Xavier GF. Contextual, but not auditory, fear conditioning is disrupted by neurotoxic selective lesion of the basal nucleus of amygdala in rats. Neurobiol Learn Mem. 2010;93:165–174. doi: 10.1016/j.nlm.2009.09.007. [DOI] [PubMed] [Google Scholar]
- Pendergast JS, Yamazaki S. The mammalian circadian system is resistant to dioxin. J Biol Rhythms. 2012;27:156–163. doi: 10.1177/0748730411434405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen SL, Curran MA, Marconi SA, Carpenter CD, Lubbers LS, McAbee MD. Distribution of mRNAs encoding the arylhydrocarbon receptor, arylhydrocarbon receptor nuclear translocator, and arylhydrocarbon receptor nuclear translocator-2 in the rat brain and brainstem. J Comp Neurol. 2000;427:428–439. doi: 10.1002/1096-9861(20001120)427:3<428::aid-cne9>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- Pleasure SJ, Collins AE, Lowenstein DH. Unique expression patterns of cell fate molecules delineate sequential stages of dentate gyrus development. J Neurosci. 2000;20:6095–6105. doi: 10.1523/JNEUROSCI.20-16-06095.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollenz RS. The mechanism of AH receptor protein down-regulation (degradation) and its impact on AH receptor-mediated gene regulation. Chem Biol Interact. 2002;141:41–61. doi: 10.1016/s0009-2797(02)00065-0. [DOI] [PubMed] [Google Scholar]
- Pollenz RS, Buggy C. Ligand-dependent and -independent degradation of the human aryl hydrocarbon receptor (hAHR) in cell culture models. Chem Biol Interact. 2006;164:49–59. doi: 10.1016/j.cbi.2006.08.014. [DOI] [PubMed] [Google Scholar]
- Puga A, Tomlinson CR, Xia Y. Ah receptor signals cross-talk with multiple developmental pathways. Biochem Pharmacol. 2005;69:199–207. doi: 10.1016/j.bcp.2004.06.043. [DOI] [PubMed] [Google Scholar]
- Qin H, Powell-Coffman JA. The Caenorhabditis elegans aryl hydrocarbon receptor, AHR-1, regulates neuronal development. Dev Biol. 2004;270:64–75. doi: 10.1016/j.ydbio.2004.02.004. [DOI] [PubMed] [Google Scholar]
- Quintana FJ, Basso AS, Iglesias AH, Korn T, Farez MF, Bettelli E, Caccamo M, Oukka M, Weiner HL. Control of T(reg) and T(H)17 cell differentiation by the aryl hydrocarbon receptor. Nature. 2008;453:65–71. doi: 10.1038/nature06880. [DOI] [PubMed] [Google Scholar]
- Sahay A, Scobie KN, Hill AS, O'Carroll CM, Kheirbek MA, Burghardt NS, Fenton AA, Dranovsky A, Hen R. Increasing adult hippocampal neurogenesis is sufficient to improve pattern separation. Nature. 2011;472:466–470. doi: 10.1038/nature09817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sartor MA, Schnekenburger M, Marlowe JL, Reichard JF, Wang Y, Fan Y, Ma C, Karyala S, Halbleib D, Liu X, Medvedovic M, Puga A. Genomewide analysis of aryl hydrocarbon receptor binding targets reveals an extensive array of gene clusters that control morphogenetic and developmental programs. Environ Health Perspect. 2009:117, 1139–1146. doi: 10.1289/ehp.0800485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato S, Shirakawa H, Tomita S, Ohsaki Y, Haketa K, Tooi O, Santo N, Tohkin M, Furukawa Y, Gonzalez FJ, Komai M. Low-dose dioxins alter gene expression related to cholesterol biosynthesis, lipogenesis, and glucose metabolism through the aryl hydrocarbon receptor-mediated pathway in mouse liver. Toxicol Appl Pharmacol. 2008;229:10–19. doi: 10.1016/j.taap.2007.12.029. [DOI] [PubMed] [Google Scholar]
- Schecter A, Birnbaum L, Ryan JJ, Constable JD. Dioxins: an overview. Environ Res. 2006;101:419–428. doi: 10.1016/j.envres.2005.12.003. [DOI] [PubMed] [Google Scholar]
- Sierra A, Encinas JM, Deudero JJ, Chancey JH, Enikolopov G, Overstreet-Wadiche LS, Tsirka SE, Maletic-Savatic M. Microglia shape adult hippocampal neurogenesis through apoptosis-coupled phagocytosis. Cell Stem Cell. 2010;7:483–495. doi: 10.1016/j.stem.2010.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyder JS, Soumier A, Brewer M, Pickel J, Cameron HA. Adult hippocampal neurogenesis buffers stress responses and depressive behaviour. Nature. 2011;476:458–461. doi: 10.1038/nature10287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spaulding SW. The possible roles of environmental factors and the aryl hydrocarbon receptor in the prevalence of thyroid diseases in Vietnam era veterans. Curr Opin Endocrinol Diabetes Obes. 2011;18:315–320. doi: 10.1097/MED.0b013e32834a8764. [DOI] [PubMed] [Google Scholar]
- Stone SS, Teixeira CM, Zaslavsky K, Wheeler AL, Martinez-Canabal A, Wang AH, Sakaguchi M, Lozano AM, Frankland PW. Functional convergence of developmentally and adult-generated granule cells in dentate gyrus circuits supporting hippocampus-dependent memory. Hippocampus. 2011;21:1348–1362. doi: 10.1002/hipo.20845. [DOI] [PubMed] [Google Scholar]
- Takeda T, Taura J, Fujii M, Koga T, Ishii Y, Yamada H. The effect of maternal exposure to dioxin on fetal steroidogenesis in the steroidogenic organs. Fukuoka Igaku Zasshi. 2011;102:159–166. [PubMed] [Google Scholar]
- Tanida T, Warita K, Ishihara K, Fukui S, Mitsuhashi T, Sugawara T, Tabuchi Y, Nanmori T, Qi WM, Inamoto T, Yokoyama T, Kitagawa H, Hoshi N. Fetal and neonatal exposure to three typical environmental chemicals with different mechanisms of action: mixed exposure to phenol, phthalate, and dioxin cancels the effects of sole exposure on mouse midbrain dopaminergic nuclei. Toxicol Lett. 2009;189:40–47. doi: 10.1016/j.toxlet.2009.04.005. [DOI] [PubMed] [Google Scholar]
- Tanos R, Patel RD, Murray IA, Smith PB, Patterson AD, Perdew GH. Aryl hydrocarbon receptor regulates the cholesterol biosynthetic pathway in a dioxin response element-independent manner. Hepatology. 2012;55:1994–2004. doi: 10.1002/hep.25571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Praag H, Schinder AF, Christie BR, Toni N, Palmer TD, Gage FH. Functional neurogenesis in the adult hippocampus. Nature. 2002;415:1030–1034. doi: 10.1038/4151030a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veldhoen M, Hirota K, Westendorf AM, Buer J, Dumoutier L, Renauld JC, Stockinger B. The aryl hydrocarbon receptor links TH17-cell-mediated autoimmunity to environmental toxins. Nature. 2008;453:106–109. doi: 10.1038/nature06881. [DOI] [PubMed] [Google Scholar]
- White SS, Birnbaum LS. An overview of the effects of dioxins and dioxin-like compounds on vertebrates, as documented in human and ecological epidemiology. J Environ Sci Health C Environ Carcinog Ecotoxicol Rev. 2009;27:197–211. doi: 10.1080/10590500903310047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson MA, Gasiewicz TA, Opanashuk LA. Aryl hydrocarbon receptor expression and activity in cerebellar granule neuroblasts: implications for development and dioxin neurotoxicity. Toxicol Sci. 2005;83:340–348. doi: 10.1093/toxsci/kfi031. [DOI] [PubMed] [Google Scholar]
- Winner B, Kohl Z, Gage FH. Neurodegenerative disease and adult neurogenesis. Eur J Neurosci. 2011;33:1139–1151. doi: 10.1111/j.1460-9568.2011.07613.x. [DOI] [PubMed] [Google Scholar]
- Zhao C, Deng W, Gage FH. Mechanisms and functional implications of adult neurogenesis. Cell. 2008;132:645–660. doi: 10.1016/j.cell.2008.01.033. [DOI] [PubMed] [Google Scholar]