

Abstract
We report germline missense mutations in ETV6 segregating with the dominant transmission of thrombocytopenia and hematologic malignancy in three unrelated kindreds, defining a new hereditary syndrome featuring thrombocytopenia with susceptibility to diverse hematologic neoplasms. Two variants, p.Arg369Gln and p.Arg399Cys, reside in the highly conserved ETS DNA-binding domain. The third variant, p.Pro214Leu, lies within the internal linker domain, which regulates DNA binding. These three amino acid sites correspond to hotspots for recurrent somatic mutation in malignancies. Functional studies show that the mutations abrogate DNA binding, alter subcellular localization, decrease transcriptional repression in a dominant-negative fashion and impair hematopoiesis. These familial genetic studies identify a central role for ETV6 in hematopoiesis and malignant transformation. The identification of germline predisposition to cytopenias and cancer informs the diagnosis and medical management of at-risk individuals.
Few genes predisposing to familial myelodysplastic syndrome (MDS) and acute leukemia have been identified thus far. The genes currently known are RUNX1 (ref. 1), CEBPA2, GATA2 (refs. 3,4), ANKRD26 (refs. 5,6) and SRP72 (ref. 7) for MDS and acute myelogenous leukemia (AML) and PAX5 (refs. 8,9) and TP53 (refs. 10,11) for acute lymphoblastic leukemia (ALL). However, most cases of familial MDS- leukemia remain unexplained.
We studied a family of German and Native American ancestry (family A) with genetically undefined familial thrombocytopenia and malignancy (Fig. 1, Supplementary Fig. 1 and Supplementary Note). Exome sequencing of family members II-4, II-5, III-1, III-2 and III-3 identified five protein-altering variants—in ETV6, TOP3B, GPR144, ITGA8 and PLEC—affecting evolutionarily conserved amino acids and segregating with thrombocytopenia and malignancy under the assumption of an autosomal dominant mode of inheritance (Supplementary Table 1). Sanger sequencing of these five mutations in II-1 and II-3 showed that only one variant was absent in both unaffected individuals: a heterozygous germline ETV6 variant, c.1195C>T (NM_001987.4), encoding p.Arg399Cys (NP_001978.1) (Supplementary Fig. 2a). The proband (III-2) of family A demonstrated easy bruising in infancy and menorrhagia in her teenage years. Affected members of family A also developed diverse hematologic malignancies, including MDS in III-2 at age 17 years, pre-B cell ALL in III-1 at age 7.5 years and multiple myeloma in II-5 at age 51 years (Table 1). Additionally, subject II-5 developed stage III colorectal adenocarcinoma at age 45 years.
Figure 1.

New ETV6 germline variants encoding p.Pro214Leu, p.Arg369Gln and p.Arg399Cys in association with thrombocytopenia and hematologic malignancy. Families A, B and C have ETV6 p.Arg399Cys, p.Arg369Gln and p.Pro214Leu variants, respectively, that segregate with thrombocytopenia and hematologic malignancy in each family. WT indicates genotyped subjects with only wild-type ETV6 alleles; R399C, R369Q, and P214L indicate subjects heterozygous for the variant allele. Arrows indicate the proband in each family.
Table 1.
Clinical features of individuals in the study families
| Family | ETV6 alteration status | Individual | Cytopenias | Malignanciesa | Additional features |
|---|---|---|---|---|---|
| A | p.Arg399Cys | II-5 | Thrombocytopenia, neutropenia | Stage III colorectal carcinoma (45), multiple myeloma (51) | |
| A | p.Arg399Cys | III-1 | Thrombocytopenia | Pre-B cell ALL (7) | |
| A | p.Arg399Cys | III-2 (proband) | Thrombocytopenia, neutropenia, anemia | Refractory anemia (9), RAEB-I (21) | Myopathy, gastrointestinal dysmotility, GERD, developmental delay, seizures, degenerative dental disease, delayed puberty |
| A | p.Arg399Cys | III-3 | Thrombocytopenia, anemia | Myopathy, undefined gastrointestinal symptoms | |
| B | DNA unavailable | I-1 | Thrombocytopenia | Skin cancer, CMML (82) | |
| B | DNA unavailable | II-1 | Thrombocytopenia (initially diagnosed as ITP) | ||
| B | WT | II-2 | Skin cancer | ||
| B | p.Arg369Gln | II-3 (proband) | Thrombocytopenia | Reading disability, GERD | |
| B | p.Arg369Gln | II-5 | Thrombocytopenia | Esophageal stricture, GERD | |
| B | p.Arg369Gln (obligate carrier) | II-8 | Thrombocytopenia | Stage IV colon cancer (43) | |
| B | WT | III-3 | Skin cancer (35) | ||
| B | WT | III-7 | Reading disability | ||
| B | p.Arg369Gln | III-8 | Thrombocytopenia | Reading disability, GERD | |
| B | p.Arg369Gln | III-9 | Thrombocytopenia | Skin cancer (34) | Esophageal stricture, GERD |
| B | p.Arg369Gln | III-12 | Thrombocytopenia | ||
| C | WT | I-2 | Colon cancer (68) | ||
| C | p.Pro214Leu | II-1 (proband) | Thrombocytopenia (initially diagnosed as ITP) | Mixed-phenotype acute leukemia (50) |
ITP, immune thrombocytopenia; GERD, gastrointestinal esophageal reflux disease; ALL, acute lymphoblastic leukemia; CMML, chronic myelomonocytic leukemia; RAEB-I, refractory anemia with excess blasts type I.
Age at diagnosis in years is included in parentheses.
Targeted sequencing of ETV6 and 84 additional genes associated with bone marrow failure and MDS/AML (Supplementary Table 2)12 for an additional 55 individuals with idiopathic familial leukemia or MDS (all lacking germline GATA2, RUNX1, CEBPA and PAX5 mutations) and 153 individuals with idiopathic cytopenias and/or bone marrow failure identified 2 additional families with thrombocytopenia and hematologic malignancy harboring germline ETV6 mutations. Family B, of Scottish ancestry, harbored the heterozygous ETV6 variant c.1106G>A (p.Arg369Gln) (Fig. 1). Affected individuals in family B had thrombocytopenia with petechiae and epistaxis. Family member I-1 developed chronic myelomonocytic leukemia (CMML) at age 82 years. Family member III-8 was diagnosed with stage IV colon cancer at age 43 years. DNA sequencing of skin fibroblasts from the proband (II-1) of family C (Fig. 1), of African-American ancestry, identified a heterozygous ETV6 variant, c.641C>T (p.Pro214Leu). This proband had a long history of nosebleeds and menorrhagia. She was found to have thrombocytopenia unresponsive to standard therapies for immune thrombocytopenia. At age 50 years, she developed T cell/myeloid mixed-phenotype acute leukemia (MPAL). Following standard induction chemotherapy, she had delayed recovery of both platelets and red blood cells and remained transfusion dependent for over 5 months until undergoing allogeneic hematopoietic stem cell transplantation. During this interval, she had two bone marrow biopsies without evidence of residual leukemia.
The segregation pattern for the ETV6 variants was consistent with the dominant transmission pattern of thrombocytopenia and elevated cancer risk. All individuals who carried an ETV6 variant had thrombocytopenia, and all individuals tested who developed a hematologic malignancy and/or thrombocytopenia carried an ETV6 variant (Supplementary Table 3).
The three ETV6 variants were absent from the public databases dbSNP139, the Exome Variant Server and the 1000 Genomes Project (see URLs). We found no germline copy number changes in ETV6 in the affected family members. We also found no damaging germ-line mutations in RUNX1, CEBPA, GATA2, SRP72, ANKRD26, TP53 or PAX5 or in additional marrow failure–associated genes (Supplementary Table 2) in any of the affected individuals.
ETV6 encodes the ETS family transcriptional repressor Ets variant 6. The ETV6 protein harbors a highly conserved ETS DNA-binding domain shared by all ETS family proteins. Arg369 and Arg399 reside in the second β sheet and third α helix of the ETV6 ETS domain, respectively (Fig. 2a). Arg399 directly contacts DNA at the first guanine of the ETS binding element GGA(A/T) via bidentate hydrogen bonds (Fig. 2b)13. Molecular modeling of the p.Arg399Cys substitution predicted a weakened interaction with DNA14 (Fig. 2c). Arg369 is involved in a hydrogen bond with the backbone carbonyl oxygen of Arg414, which itself is involved in electrostatic interactions with DNA upstream of the GGA(A/T) motif13. Thus, the p.Arg369Gln alteration might reduce ETV6 DNA binding via destabilization of the ETS domain and/or by altering the Arg414-DNA interaction (Supplementary Fig. 3). Binding of DNA by mouse Etv6 at the ETS domain is autoinhibited via a C-terminal inhibitory domain (CID; amino acids 426–436)13,15,16. The p.Pro214Leu alteration resides in a linker inhibitory domain (amino acids 127–331) that indirectly promotes DNA binding by attenuating the inhibitory effects of the CID15. Thus, all three encoded alterations fall within ETV6 domains affecting DNA binding. The linker domain is additionally important for the interaction of ETV6 with transcriptional corepressor complexes17.
Figure 2.

Missense alterations in the ETS domain abrogate ETV6 DNA binding. (a) Positions of germline and somatic alterations in ETV6 relative to the PNT oligomerization and ETS DNA-binding domains. The germline alterations reported in this study are highlighted in red. Somatic alterations affecting the same amino acids as the germline alterations are boxed. Somatic alterations reported in the literature include ones associated with MDS33,35,36, AML32,33,37–39, CMML40, immature T cell ALL34, mature T cell ALL41, B cell precursor ALL42,43, hypodiploid ALL10, multiple myeloma44, colorectal adenocarcinoma30,31 and melanoma45. Truncating alterations, including nonsense, frameshift and splice-site changes, are shown in bold. (b) Hydrogen bonding (dotted lines) of Arg399 (orange) with guanine (magenta) in the ETS binding element. The protein structure of the mouse ETV6 ETS domain (Protein Data Bank (PDB), 4MHG)13 is shown. The ETS domains of the mouse and human ETV6 proteins have 100% protein sequence identity. (c) Molecular modeling of the Arg399Cys (orange) variant using SWISS-MODEL predicts loss of hydrogen bonding to DNA. (d) Coomassie-stained SDS-PAGE gel with recombinant histidine affinity (HAT)-tagged ETS domains from wild-type (WT), Arg369Gln or Arg399Cys ETV6. (e) EMSA of the ETS domains for wild-type, Arg369Gln or Arg399Cys ETV6. Biotinylated EBS DNA probe was incubated with the indicated concentrations of purified recombinant HAT-tagged ETS domain from wild-type or mutant ETV6. Open and closed triangles indicate the positions of the protein-bound and unbound probes, respectively.
We tested the effect of the ETS-domain p.Arg369Gln and p.Arg399Cys alterations on DNA binding by electrophoretic mobility shift assay (EMSA) using a DNA probe containing a consensus ETS binding site. A shift in mobility of the DNA probe was detected with 50 nM of the ETS domain from purified recombinant wild-type ETV6, whereas no shift was observed after the addition of up to 500 nM of the ETS domain from the Arg369Gln or Arg399Cys ETV6 mutant (Fig. 2d,e), demonstrating that the p.Arg369Gln and p.Arg399Cys alterations abrogate DNA binding by ETV6.
Fluorescence microscopy of EGFP-tagged ETV6 in HeLa cells showed that wild-type ETV6 concentrated in cell nuclei (Fig. 3a,b). In contrast, Pro214Leu ETV6 exhibited predominantly cytoplasmic localization and Arg369Gln and Arg399Cys ETV6 showed reduced nuclear localization (Fig. 3a,b). Concordant with these fluorescence microscopy data, fractionation of HeLa cells transiently expressing ETV6 cDNA for the wild-type protein or the Arg399Cys mutant showed increased Arg399Cys ETV6 protein levels in the cytoplasmic fraction and decreased levels in the nuclear fraction in comparison to cells expressing the wild-type protein (Supplementary Fig. 4). Thus, the p.Pro214Leu, p.Arg369Gln and p.Arg399Cys alterations change ETV6 localization. These results concur with previous reports that residues 332–452 at the C terminus of ETV6, which includes the ETS DNA-binding domain, affect ETV6 nuclear localization18. The p.Pro214Leu alteration might affect intracellular localization through indirect effects of the linker region on ETV6 DNA binding15. Mutations resulting in predominantly cytoplasmic localization might contribute to a dominant-negative effect via oligomerization with wild-type ETV6, resulting in its cytoplasmic sequestration. Although protein levels were comparable for exogenously expressed wild-type and mutant ETV6 proteins (Fig. 4b), potential effects from ETV6 overexpression cannot be ruled out. Additional studies of the molecular mechanisms regulating the intracellular localization of endogenous ETV6 are warranted.
Figure 3.
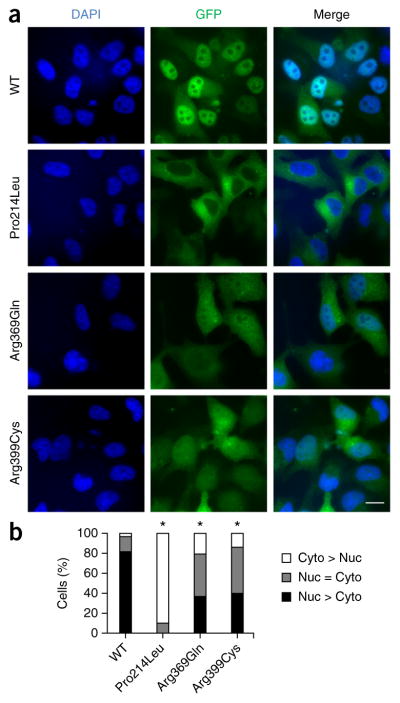
ETV6 mutation reduces nuclear localization. (a) Fluorescence images of HeLa cells transiently expressing EGFP-tagged wild-type, Pro214Leu, Arg369Gln or Arg399Cys ETV6. Scale bar, 25 μm. (b) Percentage of cells exhibiting predominantly nuclear (Nuc > Cyto), predominantly cytoplasmic (Cyto > Nuc) or equivalent nuclear and cytoplasmic (Nuc = Cyto) EGFP signal. Three individual experiments were performed, and at least 300 total cells were counted for each condition. Pairwise comparisons between wild-type protein and each mutant were performed using the χ2 test (*P < 1 × 10−48).
Figure 4.
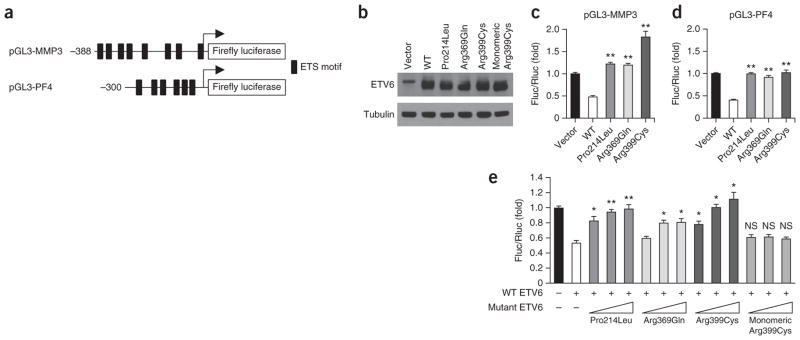
ETV6 mutants are deficient in transcriptional repression and act in a dominant-negative manner. (a) Schematic of the pGL3 reporter constructs harboring the MMP3 and PF4 promoters upstream of the firefly luciferase gene. Black rectangles represent core ETS DNA-binding motifs. (b) Protein blot analysis of ETV6 expression in HeLa whole-cell lysates. (c) HeLa cells were cotransfected with the pGL3-MMP3 reporter construct, a pHAGE expression vector (empty vector, wild-type ETV6 or mutant ETV6) and pCS2 Renilla luciferase. Firefly to Renilla luciferase ratios (Fluc/Rluc) were calculated to control for transfection efficiency. Bars show the mean (+ s.e.m.) fold change in the Fluc/Rluc ratio relative to empty vector. Data represent at least two individual experiments for each condition with duplicate measurements. Pairwise Student’s t tests were performed comparing each condition to wild type (**P < 0.0005). (d) Experiments are as in c except that the pGL3-PF4 reporter construct was used. Data represent at least three individual experiments for each condition with duplicate measurements. Pairwise Student’s t tests were performed comparing each condition to wild type (**P < 0.0005). (e) HeLa cells were cotransfected with 50 ng of wild-type ETV6 expression vector with increasing amounts (50, 150 and 250 ng) of ETV6 expression vector encoding the Pro214Leu, Arg369Gln, Arg399Cys or monomeric Arg399Cys mutant, pGL3-PF4 reporter construct and pCS2 Renilla luciferase. Bars show the mean (+ s.e.m.) fold change in the Fluc/Rluc ratio relative to empty vector. Data represent at least three individual experiments for each condition with duplicate measurements. Pairwise Student’s t tests were performed comparing each condition to wild type alone (*P < 0.005, **P < 0.0005; NS, not significant).
Because ETV6 functions as a transcriptional repressor of promoters harboring ETS binding sites (EBSs)17,19–22, we tested the effects of the ETV6 mutations on the transcriptional repression of firefly luciferase reporter constructs containing the MMP3 or PF4 promoter, which each harbor EBSs (Fig. 4a). Whereas wild-type ETV6 repressed the expression of both reporter genes, we saw no repression with the ETV6 mutants (Fig. 4c,d). Expression of an ETV6 ETS-domain deletion mutant has previously been shown to inhibit wild-type ETV6 transrepression in a dominant-negative manner20. To test whether the patient-derived missense mutations acted in a dominant-negative manner, we measured the effect of increasing the levels of mutant ETV6 cDNA, cotransfected into cells with a set quantity of wild-type ETV6 cDNA, using the PF4–firefly luciferase reporter construct. All three patient-derived mutants antagonized the repression mediated by wild-type ETV6 in a dose-dependent manner (Fig. 4e, compare bars 3–11 to bar 2).
The Pointed (PNT) domain of ETV6 mediates homo-oligomerization, a property required for stable ETV6 binding to DNA harboring tandem EBSs15. We hypothesized that the transrepression-defective ETV6 missense mutants inhibited transrepression by forming dysfunctional PNT domain–mediated heteromeric complexes with wild-type ETV6. We introduced into the Arg399Cys mutant the additional PNT-domain missense alterations p.Ala93Asp and p.Val112Glu, previously demonstrated to disrupt PNT-domain oligomerization23. In contrast to the oligomerization-competent Arg399Cys ETV6 mutant, monomeric Arg399Cys ETV6 failed to inhibit the repression mediated by wild-type ETV6 (Fig. 4e, compare bars 12–14 with bar 2). These results suggest that dominant-negative ETV6 mutants inhibit wild-type ETV6 transrepression in an oligomerization-dependent manner.
In mouse models, Etv6 is required for hematopoietic stem cell maintenance24, but hematopoiesis is unperturbed by heterozygous loss of one Etv6 allele25. To test the effect of the dominant-negative ETV6 mutants in hematopoietic stem cells, we measured the proliferation of human CD34+ hematopoietic stem/progenitor cells (HSPCs) transduced with lentiviral vectors expressing wild-type or mutant ETV6. The proliferation of CD34+ cells expressing wild-type ETV6 was similar to that of cells receiving empty vector (Fig. 5a). In contrast, the proliferation of CD34+ cells expressing any of the three ETV6 mutants was markedly reduced (Fig. 5a). We noted no increase in apoptosis.
Figure 5.
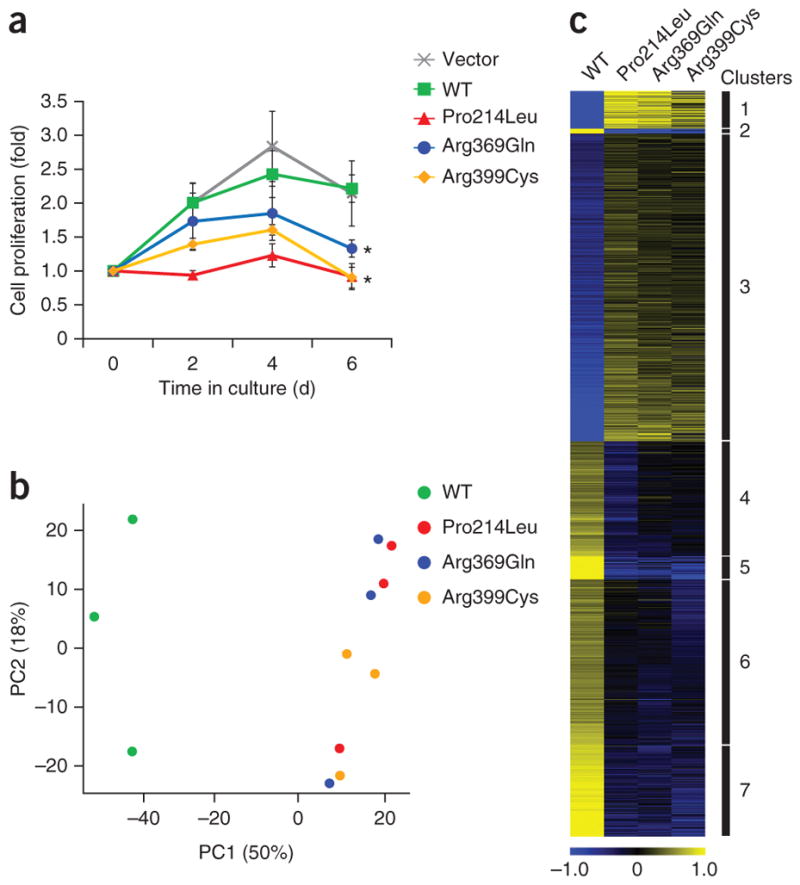
ETV6 mutants impair hematopoietic stem cell proliferation and alter the ETV6 transcriptome. (a) Proliferation of human CD34+ cells expressing wild-type, Pro214Leu, Arg369Gln or Arg399Cys ETV6 cultured under non-differentiating conditions. Viable cells were counted in triplicate every 2 d. Plotted points represent means ± s.d. Pairwise Student’s t tests were performed comparing each mutant to wild-type ETV6 on day 6 (*P < 0.01). (b) Genome-wide mRNA expression profiling with wild-type or mutant ETV6. PCA plot of the first two principal components representing 68% of the total variance in the transcriptome data set from K562 cells expressing wild-type protein or the indicated mutant ETV6 species. The data from three independent experiments are shown. (c) Heat map showing the log2-transformed and mean-centered transcript levels for differentially expressed genes in K562 cells expressing wild-type ETV6 or the indicated mutant ETV6 species. Differentially expressed genes were partitioned into seven distinct clusters by k-means clustering using Euclidian distance. Yellow and blue indicate higher and lower expression, respectively.
To further compare the functional consequences of the three ETV6 mutations, we performed RNA sequence (RNA-seq) profiling of the K562 myeloid cell line expressing wild-type, Pro214Leu, Arg369Glu or Arg399Cys ETV6. Principal-component analysis (PCA) and k-means clustering identified similar gene signature patterns for cells expressing any of the three missense mutants, which distinctly differed from the expression profiles of cells expressing wild-type ETV6 (Fig. 5b,c). There were 311 genes whose expression was reduced by all 3 ETV6 mutants in comparison to wild-type ETV6 (Supplementary Table 4) and 349 genes whose expression was increased by all 3 ETV6 mutants in comparison to wild-type ETV6 (Supplementary Table 5). Gene Ontology (GO) analysis with GOseq identified platelet-associated gene sets that were robustly expressed with wild-type ETV6 but showed reduced expression with all three missense mutants (Supplementary Tables 6 and 7). These data are consistent with the notion that all three ETV6 mutations result in similar impairment of ETV6 function.
To identify mutations acquired during malignant progression in the context of germline ETV6 mutation, we examined paired tumor and fibroblast samples from family A for mutations in 194 cancer-related genes using a targeted gene capture panel26. No deletion or mutation of the remaining wild-type ETV6 allele was observed in any of the neoplasms (Supplementary Fig. 2b and Supplementary Table 8).
In individual II-5, different sets of somatic mutations were identified in the colon adenocarcinoma sample (BRAF, CTNNB1, GNAS, PTEN and TP53) than in the multiple myeloma sample (CDK8 and KMT2A) (Supplementary Table 8). In the colon cancer sample, the BRAF mutation encoding p.Val600Glu and CTNNB1 mutations were early events, followed by mutations in GNAS and PTEN. The acquisition of multiple distinct mutations of different variant allele fractions within both GNAS and PTEN was suggestive of convergent subclonal evolution. In individual III-2, sequencing of an MDS sample upon progression to refractory anemia with excess blasts 1 (RAEB-1) identified acquired truncating mutations in BCOR and RUNX1 and an activating mutation in KRAS, all present in the same dominant clone (Supplementary Table 9). Sequencing of these mutations in an earlier sample taken before the development of excess blasts identified the BCOR and RUNX1 mutations, but the KRAS mutation was absent (Supplementary Fig. 5). This indicated that the KRAS mutation arose during progression to high-grade MDS.
Autosomal dominant transmission of thrombocytopenia and predisposition to MDS/acute leukemia caused by germline ETV6 mutations is reminiscent of phenotypes associated with mutations in RUNX1 (ref. 1) and ANKRD26 (refs. 6,27), respectively. ETV6, RUNX1 and ANKRD26 are all highly expressed in hematopoietic stem cells and megakaryocyte-erythroid progenitors28. Recent evidence suggests that ANKRD26 is transcriptionally regulated by RUNX1 and the ETS family transcription factor FLI1 and that autosomal dominant thrombocytopenia (THC2)-associated mutations in the 5′ UTR of ANKRD26 alter RUNX1- and FLI1-mediated regulation of ANKRD26 (ref. 29). The potential intersection of pathways regulated by ANKRD26, RUNX1 and ETS family transcription factors in megakaryopoiesis and hematopoietic transformation warrants further study.
Somatic point mutations in ETV6 have been recurrently observed by recent large-scale cancer genome sequencing efforts (Fig. 2a)30–35, but the role of ETV6 mutations in malignant transformation remained unclear. We identified germline missense ETV6 mutations affecting amino acids recurrently altered across diverse malignancies (Fig. 2a). The association of these mutations with cancer predisposition supports a role for ETV6 point mutations as initiating events in the early steps of malignant transformation. The study of familial cancer syndromes thus complements cancer genome sequencing approaches to identify driver mutations in malignancy.
URLs
dbSNP139, http://www.ncbi.nlm.nih.gov/projects/SNP/; National Heart, Lung, and Blood Institute (NHLBI) Exome Sequencing Project, http://evs.gs.washington.edu/EVS/; 1000 Genomes Project, http://www.1000genomes.org/.
Methods
Methods and any associated references are available in the online version of the paper.
Accession codes
The ETV6 mutations encoding p.Pro214Leu, p.Arg369Gln and p.Arg399Cys have been deposited in the NCBI ClinVar database under accessions SCV000195553, SCV000195554 and SCV000195555, respectively. The RNA-seq data have been deposited in the NCBI Sequence Read Archive (SRA) under accession SRP048957.
ONLINE METHODS
Subjects and samples
Subjects provided written informed consent in accordance with protocols approved by the institutional review boards of the Fred Hutchinson Cancer Research Center and Seattle Children’s Hospital for family A and the University of Chicago for families B and C.
Exome sequencing
Individuals II-4, II-5, III-1, III-2 and III-3 from family A were subjected to exome sequencing, as previously described46,47. Briefly, paired-end libraries with 250-bp inserts were hybridized to the SeqCap EZ Human Exome Library v2.0 (NimbleGen). Sequencing was performed with 2 × 101-bp reads using SBS v3 on a HiSeq 2000 instrument (Illumina). Rare and private variants were classified by predicted function to include all missense, nonsense, frameshift and splice-site alleles. Variants were filtered on the basis of an autosomal dominant mode of inheritance.
Targeted gene panel sequencing
For ETV6 mutational screening, capture probes were designed to target all coding exons and 20 bp of flanking sequence for ETV6 and 84 other genes involved in inherited bone marrow failure and MDS/AML (Supplementary Table 2). Targeted capture, sequencing and bio-informatics analysis were performed as previously described48. Identification of somatic alterations in a panel of 194 cancer-related genes was performed on paired tumor and fibroblast samples as previously described26. Cis or trans relationships between variants were determined using the Integrated Genomics Viewer.
Plasmids
Human ETV6 cDNA (NM_001987.4) was cloned into pHAGE-CMV-MCS-IRES-ZsGreen (pHAGE)49, and the resultant plasmid was used for the generation of the ETV6 mutants (p.Pro214Leu, p.Arg369Gln, p.Arg399Cys, p.[Ala93Asp; p.Val112Glu; p.Arg399Cys]) by QuikChange site-directed mutagenesis (Agilent Technologies). The cDNAs for wild-type and mutant human ETV6 were cloned into the pEGFP-N3 vector (Clontech). The promoters for human MMP3 and PF4 were cloned into pGL3-Basic (Agilent Technologies). The sequence encoding the ETS domain of human ETV6 (amino acids 335–430) was cloned into pHAT10 (Clontech) for bacterial expression (Supplementary Table 10).
Cell culture
HeLa cells (a gift from D. Pellman; Dana-Farber Cancer Institute) were cultured in DMEM supplemented with 10% FBS, 1% glutamine and 1% penicillin-streptomycin. CD34+ cells were isolated from anonymous discarded full-term human umbilical cord blood using the CD34 Microbead kit (Miltenyi Biotec) as previously described50. Cells were cultured in StemSpan SFEM II (StemCell Technologies) supplemented with penicillin-streptomycin and 100 ng/ml each of human stem cell factor, thrombopoietin, interleukin (IL)-6 and Flt-3 ligand (PeproTech). K562 cells (a gift from B. Torok-Storb; Fred Hutchinson Cancer Research Center) were grown in RPMI-1640 with 10% FBS, 1% glutamine, 1% penicillin-streptomycin and 1 mM sodium pyruvate. All cell lines in the laboratory were routinely tested for mycoplasma.
RNA sequencing expression analysis
K562 cells were electroporated with the pHAGE ETV6 constructs using Cell Line Nucleofector Kit V (Lonza) according to the manufacturer’s instructions, maintained in growth medium for 48 h and sorted by flow cytometry for ZsGreen positivity. Positive cells were grown for another 24 h before lysis in TRIzol reagent (Invitrogen). RNA was extracted with the RNeasy Total RNA cleanup kit (Qiagen). RNA integrity was measured using an Agilent 2200 TapeStation (Agilent Technologies). RNA-seq libraries were prepared from total RNA using the TruSeq RNA Sample Prep kit (Illumina) and a Sciclone NGSx Workstation (PerkinElmer). Sequencing was performed using an Illumina HiSeq 2500 instrument in rapid-output mode, employing a paired-end, 50-base read length sequencing strategy. Image analysis and base calling were performed using Illumina Real-Time Analysis software.
RNA sequencing data analysis
Reads of low quality were filtered out before alignment to the reference genome (UCSC hg19 assembly) using TopHat v2.0.12 (ref. 51). Counts were generated from TopHat alignments for each gene using the Python package HTSeq v0.6.1 (ref. 52). Genes with low counts in more than three samples were removed before the identification of differentially expressed genes using the Bioconductor package edgeR v3.4.2 (ref. 53). A false discovery rate (FDR) method was employed to correct for multiple testing54. Differential expression was defined as |log2 (ratio) | ≥ 0.585 (±1.5-fold) with the FDR set to 5%. k-means cluster analysis was performed for the genes found to be differentially expressed in one or more comparisons. Normalized log2 signal intensities were mean centered at the gene level, and replicate samples were averaged before clustering. The number of clusters was selected using the figure of merit (FOM) method55. k-means clustering and cluster number estimation were performed using the TM4 microarray software suite MultiExperimental Viewer (MeV)56. Over-represented GO biological process terms that comprised the genes found in the k-means clusters were identified using the Bioconductor package GOseq57. PCA plots were generated using R.
Lentiviral transduction
Lentiviruses were produced by transient transfection of HEK293T cells (a gift from A. Scharenberg; Seattle Children’s Hospital Research Institute) using polyethylenimine and concentrated by low-speed centrifugation. CD34+ cells were transduced with pHAGE lentiviral bicistronic vectors encoding wild-type or mutant ETV6 cDNA and a ZsGreen marker at a multiplicity of infection (MOI) of 10 in the presence of 8 μg/ml hexadimethrine bromide (Sigma-Aldrich)58. ZsGreen-positive cells were selected by flow cytometry.
Protein blotting
Whole-cell extracts were obtained by lysing cells in RIPA buffer (1% NP-40, 0.5% sodium deoxycholate and 0.2% SDS in PBS) with 1 mg/ml Pefabloc (Sigma-Aldrich), 1 μg/ml pepstatin (Sigma-Aldrich) and 1× Complete EDTA-free protease inhibitor cocktail (Roche). Cell fractionation was performed using NE-PER Nuclear and Cytoplasmic Extraction Reagents (Pierce). Samples were separated by 10% SDS-PAGE, transferred onto nitro-cellulose and probed with antibodies against ETV6 (N-19X (1:2,000 dilution) or H-214 (1:200 dilution), Santa Cruz Biotechnology), α-tubulin (1:10,000 dilution; DM1A, Sigma-Aldrich), NPM1 (1:10,000 dilution; FC82291, Abcam) or GAPDH (1:5,000 dilution; ab9485, Abcam). Western Lightning Plus ECL (PerkinElmer) was used for signal detection.
Immunofluorescence
HeLa cells were plated on poly-L lysine–coated coverslips and transfected with pEGFP constructs using Attractene Transfection Reagent (Qiagen). After 48 h, cells were fixed in 4% paraformaldehyde in PBS, mounted with VECTASHIELD Mounting Medium with DAPI (Vector Laboratories) and visualized using a Nikon ECLIPSE E800 microscope.
Recombinant protein expression and purification
Recombinant proteins were expressed and purified as previously described58. Purified proteins were dialyzed overnight into 20 mM sodium citrate, pH 5.3, 500 mM KCl, 1 mM EDTA, 1 mM DTT, 0.2 mM phenylmethylsulfonyl fluoride and 10% glycerol.
EMSA probes
DNA probes were modified from Green et al.15 (Supplementary Table 10). Probes were labeled by 3 ′biotinylation of the forward strand. Probes were annealed by incubation at 95 °C for 1 min and slow cooling to room temperature for 2 h.
Gel shift assays
Protein and probes were incubated in EMSA buffer (25 mM Tris, pH 8.0, 50 mM KCl, 1 mM DTT, 10% glycerol, 6 mM MgCl2, 1 mM EDTA, 50 ng/μl poly(dI-dC) and 0.1 mg/ml BSA) for 20 min at room temperature. Samples were then separated in a 6% acrylamide, 0.5% Tris-borate-EDTA (TBE) native gel for 70 min at 100 V and 4 °C. Protein–nucleic acid complexes were transferred to a nylon membrane for 35 min at 380 mA. Nucleic acids were cross-linked to the nylon membrane by ultraviolet (UV) light at 120 mJ/cm2. Biotin-labeled probes were detected on the membrane using the Chemiluminescent Nucleic Acid Detection Module (Pierce).
Luciferase assays
HeLa cells were cotransfected with pGL3 reporter construct, pHAGE expression construct and pCS2 Renilla luciferase construct using Attractene Transfection Reagent (Qiagen). Empty pHAGE plasmid was added to maintain a constant DNA concentration per transfection. Cells were collected 48 h after transfection using Passive Lysis Buffer (Promega). Renilla and firefly luciferase levels were assayed with the Dual-Luciferase Reporter Assay System (Promega) using a GloMax Microplate Luminometer with Dual Injectors (Promega). pCS2 Renilla luciferase was used to normalize for transfection efficiency.
CD34+ cell proliferation assays
Cord blood–derived CD34+ cells were purified, transduced and cultured as described above. Cells were cultured in triplicate. Viable cells, as determined by trypan blue staining, were counted every 2 d.
Supplementary Material
Acknowledgments
We thank all patients and their families for participation in this research study. We thank M. Chin (University of Washington), B. Turok-Storb (Fred Hutchinson Cancer Research Center) and S. Tapscott (Fred Hutchinson Cancer Research Center) for luciferase plasmids and reagents. We thank H. Hock, B. Stoddard, S. Meshinchi, G. Smith, A. Kumar, C. Toledo, S. Yu, A. Fong and K. MacQuarrie for helpful discussions. We thank S. Castro for clinical sample processing. This work was supported by US National Institutes of Health grants R24DK093425 and R24DK099808-01 to A.S., M.-C.K. and J.L.A.; by the Ghiglione Aplastic Anemia Fund and Julian’s Dinosaur Guild from Seattle Children’s Hospital to A.S.; by Medical Scientist Training Program Training grant T32GM007266 and Genetic Approaches to Aging Training grant T32AG000057 to M.Y.Z.; and by grants from the US National Institutes of Health (K12CA139160) and the Cancer Research Foundation to J.E.C. M.-C.K. is an American Cancer Society professor.
Footnotes
AUTHOR CONTRIBUTIONS
M.Y.Z., J.E.C., S.B.K., T.W., J.L.A., M.-C.K., L.A.G. and A.S. conceived and designed the experiments. M.Y.Z., S.B.K., T.W., C.C.P., M.S.-B., C.J.M. and S.A.C. performed the experiments. M.Y.Z., S.B.K., T.W., M.K.L., K.R.L., S.G., C.C.P., J.J.D., R.S.B., R.C.L., M.-C.K. and A.S. analyzed the data. J.E.C., S.B.K., M.F., B.G., B.S.S., B.N., R.M., I.H., D.A.W., M.S.H., L.A.G. and A.S. identified study subjects, performed clinical phenotyping and contributed biological samples. M.Y.Z., J.E.C., T.W., J.J.D., L.A.G., M.-C.K. and A.S. wrote the manuscript. A.S. and M.-C.K. jointly supervised the research.
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
Note: Any Supplementary Information and Source Data files are available in the online version of the paper.
References
- 1.Song WJ, et al. Haploinsufficiency of CBFA2 causes familial thrombocytopenia with propensity to develop acute myelogenous leukaemia. Nat Genet. 1999;23:166–175. doi: 10.1038/13793. [DOI] [PubMed] [Google Scholar]
- 2.Smith ML, Cavenagh JD, Lister TA, Fitzgibbon J. Mutation of CEBPA in familial acute myeloid leukemia. N Engl J Med. 2004;351:2403–2407. doi: 10.1056/NEJMoa041331. [DOI] [PubMed] [Google Scholar]
- 3.Hahn CN, et al. Heritable GATA2 mutations associated with familial myelodysplastic syndrome and acute myeloid leukemia. Nat Genet. 2011;43:1012–1017. doi: 10.1038/ng.913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kazenwadel J, et al. Loss-of-function germline GATA2 mutations in patients with MDS/AML or MonoMAC syndrome and primary lymphedema reveal a key role for GATA2 in the lymphatic vasculature. Blood. 2012;119:1283–1291. doi: 10.1182/blood-2011-08-374363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pippucci T, et al. Mutations in the 5′ UTR of ANKRD26, the ankirin repeat domain 26 gene, cause an autosomal-dominant form of inherited thrombocytopenia, THC2. Am J Hum Genet. 2011;88:115–120. doi: 10.1016/j.ajhg.2010.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Noris P, et al. Mutations in ANKRD26 are responsible for a frequent form of inherited thrombocytopenia: analysis of 78 patients from 21 families. Blood. 2011;117:6673–6680. doi: 10.1182/blood-2011-02-336537. [DOI] [PubMed] [Google Scholar]
- 7.Kirwan M, et al. Exome sequencing identifies autosomal-dominant SRP72 mutations associated with familial aplasia and myelodysplasia. Am J Hum Genet. 2012;90:888–892. doi: 10.1016/j.ajhg.2012.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shah S, et al. A recurrent germline PAX5 mutation confers susceptibility to pre-B cell acute lymphoblastic leukemia. Nat Genet. 2013;45:1226–1231. doi: 10.1038/ng.2754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Auer F, et al. Inherited susceptibility to pre B-ALL caused by germline transmission of PAX5 c. 547G>A. Leukemia. 2014;28:1136–1138. doi: 10.1038/leu.2013.363. [DOI] [PubMed] [Google Scholar]
- 10.Holmfeldt L, et al. The genomic landscape of hypodiploid acute lymphoblastic leukemia. Nat Genet. 2013;45:242–252. doi: 10.1038/ng.2532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Powell BC, et al. Identification of TP53 as an acute lymphocytic leukemia susceptibility gene through exome sequencing. Pediatr Blood Cancer. 2013;60:E1–E3. doi: 10.1002/pbc.24417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang MY, et al. Genomic analysis of bone marrow failure and myelodysplastic syndromes reveals phenotypic and diagnostic complexity. Haematologica. 2014 Sep 19; doi: 10.3324/haematol.2014.113456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.De S, et al. Steric mechanism of auto-inhibitory regulation of specific and non-specific DNA binding by the ETS transcriptional repressor ETV6. J Mol Biol. 2014;426:1390–1406. doi: 10.1016/j.jmb.2013.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Biasini M, et al. SWISS-MODEL: modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res. 2014;42:W252–W258. doi: 10.1093/nar/gku340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Green SM, Coyne HJ, III, McIntosh LP, Graves BJ. DNA binding by the ETS protein TEL (ETV6) is regulated by autoinhibition and self-association. J Biol Chem. 2010;285:18496–18504. doi: 10.1074/jbc.M109.096958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Coyne HJ, et al. Autoinhibition of ETV6 (TEL) DNA binding: appended helices sterically block the ETS domain. J Mol Biol. 2012;421:67–84. doi: 10.1016/j.jmb.2012.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chakrabarti SR, Nucifora G. The leukemia-associated gene TEL encodes a transcription repressor which associates with SMRT and mSin3A. Biochem Biophys Res Commun. 1999;264:871–877. doi: 10.1006/bbrc.1999.1605. [DOI] [PubMed] [Google Scholar]
- 18.Park H, Seo Y, Kim JI, Kim W, Choe SY. Identification of the nuclear localization motif in the ETV6 (TEL) protein. Cancer Genet Cytogenet. 2006;167:117–121. doi: 10.1016/j.cancergencyto.2006.01.006. [DOI] [PubMed] [Google Scholar]
- 19.Fenrick R, et al. Both TEL and AML-1 contribute repression domains to the t(12;21) fusion protein. Mol Cell Biol. 1999;19:6566–6574. doi: 10.1128/mcb.19.10.6566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fenrick R, et al. TEL, a putative tumor suppressor, modulates cell growth and cell morphology of Ras-transformed cells while repressing the transcription of stromelysin-1. Mol Cell Biol. 2000;20:5828–5839. doi: 10.1128/mcb.20.16.5828-5839.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lopez RG, et al. TEL is a sequence-specific transcriptional repressor. J Biol Chem. 1999;274:30132–30138. doi: 10.1074/jbc.274.42.30132. [DOI] [PubMed] [Google Scholar]
- 22.Kwiatkowski BA, et al. The ets family member Tel binds to the Fli-1 oncoprotein and inhibits its transcriptional activity. J Biol Chem. 1998;273:17525–17530. doi: 10.1074/jbc.273.28.17525. [DOI] [PubMed] [Google Scholar]
- 23.Kim CA, et al. Polymerization of the SAM domain of TEL in leukemogenesis and transcriptional repression. EMBO J. 2001;20:4173–4182. doi: 10.1093/emboj/20.15.4173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang LC, et al. The TEL/ETV6 gene is required specifically for hematopoiesis in the bone marrow. Genes Dev. 1998;12:2392–2402. doi: 10.1101/gad.12.15.2392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hock H, et al. Tel/Etv6 is an essential and selective regulator of adult hematopoietic stem cell survival. Genes Dev. 2004;18:2336–2341. doi: 10.1101/gad.1239604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pritchard CC, et al. Validation and implementation of targeted capture and sequencing for the detection of actionable mutation, copy number variation, and gene rearrangement in clinical cancer specimens. J Mol Diagn. 2014;16:56–67. doi: 10.1016/j.jmoldx.2013.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Marquez R, et al. A new family with a germline ANKRD26 mutation and predisposition to myeloid malignancies. Leuk Lymphoma. 2014 Apr 22; doi: 10.3109/10428194.2014.903476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Novershtern N, et al. Densely interconnected transcriptional circuits control cell states in human hematopoiesis. Cell. 2011;144:296–309. doi: 10.1016/j.cell.2011.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bluteau D, et al. Thrombocytopenia-associated mutations in the ANKRD26 regulatory region induce MAPK hyperactivation. J Clin Invest. 2014;124:580–591. doi: 10.1172/JCI71861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487:330–337. doi: 10.1038/nature11252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Seshagiri S, et al. Recurrent R-spondin fusions in colon cancer. Nature. 2012;488:660–664. doi: 10.1038/nature11282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Welch JS, et al. The origin and evolution of mutations in acute myeloid leukemia. Cell. 2012;150:264–278. doi: 10.1016/j.cell.2012.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Walter MJ, et al. Clonal diversity of recurrently mutated genes in myelodysplastic syndromes. Leukemia. 2013;27:1275–1282. doi: 10.1038/leu.2013.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang J, et al. The genetic basis of early T-cell precursor acute lymphoblastic leukaemia. Nature. 2012;481:157–163. doi: 10.1038/nature10725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bejar R, et al. Clinical effect of point mutations in myelodysplastic syndromes. N Engl J Med. 2011;364:2496–2506. doi: 10.1056/NEJMoa1013343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xu L, et al. Genomic landscape of CD34+ hematopoietic cells in myelodysplastic syndrome and gene mutation profiles as prognostic markers. Proc Natl Acad Sci USA. 2014;111:8589–8594. doi: 10.1073/pnas.1407688111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ding L, et al. Clonal evolution in relapsed acute myeloid leukaemia revealed by whole-genome sequencing. Nature. 2012;481:506–510. doi: 10.1038/nature10738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yoshida K, et al. Frequent pathway mutations of splicing machinery in myelodysplasia. Nature. 2011;478:64–69. doi: 10.1038/nature10496. [DOI] [PubMed] [Google Scholar]
- 39.Dolnik A, et al. Commonly altered genomic regions in acute myeloid leukemia are enriched for somatic mutations involved in chromatin remodeling and splicing. Blood. 2012;120:e83–e92. doi: 10.1182/blood-2011-12-401471. [DOI] [PubMed] [Google Scholar]
- 40.Padron E, et al. ETV6 and signaling gene mutations are associated with secondary transformation of myelodysplastic syndromes to chronic myelomonocytic leukemia. Blood. 2014;123:3675–3677. doi: 10.1182/blood-2014-03-562637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Griesinger F, Janke A, Podleschny M, Bohlander SK. Identification of an ETV6-ABL2 fusion transcript in combination with an ETV6 point mutation in a T-cell acute lymphoblastic leukaemia cell line. Br J Haematol. 2002;119:454–458. doi: 10.1046/j.1365-2141.2002.03850.x. [DOI] [PubMed] [Google Scholar]
- 42.Zhang J, et al. Key pathways are frequently mutated in high-risk childhood acute lymphoblastic leukemia: a report from the Children’s Oncology Group. Blood. 2011;118:3080–3087. doi: 10.1182/blood-2011-03-341412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang Q, et al. ETV6 mutation in a cohort of 970 patients with hematologic malignancies. Haematologica. 2014;99:e176–e178. doi: 10.3324/haematol.2014.104406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lohr JG, et al. Widespread genetic heterogeneity in multiple myeloma: implications for targeted therapy. Cancer Cell. 2014;25:91–101. doi: 10.1016/j.ccr.2013.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hodis E, et al. A landscape of driver mutations in melanoma. Cell. 2012;150:251–263. doi: 10.1016/j.cell.2012.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Walsh T, et al. Whole exome sequencing and homozygosity mapping identify mutation in the cell polarity protein GPSM2 as the cause of non-syndromic hearing loss DFNB82. Am J Hum Genet. 2010;87:90–94. doi: 10.1016/j.ajhg.2010.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gulsuner S, et al. Spatial and temporal mapping of de novo mutations in schizophrenia to a fetal prefrontal cortical network. Cell. 2013;154:518–529. doi: 10.1016/j.cell.2013.06.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Walsh T, et al. Mutations in 12 genes for inherited ovarian, fallopian tube, and peritoneal carcinoma identified by massively parallel sequencing. Proc Natl Acad Sci USA. 2011;108:18032–18037. doi: 10.1073/pnas.1115052108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mostoslavsky G, Fabian AJ, Rooney S, Alt FW, Mulligan RC. Complete correction of murine Artemis immunodeficiency by lentiviral vector–mediated gene transfer. Proc Natl Acad Sci USA. 2006;103:16406–16411. doi: 10.1073/pnas.0608130103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Delaney C, Varnum-Finney B, Aoyama K, Brashem-Stein C, Bernstein ID. Dose-dependent effects of the Notch ligand Delta1 on ex vivo differentiation and in vivo marrow repopulating ability of cord blood cells. Blood. 2005;106:2693–2699. doi: 10.1182/blood-2005-03-1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25:1105–1111. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Anders S, Pyl PT, Huber W. HTSeq—a Python framework to work with high-throughput sequencing data. Bioinformatics. 2014 Sep 25; doi: 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Reiner A, Yekutieli D, Benjamini Y. Identifying differentially expressed genes using false discovery rate controlling procedures. Bioinformatics. 2003;19:368–375. doi: 10.1093/bioinformatics/btf877. [DOI] [PubMed] [Google Scholar]
- 55.Yeung KY, Haynor DR, Ruzzo WL. Validating clustering for gene expression data. Bioinformatics. 2001;17:309–318. doi: 10.1093/bioinformatics/17.4.309. [DOI] [PubMed] [Google Scholar]
- 56.Saeed AI, et al. TM4: a free, open-source system for microarray data management and analysis. Biotechniques. 2003;34:374–378. doi: 10.2144/03342mt01. [DOI] [PubMed] [Google Scholar]
- 57.Young MD, Wakefield MJ, Smyth GK, Oshlack A. Gene ontology analysis for RNA-seq: accounting for selection bias. Genome Biol. 2010;11:R14. doi: 10.1186/gb-2010-11-2-r14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Burwick N, Coats SA, Nakamura T, Shimamura A. Impaired ribosomal subunit association in Shwachman-Diamond syndrome. Blood. 2012;120:5143–5152. doi: 10.1182/blood-2012-04-420166. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.