

Abstract
Transcriptional regulation plays a significant role in the biological response of bacteria to changing environmental conditions. Therefore, mapping transcriptional regulatory networks is an important step not only in understanding how bacteria sense and interpret their environment but also to identify the functions involved in biological responses to specific conditions. Recent experimental and computational developments have facilitated the characterization of regulatory networks on a genome-wide scale in model organisms. In addition, the multiplication of complete genome sequences has encouraged comparative analyses to detect conserved regulatory elements and infer regulatory networks in other less well-studied organisms. However, transcription regulation appears to evolve rapidly, thus, creating challenges for the transfer of knowledge to nonmodel organisms. Nevertheless, the mechanisms and constraints driving the evolution of regulatory networks have been the subjects of numerous analyses, and several models have been proposed. Overall, the contributions of mutations, recombination, and horizontal gene transfer are complex. Finally, the rapid evolution of regulatory networks plays a significant role in the remarkable capacity of bacteria to adapt to new or changing environments. Conversely, the characteristics of environmental niches determine the selective pressures and can shape the structure of regulatory network accordingly.
1. INTRODUCTION
Biological processes are constituted of a number of reactions forming pathways that transform chemical species into useful products. These processes may create complex biomolecules, transform energy from one form to another, or direct the assembly of complex multicellular systems. For example, photosynthesis is a biological process that transforms light energy into chemical bond energy, which can be used subsequently to drive other thermodynamically unfavorable reactions. Biological systems depend on the combination of a large number of processes, which are organized to fulfill synergistic roles. In addition, to survive in nature, biological systems need to be robust to environmental fluctuations. Therefore, the existence and function of networks to sense external or internal conditions and thereby regulate the fluxes of molecules through different pathways are essential to the survival of biological systems.
Because cellular pathways can be controlled at many levels, mapping regulatory networks can also potentially help us understand and identify additional components of critical, yet incompletely characterized, biological processes. For example, one way of regulating flux through a metabolic pathway is to control the abundance of proteins that catalyze individual reactions. Accordingly, if genes are targets of a regulator that controls their expression, then it is possible that these genes encode proteins that somehow contribute to the pathway. The blueprints for proteins and regulatory elements are found in deoxyribonucleic acid (DNA), so it is not surprising that regulation of gene expression is central to the function of biological systems. Consequently, characterizing the architecture of gene regulatory networks can reveal much about the function and the organization of biological processes.
Mapping the gene regulatory networks that control the transcriptional responses of bacteria to various environmental cues has been an ongoing effort for several decades. However, with the complete sequencing of a rapidly growing number of organisms, new experimental and computational methods have been developed to accelerate this process. As a result, large portions of the regulatory networks of model organisms have been reconstructed, spurring new studies on the processes that shape their function. Because living systems need to respond to their environment in a sensible manner, the structure of regulatory networks is subject to selective pressures; thus, they constantly evolve driven by mutations to adapt to the characteristics of internal signaling pathways, or environmental signals, or community interactions. Several recent studies predict that transcriptional regulatory networks evolve faster than the functions of the genes they regulate. Indeed, orthologous genes are not always regulated by orthologous regulators (Luscombe et al., 2004; Madan Babu, Teichmann, & Aravind, 2006; Perez & Groisman, 2009a; Price, Dehal, & Arkin, 2007). Therefore, the processes driving evolution of transcriptional regulatory networks are likely to be different from those shaping evolution of other cellular functions and are currently not well understood.
Here, we review recent studies that begin to elucidate the evolution of transcriptional regulatory networks across bacterial species. We first review the biological concepts that shape these networks and then experimental and computational approaches that can be combined to study their organization and function. We then illustrate how extensive comparative genomics analyses of both transcription factors and their regulons provide new information about the patterns of conservation or divergence of transcriptional regulatory networks and the evolutionary processes that determine the functional composition of regulons. A theme that is emerging from these studies is that the formation and evolution of transcriptional regulatory networks often directly capture the relationship between the different factors that characterize the ecological niches occupied by different bacterial species.
2. GENE EXPRESSION REGULATION AS AN OUTPUT OF SIGNAL TRANSDUCTION PATHWAYS
2.1. Relative timescales of environmental fluctuations and biological responses
Because very few environments on the planet provide a stable set of conditions, it is vital for biological systems to sense and adapt to changes. Consequently, in parallel to the evolution of energetic and metabolic processes, biological systems have evolved elaborate signaling and regulatory systems that program responses to changing conditions. Individual organisms or communities are exposed to environmental fluctuations that can happen on very different timescales. For example, fluctuations of glucose concentration may be very unpredictable in the human gut, while sunlight exposure in the open environment follows a highly predictable diurnal cycle. The time-scales of change illustrated in these two examples pose very different challenges for microbes. In the face of rapid stochastic fluctuations of metabolite concentrations, biological systems have evolved mechanisms that provide fast and dynamic responses in order to properly balance fluxes in the metabolic networks and avoid the buildup of toxic intermediates. Allosteric inhibition of enzymatic activity is a good example of rapid, real-time regulation that is integrated into metabolic pathways (Changeux & Edelstein, 2005). On the other hand, rapid response may not be optimal for systems that change on longer timescales when long-term investments, such as the assembly of large, multienzyme bioenergetic pathways, like the photosynthetic apparatus, need to be robust to short perturbations in light or other environmental cues.
Because the genome is the source of cellular genetic information, regulation at the transcriptional level is both sensible and efficient for medium to long-term regulation. Indeed, repressing the transcription of unnecessary genes saves resources for other necessary functions. However, transcription and translation, which are coupled in bacteria, are processes that require major investments of energy and occur on the timescale of minutes. Therefore, this mode of regulation is only appropriate for adaptation to fluctuations that occur on similar or slower timescales. In bacteria, shifts in metabolic regimes or some stress responses have an important regulatory component at the transcriptional level. Accordingly, characterizing the relevant transcriptional regulatory circuits is expected to help identify genes and functions involved in these processes.
2.2. Three main classes of protein transcription factors
Biological systems have developed a wide array of sensors and effectors to gather information and regulate cellular processes accordingly. At the same time, because regulatory networks are intimately intertwined with the biological processes they regulate, sensory, regulatory, or enzymatic activities can often be combined within one protein. Transcription factors are examples of proteins that can have multiple activities combined within one entity. These DNA-binding proteins are important elements in the control of gene expression and are integral parts of signal transduction pathways. Today, we know of three main classes of bacterial signal transduction pathways that regulate transcription: one- and two-component systems, and alternative sigma factors (Fig. 1.1).
Figure 1.1.

Three main classes of DNA-binding transcription factors. Diagrammatic representation of the three known classes of DNA-binding transcription regulators in bacteria, one-component (A), two-component (B), and group IV sigma factor (C). Transcription factors are depicted to bind DNA in the presence of an activating signal, but in some cases, the regulation is reversed and the transcription factors bind DNA only in the absence of the specific signal.
2.2.1 One-component transcriptional regulatory systems
One-component transcriptional regulatory systems are defined as those containing both a direct environmental input domain and a DNA-binding output domain in one polypeptide chain (Fig. 1.1A) (Ulrich, Koonin, & Zhulin, 2005). The input domain can sense signals through direct binding of a ligand (e.g., cyclic adenosine monophosphate) or cofactor (e.g., iron–sulfur cluster) and affects the activity of the output domain often through changes in conformation or oligomeric state. In their active state, one-component transcription factors often form dimers or higher-order oligomers that are able to bind a specific DNA sequence to control the transcriptional activity of targeted genes. The CRP, FNR, and LacI transcription factors of Escherichia coli are classic examples of one-component systems. The vast majority of identified one-component regulators are cytosolic proteins with no transmembrane domains; thus, they are apparently limited to sense mostly intracellular signals. One-component regulators are often the most abundant types of protein regulators found in bacterial genomes and are believed to be the evolutionary precursors of the more complex two-component systems (Ulrich et al., 2005).
2.2.2 Two-component systems
In classic two-component systems, the sensory and regulatory domains are divided between a histidine kinase sensor and a response regulator (Fig. 1.1B) (Mascher, Helmann, & Unden, 2006; Stock, Robinson, & Goudreau, 2000; Szurmant, White, & Hoch, 2007). The histidine sensor kinase is often a transmembrane protein that consists of an input domain embedded in the cell membrane and a cytoplasmic histidine kinase domain. Similar to the input domain of one-component systems, the input domain of the sensor histidine kinase often senses signals through binding of ligands or via cofactors. The state of the input domain is transmitted via a conformational change to control the activity of the histidine kinase domain, which when active, autophosphorylates a conserved histidine residue. The phosphoryl group is then transferred from the histidine sensor kinase to a conserved aspartate residue on the cognate response regulator. The interaction between histidine kinases and response regulators is determined by specific side chain interactions to provide accurate recognition of cognate response regulators (Skerker et al., 2008). For those response regulators, which act as transcription factors, their phosphorylation state determines whether the protein can oligomerize and bind a target DNA sequence to control gene expression.
The modular design of two-component systems has undoubtedly facilitated evolution of novel regulatory circuits through the recombination of sensory and regulatory domains. In addition, the catalytic nature of the signal transduction by the histidine kinase domain enables the development of complex and dynamical information processing. One remarkable example of complex information processing is the adaptation to signal variation displayed by the chemotaxis system that allows E. coli cells to orient themselves in chemical gradients (Hazelbauer, Falke, & Parkinson, 2008).
2.2.3 Alternative sigma factors
Bacterial sigma factors differ operationally from other transcription factors in several ways: they (i) are dissociable subunits of ribonucleic acid (RNA) polymerase, (ii) direct RNA polymerase to recognize specific bipartite promoter DNA sequences, and (iii) actively promote the process of transcription initiation (Helmann, 2002; Paget & Helmann, 2003; Wösten, 1998). Bacteria possess a main housekeeping sigma subunit that is responsible for transcription of most promoters (similar to E. coli σ70), but various alternative sigma factors have evolved to direct RNA polymerase toward particular sets of promoters (Gruber & Gross, 2003; Kazmierczak, Wiedmann, & Boor, 2005). That is, alternative sigma factors provide an additional strategy to regulate gene transcription activity by altering RNA polymerase affinity for promoter sequences and thereby inducing major changes in global transcription patterns. Alternative sigma factors are distributed into two main families: the σ70-family, a group that exhibits high structural homology to housekeeping sigma factors (Helmann, 2002; Paget & Helmann, 2003; Staroń et al., 2009), and the unrelated σ54-family, which relies on an additional ATP-dependent transcription factor to initiate transcription at the promoter (Buck, Gallegos, Studholme, Guo, & Gralla, 2000; Merrick, 1993). The σ70-family of sigma factors has been divided into four groups based on protein domain structure and amino acid sequence conservation (Paget & Helmann, 2003). Group I sigma factors consist of the housekeeping sigma factors, which, when tested, are known to be essential for viability. Alternative sigma factors in Groups II and III are very similar in amino acid sequence to those in Group I but are often dispensable for growth in laboratory conditions. Nevertheless, the Group II and III alternative sigma factors are involved in various cellular processes, such as development, general stress response, or virulence. Sigma factors in Group IV were only recognized some 20 years ago, but they are now known to be the largest and most diverse group of sigma factors with more than 40 subgroups identified by phylogenetic analysis of sequenced bacterial genomes (Staroń et al., 2009). Group IV sigma factors appear to have limited and specific functions, which often relate to extracytoplasmic stresses (Group IV sigma factors are also referred to as extracytoplasmic factors). In this role, Group IV sigma factors are important components of bacterial signal transduction networks (Staroń et al., 2009).
As in other signal transduction pathways, mechanisms exist to control the activity of Group IV alternative sigma factors. These sigma factors are most often controlled by cognate anti-sigma factor proteins that bind to and sequester the sigma factors until a signal triggers release (Helmann, 1999; Hughes & Mathee, 1998) (Fig. 1.1C). Anti-sigma factors often contain a signal input domain that senses a signal through binding of ligands, interactions with other proteins, or side chain chemistry, and an anti-sigma factor domain that interacts specifically with its cognate sigma factor (Helmann, 1999; Hughes & Mathee, 1998). Anti-sigma factors may also contain transmembrane domains, presumably to transmit an extracellular signal and control activity of their cognate Group IV sigma factor in the cytoplasm. The general design of the sigma/anti-sigma factor system is analogous to the two-component system design except that the signal is somehow transmitted via protein–protein interactions instead of a phosphorylation cascade. Like two-component systems, the modular organization of the different protein domains creates a large combinatorial space accessible through protein domain recombination (Staroń et al., 2009).
2.3. Other regulatory systems
Transcriptional regulation can take many additional forms in bacteria. Every step of the transcription process, as well as the protein translation process, can be regulated. Additional factors that can regulate gene expression at the transcriptional level are DNA methylation patterns (Low, Weyand, & Mahan, 2001), riboswitches (Winkler & Breaker, 2005), chromosome structure (McLeod & Johnson, 2001), small ligands, small RNAs, and RNA-binding proteins (Massé, Majdalani, & Gottesman, 2003). However, these modes of regulation will not be discussed further here.
2.4. Signal integration at the gene promoters
Gene transcription by the RNA polymerase is carried out in three general phases: initiation, elongation, and termination. Regulation of gene expression occurs principally during initiation although regulation in subsequent phases can be significant in many systems (Landick, 2006; Winkler & Breaker, 2005). Initiation is a highly regulated process and constitutes a point where many environmental and cellular signals are integrated as inputs to control RNA polymerase activity (Browning & Busby, 2004). The promoter region contains key sequence elements that determine the molecular dynamics and regulatory logic of transcription initiation. In particular, the organization of these sequence elements relative to each other is critical because the same set of sequence elements can result in opposite regulatory logic if arranged differently (van Hijum, Medema, & Kuipers, 2009).
Many bacterial transcription factors bind DNA near the promoter to interact directly or indirectly with RNA polymerase and modulate the transcriptional output of genes. Known transcription factors recognize specific target DNA sequences between 12 and 30 nucleotides long that often exist as direct sequence repeats or palindromes because many transcription factors bind DNA as homodimers (Rodionov, 2007). The binding affinity of a transcription factor to a particular region of DNA depends on the sum of all interactions with DNA or other proteins. The binding thermodynamic equilibrium to a particular DNA sequence can be approximated relatively well by the sum of the independent contributions of the binding interactions to each of the sequence nucleotides. Therefore, the DNA sequence of a particular binding site can often be translated into quantitative information about the affinity of a transcription factor (van Hijum et al., 2009). However, interactions with other proteins that are localized near the DNA-binding site, such as RNA polymerase or other transcription factors, can have a significant contribution and compensate for weak interactions with the target DNA sequence (Barnard, Wolfe, & Busby, 2004).
The location of the transcription factor binding site relative to the promoter often determines its effect on gene expression. The same transcription factor can stimulate the activity of one promoter while repressing the activity of another (Browning & Busby, 2004; van Hijum et al., 2009). Some well-studied activation mechanisms are driven by protein–protein interactions. In these cases, the transcription factor binding helps recruit and stabilize RNA polymerase at the promoter to initiate transcription. Conversely, if the transcription factor binding site overlaps with the promoter region, the competition between the transcription factor and RNA polymerase for binding DNA can reduce gene expression dramatically. The same negative effect can be achieved if the transcription factor binds downstream of the promoter to block transcription elongation. Therefore, by arranging promoter elements, transcription factor binding sites, and adjusting binding affinities, complex logical operations can be developed to adjust transcriptional output to one or multiple signals. The diversity of functions that can be created by mixing a relatively small number of promoter elements is illustrated by the elaborate regulation of genes involved in E. coli sugar metabolism (Kaplan, Bren, Zaslaver, Dekel, & Alon, 2008).
Many more complex processes have been described in both bacteria and other cells, such as DNA looping or structural remodeling of the promoter, but for many systems, the position of the transcription factor binding site relative to the promoter has been used as an indicator for its effect on gene expression (Browning & Busby, 2004; van Hijum et al., 2009). The emerging consensus from these studies is that repressor sites are often found within 60 base pairs upstream or downstream of the transcription initiation site. In contrast, binding sites for transcriptional activators tend to be found between 95 and 35 base pairs upstream of the transcription initiation site. Therefore, this information can be used to infer the control logic of many promoters.
3. MAPPING TRANSCRIPTIONAL REGULATORY NETWORKS
3.1. Regulons and transcriptional regulatory networks
The global patterns of gene expression play a major role in determining the protein content of cells and, ultimately, active cellular processes. Because cellular processes reflect the sum of the activity of many proteins, the regulation of the corresponding sets of genes also requires extensive coordination. Consequently, biological systems rely on extensive regulatory networks that integrate information and directly control the output of gene transcription. Thus it is not surprising that a significant part of the response of biological systems to changes in their environment occurs at the transcriptional level. These regulatory networks need to be characterized to understand how biological systems function in their environment.
Most transcription factors within an organism interact with multiple promoters; thus, they are able to regulate transcription of many target genes. The set of target genes of a particular transcription factor is defined as its regulon. Because genes can be regulated by multiple transcription factors, the regulons of different transcription factors often contain overlapping members. Nevertheless, regulons may be considered as higher-order functional units because the members of a particular regulon eventually relate to the particular signal that controls activity of the transcription factor. Therefore, it is useful to characterize the regulons of transcription factors to understand the set functions that are regulated in response to particular environmental signals.
3.2. Experimental characterization of regulons
Characterizing the components of one or more regulatory networks can be a tedious process. However, the recent advances in DNA sequencing technology that made available full genome sequences for many organisms triggered the development of a collection of genome-based approaches to identify these networks. These approaches, such as whole-genome gene expression analysis, allow researchers to probe the effects of biological perturbations on global transcript levels, hence accelerating the collection of data necessary to map transcriptional regulatory networks (Blais & Dynlacht, 2005; Zhou & Yang, 2006).
3.2.1 Global transcription profiling
The abundance of messenger RNAs can be monitored genome wide using either microarrays of DNA probes, where each labeled transcript hybridizes to a specific set of probes, or high-throughput deep sequencing analysis of transcript-derived cDNA molecules. Using these approaches, researchers can characterize the effect of biological perturbations, such as gene deletion or environmental changes, on the global gene expression pattern of an organism. For example, the regulon of a particular transcription factor may be inferred by identifying genes that have altered transcript levels in conditions where the transcription factor is active versus conditions where it is not active. However, these types of comparisons do not allow researchers to distinguish primary effects from secondary effects on gene regulation since perturbations often have pleiotropic effects.
With the accumulation of datasets from various experimental treatments, more advanced computational approaches were developed to infer networks by clustering genes whose expression patterns were coregulated. For example, clustering techniques were used to compare gene expression profiles across multiple conditions and identify groups of genes that are coexpressed and likely to be coregulated (Quackenbush, 2001). In addition, if a sufficiently large amount of data are used for clustering analysis, it is possible to predict primary and secondary effects on gene expression triggered by experimental treatments. Methods that have been successfully used to discover coregulated genes from expression profiles include principal component analysis, hierarchical clustering, self-organization maps, and K-means clustering (Slonim, 2002).
However, these techniques only offer indirect evidence for the direct regulation of target genes by specific transcription factors. Therefore, the resulting hypotheses need to be validated with additional methods such as in vitro transcription assays, promoter fusions with a reporter gene, or chromatin immunoprecipitation. To determine components and potential overlaps among regulatory networks, high-throughput technologies offer the significant advantage of obtaining genome-wide datasets.
3.2.2 Chromatin immunoprecipitation for protein binding site localization
Chromatin immunoprecipitation followed by hybridization to a chip or high-throughput DNA sequencing (chromatin immunoprecipitation on an oligo microarray chip (ChIP-chip) and chromatin immunoprecipitation followed by high-throughput sequencing (ChIP-seq)) have been used to detect genome-wide protein–DNA interactions in vivo and provide direct evidence for the regulation of genes by a transcription factor (Buck & Lieb, 2004; Mardis, 2007). The result of a ChIP-chip or ChIP-seq experiment consists of a series of enrichment signals distributed over the genomic locations where proteins bind DNA (Buck & Lieb, 2004). Therefore, a single experiment can identify in principle all the binding sites bound by a particular protein under the chosen experimental conditions. The precision in locating binding sites depends on the length distribution of the sheared DNA, the spacing between consecutive probes on the microarray in a ChIP-chip assay, or the depth or coverage of sequence information derived in a ChIP-seq experiment. ChIP analyses are also subject to false-positive (binding events that do not result in changes in gene expression) or false-negative events (the failure to observe binding if growth conditions are not optimized to observe all such events). Therefore, it is also necessary to complement the ChIP data analysis with computational sequence analysis to determine the exact sequence recognized by the targeted DNA-binding protein. On the other hand, ChIP data do not often inform about the biological role of the targeted protein. For example, if we consider a transcription factor, binding to a promoter region may activate, repress, or not affect the transcription activity of the downstream genes depending on the promoter configuration. Therefore, additional experiments, such as genome-wide expression profiling or more traditional in vivo or in vitro analysis of candidate target genes, are necessary to determine the function of a transcription factor at each binding site.
3.3. Reverse engineering transcriptional regulatory networks
The combination of binding site localization and global expression profiling experiments with computational sequence analysis can be used to characterize the regulons of targeted transcription factors. If performed systematically, this approach can help reconstruct large portions of the transcriptional regulatory networks of well-studied model organisms (Blais & Dynlacht, 2005; Bonneau et al., 2007; Yoon, McDermott, Porwollik, McClelland, & Heffron, 2009).
However, making correct predictions about regulatory networks still face many obstacles, even in the best-studied model organisms such as E. coli and Saccharomyces cerevisiae. First, despite the availability of complete genome sequences, a comprehensive list of all possible components of regulatory networks is unavailable because many gene products of unknown functions may participate in signal transduction or gene regulation. For example, elements, such as small regulatory RNAs, have been identified only recently as significant players in global gene expression regulation. These small RNAs can affect messenger RNA transcription, translation, or stability on limited sets of genes or act at the global level (Massé et al., 2003). Therefore, further biochemical and genetic experiments on uncharacterized gene products are necessary to identify all the elements involved in regulatory networks. Second, interactions among cellular components need to be characterized because these interactions determine the network topology, which ultimately controls information flow within cells. However, interactions between components of regulatory networks can take many forms (e.g., protein–protein, DNA–protein, RNA–DNA, small molecules–protein), which make it difficult to develop standardized and automated experimental approaches. Third, the logic of each interaction needs to be determined to understand how the information is processed at each node of the networks (Veiga, Dutta, & Balázsi, 2010). For example, as discussed earlier, the regulatory logic of gene transcription depends on the positions of transcription factor binding sites relative to other promoter elements. Finally, the dynamical behaviors of all the network component interactions represent a fourth layer of information that determines the overall performance of the regulatory networks. At this level, our knowledge is limited to very few well characterized and small systems. For all of the above reasons, the reconstruction of transcriptional regulatory networks remains a substantial undertaking that requires extensive resources.
The development of computational tools has greatly contributed in efforts to reconstruct regulatory networks. For example, databases and visualization tools are important assets to store, manage, explore, and retrieve the rapidly growing amount of data resulting from the systematic use of high-throughput experiments. The Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo/) offers a public repository for data generated by array-based gene expression profiling experiments and provides basic tools to explore and retrieve information. Another database, RegulonDB (http://regulondb.ccg.unam.mx/), aims at gathering and organizing all the information scattered in the published scientific literature that is relevant to E. coli transcriptional regulatory networks. A second task that greatly benefits from computational tools is the recognition of patterns in complex datasets. Examples of algorithms that perform pattern recognition were presented earlier when discussing the discovery of transcription factor binding sites in sets of promoter sequences or clustering of gene expression profiles. Third, computational systems can build quantitative models of biological systems and perform simulations to explore rapidly the outcome of different hypotheses (Sauer, Heinemann, & Zamboni, 2007). Despite these advances, many existing problems, such as the global reconstruction of transcriptional regulatory networks in biological systems or the accurate annotation of protein functions, would greatly benefit from the development of more advanced computational tools.
3.4. Characterization of conserved regulatory networks using comparative genomics
Another source of information, which can potentially facilitate the reconstruction of regulatory networks, is the known or predicted evolutionary relationships between organisms. Indeed, it appears that comparative genomics has utility in such reconstructions since some components and the topology of regulatory networks may be conserved among organisms performing similar biological functions (Rajewsky, Socci, Zapotocky, & Siggia, 2002; Rodionov, 2007). Consequently, comparative genomics can be used to take advantage of the growing number of fully sequenced bacterial genomes to infer or identify regulatory network patterns shared among closely or distantly related species. As of May 2012, there are over 3000 publically available bacterial genome sequences (http://img.jgi.doe.gov/). In addition, large-scale comparative genomic studies may shed some light on the relationship between cellular functions and ecology, as well as the evolutionary mechanisms shaping the architecture of transcriptional regulatory networks (Haft, Selengut, Brinkac, Zafar, & White, 2005; Hughes Martiny & Field, 2005; van Hijum et al., 2009).
3.4.1 Homologues, orthologs, and paralogs
A significant challenge to overcome when performing comparative genomics studies is to identify across genomes which genes share common ancestry (Kuzniar, van Ham, Pongor, & Leunissen, 2008). This task is not trivial because bacteria have relatively high rates of mutation, recombination, gene duplication, or horizontal DNA transfer between species (Boto, 2010; Didelot & Maiden, 2010; Hudson, Bergthorsson, & Ochman, 2003; Tago et al., 2005). Nevertheless, because protein coding and regulatory sequences are under selective pressure to maintain their biological functions, genes sharing common histories can often be identified based on sequence similarity either at the DNA or at the deduced amino acid level. Genes that have descended from a common ancestor are defined as homologues. Orthologs are homologues that were separated by speciation events, whereas paralogs are homologues generated by gene duplication. Homologues do not necessarily maintain the same biological function depending on the selective pressures experienced by different species, while it is generally believed that orthologs are more likely to maintain their functions if their roles are essential to the cell. Paralogs are not under the same selective pressure because with two copies of the same gene per organism, one copy is likely to diverge, while divergence of the other copy is limited to ensure it fulfills the required function. It is extremely difficult to distinguish between orthologs and paralogs using sequence information only because, in addition to functional divergence, multiple gene duplications, gene losses, or horizontal transfers may have occurred since the original gene duplication event.
All sequence comparisons rely on alignments that calculate the degree of similarity between sequences. Different approaches have been developed to detect sequence homology and build accurate sequence alignments (e.g., BLAST, Altschul, Gish, Miller, Myers, & Lipman, 1990; HMMER, Eddy, 1998; CLUSTAL W, Thompson, Higgins, & Gibson, 1994; MUSCLE, Edgar, 2004). Once the distance between sequences has been calculated, different algorithms can be used to organize genes in functionally related groups. One of the simplest ways to construct such families group genes that are best reciprocal matches between pairs of genomes (reciprocal best Blast hits) (Moreno-Hagelsieb & Latimer, 2008). Unfortunately, this method performs poorly when comparing distantly related organisms because reciprocity breaks down if gene duplication is followed by gene loss. A similar but more stringent approach considers the triangular relationship between genes from two or more species (Cluster of Orthologous Groups) (Tatusov, Koonin, & Lipman, 1997). Alternatively, more sophisticated approaches have been developed to represent the relationship between sequences using a graph structure, which is then analyzed to detect densely connected subgraphs representing functionally related gene families (OrthoMCL, Li, Stoeckert, & Roos, 2003; TribeMCL, Enright, van Dongen, & Ouzounis, 2002). When comparing different approaches, it appears that groups of orthologs determined by OrthoMCL currently achieve the best balance between sensitivity and specificity (Chen, Mackey, Vermunt, & Roos, 2007).
To date, algorithms that aim to detect orthologs across species use only information from protein sequences, but additional information may be relevant. For example, synteny (the physical colocation of genes in the genome) may provide information about the gene’s evolutionary history, as well as its function in the context of other genes, because genes participating to the same pathway are often organized in operons (Rocha, 2008). In addition, the regulation of a particular gene is an integral part of the gene function; therefore, homologous genes that perform identical functions across species are expected to be regulated similarly (Rodionov, 2007). Therefore, supplementing sequence similarity data with information from genomic and regulatory contexts may improve the prediction of orthologs and paralogs across genomes. Conversely, more accurate predictions of orthologs could benefit analyses aimed at reconstructing the evolutionary history of transcriptional regulatory networks with respect to biological functions (Francke, Siezen, & Teusink, 2005).
3.4.2 Detecting conserved regulatory sequences
Selective pressure to maintain function also applies to regulatory DNA sequences found in the promoter regions of genes or operons. Consequently, it has been observed that regulatory DNA sequences are more likely to be conserved across related species than the surrounding nonfunctional sequences (Cliften et al., 2003). This observation prompted efforts to use collections of homologous promoter regions across species to detect functional sequence elements (also called phylogenetic footprinting) (Blanchette & Tompa, 2002). This approach is similar to the detection of shared regulatory sequences in groups of coregulated genes within one genome, but it has the advantage that if only a few target genes are known in one organism the collection of promoter sequences containing a particular regulatory sequence can be supplemented by the promoter sequences of orthologous genes across genomes. As expression of genes for transcription factors is often autoregulated in bacteria, this approach also has the ability to help assign binding sequences to their corresponding transcription factor.
Phylogenetic footprinting algorithms aim to detect overrepresented sequence elements in a collection of promoter sequences. However, if the evolutionary distance between homologous promoter sequences is short and only few mutations occurred, then it is difficult to distinguish between functional and nonfunctional sequences. Conversely, if the evolutionary distance is too great, then it is possible that the regulatory network changed and nonhomologous transcription factors regulate homologous target genes or that homologous transcription factors recognize different binding sequences. Either of these events would limit the ability of phylogenetic footprint to identify correctly the components and architecture of a given regulatory network (Baumbach, 2010). For example, the transcription factor LexA, which is broadly conserved across bacteria, has evolved to recognize completely unrelated sequences between Bacillus subtilis and E. coli, even though LexA still regulates functions related to DNA damage in both species (Erill et al., 2004). Therefore, to accommodate evolutionary history, sequence detection algorithms have incorporated phylogenetic information and evolution models to increase sensitivity and specificity (Micro-FootPrinter, Neph & Tompa, 2006; PhyME, Sinha, 2007; and PhyloGibbs, Siddharthan, Siggia, & van Nimwegen, 2005).
3.4.3 Operon predictions
In bacteria, genes are often transcribed in polycistronic messenger RNA; thus, several consecutive genes can be under the control of only one promoter. A set of cotranscribed genes is defined as an operon. The existence of operons provides a way for bacteria to ensure that expression of genes participating to the same biological process is coordinated (Price, Huang, Arkin, & Alm, 2005). While the existence of operons can help researchers identify related functions (Overbeek, Fonstein, D’Souza, Pusch, & Maltsev, 1999), the inability to predict correctly operons can pose a problem when trying to computationally predict promoter regions in genome sequences. Indeed, large regions containing other coding or transcribed sequences (small RNA, etc.) may separate a gene from its promoter. In addition, the systematic experimental determination of the operon structure of one genome is not trivial. Therefore, this information is not available for most sequenced bacterial genomes (the most extensive datasets available are for E. coli http://regulondb.ccg.unam.mx/ and B. subtilis http://dbtbs.hgc.jp/).
Computational tools to predict operons in genomic sequences have been developed to resolve this problem (Brouwer, Kuipers, & van Hijum, 2008). The main sources of information used by these algorithms are experimental evidence, regulatory sequences, intergenic distances, functional relation, or phylogenetic conservation. However, it appears that a small intergenic distance is by far the best indicator to predict if two consecutive genes are cotranscribed (Brouwer et al., 2008). Operon predictions for many sequenced bacterial genomes are available (http://csbl1.bmb.uga.edu/OperonDB/DOOR.php, Mao, Dam, Chou, Olman, & Xu, 2009, http://www.microbesonline.org/operons/, Price et al., 2005).
4. FACTORS SHAPING THE FUNCTIONAL COMPOSITION OF REGULONS
The selective pressures shaping the composition of regulons are a priori rather clear. Genes encoding functions that are part of the same pathways or structures often need to be coregulated, and in bacteria, these genes are often organized in operons to ensure coordinate transcription (Price et al., 2005). In addition, it is essential for cells to regulate functions according to the appropriate environmental signals. Therefore, regulatory networks have evolved to connect genes and the functions they encode to the appropriate environmental signals. This notion has been confirmed numerous times by characterizing transcriptional regulatory networks and target genes. However, the relationships between signals and cellular functions are not always simple and direct because several additional factors may play a role in the evolution of regulatory networks. For example, epistatic interactions between genetic mutations can create unpredictable phenotypes and complicate the relationship between the selective pressure and the structure of regulatory networks. A study analyzed the global gene expression profiles after the deletion of the crp gene, which encodes a global regulator in E. coli, in two strains that were evolved independently in a controlled environment and in their common ancestor. The results revealed that even though new parallel epistatic interactions evolved as a result of the defined environmental conditions, 20,000 generations were sufficient to observe significant divergence in the composition of the CRP regulon with no mutations found in the crp gene itself (Cooper, Remold, Lenski, & Schneider, 2008).
4.1. Signal integration
It is often beneficial for organisms to regulate expression of proteins according to multiple signals. For example, the E. coli catabolite repression system induces the expression of the lactose operon only if lactose is present and glucose is absent from the environment, presumably because glucose is easier to metabolize than lactose (Deutscher, 2008). Catabolite repression requires the coordinated activity of two transcriptions factors, CRP and LacI, at the promoter of the lac operon. Therefore, the lac operon is connected to two signaling pathways that respond to the absence of glucose and the presence of lactose.
The control logic of gene expression has been extensively studied at many bacterial promoters. From these studies, it appears that different logical functions can be achieved from a variety of mechanisms, but as a result, genes often belong to more than one regulon (see reviews Alon, 2007; Browning & Busby, 2004; Cases & de Lorenzo, 2005; Janga & Collado-Vides, 2007; van Hijum et al., 2009). Overall, the benefits of integrating multiple signals drives the elaboration of intertwined regulatory networks resulting in regulon overlap and regulatory cascades that can make it difficult to identify a direct relationship between the signals and the regulated functions.
In addition, free-living bacteria found in rich and fluctuating environments are presumably expected to be able to sense many signals to respond accordingly to numerous sources of stress. The analysis of protein families in bacterial genomes indicates that the number of regulators scales with the square of the total number of genes found in the genome in general (Nimwegen, 2006). However, the genome of bacteria living in complex environments, such as soil, seems to be enriched for transcription factors beyond what is expected from the general correlation (Cases, de Lorenzo, & Ouzounis, 2003).
4.2. Signal correlation
In some environments, signals or physical factors may vary in a correlated manner, and thus, two or more signals may convey equivalent information about changes in the environment. Actually, correlations between signals can be exploited by organisms to infer the state of factors in the environment that they may not be able to sense. For example, bacteria are not able to see and count the number of sister cells present in their immediate surroundings, but they can estimate the concentrations of diffusible small metabolites. Therefore, as the concentration of excreted metabolites increases with population density, bacteria evolved mechanisms to use the concentration of particular metabolites as a proxy to infer the number of neighboring cells (Miller & Bassler, 2001).
Predicting changes in the environment can be another way to take advantage of signal correlation. For example, when E. coli experiences an elevation in temperature, genes encoding for functions involved in aerobic respiration were downregulated (Tagkopoulos, Liu, & Tavazoie, 2008). The connection between temperature- and oxygen-related functions presumably reflects the covariation of these two factors when E. coli makes the transition between the outside world and the mammalian gut. Therefore, E. coli cells are able to predict changes in oxygen levels by monitoring temperature variations. This hypothesis is supported by the fact that the coregulation of these two different functions is rapidly lost when E. coli cells evolve in a novel-controlled environment (Tagkopoulos et al., 2008). Similarly, E. coli induces genes necessary for maltose metabolism in the presence of lactose, but not vice versa. The induction of the maltose operon is lost when E. coli is evolved in an environment constantly high in lactose with no maltose (Mitchell et al., 2009). A follow-up mathematical modeling analysis demonstrates the benefit of this type of unidirectional predictive regulation in certain environmental conditions (Mitchell& Pilpel, 2011). Anticipatory behavior has been observed in several other organisms. For example, Vibrio cholerae expresses genes associated with fitness in aquatic environments while still in the late stage of infection of the human host (Schild et al., 2007). Candida albicans induces genes involved in stress responses in the presence of glucose, which is presumably an indication that the cell successfully infected the blood stream. In Caulobacter crescentus, gene profiling analysis revealed that addition of xylose in the growth medium induced the expression of exoenzymes associated with plant polymer degradation, indicating that the presence of xylose is associated with the presence of other plant material (Hottes et al., 2004).
The regulatory mechanisms evolved by cells to achieve predictive behavior have not been completely elucidated yet, but in Rhodobacter sphaeroides, genes involved in aerobic respiration and genes involved in photosynthesis are directly regulated by the same regulator, FnrL (Dufour, Kiley, & Donohue, 2010). The presence of photosynthetic functions under the control of a common transcription factor whose activity is regulated in an oxygen-dependent manner may reflect the covariation of oxygen and light in the natural environment of R. sphaeroides. This is another example of direct regulation of distinct functions by a common regulator that likely represents an effective and easy to evolve strategy to achieve associative learning. In conclusion, signal correlation may cause the placement of different cellular functions under the control of the same transcription factor and eventually confound the direct functional relationship between signals and regulated genes.
4.3. The concept of core and extended regulons
As discussed above, selective pressures tend to force a direct functional relationship between signals and regulated genes while particular environmental factors may complicate this relationship. Therefore, the regulon composition of an individual regulator is expected to be specific to environmental conditions and, thus, vary across bacterial species with different lifestyles. Indeed, the results of the comparative genomics analysis of the FnrL regulon across ecologically diverse bacteria support this model (Dufour et al., 2010). For example, the R. sphaeroides FnrL regulon includes functions involved in aerobic respiration and photosynthesis. However, only functions related to aerobic or anaerobic respiration are conserved in the regulon of FnrL orthologs across species, even among other photosynthetic α-proteobacteria. Interestingly, the conserved functions found in the so-called core FnrL regulon are directly related to oxygen availability, which is the signal regulating FnrL activity. On the other hand, the functions present in the extended part of the FnrL regulon in R. sphaeroides reflect a specific adaptation of this bacterium to its environment.
Another example of this concept is provided by studying the E. coli alternative sigma factor RpoE that is activated by cell envelope stress. A comparative analysis of the E. coli σE regulon in nine γ-proteobacteria revealed the existence of a core regulon that contains functions involved in the synthesis and maintenance of lipopolysaccharide and outer membrane porins, which are functions directly related to the inducing stress (Rhodius, Suh, Nonaka, West, & Gross, 2005). At the same time, the extended RpoE regulon in E. coli comprises functions related to pathogenesis or symbiosis, indicating that envelope stress is also an indicator of host interactions for this bacterium. In addition, in several species of the Enterobacteriaceae family, the PhoP transcription factor, which responds to Mg2+ levels, regulates not only genes necessary for adaptation to limiting levels of Mg2+ but also, in only some species, genes involved in pathogenesis functions (Perez et al., 2009). Other comparative genomics studies uncovered similar patterns for regulon conservation across related species (Nonaka, Blankschien, Herman, Gross, & Rhodius, 2006; Oliver, Orsi, Wiedmann, & Boor, 2010; Perez & Groisman, 2009b; Swingle et al., 2008).
In conclusion, several comparative genomics studies of transcription factor regulons support the idea that regulons comprise different sets of functions adapted to correlated signals, which are specific to ecological niches. However, only functions directly related to the signal relayed by the transcription factor are conserved across related species because correlated signals are different in each ecological niche (Fig. 1.2).
Figure 1.2.
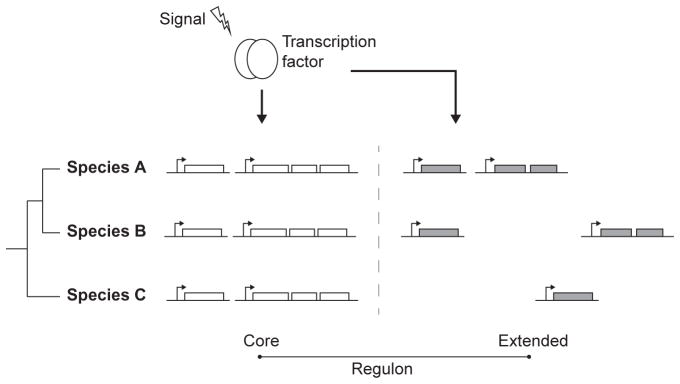
The core and extended regulon structure of orthologous transcription factors. Diagrammatic representation of three species sharing orthologous transcription factors and variable sets of target genes. Target genes that are conserved across most or all species constitute the so-called core regulon. The remaining target genes that are variably conserved across species constitute the extended regulon. Functions encoded in the core regulon are usually directly related to the signal that activates the transcription factor. Functions in the extended regulon are likely to represent particular adaptation of species to their ecological niche.
4.4. The dynamics of transcription factor binding sites
Several recent analyses suggest that transcriptional regulatory networks evolve more rapidly than the functions they control (Dufour et al., 2010; Lozada-Chavez, Janga, & Collado-Vides, 2006; Madan Babu et al., 2006; Price et al., 2007). Indeed, as discussed above, the extended part of different regulons is often not conserved even between very closely related species. Several factors may underlie the capacity of bacteria to rewire their transcriptional regulatory networks over a short evolutionary time (van Hijum et al., 2009; Wang, Wang, & Qian, 2011). For example, analyses of gene promoters revealed that, even in the absence of apparent changes in the regulatory network architecture, transcription factor binding sites still experience significant turnover (Doniger & Fay, 2007; Huang, Nevins, & Ohler, 2007). Binding site turnover occurs when a new transcription factor target site appears next to the original binding site because of random mutations; then, elements of the original binding site evolve to a point where it is no longer recognized by the original protein. This observation indicates that a relatively high rate of mutation in noncoding genomic regions (estimated to be ~10−9–10−10 mutation per cell per generation; Hudson et al., 2003; Tago et al., 2005) creates and destroys transcription factor binding sites frequently. Additional studies showed that spurious binding sites in promoter regions appear frequently but are under strong selection (Froula & Francino, 2007; Hahn, Stajich, & Wray, 2003). Finally, the high rates of duplication, recombination, and transposition in bacterial genome may also contribute significantly to rapid changes in the distribution of existing transcription factor binding sites throughout the genome. For example, the insertion of IS elements into the promoter of flhDC, which encodes for the master regulator of motility in E. coli, occurs at high frequency and allows the adaptive evolution of higher motility in soft agar by disrupting promoter repression (Barker, Prüß, & Matsumura, 2004). Other examples of adaptive evolution resulting from promoter modification by transposable DNA sequences have been documented (Jaurin & Normark, 1983; Podglajen, Breuil, & Collatz, 1994).
4.5. Regulons evolve rapidly
The rapid evolution of transcription factor binding sites discussed above, together with the possibility that some transcription factors may control genes involved in distinct functions because of correlations in environmental factors, creates circumstances that can allow rapid adaptive or nonadaptive evolution of transcriptional regulatory networks. For example, experiments demonstrated that great changes in the CRP-dependent expression profiles can be observed in E. coli after only 20,000 generations of directed evolution (Cooper et al., 2008). Therefore, it is not surprising that many comparative analyses found that orthologous bacterial genes are rarely regulated by orthologous regulators (Lozada-Chavez et al., 2006; Madan Babu et al., 2006; Price et al., 2007). However, many analyses have not considered two important aspects of transcriptional regulatory network evolution. First, when assessing the conservation of regulon across species, a distinction must be made between genes in the core regulon versus genes in the extended regulon. Indeed, the composition of the core regulon may evolve more slowly or in parallel with the function of the transcription factor because of the direct functional connection between the core functions and the regulating signal. On the other hand, the composition of the extended regulon may evolve more rapidly to reflect particular correlations in environmental factors. The rapid changes in the extended regulon may underlie the capacity of bacteria for rapid integration of laterally acquired functions and adaptation to new conditions. Second, because transcription factors can have complex evolutionary histories and may evolve to sense different signals, attempting to infer transcription factor functions from comparative sequence analyses is often difficult or can lead to inaccurate predictions when considering distantly related species. Indeed, regulators that appear to be orthologous may in fact have different functions (Price et al., 2007). For example, the CRP–FNR family has many subfamilies represented in α-proteobacteria species that may be hard to differentiate using protein sequence information only without specific knowledge of their biochemistry (Dufour et al., 2010). Therefore, the simultaneous analysis of transcription factors and their associated core regulon may help identify functional divergence more reliably.
5. EVOLUTION OF TRANSCRIPTION FACTOR FUNCTIONS
Although transcription factors are functionally diverse, sequence and structural domain analyses suggest that these proteins can be classified into relatively few homologous groups, indicating that transcription factors with different functions share common origins (Gelfand, 2006; Rodionov, 2007). For example, a phylogenetic analysis of the CRP/FNR family of transcription factors across 87 α-proteobacteria revealed that this superfamily is composed of 19 distinct subfamilies (Dufour et al., 2010). Further analysis of the FNR, FixK, and DNR subfamilies showed that transcription factors from each family have different functions. These results illustrate that transcription factors that share common ancestry can diverge to acquire new functions. Intuitively, several mechanisms can contribute to the evolution of new transcription factor functions, such as point mutations or domain recombination. However, the precise processes underlying the evolution of new transcription factor functions are not well understood yet. Recent studies attempting to shed light on this question using comparative genomics analyses have led to two alternative models to explain the functional divergence of transcription factors (Price, Dehal, & Arkin, 2008; Teichmann & Babu, 2004).
5.1. The duplication and divergence model
The first widely accepted model addressing the functional divergence of transcription factors was proposed to rely on gene duplication followed by functional divergence (Teichmann & Babu, 2004). Proteins encoded in the E. coli genome were grouped into families of homologues based on sequence similarity to identify presumed duplicated genes. Then, the analysis of the network of known regulatory interactions revealed that about a third of all known regulatory interactions between transcription factors and target genes in E. coli were constituted by homologous transcription factors regulating at least one common target gene or one regulator regulating two homologous target genes. In addition, approximately 6% of these regulatory interactions are represented by two homologous transcription factors regulating two homologous target genes, indicating that these homologous regulatory interactions may have been inherited from the simultaneous duplication of the transcription factor and its target genes. For example, in E. coli, the regulators ZntR and CueR, two paralogs of the MerR family, independently regulate transcription of the homologous genes zntA and copA, which encode respectively for zinc and copper transporters (Yamamoto & Ishihama, 2005a, 2005b). Similarly in Salmonella enterica, the related transcription factors, CueR and GolS, show very specific regulation in response to copper and gold stress despite sharing almost identical DNA-binding sites. Only two mutations in the DNA recognition sequence are sufficient to switch specificity between CueR and GolS (Pérez Audero et al., 2010). This example illustrates how very few evolutionary steps can create regulators of different functions. In conclusion, these results support that duplication of transcription factors or target genes, followed by the gain and loss of new regulatory interactions, contributes significantly to the evolution and growth of transcriptional regulatory networks in bacteria (Teichmann & Babu, 2004). In R. sphaeroides, the properties of two homologues of the heat shock sigma factor, RpoHI and RpoHII, may be another example of this evolutionary model. Indeed, the phylogenetic trees of these two homologues suggest that α-proteobacteria possessing two RpoH homologues inherited these two regulators by vertical descent after duplication of an ancestral rpoH gene (Green & Donohue, 2006). Since the proposed duplication event, RpoHI and RpoHII have evolved to fulfill regulatory functions in different stress response pathways, heat shock and singlet oxygen, respectively (Dufour, Landick, & Donohue, 2008; Nuss, Glaeser, & Klug, 2009). Overall, however, the gene duplication and divergence analysis do not take into account the possibility that presumed paralogs may in fact be acquired from lateral gene transfer, which is frequent among bacteria (Boto, 2010), thus, overestimating the contribution from gene duplication.
5.2. The role of lateral gene transfer
A second evolutionary model was proposed by Price et al. (2008) to account for the contribution of lateral gene transfer to the evolution of transcriptional regulatory networks. In this analysis, the authors conducted a phylogenetic analysis of transcription factors found in E. coli and other related γ-proteobacteria to distinguish homologues that were created by gene duplication from homologues acquired through lateral gene transfer (Price et al., 2008). Their results revealed that very few transcription factors have been duplicated in the E. coli lineage, but that transcription factors had a complex history of lateral gene transfers. Furthermore, an analysis of the regulatory interactions suggested that similarities in the regulation of homologous target genes by homologous transcription factors are likely to have arisen by convergent evolution rather than being inherited. This analysis and a more recent protein family analysis pipeline concluded that only a minor part of the E. coli transcriptional regulatory network was created by gene duplication (Price et al., 2008; Treangen & Rocha, 2011). Their model for the evolution of transcription factors proposed that an ancestral transcription factor is transferred to different species in which it acquires a new function; then, the inherited protein is reacquired by the first species through lateral gene transfer, potentially with some associated target genes to facilitate the integration of the xenogenic genes in the recipient regulatory network. In general, horizontal gene transfer appears to be a major contributor to the diversity of function found in bacterial genomes and a significant driver for the adaptation to new ecological niches (Ochman, Lawrence, & Groisman, 2000; Wiedenbeck & Cohan, 2011).
5.3. Extended regulons may facilitate the evolution of new transcription factor functions
The above models do not address the exact process by which a transcription factor may acquire a new function. Because a transcription factor constitutes a link between a signal and a set of biological functions, the evolution of a new function requires that the transcription factor respond to a new signal and that the composition of the associated regulon adapt to the new signal. These steps are unlikely to happen simultaneously; thus, the evolutionary path taken by transcription factors needs to be orchestrated carefully. For example, if the input domain of a transcription factor evolves to respond to a new signal, either from a point mutation or from a domain recombination, then the functions encoded in the original regulon will be regulated by a new signal that may not be relevant. Conversely, if the output domain evolves before the transcription factor acquires a new regulon, a problem still exists. In both cases, it is apparent that independent changes in the input or output domains of transcription factors can lead to a state that does not offer any benefit to the cell; thus, these changes will not be fixed in the population. Therefore, a transcription factor can successfully evolve a new and beneficial function to the cell only if two relatively rare events occur either simultaneously or if the cell can tolerate a transitional transcription factor that does not function properly long enough to allow more genetic changes to occur and reestablish a functional connection between signal and target genes.
Several phylogenetic analyses revealed some aspects of the regulon organization that may play a significant role in the processes underlying the evolution of new transcription factor functions (Dufour et al., 2010, 2008; Perez et al., 2009; Rhodius et al., 2005; Rodionov, Dubchak, Arkin, Alm, & Gelfand, 2004, 2005; Turkarslan et al., 2011). Indeed, the existence of functionally distinct core and extended regulons may provide an evolutionary path that avoids the transition through a nonfunctional state. In this model, correlation between two environmental signals may cause a transcription factor, which is able to sense one signal, to acquire an extended regulon encoding for functions that are relevant to the second signal. In this situation, the two signals convey the same information about the state of the environment and the two sets of functions are coregulated; thus, changes in the input domain that cause the transcription factor to sense the second signal are functionally neutral. Therefore, changes that allow the transcription factor to sense the second signal do not affect the overall function of the transcriptional regulatory network. As a result, the extended part of the regulon becomes the core regulon, and vice versa (Fig. 1.3). Through this process, a transcription factor may acquire a new input signal and a new set of target genes in a stepwise manner without transitioning through a nonfunctional state, which may be subject to purifying selection. It is also conceivable that a duplication of the transcription factor precedes functional divergence. In this scenario, one of the duplicate regulators evolves to sense the second signal and the two regulators are free to split the original composite regulon according to the two distinct sets of functions. Although studies are needed to test this model, analyses of the phylogenetic conservation of members of the CRP/FNR transcription factor family suggest that this process may have occurred in some bacterial species (Dufour et al., 2010). Indeed, a few bacteria, such as Oceanicola batsensis, appear to possess the core regulon associated with FNR-type regulators but no gene encoding for an FNR-type regulator. Instead, these species possess genes encoding for homologues of FNR that are predicted to be unable to sense oxygen because they lack one or more conserved cysteine residues that are required to coordinate an iron–sulfur cluster. The signal sensed by these regulators is unknown, but the transcription networks may have acquired the ability to sense a new signal, thus, a new function, while still maintaining in their regulons genes that define the FNR core regulon. Another phylogenetic analysis done in Archaea revealed that the unusually large expansion in number of the transcription factor B (TFB) family may underlie the rapid adaptation of halophilic species to the very diverse ecological niches found in hypersaline ecosystems (Turkarslan et al., 2011). Members of the TFB family in Halobacterium salinarum have distinct functions but still show large overlaps in their contributions to gene expression profiles under various experimental conditions (Facciotti et al., 2007; Turkarslan et al., 2011). These observations may represent an interesting illustration of the proposed model for the evolution of new transcription factor functions.
Figure 1.3.

Evolution of new transcription factor functions through the extended regulon. Diagrammatic representation of the successive steps in the evolution of new transcription factor functions using the extended regulon as an evolutionary bridge. (A) Selective pressures drive the evolution of regulatory connections between a transcription factor and functions that are necessary for the biological response to the activating signal. (B) Correlation between signals (signals A and B) in a particular environment allows the incorporation into the regulon of additional functions that are relevant to the biological response to signal B. (C) The signal correlation and the presence of an extended regulon allow the transcription factor to evolve a new function without affecting the function of the transcriptional regulatory network. (D) The transcription factor has acquired a new function and is associated with a new core regulon.
6. CONCLUSIONS
One theme that emerged from early comparative genomics studies of transcription factors and their targets was that transcriptional regulatory networks are not well conserved even across closely related species. Accordingly, the evolution of regulatory networks is a very rapid and dynamical process that contributes significantly to the remarkable capacity of bacteria to adapt to new environments. Comparative studies also revealed that this rapid evolution may be driven by various factors, such as gene duplication, genome recombination, horizontal gene transfer, or transcription factor binding site turn over.
Fortunately, the availability of a growing number of genome sequences provides us with more resolving power to generate specific hypothesis about the structure of regulatory networks and their conservation across organisms. Furthermore, technological advances leading to the creation of high-throughput experimental tools have accelerated the validation of network structures in model organisms. However, the rapid evolution of gene regulation poses challenges because knowledge acquired from studies in model organisms may not be directly transferrable to related species.
Another factor that appears to be important in shaping regulatory networks is the nature of the relationship between the environmental variables that characterize the ecological niche of specific organisms. Indeed, covariations of signals may result in networks where different functions are placed under the regulation of a common regulator, thus, confounding the expected connection between one signal, one regulator, and one function. Therefore, it will be critical to integrate ecological information with phylogenetic data to improve the predictive power of transcriptional regulatory network reconstruction.
Finally, it is often not possible to test evolutionary theory in a laboratory or in populations of complex organisms or communities because of the long-timescale on which evolution operates. However, some studies in bacteria have shown that the evolution of transcriptional networks can occur in a few thousand generations. Therefore, directed evolution experiments could be proposed to test the effects of environmental conditions on the architecture of transcriptional regulatory networks. In addition, the rapid progress in DNA sequencing technologies makes monitoring changes in the genome sequence to detect genetic basis of adaptation practically feasible.
Acknowledgments
The work cited in this chapter from the authors’ was supported by grants from the National Institutes of General Medical Sciences (GM075273 to T. J. D.). The Great Lakes Bioenergy Research Center is supported by the Office of Science, Department of Energy (DE-FG02-07ER64495). Y. S. D. was a previous fellow on the Department of Energy BACTER training grant (ER63232-1018220-0007203, DE-FG02-05ER15653), a recipient of a Wisconsin Distinguished Graduate Fellowship from the UW-Madison College of Agricultural and Life Sciences, and the William H. Peterson Predoctoral Fellowship from the UW-Madison Department of Bacteriology. The authors thank Saheed Imam for his comments on this chapter as it was being prepared for publication.
ABBREVIATIONS
- DNA
deoxyribonucleic acid
- RNA
ribonucleic acid
- ChIP-chip
chromatin immunoprecipitation on an oligo microarray chip
- ChIP-seq
chromatin immunoprecipitation followed by high-throughput sequencing
References
- Alon U. Network motifs: Theory and experimental approaches. Nature Reviews. Genetics. 2007;8:450–461. doi: 10.1038/nrg2102. [DOI] [PubMed] [Google Scholar]
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. Journal of Molecular Biology. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- Barker CS, Prüß BM, Matsumura P. Increased motility of Escherichia coli by insertion sequence element integration into the regulatory region of the flhD operon. Journal of Bacteriology. 2004;186:7529–7537. doi: 10.1128/JB.186.22.7529-7537.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnard A, Wolfe A, Busby S. Regulation at complex bacterial promoters: How bacteria use different promoter organizations to produce different regulatory outcomes. Current Opinion in Microbiology. 2004;7:102–108. doi: 10.1016/j.mib.2004.02.011. [DOI] [PubMed] [Google Scholar]
- Baumbach J. On the power and limits of evolutionary conservation—Unraveling bacterial gene regulatory networks. Nucleic Acids Research. 2010;38:7877. doi: 10.1093/nar/gkq699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blais A, Dynlacht BD. Constructing transcriptional regulatory networks. Genes & Development. 2005;19:1499. doi: 10.1101/gad.1325605. [DOI] [PubMed] [Google Scholar]
- Blanchette M, Tompa M. Discovery of regulatory elements by a computational method for phylogenetic footprinting. Genome Research. 2002;12:739–748. doi: 10.1101/gr.6902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonneau R, Facciotti MT, Reiss DJ, Schmid AK, Pan M, Kaur A, et al. A predictive model for transcriptional control of physiology in a free living cell. Cell. 2007;131:1354–1365. doi: 10.1016/j.cell.2007.10.053. [DOI] [PubMed] [Google Scholar]
- Boto L. Horizontal gene transfer in evolution: Facts and challenges. Proceedings of the Royal Society B: Biological Sciences. 2010;277:819. doi: 10.1098/rspb.2009.1679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brouwer RWW, Kuipers OP, van Hijum SAFT. The relative value of operon predictions. Briefings in Bioinformatics. 2008;9:367–375. doi: 10.1093/bib/bbn019. [DOI] [PubMed] [Google Scholar]
- Browning DF, Busby SJW. The regulation of bacterial transcription initiation. Nature Reviews. Microbiology. 2004;2:57–65. doi: 10.1038/nrmicro787. [DOI] [PubMed] [Google Scholar]
- Buck M, Gallegos MT, Studholme DJ, Guo Y, Gralla JD. The bacterial enhancer-dependent σ54 (σN) transcription factor. Journal of Bacteriology. 2000;182:4129–4136. doi: 10.1128/jb.182.15.4129-4136.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buck MJ, Lieb JD. ChIP-chip: Considerations for the design, analysis, and application of genome-wide chromatin immunoprecipitation experiments. Genomics. 2004;83:349–360. doi: 10.1016/j.ygeno.2003.11.004. [DOI] [PubMed] [Google Scholar]
- Cases I, de Lorenzo V. Promoters in the environment: Transcriptional regulation in its natural context. Nature Reviews. Microbiology. 2005;3:105–118. doi: 10.1038/nrmicro1084. [DOI] [PubMed] [Google Scholar]
- Cases I, de Lorenzo V, Ouzounis CA. Transcription regulation and environmental adaptation in bacteria. Trends in Microbiology. 2003;11:248–253. doi: 10.1016/s0966-842x(03)00103-3. [DOI] [PubMed] [Google Scholar]
- Changeux JP, Edelstein SJ. Allosteric mechanisms of signal transduction. Science. 2005;308:1424–1428. doi: 10.1126/science.1108595. [DOI] [PubMed] [Google Scholar]
- Chen F, Mackey AJ, Vermunt JK, Roos DS. Assessing performance of orthology detection strategies applied to eukaryotic genomes. PLoS One. 2007;2:e383. doi: 10.1371/journal.pone.0000383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cliften P, Sudarsanam P, Desikan A, Fulton L, Fulton B, Majors J, et al. Finding functional features in Saccharomyces genomes by phylogenetic footprinting. Science. 2003;301:71–76. doi: 10.1126/science.1084337. [DOI] [PubMed] [Google Scholar]
- Cooper TF, Remold SK, Lenski RE, Schneider D. Expression profiles reveal parallel evolution of epistatic interactions involving the CRP regulon in Escherichia coli. PLoS Genetics. 2008;4:e35. doi: 10.1371/journal.pgen.0040035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deutscher J. The mechanisms of carbon catabolite repression in bacteria. Current Opinion in Microbiology. 2008;11:87–93. doi: 10.1016/j.mib.2008.02.007. [DOI] [PubMed] [Google Scholar]
- Didelot X, Maiden MCJ. Impact of recombination on bacterial evolution. Trends in Microbiology. 2010;18:315–322. doi: 10.1016/j.tim.2010.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doniger SW, Fay JC. Frequent gain and loss of functional transcription factor binding sites. PLoS Computational Biology. 2007;3:e99. doi: 10.1371/journal.pcbi.0030099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dufour YS, Kiley PJ, Donohue TJ. Reconstruction of the core and extended regulons of global transcription factors. PLoS Genetics. 2010;6:e1001027. doi: 10.1371/journal.pgen.1001027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dufour YS, Landick R, Donohue TJ. Organization and evolution of the biological response to singlet oxygen stress. Journal of Molecular Biology. 2008;383:713–730. doi: 10.1016/j.jmb.2008.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eddy SR. Profile hidden Markov models. Bioinformatics. 1998;14:755–763. doi: 10.1093/bioinformatics/14.9.755. [DOI] [PubMed] [Google Scholar]
- Edgar RC. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Research. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enright AJ, van Dongen S, Ouzounis CA. An efficient algorithm for large-scale detection of protein families. Nucleic Acids Research. 2002;30:1575–1584. doi: 10.1093/nar/30.7.1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erill I, Jara M, Salvador N, Escribano M, Campoy S, Barbé J. Differences in LexA regulon structure among proteobacteria through in vivo assisted comparative genomics. Nucleic Acids Research. 2004;32:6617–6626. doi: 10.1093/nar/gkh996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Facciotti MT, Reiss DJ, Pan M, Kaur A, Vuthoori M, Bonneau R, et al. General transcription factor specified global gene regulation in archaea. Proceedings of the National Academy of Sciences. 2007;104:4630. doi: 10.1073/pnas.0611663104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francke C, Siezen RJ, Teusink B. Reconstructing the metabolic network of a bacterium from its genome. Trends in Microbiology. 2005;13:550–558. doi: 10.1016/j.tim.2005.09.001. [DOI] [PubMed] [Google Scholar]
- Froula JL, Francino MP. Selection against spurious promoter motifs correlates with translational efficiency across bacteria. PLoS One. 2007;2:e745. doi: 10.1371/journal.pone.0000745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelfand MS. Evolution of transcriptional regulatory networks in microbial genomes. Current Opinion in Structural Biology. 2006;16:420–429. doi: 10.1016/j.sbi.2006.04.001. [DOI] [PubMed] [Google Scholar]
- Green HA, Donohue TJ. Activity of Rhodobacter sphaeroides RpoHII, a second member of the heat shock sigma factor family. Journal of Bacteriology. 2006;188:5712. doi: 10.1128/JB.00405-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruber TM, Gross CA. Multiple sigma subunits and the partitioning of bacterial transcription space. Annual Review of Microbiology. 2003;57:441–466. doi: 10.1146/annurev.micro.57.030502.090913. [DOI] [PubMed] [Google Scholar]
- Haft DH, Selengut JD, Brinkac LM, Zafar N, White O. Genome properties: A system for the investigation of prokaryotic genetic content for microbiology, genome annotation and comparative genomics. Bioinformatics. 2005;21:293–306. doi: 10.1093/bioinformatics/bti015. [DOI] [PubMed] [Google Scholar]
- Hahn MW, Stajich JE, Wray GA. The effects of selection against spurious transcription factor binding sites. Molecular Biology and Evolution. 2003;20:901–906. doi: 10.1093/molbev/msg096. [DOI] [PubMed] [Google Scholar]
- Hazelbauer GL, Falke JJ, Parkinson JS. Bacterial chemoreceptors: High-performance signaling in networked arrays. Trends in Biochemical Sciences. 2008;33:9–19. doi: 10.1016/j.tibs.2007.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helmann JD. Anti-sigma factors. Current Opinion in Microbiology. 1999;2:135–141. doi: 10.1016/S1369-5274(99)80024-1. [DOI] [PubMed] [Google Scholar]
- Helmann JD. The extracytoplasmic function (ECF) sigma factors. Advances in Microbial Physiology. 2002;46:47–110. doi: 10.1016/s0065-2911(02)46002-x. [DOI] [PubMed] [Google Scholar]
- Hottes AK, Meewan M, Yang D, Arana N, Romero P, McAdams HH, et al. Transcriptional profiling of Caulobacter crescentus during growth on complex and minimal media. Journal of Bacteriology. 2004;186:1448–1461. doi: 10.1128/JB.186.5.1448-1461.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W, Nevins JR, Ohler U. Phylogenetic simulation of promoter evolution: Estimation and modeling of binding site turnover events and assessment of their impact on alignment tools. Genome Biology. 2007;8:R225. doi: 10.1186/gb-2007-8-10-r225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson RE, Bergthorsson U, Ochman H. Transcription increases multiple spontaneous point mutations in Salmonella enterica. Nucleic Acids Research. 2003;31:4517–4522. doi: 10.1093/nar/gkg651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes Martiny JB, Field D. Ecological perspectives on the sequenced genome collection. Ecology Letters. 2005;8:1334–1345. [Google Scholar]
- Hughes KT, Mathee K. The anti-sigma factors. Annual Review of Microbiology. 1998;52:231–286. doi: 10.1146/annurev.micro.52.1.231. [DOI] [PubMed] [Google Scholar]
- Janga SC, Collado-Vides J. Structure and evolution of gene regulatory networks in microbial genomes. Research in Microbiology. 2007;158:787–794. doi: 10.1016/j.resmic.2007.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaurin B, Normark S. Insertion of IS2 creates a novel ampC promoter in Escherichia coli. Cell. 1983;32:809–816. doi: 10.1016/0092-8674(83)90067-3. [DOI] [PubMed] [Google Scholar]
- Kaplan S, Bren A, Zaslaver A, Dekel E, Alon U. Diverse two-dimensional input functions control bacterial sugar genes. Molecular Cell. 2008;29:786–792. doi: 10.1016/j.molcel.2008.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazmierczak MJ, Wiedmann M, Boor KJ. Alternative sigma factors and their roles in bacterial virulence. Microbiology and Molecular Biology Reviews. 2005;69:527–543. doi: 10.1128/MMBR.69.4.527-543.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuzniar A, van Ham RCHJ, Pongor S, Leunissen JAM. The quest for orthologs: Finding the corresponding gene across genomes. Trends in Genetics. 2008;24:539–551. doi: 10.1016/j.tig.2008.08.009. [DOI] [PubMed] [Google Scholar]
- Landick R. The regulatory roles and mechanism of transcriptional pausing. Biochemical Society Transactions. 2006;34:1062–1066. doi: 10.1042/BST0341062. [DOI] [PubMed] [Google Scholar]
- Li L, Stoeckert CJ, Jr, Roos DS. OrthoMCL: Identification of ortholog groups for eukaryotic genomes. Genome Research. 2003;13:2178–2189. doi: 10.1101/gr.1224503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Low DA, Weyand NJ, Mahan MJ. Roles of DNA adenine methylation in regulating bacterial gene expression and virulence. Infection and Immunity. 2001;69:7197–7204. doi: 10.1128/IAI.69.12.7197-7204.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lozada-Chavez I, Janga SC, Collado-Vides J. Bacterial regulatory networks are extremely flexible in evolution. Nucleic Acids Research. 2006;34:3434. doi: 10.1093/nar/gkl423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luscombe NM, Babu MM, Yu H, Snyder M, Teichmann SA, Gerstein M. Genomic analysis of regulatory network dynamics reveals large topological changes. Nature. 2004;431:308–312. doi: 10.1038/nature02782. [DOI] [PubMed] [Google Scholar]
- Madan Babu M, Teichmann SA, Aravind L. Evolutionary dynamics of prokaryotic transcriptional regulatory networks. Journal of Molecular Biology. 2006;358:614–633. doi: 10.1016/j.jmb.2006.02.019. [DOI] [PubMed] [Google Scholar]
- Mao F, Dam P, Chou J, Olman V, Xu Y. DOOR: A database for prokaryotic operons. Nucleic Acids Research. 2009;37:D459–D463. doi: 10.1093/nar/gkn757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mardis ER. ChIP-seq: Welcome to the new frontier. Nature Methods. 2007;4:613–614. doi: 10.1038/nmeth0807-613. [DOI] [PubMed] [Google Scholar]
- Mascher T, Helmann JD, Unden G. Stimulus perception in bacterial signal-transducing histidine kinases. Microbiology and Molecular Biology Reviews. 2006;70:910–938. doi: 10.1128/MMBR.00020-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massé E, Majdalani N, Gottesman S. Regulatory roles for small RNAs in bacteria. Current Opinion in Microbiology. 2003;6:120–124. doi: 10.1016/s1369-5274(03)00027-4. [DOI] [PubMed] [Google Scholar]
- McLeod SM, Johnson RC. Control of transcription by nucleoid proteins. Current Opinion in Microbiology. 2001;4:152–159. doi: 10.1016/s1369-5274(00)00181-8. [DOI] [PubMed] [Google Scholar]
- Merrick M. In a class of its own—The RNA polymerase sigma factor σ54 (σN) Molecular Microbiology. 1993;10:903–909. doi: 10.1111/j.1365-2958.1993.tb00961.x. [DOI] [PubMed] [Google Scholar]
- Miller MB, Bassler BL. Quorum sensing in bacteria. Annual Review of Microbiology. 2001;55:165–199. doi: 10.1146/annurev.micro.55.1.165. [DOI] [PubMed] [Google Scholar]
- Mitchell A, Pilpel Y. A mathematical model for adaptive prediction of environmental changes by microorganisms. Proceedings of the National Academy of Sciences. 2011;108:7271. doi: 10.1073/pnas.1019754108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell A, Romano GH, Groisman B, Yona A, Dekel E, Kupiec M, et al. Adaptive prediction of environmental changes by microorganisms. Nature. 2009;460:220–224. doi: 10.1038/nature08112. [DOI] [PubMed] [Google Scholar]
- Moreno-Hagelsieb G, Latimer K. Choosing BLAST options for better detection of orthologs as reciprocal best hits. Bioinformatics. 2008;24:319–324. doi: 10.1093/bioinformatics/btm585. [DOI] [PubMed] [Google Scholar]
- Neph S, Tompa M. MicroFootPrinter: A tool for phylogenetic footprinting in prokaryotic genomes. Nucleic Acids Research. 2006;34:W366–W368. doi: 10.1093/nar/gkl069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nimwegen E. Scaling laws in the functional content of genomes. In: Koonin WY, EV, Karev GP, editors. Power Laws, Scale-Free Networks and Genome Biology. New York, NY, USA: Springer; 2006. pp. 236–253. [Google Scholar]
- Nonaka G, Blankschien M, Herman C, Gross CA, Rhodius VA. Regulon and promoter analysis of the E. coli heat-shock factor, σ32, reveals a multifaceted cellular response to heat stress. Genes & Development. 2006;20:1776. doi: 10.1101/gad.1428206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nuss AM, Glaeser J, Klug G. RpoHII activates oxidative-stress defense systems and is controlled by RpoE in the singlet oxygen-dependent response in Rhodobacter sphaeroides. Journal of Bacteriology. 2009;191:220–230. doi: 10.1128/JB.00925-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochman H, Lawrence JG, Groisman EA. Lateral gene transfer and the nature of bacterial innovation. Nature. 2000;405:299–304. doi: 10.1038/35012500. [DOI] [PubMed] [Google Scholar]
- Oliver H, Orsi R, Wiedmann M, Boor K. Listeriamonocytogenes σB has a small core regulon and a conserved role in virulence but makes differential contributions to stress tolerance across a diverse collection of strains. Applied and Environmental Microbiology. 2010;76:4216. doi: 10.1128/AEM.00031-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overbeek R, Fonstein M, D’Souza M, Pusch GD, Maltsev N. The use of gene clusters to infer functional coupling. Proceedings of the National Academy of Sciences. 1999;96:2896. doi: 10.1073/pnas.96.6.2896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paget M, Helmann JD. The sigma70family of sigma factors. Genome Biology. 2003;4:203. doi: 10.1186/gb-2003-4-1-203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pérez Audero ME, Podoroska BM, Ibáñez MM, Cauerhff A, Checa SK, Soncini FC. Target transcription binding sites differentiate two groups of MerR monovalent metal ion sensors. Molecular Microbiology. 2010;78:853–865. doi: 10.1111/j.1365-2958.2010.07370.x. [DOI] [PubMed] [Google Scholar]
- Perez JC, Groisman EA. Evolution of transcriptional regulatory circuits in bacteria. Cell. 2009a;138:233–244. doi: 10.1016/j.cell.2009.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez JC, Groisman EA. Transcription factor function and promoter architecture govern the evolution of bacterial regulons. Proceedings of the National Academy of Sciences. 2009b;106:4319. doi: 10.1073/pnas.0810343106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez JC, Shin D, Zwir I, Latifi T, Hadley TJ, Groisman EA. Evolution of a bacterial regulon controlling virulence and Mg2+ homeostasis. PLoS Genetics. 2009;5:e1000428. doi: 10.1371/journal.pgen.1000428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Podglajen I, Breuil J, Collatz E. Insertion of a novel DNA sequence, IS 1186, upstream of the silent carbapenemase gene cfiA, promotes expression of carbapenem resistance in clinical isolates of Bacteroides fragilis. Molecular Microbiology. 1994;12:105–114. doi: 10.1111/j.1365-2958.1994.tb00999.x. [DOI] [PubMed] [Google Scholar]
- Price MN, Dehal PS, Arkin AP. Orthologous transcription factors in bacteria have different functions and regulate different genes. PLoS Computational Biology. 2007;3:e175. doi: 10.1371/journal.pcbi.0030175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price MN, Dehal PS, Arkin AP. Horizontal gene transfer and the evolution of transcriptional regulation in Escherichia coli. Genome Biology. 2008;9:R4. doi: 10.1186/gb-2008-9-1-r4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price MN, Huang KH, Arkin AP, Alm EJ. Operon formation is driven by co-regulation and not by horizontal gene transfer. Genome Research. 2005;15:809–819. doi: 10.1101/gr.3368805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quackenbush J. Computational analysis of microarray data. Nature Reviews. Genetics. 2001;2:418–427. doi: 10.1038/35076576. [DOI] [PubMed] [Google Scholar]
- Rajewsky N, Socci ND, Zapotocky M, Siggia ED. The evolution of DNA regulatory regions for proteo-gamma bacteria by interspecies comparisons. Genome Research. 2002;12:298–308. doi: 10.1101/gr.207502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhodius VA, Suh WC, Nonaka G, West J, Gross CA. Conserved and variable functions of the σE stress response in related genomes. PLoS Biology. 2005;4:e2. doi: 10.1371/journal.pbio.0040002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocha EPC. Evolutionary patterns in prokaryotic genomes. Current Opinion in Microbiology. 2008;11:454–460. doi: 10.1016/j.mib.2008.09.007. [DOI] [PubMed] [Google Scholar]
- Rodionov DA. Comparative genomic reconstruction of transcriptional regulatory networks in bacteria. Chemical Reviews. 2007;107:3467–3497. doi: 10.1021/cr068309+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodionov DA, Dubchak I, Arkin A, Alm E, Gelfand MS. Reconstruction of regulatory and metabolic pathways in metal-reducing delta-proteobacteria. Genome Biology. 2004;5:R90. doi: 10.1186/gb-2004-5-11-r90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodionov DA, Dubchak IL, Arkin AP, Alm EJ, Gelfand MS. Dissimilatory metabolism of nitrogen oxides in bacteria: Comparative reconstruction of transcriptional networks. PLoS Computational Biology. 2005;1:e55. doi: 10.1371/journal.pcbi.0010055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauer U, Heinemann M, Zamboni N. GENETICS: Getting closer to the whole picture. Science’s STKE. 2007;316:550. doi: 10.1126/science.1142502. [DOI] [PubMed] [Google Scholar]
- Schild S, Tamayo R, Nelson EJ, Qadri F, Calderwood SB, Camilli A. Genes induced late in infection increase fitness of Vibrio cholerae after release into the environment. Cell Host & Microbe. 2007;2:264–277. doi: 10.1016/j.chom.2007.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siddharthan R, Siggia ED, van Nimwegen E. PhyloGibbs: A Gibbs sampling motif finder that incorporates phylogeny. PLoS Computational Biology. 2005;1:e67. doi: 10.1371/journal.pcbi.0010067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinha S. PhyME: A software tool for finding motifs in sets of orthologous sequences. Methods in Molecular Biology (Clifton, NJ) 2007;395:309. [PubMed] [Google Scholar]
- Skerker JM, Perchuk BS, Siryaporn A, Lubin EA, Ashenberg O, Goulian M, et al. Rewiring the specificity of two-component signal transduction systems. Cell. 2008;133:1043–1054. doi: 10.1016/j.cell.2008.04.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slonim DK. From patterns to pathways: Gene expression data analysis comes of age. Nature Genetics. 2002;32:502–508. doi: 10.1038/ng1033. [DOI] [PubMed] [Google Scholar]
- Staroń A, Sofia HJ, Dietrich S, Ulrich LE, Liesegang H, Mascher T. The third pillar of bacterial signal transduction: Classification of the extracytoplasmic function (ECF) σ factor protein family. Molecular Microbiology. 2009;74:557–581. doi: 10.1111/j.1365-2958.2009.06870.x. [DOI] [PubMed] [Google Scholar]
- Stock AM, Robinson VL, Goudreau PN. Two-component signal transduction. Annual Review of Biochemistry. 2000;69:183–215. doi: 10.1146/annurev.biochem.69.1.183. [DOI] [PubMed] [Google Scholar]
- Swingle B, Thete D, Moll M, Myers CR, Schneider DJ, Cartinhour S. Characterization of the PvdS-regulated promoter motif in Pseudomonas syringae pv. tomato DC3000 reveals regulon members and insights regarding PvdS function in other pseudomonads. Molecular Microbiology. 2008;68:871–889. doi: 10.1111/j.1365-2958.2008.06209.x. [DOI] [PubMed] [Google Scholar]
- Szurmant H, White RA, Hoch JA. Sensor complexes regulating two-component signal transduction. Current Opinion in Structural Biology. 2007;17:706–715. doi: 10.1016/j.sbi.2007.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tagkopoulos I, Liu YC, Tavazoie S. Predictive behavior within microbial genetic networks. Science. 2008;320:1313. doi: 10.1126/science.1154456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tago Y, Imai M, Ihara M, Atofuji H, Nagata Y, Yamamoto K. Escherichia coli mutator ΔpolA is defective in base mismatch correction: The nature of in vivo DNA replication errors. Journal of Molecular Biology. 2005;351:299–308. doi: 10.1016/j.jmb.2005.06.014. [DOI] [PubMed] [Google Scholar]
- Tatusov RL, Koonin EV, Lipman DJ. A genomic perspective on protein families. Science. 1997;278:631–637. doi: 10.1126/science.278.5338.631. [DOI] [PubMed] [Google Scholar]
- Teichmann SA, Babu MM. Gene regulatory network growth by duplication. Nature Genetics. 2004;36:492–496. doi: 10.1038/ng1340. [DOI] [PubMed] [Google Scholar]
- Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Research. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treangen TJ, Rocha EPC. Horizontal transfer, not duplication, drives the expansion of protein families in prokaryotes. PLoS Genetics. 2011;7:e1001284. doi: 10.1371/journal.pgen.1001284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turkarslan S, Reiss DJ, Gibbins G, Su WL, Pan M, Bare JC, et al. Niche adaptation by expansion and reprogramming of general transcription factors. Molecular Systems Biology. 2011;7:554. doi: 10.1038/msb.2011.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulrich LE, Koonin EV, Zhulin IB. One-component systems dominate signal transduction in prokaryotes. Trends in Microbiology. 2005;13:52–56. doi: 10.1016/j.tim.2004.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Hijum SAFT, Medema MH, Kuipers OP. Mechanisms and evolution of control logic in prokaryotic transcriptional regulation. Microbiology and Molecular Biology Reviews. 2009;73:481. doi: 10.1128/MMBR.00037-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veiga DFT, Dutta B, Balázsi G. Network inference and network response identification: Moving genome-scale data to the next level of biological discovery. Molecular BioSystems. 2010;6:469–480. doi: 10.1039/b916989j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Wang FF, Qian W. Evolutionary rewiring and reprogramming of bacterial transcription regulation. Journal of Genetics and Genomics. 2011;38:279–288. doi: 10.1016/j.jgg.2011.06.001. [DOI] [PubMed] [Google Scholar]
- Wiedenbeck J, Cohan FM. Origins of bacterial diversity through horizontal genetic transfer and adaptation to new ecological niches. FEMS Microbiology Reviews. 2011;35:957–976. doi: 10.1111/j.1574-6976.2011.00292.x. [DOI] [PubMed] [Google Scholar]
- Winkler WC, Breaker RR. Regulation of bacterial gene expression by riboswitches. Annual Review of Microbiology. 2005;59:487–517. doi: 10.1146/annurev.micro.59.030804.121336. [DOI] [PubMed] [Google Scholar]
- Wösten M. Eubacterial sigma-factors. FEMS Microbiology Reviews. 1998;22:127–150. doi: 10.1111/j.1574-6976.1998.tb00364.x. [DOI] [PubMed] [Google Scholar]
- Yamamoto K, Ishihama A. Transcriptional response of Escherichia coli to external copper. Molecular Microbiology. 2005a;56:215–227. doi: 10.1111/j.1365-2958.2005.04532.x. [DOI] [PubMed] [Google Scholar]
- Yamamoto K, Ishihama A. Transcriptional response of Escherichia coli to external zinc. Journal of Bacteriology. 2005b;187:6333. doi: 10.1128/JB.187.18.6333-6340.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon H, McDermott JE, Porwollik S, McClelland M, Heffron F. Coordinated regulation of virulence during systemic infection of Salmonella enterica serovar Typhimurium. PLoS Pathogens. 2009;5:e1000306. doi: 10.1371/journal.ppat.1000306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou D, Yang R. Global analysis of gene transcription regulation in prokaryotes. Cellular and Molecular Life Sciences. 2006;63:2260–2290. doi: 10.1007/s00018-006-6184-6. [DOI] [PMC free article] [PubMed] [Google Scholar]