

Abstract
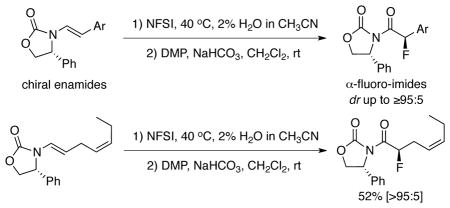
A highly π-facial selective and regioselective fluorination of chiral enamides is described. The reaction involves an enantioselective fluorination exclusively at the electron-rich enamide olefin with N-F reagents such as Selectfluor™ and N-fluoro-benzenesulfonimide [NFSI] accompanied by trapping of the β-fluoro-iminium cationic intermediate with water. The resulting N,O-hemiacetal could be oxidized using Dess-Martin periodinane, leading to an asymmetric sequence for syntheses of chiral α-fluoro-imides and optically enriched α-fluoro-ketones.
The importance of organo-fluorine compounds has been abundantly validated through a broad range of applications in medicinal chemistry and drug development1 as well as material2 and agrochemical sciences.3 The incorporation of fluorine and/or fluorine-containing groups into an organic compound has often provided agents and materials with unique chemical, physical, and biological properties.4 One of the major synthetic challenges in fluoro-organic chemistry is to asymmetrically construct fluorinated stereogenic carbon centers. Differding5 demonstrated the first stoichiometric asymmetric fluorination of β-ketoester enolates with a chiral N-F (N-fluoroamine) reagent in 1988. Given the number of impressive examples of enantioselective fluorinations being reported over the last decade,6 asymmetric fluorinations represent an area of immense interest from the synthetic community. In relevance to our work, Davis7 reported an elegant synthesis of α-fluoro-imides using enolate derived from chiral imide 1 substituted with chiral oxazolidinone auxiliary8 (1→2 in Scheme 1). Our longstanding interest in the chemistry of enamides has drawn us to develop stereoselective fluorination methods using chiral enamides,9 which has remained elusive. Such an approach in combination with an oxidative process (3→4) would represent a complementary approach to Davis’ asymmetric approach to α-fluoro-imides.
Scheme 1.

Stereoselective Synthesis of α-Fluoro-Imides
With the nitrogen atom being substituted with an electron withdrawing functionality, chiral enamides possess superior stability to their enamine counterparts, while maintaining excellent reactivities. This is evident by their recent emergence as another powerful chiral building blocks in organic synthesis.10,11 In particular, chiral enamides have been employed in highly regio- and/or stereoselective Diels–Alder cycloadditions,12 [2 + 2] cycloadditions,13 cy-clopropa-nation,14 epoxidation,15 and halo-etherification.16 We wish to report here a highly regio- and stereoselective fluorination of chiral enamides for the synthesis of chiral α-fluoro-imides and optically enriched α-fluoro-ketones.
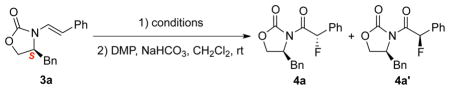
Our efforts commenced with exploring the right fluorination conditions using chiral enamide 3a as the testing enamide, and with Selectflur™,17 N-fluoropyridinium triflate [Py-F]18 and N-fluoro-benzenesulfonimide [NFSI]19 as the fluorinating agent. When reactions were carried out in CH3CN at 40 °C, we found that it was faster with Select-flur™ (entry 1 in Table 1) and much slower with Py-F (entry 2), and that NFSI was the best N-F reagent20 in terms of diastereoselectivity, leading to α-fluoro-imides 4a and 4a’ as an isomeric mixture in 57% yield after DMP oxidation (entry 3).21 We note here that attempts to directly work-up the crude fluorination mixture without any oxidation protocols failed to give us the desired N,O-hemiacetal products, or the corresponding α-fluoro-aldehydes via hydrolysis.
Table 1.
Optimization of the Fluorination Conditionsa
| ||||
|---|---|---|---|---|
| entry | solvent | reagenta | time [h] | yield [%]b [dr]c |
| 1 | CH3CN | Selectfluor | 0.5 | 29 [57:43] |
| 2 | CH3CN | Py-F | 24 | 26 [61:39] |
| 3 | CH3CN | NFSI | 9.5 | 57 [89:11] |
| 4 | anhyd CH3CNd | NFSI | 8 | <10 [85:15] |
| 5 | 2.0 equiv H2O in CH3CN | NFSI | 8 | 43 [89:11] |
| 6 | 1 % H2O in CH3CN | NFSI | 12 | 62 [91:9] |
| 7 | 2 % H2O in CH3CN | NFSI | 12 | 68 [91:9] |
| 8 | 5 % H2O in CH3CN | NFSI | 31 | 29 [91:9] |
| 9e | 2 % H2O in CH CN | NFSI | 6 | 22 [71:29] |
In all reactions, 1.1 equiv N-F reagent was added. Unless otherwise noted, reactions were run at 40 °C. DMP: Dess-Marin Periodinane; NSFI: N-fluoro-benzenesulfonimide; Py-F: N-fluoropyridinium triflate;
Yields were determined by 1HNMR analysis using mesitylene as the internal standard.
Diastereomeric ratios [dr] were determined using 1H or/and 19F NMR spectroscopy.
CH3CN distilled over CaH2.
The reaction temperature is 80 °C.
Most notably, H2O plays a significant role in this reaction. The use of anhydrous CH3CN did afforded comparable diastereoselectivity, but the yield is very low (entry 4). The addition of 2.0 equiv, 1%, 2%, and 5% H2O in the CH3CN appeared to improve the efficiency (entries 5–8) with 2% H2O being the most optimal (entry 7). On the other hand, when we increased the temperature from 40 °C to 80 °C, neither the dr ratio nor the yield was satisfactory.
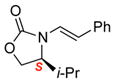
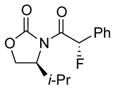
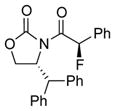
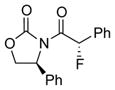
To improve the diastereoselectivity, we examined a range of chiral auxiliaries as shown in Table 2. Enamide 3b with the i-Pr-substituted Evans chiral auxiliary22 gave α-fluoro-imide 4b in 58% yield with a 93:7 dr ratio. Fluorination of chiral enamide 3c substituted with the Sibi auxiliary23 afforded 4c in 51% (entry 2). When using the Ph-substituted Evans auxiliary, fluorinations of 3d and ent-3d with NFSI at 40 °C in 2% H2O in CH3CN led to 4d and ent-4d, respectively, in good yields and essentially as a single diastereomer (entries 3 and 4). X-Ray structures of α-fluoro-imide 4b and ent-4d allowed for unambiguous assignment of both stereochemical and structural integrity (Figure 1).
Table 2.
Effect of the Chiral Auxiliary on Selectivitya
| entry | enamides | products | time [h] | yield [%]b[dr]c |
|---|---|---|---|---|
| 1 |
 3b |
 4b |
14 | 58 [93:7] |
| 2 |
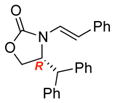 3c |
 4c |
12 | 51 [91:9] |
| 3 |
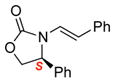 3d |
 4d |
12 | 72 [>95:5] |
| 4 |
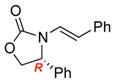 ent-3d |
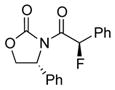 ent-4d |
12 | 63 [>95:5] |
Reaction condition: 1.1 equiv NFSI, 2 % H2O in CH3CN, 40 °C; and then, DMP, NaHCO3, CH2Cl2, rt.
Isolated yields.
Diastereomeric ratios [dr] were determined by 1H or/and 19F NMR spectroscopy.
Figure 1.

X-Ray Structures of α-Fluoro-Imides 4b and ent-4d
Success in finding chiral amides that can provide a high level of diastereoselectivity allowed us to broaden the scope of this fluorination significantly as shown in Table 3. For all examples in which a variety of different R substituents were evaluated, and excellent diastereoselectivities of more than 92:8 ratio and middle to good yields were obtained. It is noteworthy that while electron-donating and electron-withdrawing substitutions groups at aryl does not influence on diastereoselectivity, with a strong electron-withdrawing group, reactions indeed took a much longer time (see entry 3). Lastly, although predictable, these asymmetric fluorinations can indeed be highly regioselective in favor of the electron-rich enamide-olefin as demonstrated with fluorinations of chiral enamides 7 and 9 (Scheme 2).
Table 3.
Stereoselective Fluorination of Chiral Enamides
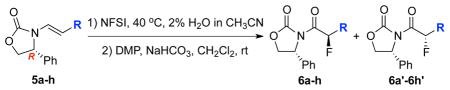
| |||
|---|---|---|---|
| entry | α–fluoro-imides: R = | time [h] | yield [%]a [dr]b |
| 1 | 4-MeC6H4 (6a) | 10 | 67 [94:4] |
| 2 | 4-MeOC6H4 (6b) | 12 | 42 [92:8] |
| 3 | 4-NO2C6H4 (6c) | 140 | 71 [>95:5] |
| 4 | 4-FC6H4 (6d) | 12 | 61 [>95:5] |
| 5 | 4-ClC6H4 (6e) | 12 | 65 [>95:5] |
| 6 | 4-BrC6H4 (6f) | 14 | 69 [95:5] |
| 7 | 2-ClC6H4 (6g) | 12 | 59 [>95:5] |
| 8 | n-Pent (6h) | 11 | 53 [>95:5] |
Isolated yields.
The diastereomeric ratio [dr] was determined by 1H or/and 19F NMR spectroscopy.
Scheme 2.
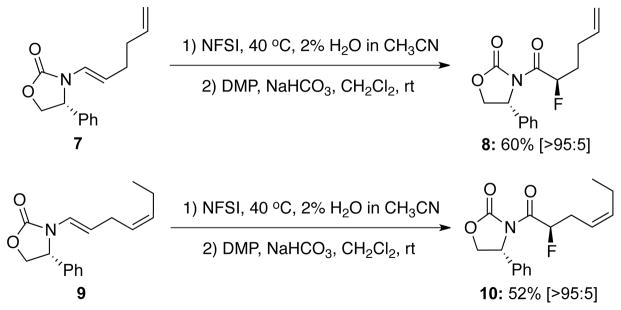
A Regioselective Fluorination
While synthetically this method represents a stereoselective approach for constructing chiral α-fluoro-imides, mechanistically, this fluorination provides some insight to the chemistry of chiral enamides. On the basis of the observed stereo-chemical outcome, a proposed mechanistic model is shown in Scheme 3. We had calculated that the most stable conformation of these chiral enamides being that the chiral oxazolidinone ring is essentially coplanar with alkene to maximize the delocalization of nitrogen lone pair into the olefin.24 With this conformational preference, the chiral oxazolidinone auxiliary plays a distinct role in providing a key facial bias for the fluorine to approach from the Si-face as represented in ent-3d. Subsequent trapping of the N-acyl iminium ion A with water can in principle be stereoselective, but this stereochemical information is lost in the ensuing oxidation.
Scheme 3.
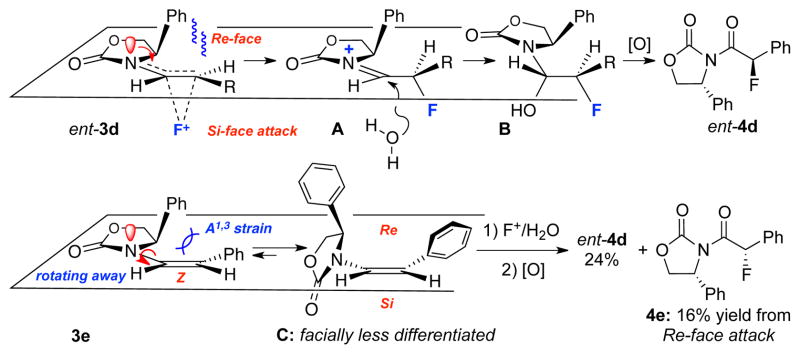
A Proposed Stereochemical Model
On the other hand, for Z-enamides such as 3e, predicted selectivity would be poor based on this conformational model. To alleviate the allylic strain shown in 3e, the chiral oxazolidinone ring needs to rotate away and can no longer be coplanar with the alkene, thereby leading much less differentiated π-faces [see C]. To support this model, we prepared Z-enamide 3e and found that not only is the yield of its fluorination inferior to those of E-enamides, but also diastereoselectivity is diminished significantly.
Lastly, as a useful synthetic application, we examined asymmetric fluorinations of trisubstituted enamides. As shown in Scheme 4, optically enriched 2-fluorocyclohexanone 1325 could be obtained directly from trisubstituted enamide 11 after a simple work up with the fluorinated N,O-hemiacetal intermediate 12 being insufficiently stable for isolation. Likewise, the use of enamide 14 led to α-fluorinated aryl ketone 15 in 78% yield with an er of ≥95:5. This is in direct contrast with our earlier attempts to isolate α-fluoro-aldehydes via hydrolysis of the corresponding N,O-hemiacetal intermediate from fluorinations of disubstituted enamides.
Scheme 4.
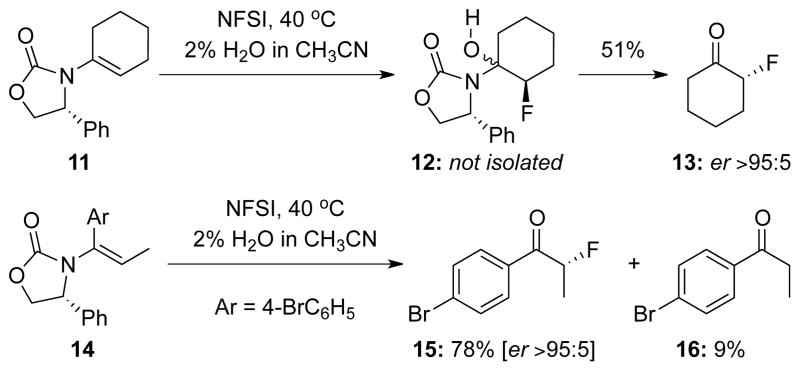
Fluorinations of Trisubstituted Enamides
We have reported an asymmetric synthesis of α-fluoro-imides via fluorinations of chiral enamides. The reaction involves selectively fluorinating the electron-rich enamide olefin using Selectfluor™ and N-fluoro-benzenesulfonimide [NFSI] followed by trapping of β-fluoro-iminium cationic intermediate with water and oxidation of resulting N,O-hemiacetal. From a practical perspective, the reaction is operationally simple, requires inexpensive reagents and mild conditions, and provides chiral α-fluoro-imides and optically enriched α-fluoro-ketones in good yields and high selectivities.
Supplementary Material
Acknowledgments
RPH thanks NIH (GM66055) for funding. YT thanks National Natural Science Foundation of China (Nos. 21172169 and 21172168) and The National Basic Research Project (No. 2014CB932201) for generous funding. We also thank Dr. Hai-Bin Song of the Nankai University for providing X-ray structure and data analysis.
Footnotes
Notes
The authors declare no competing financial interest.
Experimental procedures as well as NMR spectra, characterizations, and X-ray structural files (CIF). This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Liang T, Neumann CN, Ritter T. Angew Chem Int Ed. 2013;52:8214. doi: 10.1002/anie.201206566. [DOI] [PubMed] [Google Scholar]; (b) Böhm HJ, Banner D, Bendels S, Kansy M, Kuhn B, Muller K, Obst-Sander U, Stahl M. ChemBioChem. 2004;5:637. doi: 10.1002/cbic.200301023. [DOI] [PubMed] [Google Scholar]; (c) Wang J, Sánchez-Roselló M, Pozo AD, Sorochinsky AE, Fustero S, Soloshonok VA, Liu H. Chem Rev. 2014;114:2432. doi: 10.1021/cr4002879. [DOI] [PubMed] [Google Scholar]; (d) Purser S, Moore PR, Swallowb S, Gouverneur V. Chem Soc Rev. 2008;37:320. doi: 10.1039/b610213c. [DOI] [PubMed] [Google Scholar]; (e) Isanbor C, O’Hagan D. J Fluorine Chem. 2006;127:303. [Google Scholar]; (f) Hagmann WK. J Med Chem. 2008;51:4359. doi: 10.1021/jm800219f. [DOI] [PubMed] [Google Scholar]; (g) Kirk KL. J Fluorine Chem. 2006;127:1013. [Google Scholar]
- 2.Babudri F, Farinola GM, Naso F, Ragni R. Chem Commun. 2007:1003. doi: 10.1039/b611336b. [DOI] [PubMed] [Google Scholar]; (b) Cametti M, Crousse B, Metrangolo P, Milanicd R, Resnati G. Chem Soc Rev. 2012;41:31. doi: 10.1039/c1cs15084g. [DOI] [PubMed] [Google Scholar]; (c) Kassis CM, Steehler JK, Betts DE, Guan Z, Romack TJ, DeSimone JM, Linton RW. Macromolecules. 1996;29:3247. [Google Scholar]; (d) Li Y. Acc Chem Res. 2012;45:723. doi: 10.1021/ar2002446. [DOI] [PubMed] [Google Scholar]; (e) Kirsch P, Hahn A. Eur J Org Chem. 2005:3095. [Google Scholar]
- 3.(a) Jeschke P. ChemBioChem. 2004;5:570. doi: 10.1002/cbic.200300833. [DOI] [PubMed] [Google Scholar]; (b) Maienfisch P, Hall RG. Chimia. 2004;58:93. [Google Scholar]
- 4.(a) Kirsch P. Modern Fluoroorganic Chemistry. Wiley-VCH; Weinheim, Germany: 2004. [Google Scholar]; (b) Davis FA, Yi H, Sundarababu G. In: Enantiocontrolled Synthesis of Fluoro-Organic Compounds. Soloshonok VA, editor. Wiley; Chichester, U.K: 1999. p. 1. [Google Scholar]; (c) Taylor SD, Kotoris CC, Hum G. Tetrahedron. 1999;55:12431. [Google Scholar]; (d) Smart BE. J Fluorine Chem. 2001;109:3. [Google Scholar]; (e) Schlosser M. Tetrahedron. 1978;34:3. [Google Scholar]
- 5.Differding E, Lang RW. Tetrahedron Lett. 1988;29:6087. [Google Scholar]
- 6.For reviews, see: Ma J, Cahard D. Chem Rev. 2004;104:6119. doi: 10.1021/cr030143e.Rozen S. Acc Chem Res. 2005;38:803. doi: 10.1021/ar040270c.For recent development of catalytic enantioselective fluorination, see: Kwiatkowski P, Beeson TD, Conrad JC, MacMillan DWC. J Am Chem Soc. 2011;133:1738. doi: 10.1021/ja111163u.Beeson TD, MacMillan DWC. J Am Chem Soc. 2005;127:8826. doi: 10.1021/ja051805f.Enders D, Huttl MRM. Synlett. 2005;6:991.Wang F, Li J, Hu Q, Yang X, Wu XY, He H. Eur J Org Chem. 2014:3607.Hamashima Y, Suzuki T, Takano H, Shimura Y, Sodeoka M. J Am Chem Soc. 2005;127:10164. doi: 10.1021/ja0513077.Honjo T, Phipps RJ, Rauniyar V, Toste FD. Angew Chem Int Ed. 2012;51:9684. doi: 10.1002/anie.201205383.Rauniyar V, Lackner AD, Hamilton GL, Toste FD. Science. 2011;334:1681. doi: 10.1126/science.1213918.Yang X, Phipps RJ, Toste FD. J Am Chem Soc. 2014;136:5225. doi: 10.1021/ja500882x.Paull DH, Scerba MT, Alden-Danforth E, Widger LR, Lectka T. J Am Chem Soc. 2008;130:17260. doi: 10.1021/ja807792c.Steiner DD, Mase N, Barbas CF. Angew Chem Int Ed. 2005;117:3772. doi: 10.1002/anie.200500571.Marigo M, Fielenbach D, Braunton A, Kjærsgaard A, Jørgensen KA. Angew Chem Int Ed. 2005;117:3769. doi: 10.1002/anie.200500395.
- 7.(a) Davis FA, Han W. Tetrahedron Lett. 1992;33:1153. [Google Scholar]; (b) Davis FA, Kasu PVN. Tetrahedron Lett. 1998;39:6135. [Google Scholar]
- 8.(a) Brunet VA, O’Hagan D, Slawin AMZ. J Fluorine Chem. 2007;128:1271. [Google Scholar]; (b) Lubin H, Dupuis C, Pytkowicz J, Brigaud T. J Org Chem. 2013;78:3487. doi: 10.1021/jo400210j. [DOI] [PubMed] [Google Scholar]; (c) Nakayama K, Browne DL, Baxendale IR, Ley SV. Synlett. 2013;24:1298. [Google Scholar]; (d) Alvarado J, Herrmann AT, Zakarian A. J Org Chem. 2014;79:6206. doi: 10.1021/jo500957d. [DOI] [PubMed] [Google Scholar]
- 9.Phipps RJ, Hiramatsu K, Toste FD. J Am Chem Soc. 2012;134:8376. doi: 10.1021/ja303959p. [DOI] [PubMed] [Google Scholar]
- 10.For a recent review on chemistry of enamides, see: Carbery DR. Org Biomol Chem. 2008;9:3455. doi: 10.1039/b809319a.. Also see: Rappoport Z. The Chemistry of Enamines in The Chemistry of Functional Group. John Wiley and Sons; New York: 1994.
- 11.For a review on the synthesis of enamides, see: Tracey MR, Hsung RP, Antoline J, Kurtz KCM, Shen L, Slafer BW, Zhang Y. In: Science of Synthesis, Houben-Weyl Methods of Molecular Transformations. Weinreb SM, editor. Chapter 21.4. Georg Thieme Verlag KG; 2005. For leading examples of enamide syntheses, see: Bolshan Y, Batey RA. Tetrahedron. 2010;66:5283.Zhang X, Zhang Y, Huang J, Hsung RP, Kurtz KCM, Oppenheimer J, Petersen ME, Sagamanova IK, Tracey MR. J Org Chem. 2006;71:4170. doi: 10.1021/jo060230h.Chechik-Lankin H, Livshin S, Marek I. Synlett. 2005:2098.Han C, Shen R, Su S, Porco JA., Jr Org Lett. 2004;6:27. doi: 10.1021/ol0360041.Pan X, Cai Q, Ma D. Org Lett. 2004;6:1809. doi: 10.1021/ol049464i.Langner M, Bolm C. Angew Chem, Int Ed. 2004;43:5984. doi: 10.1002/anie.200460953.Jiang L, Job GE, Klapars A, Buchwald SL. Org Lett. 2003;5:3667. doi: 10.1021/ol035355c.
- 12.For Diels-Alder cycloadditions of chiral enamides, see: Crawly SL, Funk RL. Org Lett. 2006;8:3995. doi: 10.1021/ol061461d.Gohier F, Bouhadjera K, Faye D, Gaulon C, Maisonneuve V, Dujardin G, Dhal R. Org Lett. 2007;9:211. doi: 10.1021/ol062626l.Fang LC, Hsung RP, Ma ZX, Presser WR. Org Lett. 2013;15:4842. doi: 10.1021/ol402254p.Fang LC, Hsung RP. Org Lett. 2014;16:1826. doi: 10.1021/ol500390a.
- 13.For [2 + 2] cycloadditions of chiral enamides, see: Ma ZX, Feltenberger JB, Hsung RP. Org Lett. 2012;14:2742. doi: 10.1021/ol300967a.Feltenberger JB, Hayashi R, Tang Y, Babiash ESC, Hsung RP. Org Lett. 2009;11:3666. doi: 10.1021/ol901434g.Riches AG, Wernersbach LA, Hegedus LS. J Org Chem. 1998;63:4691.Hegedus LS, Bates RW, Soderberg BC. J Am Chem Soc. 1991;113:923.
- 14.For cyclopropanations, see: Lu T, Song ZL, Hsung RP. Org Lett. 2008;10:541. doi: 10.1021/ol702824s.Song ZL, Lu T, Hsung RP, Al-Rashid ZF, Ko CH, Yu T. Angew Chem Int Ed. 2007;46:4096. doi: 10.1002/anie.200700681.Voigt J, Noltemeyer M, Reiser O. Synlett. 1997:202.
- 15.For epoxidations, see: Xiong H, Hsung RP, Shen L, Hahn JM. Tetrahedron Lett. 2002;43:4449.Adam W, Bosio SG, Wolff BT. Org Lett. 2003;5:819. doi: 10.1021/ol0273194.Sivaguru J, Saito H, Poon T, Omonuwa T, Franz R, Jockusch S, Hooper C, Inoue Y, Adam W, Turro NJ. Org Lett. 2005;7:2089. doi: 10.1021/ol0502230.Koseki Y, Kusano S, Ichi D, Yoshida K, Nagasaka T. Tetrahedron. 2000;56:8855.
- 16.Ko CH, Hsung RP, Al-Rashid ZF, Feltenberger JB, Lu T, Yang JH, Wei YG, Zificsak CA. Org Lett. 2007;9:4459. doi: 10.1021/ol701768n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Banks RE, Mohialdin-Khaffaf SN, Lal GS, Sharifa L, Syvret RG. J Chem Soc Chem Commun. 1992:595. [Google Scholar]
- 18.(a) Umemoto T, Kawada K, Tomita K. Tetrahedron Lett. 1986;27:4465. [Google Scholar]; (b) Umemoto T, Fukami S, Tomizawa G, Harasawa K, Kawada K, Tomita K. J Am Chem Soc. 1990;112:8563. [Google Scholar]; (d) Umemoto T, Nagayoshi M, Adachi K, Tomizawa G. J Org Chem. 1998;63:3379. [Google Scholar]
- 19.Differding E, Ofner H. Synlett. 1991:187. [Google Scholar]
- 20.(a) Barton DHR, Ganguly AK, Hesse RH, Loo SN, Pechet MM. Chem Commun. 1968:806. [Google Scholar]; (b) Barton DHR, Westcott ND, Toh HT, Tarzia G, Hesse RH, Pechet MM. J Chem Soc Chem Commun. 1972:122. [Google Scholar]; (c) Airey J, Barton DHR, Ganguly AK, Hesse RH, Pechet MM. An Quim. 1974;70:871. [Google Scholar]
- 21.See Supporting Information.
- 22.Evans DA. Aldrichimica Acta. 1982;15:23. [Google Scholar]
- 23.Sibi MP, Porter NA. Acc Chem Res. 1999;32:163. [Google Scholar]
- 24.(a) Lu T, Song Z, Hsung RP. Org Lett. 2008;10:541. doi: 10.1021/ol702824s. [DOI] [PubMed] [Google Scholar]; (b) Song Z, Lu T, Hsung RP, Al-Rashid ZF, Ko C, Tang Y. Angew Chem Int Ed. 2007;46:4069. doi: 10.1002/anie.200700681. [DOI] [PubMed] [Google Scholar]
- 25.The absolute stereochemistry of 2-fluorocyclohexanone 13 was confirmed through comparison of its optical rotation ([α]D23 = +45.7 [c 0.73, CHCl3]) with reported values (a) [α]D23 = +54.8 [c 1.0, C6H6]: Enders D, Faure S, Potthoff M, Runsink J. Synthesis. 2001:2307.[α]D23 = +49.6 [c 0.9, C6H6]: Kwiatkowski P, Beeson TD, Conrad JC, MacMillan DWC. J Am Chem Soc. 2011;133:1738. doi: 10.1021/ja111163u.. For ketone 15, it is assigned based on mechanistic analogy with only the racemic form having been reported, and that the assignment of 13 is in good agreement with our stereochemical assessment for all other α–fluorinated-imides.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.