

Abstract
Objective
We investigated whether, and the extent to which, vascular and degenerative lesions in the brain mediate the association of diabetes with poor cognitive performance.
Methods
This cross-sectional study included 4,206 participants (age>65 years; 57.8% women) of the Age, Gene/Environment Susceptibility–Reykjavik Study. Data were collected through interview, clinical examination, psychological testing, and laboratory tests. The composite scores on memory, information-processing speed, and executive function were derived from a cognitive test battery. Markers of cerebral macrovascular (cortical infarcts), microvascular (subcortical infarcts, cerebral microbleeds, and higher white matter lesion volume), and neurodegenerative (lower gray matter, normal white matter, and total brain tissue volumes) processes were assessed on magnetic resonance images. Mediation models were employed to test the mediating effect of brain lesions on the association of diabetes with cognitive performance controlling for potential confounders.
Results
There were 462 (11.0%) persons with diabetes. Diabetes was significantly associated with lower scores on processing speed and executive function, but not with memory function. Diabetes was significantly associated with all markers of brain pathology. All of these markers were significantly associated with lower scores on memory, processing speed, and executive function. Formal mediation tests suggested that markers of cerebrovascular and degenerative pathology significantly mediated the associations of diabetes with processing speed and executive function.
Interpretation
Diabetes is associated with poor performance on cognitive tests of information-processing speed and executive function. The association is largely mediated by markers of both neurodegeneration and cerebrovascular disease. Older people with diabetes should be monitored for cognitive problems and brain lesions.
Previous research has provided robust evidence linking type 2 diabetes to late-life cognitive impairment, dementia, and Alzheimer disease.1–3 However, the structural brain damage underlying the association is not well understood. Diabetes leads to cerebral macro- and microvascular diseases such as stroke, white matter lesions (WMLs), and cerebral microbleeds (CMBs).4–6 Population-based neuroimaging studies also have suggested an association of diabetes in middle age or later in life with markers of neurodegeneration such as global and medial temporal lobe atrophy,7–9 although clinicopathologic research has been inconsistent in linking diabetes to the burden of neurodegenerative pathology (amyloid plaques and neurofibrillary tangles).10 The diabetes-related pathophysiological mechanisms, such as inflammation, oxidative stress, hyperglycemia, and insulin resistance, have been implicated in atherosclerosis, cerebrovascular disease, and neurodegeneration.11,12 Although brain infarcts and neurodegeneration are associated with dementia, evidence has emerged that cerebral microvascular lesions also are involved in the development, progression, and expression of cognitive impairment and dementia. For instance, CMBs and disruption of white matter integrity have been associated with slowing of processing speed and poor executive function.13,14 Furthermore, follow-up studies have shown that diabetes is associated with accelerated progression of brain atrophy, accompanied by decline in processing speed and executive function.15,16
Collectively, a plausible hypothesis can be generated from extant studies that diabetes is associated with poor cognitive performance, and that the association is mediated by mixed cerebral vascular and degenerative pathology that are associated with diabetes. We examine this hypothesis in the Age, Gene/Environment Susceptibility (AGES)–Reykjavik Study, where brain structure and function are well characterized in a large population-based cohort of older men and women.
Subjects and Methods
Study Population
The study population included participants in the AGES–Reykjavik Study as fully described elsewhere.17 Briefly, the AGES–Reykjavik Study is a continuation of the Reykjavik Study in Iceland. The Reykjavik Study was initiated in 1967 by the Icelandic Heart Association and included men and women born in 1907–1935 and living in the Reykjavik area. During 2002–2006, 5,764 persons randomly chosen from survivors of the Reykjavik Study cohort were re-examined for the AGES–Reykjavik Study. The AGES–Reykjavik Study aimed to investigate genetic and environmental factors and biological mechanisms leading to major clinical and subclinical disorders in old age. As part of the comprehensive assessments at the Reykjavik center, participants completed a questionnaire, undertook a clinical examination, completed a cognitive test battery, and received a structural brain magnetic resonance imaging (MRI) scan.
The AGES–Reykjavik Study was approved by the National Bioethics Committee (VSN-00-063), which acts as the institutional review board for the Icelandic Heart Association, the Icelandic Data Protection Authority, Iceland, and by the institutional review board for the NIH National Institute on Aging in the USA. Written informed consent was obtained from all participants.
MRI Acquisition and Reading Protocol
All eligible participants without contraindications had high-resolution MRI scans on a single research-dedicated 1.5T Signa TwinSpeed System (General Electric Medical Systems, Waukesha, WI).18 The structural image protocol included a T1-weighted 3D spoiled gradient-echo sequence (time to echo [TE]=8 milliseconds; repetition time [TR]=21 milliseconds; flip angle [FA]=30°; field of view [FOV]=240mm; matrix=256 × 256; slice thickness=1.5mm), a proton density/T2-weighted fast spin-echo sequence (TE1=22 milliseconds; TE2=90 milliseconds; TR=3,220 milliseconds; echo train length=8; FA=90°; FOV=220mm; matrix=256 × 256; slice thickness=3mm), a fluid-attenuated inversion recovery (FLAIR) sequence (TE=100 milliseconds; TR=8,000 milliseconds; inversion time=2,000 milliseconds; FA=90°; FOV=220mm; matrix=256 × 256), and a T2*-weighted gradient-echo type echo-planar sequence (TE=50 milliseconds; TR=3,050 milliseconds; FA=90°; FOV=220mm; matrix=256 × 256; slice thickness=3mm). All images were acquired to give full-brain coverage.
Brain images were processed with the validated AGES/Montreal Neurologic Institute pipeline, as fully described elsewhere.18 Briefly, the algorithm segmented the whole brain into gray matter, normal white matter, WMLs, and cerebrospinal fluid (CSF). Total brain tissue volume was computed as the sum of gray matter, normal white matter, and WML volumes; the total intracranial volume was computed as the sum of total brain tissue and CSF volumes.
Brain infarcts, manually assessed by trained readers, were defined as defects of the brain parenchyma with a signal intensity that was isointense to that of CSF on FLAIR, T2-, and proton density (PD)-weighted sequences surrounded by a rim of high signal intensity on FLAIR and PD images. Furthermore, brain infarcts had to have a minimal diameter of 4mm, except for infarcts in the cerebellum, brainstem, and cortex, which had no size criteria. The average inter-rater reliability (weighted kappa) was 0.7 for brain infarcts, and the intrarater reliability was 0.9 in 5% of all MRI scans reread without knowledge of the previous reading.
Cognitive Function Assessment
The cognitive test battery included multiple tests for each of 3 cognitive domains as previously described.19 Briefly, memory function was assessed with a modified version of the California Verbal Learning Test that includes subtests of immediate and delayed recall. Information-processing speed was measured with the Digit Symbol Substitution Test, the Salthouse Figure Comparison Test, and the Stroop Test parts 1 and 2. Executive function was assessed with the Digits Backward test, a shortened version of the Cambridge Neuropsychological Test Automated Battery spatial working memory test, and the Stroop Test part 3. The composite score for each of the 3 cognitive domains was constructed according to a theoretical grouping of tests, and was computed by converting raw scores to standardized z scores and averaging them across all tests for the domain. The composite scores were derived based on the distribution of cognitive tests in the total study sample. The inter-rater reliability for all tests was excellent (Spearman correlation coefficients range=0.96–0.99).
Assessments of Diabetes and Confounders
At baseline (2002–2006), diabetes was ascertained according to self-reported history of diabetes, use of oral blood glucose-lowering drugs, insulin injection, or fasting blood glucose ≥7.0 mmol/l. Fasting blood samples were taken, and blood glucose was measured in the laboratory of the Icelandic Heart Association. Demographics (age, gender, and education), lifestyle factors (eg, smoking), and medical history (eg, diabetes) were assessed via a questionnaire. Use of medications (eg, antihypertensive and blood glucose-lowering drugs) was ascertained from medication vials brought to the research center. In addition, arterial blood pressure at middle age was measured following standard protocols, and midlife hypertension was defined as blood pressure ≥140/90mmHg or use of antihypertensive drugs.
Analytical Sample
Of the 5,764 participants in the AGES–Reykjavik Study, 761 (13.2%) did not have an MRI scan due to contraindications, refusal, claustrophobia, or only participating in a home visit. An additional 410 subjects did not have the specific images needed for volumetric segmentation or for assessment of brain infarcts. Among those with complete MRI data (n=4,593), we further excluded 387 subjects with missing cognitive data, leaving 4,206 (73.0%) subjects for the current analysis. Compared with persons who were included, those excluded (n=1,558) were older (79.2±6.6 vs 76.2±5.4 years, p<0.01), and more likely to smoke (14.7% vs 11.5%, p<0.01) and to have diabetes (18.4% vs 11.0%, p<0.01), but the 2 groups did not differ significantly by sex, education, midlife hypertension, or mean total cholesterol.
Statistical Analysis
General linear or logistic regression analysis was used to compare characteristics of participants by diabetes status, adjusting for age. We employed mediation models to test the mediating effects of brain pathology variables on the association of diabetes with cognitive function, controlling for age, sex, education, smoking, and midlife hypertension. The mediation model quantifies the degree to which a mediator (B) statistically mediates the association of an independent variable (A) with a dependent variable (C; Fig 1), and provides a formal statistical test of whether B is a significant mediator for the association of A with C.20 The bootstrapping method was used to estimate the 95% confidence interval (CI) of the mediating effect. Seven MRI markers of brain pathology were examined as mediators: 4 markers for cerebrovascular diseases (cortical infarcts, subcortical infarcts, WMLs, and CMBs), and 3 markers for brain degeneration (gray matter, normal white matter, and total brain tissue volumes). We tested the mediating effects in 3 steps; in step 1 the mediating effect of each of the 7 individual MRI markers was examined, in step 2 we examined the mediating effect when either 4 vascular lesion markers or 2 degenerative brain markers (ie, gray matter and normal white matter volumes, but not total brain tissue volume, due to its high multicollinearity with gray matter and white matter volumes) were simultaneously entered into the mediation model, and in step 3 all 6 markers of brain vascular and degenerative lesions were examined together. We used SAS v9.3 for Windows (SAS Institute Inc, Cary, NC) and IBM SPSS Statistics 20 for Windows (IBM SPSS, Chicago, IL) for all analyses.
FIGURE 1.
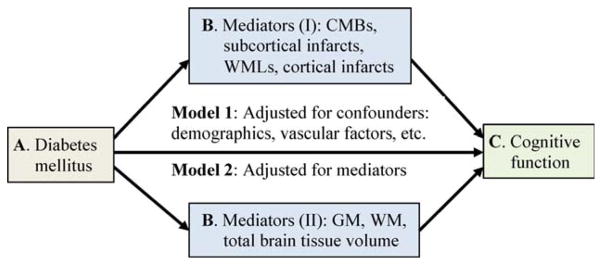
Mediation model. Markers of cerebrovascular disease (cortical infarcts, cerebral microbleeds [CMBs], subcortical infarcts, and white matter lesion [WMLs]) and neurodegenerative lesions (small volumes of gray matter [GM], normal white matter [WM], and total brain tissue) as mediators (B) are hypothesized to mediate the association of diabetes (A) with cognitive function (memory, processing speed, and executive function; C). [Color figure can be viewed in the online issue, which is available at www.annalsofneurology.org.]
Results
Of the 4,206 participants, 462 (11.0%) subjects were ascertained to have diabetes. Controlling for age, people with diabetes were more likely than those without diabetes to be male and to have more years of education, midlife hypertension, brain infarcts, higher WML volume, and lower volumes of gray matter, normal white matter, and total brain tissue, but the 2 groups did not significantly differ in mean age and current smoking (Table 1).
TABLE 1.
Characteristics of Study Participants by Diabetes Status
| Characteristics | Diabetes Mellitus | pa | |
|---|---|---|---|
| No, n =3,744 | Yes, n =462 | ||
| Age, yr | 76.2 (5.4) | 76.0 (5.1) | 0.30 |
| Female sex, No. (%) | 2,249 (60.1) | 207 (44.8) | <0.01 |
| Educational level,b No. (%) | |||
| Elementary school | 886 (23.7) | 86 (18.6) | |
| Middle or high school | 1,856 (49.7) | 244 (52.8) | |
| University or above | 996 (26.6) | 132 (28.6) | 0.05 |
| Current smoking,b No. (%) | 434 (11.6) | 49 (10.6) | 0.45 |
| Midlife hypertension, No. (%) | 943 (25.2) | 215 (46.5) | <0.01 |
| Cortical infarcts, No. (%) | 375 (10.1) | 79 (17.1) | <0.01 |
| Subcortical infarcts, No. (%) | 389 (10.4) | 87 (18.8) | <0.01 |
| Cerebral microbleeds, No. (%) | |||
| No | 3,322 (88.7) | 399 (86.4) | |
| A single | 264 (7.1) | 31 (6.7) | |
| Multiple, ≥2 | 158 (4.2) | 32 (6.9) | 0.02 |
| White matter lesion volumec | 1.3 (1.3) | 1.5 (1.3) | <0.01 |
| Gray matter volumec | 45.2 (3.3) | 44.6 (3.3) | <0.01 |
| Normal white matter volumec | 25.7 (1.9) | 25.1 (1.9) | <0.01 |
| Total brain tissue volumec | 72.2 (3.9) | 71.1 (3.9) | <0.01 |
| Cerebrospinal fluid volumec | 27.8 (3.9) | 28.9 (3.9) | <0.01 |
| Total intracranial volume, ml | 1,498.8 (147.1) | 1,518.4 (151.7) | <0.01 |
Data are mean (standard deviation), unless otherwise indicated.
Probability value is for test of difference between subjects with and without diabetes controlling for age.
Data were missing in 6 subjects for education and 2 subjects for smoking. In subsequent analyses, subjects with missing values were in the referent group.
Data were corrected by total intracranial volume (a percentage of total intracranial volume).
Controlling for age, sex, education, current smoking, and midlife hypertension, diabetes was significantly associated with an increased likelihood of cortical infarcts, subcortical infarcts, multiple (≥2) CMBs, higher WML volume, and lower volumes of gray matter, normal white matter, and total brain tissue (Fig 2A). Furthermore, cortical infarcts, subcortical infarcts, multiple CMBs, and higher WML volume were significantly associated with lower scores on memory, processing speed, and executive function, and higher gray matter, normal white matter, and total brain tissue volumes were significantly associated with higher scores in all 3 cognitive domains (see Fig 2A–C).
FIGURE 2.

Associations of diabetes, mediators of brain pathology markers, and cognitive function. (A) Memory function; (B) processing speed; (C) executive function. β-Coefficient (standard error) was derived from the mediation models controlling for age, sex, education, current smoking, and midlife hypertension. CMB=cerebral microbleeds; GM=gray matter; WM=white matter; WML=WM lesion. *p < 0.05, †p < 0.01. [Color figure can be viewed in the online issue, which is available at www.annalsofneurology.org.]
In the models of formal mediation testing, controlling for multiple confounders, diabetes was significantly associated with lower scores on processing speed and executive function, but not memory function (Table 2, total effect of diabetes).
TABLE 2.
Mediating Effects of Individual Markers of Brain Lesions on the Associations of Diabetes with Cognitive Function
| Diabetes and Mediators | Memory Function, β (95% CI)a,b | Processing Speed, β (95% CI)a,b | Executive Function, β (95% CI)a,b |
|---|---|---|---|
| Total effect of diabetes | −0.024 (−0.101 to 0.053) | −0.097 (−0.166 to −0.028) | −0.077 (−0.138 to −0.015) |
| Mediator, cortical infarcts | |||
| Direct effect of diabetes | −0.011 (−0.087 to 0.066) | −0.083 (−0.151 to −0.014) | −0.069 (−0.130 to −0.008) |
| Mediating effect | −0.013 (−0.023 to −0.005) | −0.015 (−0.026 to −0.005) | −0.008 (−0.015 to −0.003) |
| Mediator, subcortical infarcts | |||
| Direct effect of diabetes | −0.012 (−0.089 to 0.065) | −0.084 (−0.153 to −0.015) | −0.068 (−0.129 to −0.007) |
| Mediating effect | −0.012 (−0.021 to −0.005) | −0.013 (−0.024 to −0.005) | −0.009 (−0.016 to −0.003) |
| Mediator, cerebral microbleeds | |||
| Direct effect of diabetes | −0.022 (−0.099 to 0.055) | −0.093 (−0.162 to −0.024) | −0.073 (−0.135 to −0.012) |
| Mediating effect | |||
| A single cerebral microbleed | 0.001 (−0.001 to 0.004) | 0.001 (−0.001 to 0.003) | 0.000 (−0.001 to 0.002) |
| Multiple cerebral microbleeds | −0.003 (−0.008 to 0.001) | −0.005 (−0.011 to 0.001) | −0.004 (−0.009 to 0.001) |
| Mediator, white matter lesion volume | |||
| Direct effect of diabetes | −0.015 (−0.092 to 0.061) | −0.089 (−0.158 to −0.020) | −0.070 (−0.131 to −0.009) |
| Mediating effect | −0.009 (−0.018 to −0.001) | −0.008 (−0.018 to −0.001) | −0.007 (−0.014 to −0.001) |
| Mediator, gray matter volume | |||
| Direct effect of diabetes | −0.012 (−0.088 to 0.065) | −0.076 (−0.144 to −0.008) | −0.058 (−0.118 to 0.002) |
| Mediating effect | −0.012 (−0.022 to −0.004) | −0.021 (−0.037 to −0.008) | −0.019 (−0.032 to −0.006) |
| Mediator, normal white matter volume | |||
| Direct effect of diabetes | −0.015 (−0.092 to 0.062) | −0.063 (−0.132 to 0.005) | −0.043 (−0.104 to 0.017) |
| Mediating effect | −0.009 (−0.017 to −0.002) | −0.034 (−0.048 to −0.021) | −0.034 (−0.046 to −0.021) |
| Mediator, total brain tissue volume | |||
| Direct effect of diabetes | −0.014 (−0.091 to 0.062) | −0.071 (−0.139 to −0.002) | −0.052 (−0.112 to 0.008) |
| Mediating effect | −0.010 (−0.017 to −0.004) | −0.026 (−0.041 to −0.014) | −0.025 (−0.037 to −0.013) |
β and 95% confidence interval (CI) were derived from the mediation models, controlling for age, sex, education, smoking, and midlife hypertension.
β of direct effect of diabetes was the coefficient for an association of diabetes with cognition, after taking into account the effect of a mediator; β of mediating effect was the coefficient for an association of diabetes with cognition that was mediated by the mediator. β of direct effect of diabetes +β of mediating effect =β of total effect of diabetes.
First, when each of the 7 brain MRI markers was, separately, included in the mediation model, there was attenuation of the association of diabetes with memory, processing speed, and executive function (see Table 2). For all MRI markers, except CMBs, there was evidence of significant mediation of the association of diabetes with each of the 3 cognitive domains. When each of the MRI markers, except normal white matter volume, was added to the mediation model, the direct association of diabetes with a lower score on processing speed remained significant. When the 4 vascular markers were entered into separate mediation models, the direct association of diabetes with a lower score on executive function remained significant, but the direct association became statistically nonsignificant when each of the 3 brain atrophic markers (gray matter, normal white matter, and total brain tissue volumes) was entered into separate models.
Second, simultaneous entry of the 4 markers for cerebrovascular lesions showed that, aside from CMBs, all markers were significant mediators in the associations of diabetes with all 3 cognitive domains (Table 3). The direct associations of diabetes with processing speed and executive function became statistically marginal or nonsignificant. When the 2 brain atrophic markers (gray matter and normal white matter volumes) together were included in the mediation model, both were significant mediators in the associations of diabetes with processing speed and executive function. The direct associations of diabetes with processing speed and executive function became nonsignificant. Gray matter volume also played a significant mediating effect on the association of diabetes with memory function.
TABLE 3.
Mediating Effect of Multiple Cerebrovascular or Degenerative Lesions on the Association of Diabetes with Cognitive Function
| Diabetes and Mediators | Memory Function, β (95% CI)a,b | Processing Speed, β (95% CI)a,b | Executive Function, β (95% CI)a,b |
|---|---|---|---|
| Markers of cerebrovascular disease as mediatorsc | |||
| Direct effect of diabetes | 0.001 (−0.075 to 0.078) | −0.068 (−0.137 to 0.000) | −0.059 (−0.120 to 0.002) |
| Mediating effects | |||
| Cortical infarcts | −0.011 (−0.020 to −0.004) | −0.012 (−0.023 to −0.004) | −0.006 (−0.012 to −0.002) |
| Subcortical infarcts | −0.007 (−0.014 to −0.002) | −0.008 (−0.017 to −0.002) | −0.005 (−0.010 to −0.000) |
| Cerebral microbleeds | |||
| A single cerebral microbleed | 0.000 (−0.001 to 0.003) | 0.000 (−0.001 to 0.003) | 0.000 (−0.001 to 0.002) |
| Multiple cerebral microbleeds | 0.000 (−0.004 to 0.003) | −0.002 (−0.007 to 0.001) | −0.002 (−0.006 to 0.001) |
| White matter lesion volume | −0.007 (−0.015 to −0.001) | −0.006 (−0.013 to −0.001) | −0.006 (−0.012 to −0.001) |
| Markers of brain atrophic lesions as mediatorsd | |||
| Direct effect of diabetes | −0.016 (−0.093 to 0.061) | −0.065 (−0.133 to 0.003) | −0.045 (−0.105 to 0.016) |
| Mediating effects | |||
| Gray matter volume | −0.014 (−0.026 to −0.004) | −0.017 (−0.030 to −0.006) | −0.013 (−0.024 to −0.004) |
| Normal white matter volume | 0.006 (−0.002 to 0.017) | −0.016 (−0.027 to −0.007) | −0.019 (−0.030 to −0.010) |
β and 95% confidence interval (CI) were derived from the mediation models, controlling for age, sex, education, smoking, and midlife hypertension.
β of direct effect of diabetes was the coefficient for an association of diabetes with cognition, after taking into account the effects of multiple mediators; β of mediating effect was the coefficient for an association of diabetes with cognition that was mediated by the mediator.
Multiple markers of cerebrovascular disease (cortical infarcts, subcortical infarcts, cerebral microbleeds, and white matter lesions) were simultaneously entered into the mediation model.
Multiple markers of brain atrophic lesions (gray matter and normal white matter volumes) were simultaneously entered into the mediation model.
Third, when all 6 brain lesion markers (4 for cerebrovascular and 2 for atrophic lesions) were entered simultaneously into the mediation model, all markers, except CMBs, generally had a significant mediating effect on the association of diabetes with memory, processing speed, and executive function (data not shown). There remained direct associations of diabetes with processing speed and executive function, although the associations were not statistically significant (processing speed: β=−0.043, 95% CI=−0.111 to 0.025, p=0.21; executive function: β=−0.033, 95% CI=−0.093 to 0.027, p=0.28).
Discussion
The main findings from this population-based study of older men and women are summarized as follows. First, diabetes is associated with markers of cerebral macrovascular, microvascular, and degenerative diseases. Second, cerebrovascular and degenerative lesions are associated with lower scores on memory, processing speed, and executive function. Third, the associations of diabetes with slowing of processing speed and poor executive function are largely mediated by cerebrovascular and degenerative pathology. Finally, the association of diabetes with a lower score on memory function is less clear. By integrating clinical and structural brain imaging data, this study provides evidence suggesting that mixed cerebrovascular and degenerative pathology may underlie cognitive impairment in persons with diabetes.
Previously, population-based studies have linked diabetes to global cognitive impairment, cognitive decline, and amnestic mild cognitive impairment.21–23 In this study, we found that older people with diabetes had lower scores specifically in tests of processing speed and executive function, consistent with several reports from the community-based studies.24–26 By contrast, studies of the association between diabetes and memory function have yielded mixed results.12,21,23 Our data suggest a potential association of diabetes with a lower memory score, which is also mediated by markers of vascular and degenerative pathology in the brain. We also found that, even after accounting for the MRI-detectable cerebrovascular and atrophic changes, diabetes was still associated with poor cognitive function. In this context, these remaining associations may be due to metabolic changes or brain lesions (eg, microinfarcts) that are not captured in the MRI scans but contribute to cognitive impairment.20,27–29
The possible role of brain lesions in the association of diabetes with cognitive function has been rarely investigated in the settings of the general population. The Women’s Health Initiative Study suggested that the association of diabetes with poor global cognitive function was partly accounted for by brain lesion markers (eg, lower gray matter volume and higher ischemic lesion volume).30 Furthermore, the clinically based studies of patients with diabetes suggest that impairments in processing speed and executive function are related to brain microstructural (eg, WMLs and silent infarcts) and neurodegenerative (eg, cortical and subcortical atrophy) lesions.31,32 The recent large-scale clinical study also revealed that the associations of diabetes with poor visual memory, planning, and speed were attenuated substantially when controlling for gray matter volume but not cerebrovascular lesions (eg, infarcts or WMLs), suggesting that neurodegeneration may play a key role in diabetes-related cognitive impairment.33 Our analysis suggests that diabetes-related cognitive impairment in old age is likely to be caused by mixed cerebral vascular and neurodegenerative pathology.
Clinicopathological studies show that subcortical infarcts, WMLs, and CMBs are imaging markers of cerebral microvascular disease,34–36 and losses of total brain tissue, gray matter, or white matter volume are markers of global and local brain neurodegeneration.37–39 However, there are several mechanisms that integrate cerebrovascular and neurodegenerative pathophysiology. First, cerebrovascular lesions could contribute to global and regional brain degeneration; thus, markers of brain atrophy or degeneration also can be indicative of cerebrovascular lesions.20,38,39 Second, Wallerian degeneration secondary to neurodegenerative lesions may lead to cerebral microvascular changes (eg, white matter integrity disruption and WMLs).36 Finally, atherosclerosis and neurodegeneration in the brain, being common pathologies as we age, are likely to converge.40 Chronic hyperglycemia, insulin resistance, inflammation, and oxidative stress owing to diabetes can cause mixed cerebrovascular and neurodegenerative lesions, which in turn may lead to cognitive consequences.11,12 Furthermore, clinicopathological research has linked silent brain infarcts and microinfarcts to cognitive impairment.20,27 Cerebral white matter consists of a complex network of nerve fiber connections; the extent to which the brain can efficiently transfer information across regions depends on the integrity and organization of the network. This is supported by studies showing that WMLs and loss of white matter integrity are associated with impaired processing speed, executive function, and global cognitive function.14,41
This study is based on a large cohort of older people living in the community who are well characterized with regard to extensive health-related factors, medical conditions, and reliable imaging makers of cerebrovascular and neurodegenerative lesions. Furthermore, the 3 cognitive domains were measured with a cognitive test battery consisting of multiple tests, which provided more robust assessments of cognitive function compared to those based only on a single cognitive test. Finally, the mediation models uniquely combine diabetes, markers of brain lesions, and cognitive function, and provide formal testing for their associations. However, limitations of this study need to be kept in mind when interpreting the results. First, the cross-sectional association is subject to biases due to selective survival or participation. Moreover, both poor cognitive function and increased brain pathology could exacerbate the severity of diabetes, which may lead to a vicious cycle.42 Finally, although MRI markers of brain atrophy are correlated with pathological indices of neurodegeneration, as described above, they are not specific for neurodegeneration.38,39 Biomarkers (eg, CSF β-amyloid and τ protein) and positron emission tomography imaging of brain β-amyloid plaques may help clarify neuropathological mechanisms of diabetes in cognitive impairment.
In summary, this study suggests that people with diabetes perform worse than those without on tests of processing speed and executive function, and this difference is largely mediated by cerebrovascular disease and neurodegeneration. Because diabetes is highly prevalent in older people, our study emphasizes the need of routine assessments of cognitive function and brain lesions for older people with diabetes. Furthermore, given that processing speed and executive function are predominantly associated with the frontal lobe, the role of brain region–specific vascular and degenerative lesions in the association of diabetes with specific cognitive domains warrants further investigation. Finally, longitudinal studies that integrate clinical and dynamic brain structural and functional data will help further understand mechanisms linking diabetes to cognitive dysfunction, thus paving the way for early therapeutic intervention against diabetes-associated cognitive decline in old age.
Acknowledgments
The AGES–Reykjavik Study was funded by NIH/National Institute on Aging (NIA) contract N01-AG-12100 and the Intramural Research Program of the NIA, USA and by the Icelandic Heart Association and the Icelandic Parliament, Iceland. C.Q. is supported by grants from the Swedish Research Council and Karolinska Institutet, Stockholm, Sweden. The funding sources had no role in the study design; in data collection, analysis, and interpretation; in writing of the report; or in the decision to submit the report for publication. We thank all participants in the AGES–Reykjavik Study and clinic staff at the Icelandic Heart Association for their invaluable contribution.
Footnotes
Authorship
C.Q. and L.J.L. drafted the manuscript, and made the final decision to submit it for publication. C.Q., Q.Z., and M.E.G. organized and analyzed the data. L.J.L., S.S., M.K.J., O.K., G.E., T.B.H., M.A.v.B., and V.G. contributed to the design of the AGES–Reykjavik Study, data collection, and assessments. All authors have made critical revisions and approved the final version of the manuscript. C.Q. and L.J.L. had full access to all the data in this study.
Potential Conflicts of Interest
Nothing to report.
References
- 1.Launer LJ. Diabetes: vascular or neurodegenerative: an epidemiologic perspective. Stroke. 2009;40(3 suppl):S53–S55. doi: 10.1161/STROKEAHA.108.533075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ritchie K, Carrière I, Ritchie CW, et al. Designing prevention programmes to reduce incidence of dementia: prospective cohort study of modifiable risk factors. BMJ. 2010;341:c3885. doi: 10.1136/bmj.c3885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barnes DE, Yaffe K. The projected effect of risk factor reduction on Alzheimer’s disease prevalence. Lancet Neurol. 2011;10:819–828. doi: 10.1016/S1474-4422(11)70072-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sarwar N, Gao P, Seshasai SR, et al. Diabetes mellitus, fasting blood glucose concentration, and risk of vascular disease: a collaborative meta-analysis of 102 prospective studies. Lancet. 2010;375:2215–2222. doi: 10.1016/S0140-6736(10)60484-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Knopman DS, Penman AD, Catellier DJ, et al. Vascular risk factors and longitudinal changes on brain MRI: the ARIC study. Neurology. 2011;76:1879–1885. doi: 10.1212/WNL.0b013e31821d753f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Qiu C, Cotch MF, Sigurdsson S, et al. Retinal and cerebral microvascular signs and diabetes: the Age, Gene/Environment Susceptibility-Reykjavik study. Diabetes. 2008;57:1645–1650. doi: 10.2337/db07-1455. [DOI] [PubMed] [Google Scholar]
- 7.den Heijer T, Vermeer SE, van Dijk EJ, et al. Type 2 diabetes and atrophy of medial temporal lobe structures on brain MRI. Diabetologia. 2003;46:1604–1610. doi: 10.1007/s00125-003-1235-0. [DOI] [PubMed] [Google Scholar]
- 8.Korf ES, White LR, Scheltens P, Launer LJ. Brain aging in very old men with type 2 diabetes: the Honolulu-Asia Aging Study. Diabetes Care. 2006;29:2268–2274. doi: 10.2337/dc06-0243. [DOI] [PubMed] [Google Scholar]
- 9.Saczynski JS, Siggurdsson S, Jonsson PV, et al. Glycemic status and brain injury in older individuals: the Age Gene/Environment Susceptibility-Reykjavik study. Diabetes Care. 2009;32:1608–1613. doi: 10.2337/dc08-2300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kalaria RN. Neurodegenerative disease: diabetes, microvascular pathology and Alzheimer disease. Nat Rev Neurol. 2009;5:305–306. doi: 10.1038/nrneurol.2009.72. [DOI] [PubMed] [Google Scholar]
- 11.Craft S, Watson GS. Insulin and neurodegenerative disease: shared and specific mechanisms. Lancet Neurol. 2004;3:169–178. doi: 10.1016/S1474-4422(04)00681-7. [DOI] [PubMed] [Google Scholar]
- 12.Strachan MW, Reynolds RM, Marioni RE, Price JF. Cognitive function, dementia and type 2 diabetes mellitus in the elderly. Nat Rev Endocrinol. 2011;7:108–114. doi: 10.1038/nrendo.2010.228. [DOI] [PubMed] [Google Scholar]
- 13.Qiu C, Cotch MF, Sigurdsson S, et al. Cerebral microbleeds, retinopathy, and dementia: the AGES-Reykjavik Study. Neurology. 2010;75:2221–2228. doi: 10.1212/WNL.0b013e3182020349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vernooij MW, Ikram MA, Vrooman HA, et al. White matter microstructural integrity and cognitive function in a general elderly population. Arch Gen Psychiatry. 2009;66:545–553. doi: 10.1001/archgenpsychiatry.2009.5. [DOI] [PubMed] [Google Scholar]
- 15.Debette S, Seshadri S, Beiser A, et al. Midlife vascular risk factor exposure accelerates structural brain aging and cognitive decline. Neurology. 2011;77:461–468. doi: 10.1212/WNL.0b013e318227b227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Falvey CM, Rosano C, Simonsick EM, et al. Macro- and microstructural magnetic resonance imaging indices associated with diabetes among community-dwelling older adults. Diabetes Care. 2013;36:677–682. doi: 10.2337/dc12-0814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Harris T, Launer LJ, Eiriksdottir G, et al. Age, Gene/Environment Susceptibility-Reykjavik Study: multidisciplinary applied phenomics. Am J Epidemiol. 2007;165:1076–1087. doi: 10.1093/aje/kwk115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sigurdsson S, Aspelund T, Forsberg L, et al. Brain tissue volumes in the general population of the elderly: the AGES-Reykjavik Study. Neuroimage. 2012;59:3862–3870. doi: 10.1016/j.neuroimage.2011.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Saczynski JS, Jónsdóttir MK, Garcia M, et al. Cognitive impairment: an increasingly important complication of type 2 diabetes: the Age, Gene/ Environment Susceptibility-Reykjavik Study. Am J Epidemiol. 2008;168:1132–1139. doi: 10.1093/aje/kwn228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Launer LJ, Hughes TM, White LR. Microinfarcts, brain atrophy, and cognitive function: the Honolulu Asia Aging Study Autopsy Study. Ann Neurol. 2011;70:774–780. doi: 10.1002/ana.22520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cukierman T, Gerstein HC, Williamson JD. Cognitive decline and dementia in diabetes—systematic overview of prospective observational studies. Diabetologia. 2005;48:2460–2469. doi: 10.1007/s00125-005-0023-4. [DOI] [PubMed] [Google Scholar]
- 22.Luchsinger JA, Reitz C, Patel B, et al. Relation of diabetes to mild cognitive impairment. Arch Neurol. 2007;64:570–575. doi: 10.1001/archneur.64.4.570. [DOI] [PubMed] [Google Scholar]
- 23.Roberts RO, Knopman DS, Geda YE, et al. Association of diabetes with amnestic and nonamnestic mild cognitive impairment. Alzheimers Dement. 2013 doi: 10.1016/j.jalz.2013.01.001. pii: S1552-5260(13)00013-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yeung SE, Fischer AL, Dixon RA. Exploring effects of type 2 diabetes on cognitive functioning in older adults. Neuropsychology. 2009;23:1–9. doi: 10.1037/a0013849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yaffe K, Falvey C, Hamilton N, et al. Diabetes, glucose control, and 9-year cognitive decline among older adults without dementia. Arch Neurol. 2012;69:1170–1175. doi: 10.1001/archneurol.2012.1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Spauwen PJ, Köhler S, Verhey FR, et al. Effects of type 2 diabetes on 12-year cognitive change: results from the Maastricht Aging Study. Diabetes Care. 2013;36:1554–1561. doi: 10.2337/dc12-0746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Arvanitakis Z, Leurgans SE, Barnes LL, et al. Microinfarct pathology, dementia, and cognitive systems. Stroke. 2011;42:722–727. doi: 10.1161/STROKEAHA.110.595082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Smith EE, Schneider JA, Wardlaw JM, Greenberg SM. Cerebral microinfarcts: the invisible lesions. Lancet Neurol. 2012;11:272–282. doi: 10.1016/S1474-4422(11)70307-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Westover MB, Bianchi MT, Yang C, et al. Estimating cerebral microinfarct burden from autopsy samples. Neurology. 2013;80:1365–1369. doi: 10.1212/WNL.0b013e31828c2f52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Espeland MA, Bryan RN, Goveas JS, et al. Influence of type 2 diabetes on brain volume and changes in brain volumes: results from the Women’s Health Initiative Magnetic Resonance Imaging studies. Diabetes Care. 2013;36:90–97. doi: 10.2337/dc12-0555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Manschot SM, Brands AM, van der Grond J, et al. Brain magnetic resonance imaging correlates of impaired cognition in patients with type 2 diabetes. Diabetes. 2006;55:1106–1113. doi: 10.2337/diabetes.55.04.06.db05-1323. [DOI] [PubMed] [Google Scholar]
- 32.Reijmer YD, Leemans A, Brundel M, et al. Disruption of the cerebral white matter network is related to slowing of information processing speed in patients with type 2 diabetes. Diabetes. 2013;62:2112–2115. doi: 10.2337/db12-1644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Moran C, Phan TG, Chen J, et al. Brain atrophy in type 2 diabetes: regional distribution and influence on cognition. Diabetes Care. 2013;36:4036–4042. doi: 10.2337/dc13-0143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Young VG, Halliday GM, Kril JJ. Neuropathologic correlates of white matter hyperintensities. Neurology. 2008;71:804–811. doi: 10.1212/01.wnl.0000319691.50117.54. [DOI] [PubMed] [Google Scholar]
- 35.Greenberg SM, Vernooij MW, Cordonnier C, et al. Cerebral microbleeds: a guide to detection and interpretation. Lancet Neurol. 2009;8:165–174. doi: 10.1016/S1474-4422(09)70013-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Erten-Lyons D, Woltjer R, Kaye J, et al. Neuropathologic basis of white matter hyperintensity accumulation with advanced age. Neurology. 2013;81:977–983. doi: 10.1212/WNL.0b013e3182a43e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gosche KM, Mortimer JA, Smith CD, et al. Hippocampal volume as an index of Alzheimer neuropathology: findings from the Nun Study. Neurology. 2002;58:1476–1482. doi: 10.1212/wnl.58.10.1476. [DOI] [PubMed] [Google Scholar]
- 38.Jack CR., Jr Alliance for aging research AD biomarkers work group: structural MRI. Neurobiol Aging. 2011;32(suppl 1):S48–S57. doi: 10.1016/j.neurobiolaging.2011.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Erten-Lyons D, Dodge HH, Woltjer R, et al. Neuropathologic basis of age-associated brain atrophy. JAMA Neurol. 2013;70:616–622. doi: 10.1001/jamaneurol.2013.1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Casserly I, Topol E. Convergence of atherosclerosis and Alzheimer’s disease: inflammation, cholesterol, and misfolded proteins. Lancet. 2004;363:1139–1146. doi: 10.1016/S0140-6736(04)15900-X. [DOI] [PubMed] [Google Scholar]
- 41.Prins ND, van Dijk EJ, den Heijer T, et al. Cerebral small-vessel disease and decline in information processing speed, executive function and memory. Brain. 2005;128:2034–2041. doi: 10.1093/brain/awh553. [DOI] [PubMed] [Google Scholar]
- 42.Punthakee Z, Miller ME, Launer LJ, et al. Poor cognitive function and risk of severe hypoglycemia in type 2 diabetes: post hoc epidemiologic analysis of the ACCORD trial. Diabetes Care. 2012;35:787–793. doi: 10.2337/dc11-1855. [DOI] [PMC free article] [PubMed] [Google Scholar]