

Abstract
Inhalation of vesicants including sulfur mustard can cause significant damage to the upper airways. This is the result of vesicant-induced modifications of proteins important in maintaining the integrity of the lung. Cytochrome P450’s are the major enzymes in the lung mediating detoxification of sulfur mustard and its metabolites. NADPH cytochrome P450 reductase is a flavin-containing electron donor for cytochrome P450. The present studies demonstrate that the sulfur mustard analog, 2-chloroethyl ethyl sulfide (CEES), is a potent inhibitor of human recombinant cytochrome P450 reductase, as well as native cytochrome P450 reductase from liver microsomes of saline and β-naphthoflavone treated rats, and cytochrome P450 reductase from type II lung epithelial cells. Using rat liver microsomes from β-naphthoflavone-treated rats, CEES was found to inhibit CYP 1A1 activity. This inhibition was overcome by microsomal cytochrome P450 reductase from saline-treated rats, which lack CYP 1A1 activity, demonstrating that the CEES inhibitory activity was selective for cytochrome P450 reductase. Cytochrome P450 reductase also generates reactive oxygen species (ROS) via oxidation of NADPH. In contrast to its inhibitory effects on the reduction of cytochrome c and CYP1A1 activity, CEES was found to stimulate ROS formation. Taken together, these data demonstrate that sulfur mustard vesicants target cytochrome P450 reductase and that this effect may be an important mechanism mediating oxidative stress and lung injury.
Keywords: vesicants, sulfur mustard, CEES, pulmonary toxicity, oxidative stress
Introduction
Sulfur mustard is a bifunctional alkylating agent that has been used in chemical warfare (Papirmeister et al., 1991). Most deaths caused by sulfur mustard are due to pulmonary toxicity and resulting secondary infections (Uhrig, 1962). In humans, inhaled sulfur mustard causes irritation in the nose and throat, cough, bronchial constriction, and, over time or at higher doses, laryngitis, aphonia, bronchitis, and pneumonia (Emad and Rezaian, 1999). In animal models, sulfur mustard and related vesicants including 2-chloroethyl ethyl sulfide (CEES) and 2-chloroethyl 4-chlorobutyl sulfide, have been shown to induce oxidative stress (O’Neill et al.; Elsayed et al., 1992; Das et al., 2003; Elsayed and Omaye, 2004). This is associated with the accumulation of oxidized glutathione (Elsayed and Omaye, 2004) and inflammatory cytokines such as tumor necrosis factor α (Chatterjee et al., 2003) in the lung and the release of glutathione peroxidase into lung and bronchoalveolar fluid (Anderson et al., 2000; Elsayed and Omaye, 2004). These changes may contribute to pulmonary fibrosis and chronic alveolitis, long term consequences of exposure to sulfur mustard (Emad and Rezaian, 1999).
NADPH cytochrome P450 reductase supplies electrons to cytochrome P450 enzymes, which constitutes a system for the metabolism of many endogenous compounds and xenobiotics (Murataliev et al., 2004). The enzyme is composed of multiple domains that transport electrons from NADPH to various acceptors via flavin cofactors (Vermilion et al., 1981). Recent studies have shown that cytochrome P450’s are important targets for vesicants, although their precise site of action is unknown (Brimfield et al., 2008). In the present studies we used CEES as a model to evaluate mechanisms underlying the effects of sulfur mustard vesicants on cytochrome P450 reductase. CEES was found to be a potent inhibitor of human recombinant, rat microsomal and type II lung epithelial cell cytochrome P450 reductase. Cytochrome P450 reductase has been termed a ‘leaky enzyme’ because of its ability to autooxidize and generate reactive oxygen species (ROS) (Aust et al., 1972; Mishin et al., 1976; Grover and Piette, 1981; Mishin et al., 2010). Interestingly, CEES was also found to stimulate cytochrome P450 reductase mediated generation of ROS. These data indicate that vesicants can target cytochrome P450 reductase and inhibit xenobiotic metabolism. Stimulation of microsomal ROS production represents a novel activity of vesicants, a reaction that can increase cellular oxidative stress and contribute to toxicity.
Materials and Methods
Reagents and Treatments
Liver microsomes from corn oil, saline or β-naphthoflavone treated rats were obtained from Xenotech (Lenexa, KS). Supersomes™ expressing human cytochrome P450 reductase with or without cytochrome P450 1A1 (CYP1A1) were purchased from BD Biosciences (San Jose, CA), and 10-acetyl-3,7-dihydroxyphenoxazine (AMPLEX-RED) from Molecular Probes (Eugene, OR). 7-ethoxyresorufin, CEES, cytochrome c, 2, -6-dichlorophenolindophenol (DCPIP), NADPH, menadione, horseradish peroxidase, and all other chemicals were purchased from Sigma-Aldrich (St. Louis, MO). CEES was prepared fresh for each experiment by dilution in ethanol and mixing by gentle inversion. For all experiments, CEES was added to aqueous solutions at 1:100 dilutions and treatments were initiated within 10 s to minimize hydrolytic degradation. Unless otherwise indicated, CEES treatment of cellular lysates from lung epithelial cells (100 μg/ml), Supersomes, or rat liver microsomes were performed in 0.1 ml reactions at 37°C in 10 mM phosphate buffer (pH 7.4). After 1 hr, reactions were adjusted to pH 7.4 using 100 mM phosphate buffer (PBS) (pH 7.4).
Cells
MLE-15 murine lung epithelial cells were kindly provided by Dr. Jacob Finkelstein (University of Rochester) and maintained as previously described (Wikenheiser et al., 1993). Cells were grown in 6 well cell culture dishes and treated with vehicle control or CEES in 1 ml PBS. After 30 min, cells were washed twice with 2 ml PBS, gently scraped from culture dishes, centrifuged (500 g, 5 min), resuspended in 0.5 ml PBS, and sonicated on ice. Disrupted cells were centrifuged at 3000 × g for 10 min to remove cellular debris and 12,000 × g for 1 hr to separate mitochondrial fractions. The resulting supernatant was used in enzyme assays. In some experiments, cells were lysed as described above prior to treatment with CEES. Protein concentrations were measured using a BCA protein assay kit (Pierce Chemical Co., Rockford, IL) with bovine serum albumin as the standard.
Enzyme assays
Spectrophotometric assays were used to quantify reduction of cytochrome c, and DCPIP and NADPH utilization. Assays were performed at 37°C in 0.5 mL quartz cuvettes using a Lambda 20 UV/visible spectrophotometer (PerkinElmer Life Sciences). Cytochrome P450 reductase-dependent reduction of cytochrome c was measured by increases in absorbance at 550 nm every 10 s for 3 min or every min for 30 min (Phillips and Langdon, 1962). Reaction mixes contained rat liver microsomes (protein amount equivalent to 10 Units of cytochrome P450 reductase/ml) or 100 μg protein/ml MLE-15 cell lysates, 0.5 mM NADPH and 60 μM horse heart cytochrome c. Reduction of DCPIP, an artificial one electron acceptor which intercepts reducing equivalents prior to electron transfer from the flavin domains of cytochrome P450 reductase to cytochrome P450’s (Smith et al., 1994), was quantified by rates of decrease in absorbance at 600 nm over 10 min. Typical reactions contained 100 μM DCPIP, 7.2 U/ml recombinant human cytochrome P450 reductase or 100 mg protein/ml of lung cell lysates, and 0.1 mM NADPH. For NADPH utilization assays, recombinant human cytochrome P450 reductase (6 Units/ml) or rat liver microsomes from β-naphthoflavone-treated rats (equivalent to 2.9 Units of cytochrome P450 reductase activity/ml) were used. Final reaction mixes for measuring NADPH utilization contained 0.1 mM NADPH and either liver microsomes and 16.6 μM 7-ethoxyresorufin, or recombinant human cytochrome P450 reductase and 10 μM menadione. Reactions were initiated by the addition of NADPH, which was quantified by decreases in absorbance at 340 nm monitored at 2.5 min intervals for 30 min. For experiments with lysates from lung epithelial cells, 10 μM menadione was added to the reactions in place of 7-ethoxyresorufin as a terminal electron acceptor for cytochrome P450 reductase.
CYP 1A1 activity was measured by ethoxyresorufin O-deethylation (EROD) of 7-ethoxyresorufin to the fluorescent product resorufin (Burke et al., 1994). Reaction mixes in 100 μl consisted of 7.1 μg rat liver microsomes from β-naphthoflavone-treated rats, 3.3 mM MgCl2, 0.5 mM NADPH, 0.5 units glucose 6-phosphate dehydrogenase, 10 mM glucose 6-phosphate, and 4.2 μM 7-ethoxyresorufin. The activity of EROD was 3710 pmol resorufin/mg microsomal protein/min. Resorufin fluorescence was recorded every 2.5 min for 30 min using an HTS 7000 Plus Fluorescent BioAssay Reader (PerkinElmer Life Sciences) fitted with a 540-nm excitation filter and a 595-nm emission filter. The reaction mix contained 1 nmol/min of cytochrome c reductase activity. For experiments assessing the ability of cytochrome P450 reductase to restore EROD activity of CEES-treated microsomes from β-naphthoflavone-treated rats, aliquots of liver microsomes from saline-treated rats containing cytochrome P450 reductase activity (3.6 or 10.8 Units of NADPH cytochrome P450 reductase activity where 1 Unit is equivalent to 1 nmol cytochrome c reduced per min), were added 1 hr after the addition of CEES.
Assays for hydrogen peroxide
The Amplex Red/horseradish peroxidase assay was used to quantify production of ROS by cytochrome P450 reductase (Gray et al., 2007). Reaction mixes contained recombinant cytochrome P450 reductase, 25 μM Amplex Red, 1 Unit/ml horseradish peroxidase and 0.25 mM NADPH. Fluorescence was monitored using an HTS 7000 Plus Fluorescent Bio Assay Reader. In some experiments, cytochrome P450 reductase was treated with 0.01–10 mM CEES prior to addition to the enzyme assay. Subsequently, an NADPH regenerating system consisting of 0.1 mM NADPH, 0.5 U/ml glucose-6-phosphate dehydrogenase, and 10 mM glucose-6-phosphate was used. The rate of hydrogen peroxide formation was calculated based on a standard curve constructed with hydrogen peroxide in a concentration range of 0.5–16 μM. Hydrogen peroxide formation was also used to quantify chemical redox cycling in the presence of menadione by cytochrome P450 reductase as a measure of electron flow from NADPH through the flavin cofactors of the enzyme. Redox cycling reactions contained recombinant cytochrome P450 reductase (equivalent to 0.032 Units/ml) or 100 μg protein/ml of lung cell lysate, 25 μM Amplex Red, 0.25 mM NADPH, 1 Unit/ml horseradish peroxidase and 10 μM menadione.
Results
CEES inhibits CYP1A1 by inhibiting cytochrome P450 reductase
In initial studies, the effects of CEES on CYP1A1 related activity were characterized. In both recombinant CYP1A1 (Supersomes constructed to co-express cytochrome P450 reductase and cytochrome P450 1A1) and microsomes from β-naphthoflavone-treated rats, which contain CYP1A1, CEES (0.1–10 mM) was found to inhibit enzyme activity (Fig. 1, panels A and panel B). Using the recombinant P450 system, both EROD activity (IC50 = 1.2 mM) (Fig. 1, panel A) and NADPH utilization (IC50 = 3.6 mM) (Fig. 1, panel C), were inhibited. We next determined if inhibition of CYP1A1 activity was due to alterations in cytochrome P450 reductase activity. In rat liver microsomes, CEES was found to cause a concentration-dependent inhibition of cytochrome P450 reductase, as measured by cytochrome c reduction (IC50 = 2.9 mM) (Fig. 1, panel D). To confirm this, we performed reconstitution studies. In these experiments, microsomes from saline-treated rats, which contain cytochrome P450 reductase activity (3.6 or 10.8 Units of cytochrome c reductase activity), but little or no CYP1A1 activity (Sesardic et al. 1990), were added to reaction mixes containing CEES-treated microsomes from β-naphthoflavone-treated rats, which originally contained cytochrome P450 reductase activity equivalent to 1.0 Units of cytochrome c reductase activity. Under these conditions, EROD activity in the CEES-treated microsomes was completely restored (Fig. 1, panel B). These results indicate that CYP1A1 activity is largely unaffected by CEES, and that cytochrome P450 reductase is a primary target for CEES.
Figure 1. Inhibition of CYP1A1 and cytochrome P450 reductase by CEES.

Panels A and B. Effects of CEES on CYP1A1 activity. In panel A, human recombinant CYP1A1 co-expressed with recombinant cytochrome P450 reductase was pre-treated with CEES (0.1–10 mM) or vehicle control for 1 h and then analyzed for CYP1A1 by EROD activity. In panel B, rat liver microsomes from β-naphthoflavone-treated rats, which contain CYP1A1, were treated with 10 mM CEES to inhibit EROD activity. After 1 hr, cytochrome P450 reductase containing microsomes (3.6 or 10.8 U) from saline treated rats, which contain low or no CYP1A1, were added to the CEES-treated microsomes to restore CYP1A1 activity. The inset compares EROD activity in control- and 10 mM CEES-treated microsomes from β-naphthoflavone-treated rats. Panel C. Effects of CEES on NADPH utilization by human recombinant cytochrome P450 reductase. NADPH utilization was monitored in enzyme assays as described in Panel A. Panel D. Rat liver microsomes from corn oil-treated rats were treated with CEES (0.1–10 mM) and then analyzed for cytochrome P450 reductase activity using the cytochrome c reduction assay. Each point is the mean ± SE, n = 3.
In further experiments we characterized the inhibitory effects of CEES on cytochrome P450 reductase using human recombinant enzyme. CEES was found to cause a concentration-dependent inhibition of cytochrome c reduction by the recombinant enzyme (IC50 = 0.38 mM) (Fig. 2, panel A). At higher concentrations, CEES also inhibited reduction of DCPIP (IC50 = 8.6 mM), a substrate that accepts electrons from the flavin cofactor of the enzyme (Vermilion et al., 1981) (Fig. 2, panel B), as well as NADPH utilization (IC50 = 3.6 mM) (Fig. 2, panel C). In a distinct reaction, cytochrome P450 reductase also mediates the one electron reduction of redox active chemicals including menadione resulting in the formation of a semiquinone radical that can reduce molecular oxygen, generating superoxide anion and hydrogen peroxide (Thor et al., 1982; Butler and Hoey, 1993). This chemical redox cycling reaction is also mediated by electron flow through flavin cofactors in cytochrome P450 reductase (Murataliev et al., 2004). We found that menadione-stimulated redox cycling by cytochrome P450 reductase was also inhibited by CEES (IC50 = 1.2 mM) (Fig. 2, panel D).
Figure 2. Inhibition of human recombinant cytochrome P450 reductase by CEES.

Recombinant human cytochrome P450 reductase was pretreated with CEES (0.1–10 mM) or vehicle control. After 1 hr, enzyme activity was assessed by reduction of cytochrome c (Panel A), reduction of DCPIP (Panel B), utilization of NADPH during redox cycling (Panel C), or production of hydrogen peroxide during redox cycling (Panel D). In panels A and C, the quinone menadione (10 μM) was used to stimulate redox cycling. Each point is the mean ± SE, n = 3.
We next tested the effects of CEES on cytochrome P450 reductase activity in type II epithelial cells. In these experiments, MLE-15 cells were treated with CEES, lysed, and then assayed for enzyme activity. Using cytochrome c as the cytochrome P450 reductase electron acceptor, CEES was found to cause a concentration-dependent inhibition of enzyme activity (IC50 = 1.9 mM) (Fig. 3, panel A). CEES also inhibited the ability of lung epithelial cell lysates to mediate menadione-driven redox cycling (IC50 = 5 mM) and to reduce DCPIP (IC50 = 3.3 mM), a substrate that accepts electrons from the flavin cofactors of the enzyme (Fig. 3, panels B and C). Treatment of lysates of MLE-15 cells with CEES also resulted in inhibition of NADPH utilization in enzyme assays (IC50 = 2.4 mM), cytochrome c reduction (IC50 = 10 mM), and menadione-stimulated redox cycling (IC50 = 21 mM) (Fig. 3, panel D and not shown). Decreased sensitivity of cytochrome P450 reductase to CEES in lysates is likely due to increased availability of sulfhydryl-reactive materials.
Figure 3. Inhibition of cytochrome P450 reductase by CEES in type II lung epithelial cells.

MLE-15 cells were treated with increasing concentrations of CEES (0.1–10 mM) or vehicle control. After 1 hr, lysates were prepared and assayed for cytochrome P450 reductase-mediated reduction of cytochrome c (Panel A), or chemical redox cycling in the presence of 10 μM menadione (Panel B). Lysates from type II cells were also treated with CEES and then assayed for chemical redox cycling (Panel C) or their ability to reduce DCPIP (Panel D).
Cytochrome P450 reductase has the ability to generate superoxide anion (Grover and Piette, 1981; Mishin et al., 2010). Dismutation of superoxide anion generates hydrogen peroxide which can readily be quantified in reaction mixes (Zhou et al., 1997). Human recombinant cytochrome P450 reductase was found to generate hydrogen peroxide in a reaction that was dependent on NADPH (Fig. 4, panel A). In contrast to the inhibitory effects of CEES on redox cycling by cytochrome P450 reductase, spontaneous hydrogen peroxide production by the enzyme was stimulated by CEES (Fig. 4, panel B). This response was concentration dependent in the range of 0.1–10 mM CEES, reaching a maximum at 0.3 mM.
Figure 4. CEES stimulates the production of ROS by cytochrome P450 reductase.
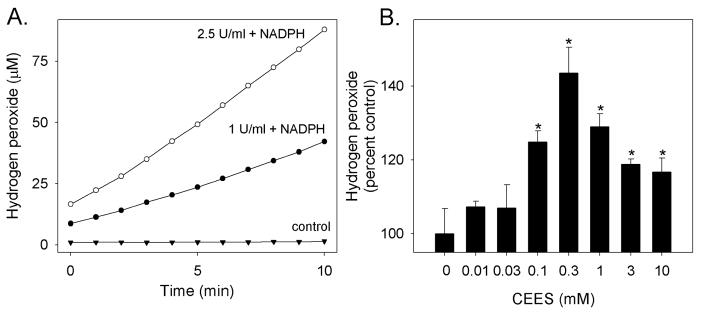
Panel A. Basal production of hydrogen peroxide. Recombinant human cytochrome P450 reductase (1 or 2.5 U/ml) was incubated with and without NADPH (100 μM). Hydrogen peroxide formation was quantified using Amplex Red. Panel B. CEES increases hydrogen peroxide formation. Recombinant NADPH cytochrome P450 reductase (1 U/ml) was treated with vehicle control or increasing concentrations of CEES (0.1–10 mM) and then analyzed for its ability to produce hydrogen peroxide. Values are expressed as percentage increase relative to control. Each point is the mean ± SE, n = 3. *Significantly different (p < 0.05) from control.
Discussion
Cytochrome P450 reductase is a ubiquitously expressed NADPH oxidoreductase that reduces a number of important cellular electron acceptors including the cytochrome P450 enzymes, cytochrome b5 and cytochrome c (Paine et al., 2005). During this reaction, electrons are shuttled from NADPH to acceptors via flavin cofactors by a two-step transfer; reduced FAD transfers electrons to FMN, and then from FMN to target acceptors. The present studies demonstrate that the model sulfur mustard vesicant, CEES, causes a concentration-dependent inhibition of recombinant and microsomal cytochrome P450 reductase activity, as well as cytochrome P450 reductase activity in type II epithelial cells. These findings are significant since this can result in inhibition of all cytochrome P450 enzymes, a process that may limit drug metabolism (Henderson et al., 2003). CEES was also found to inhibit recombinant and microsomal CYP1A1-related activity. To determine if this was due to the inhibition of cytochrome P450 reductase and/or CYP1A1, we performed reconstitution experiments in which control microsomes from saline treated rats which possess very low or no CYP1A1, were mixed with CEES-treated microsomes from β-naphthoflavone treated rats. Inhibition of CYP1A1 activity in the CEES-treated microsomes was effectively restored by the addition of control microsomes demonstrating that the half mustard primarily inactivates cytochrome P450 reductase. Inhibition of the cytochrome P450 system by vesicants such as CEES may interfere with xenobiotic metabolism in the lung, a process likely to contribute to pulmonary injury (Henderson et al., 2003).
Electron flow through cytochrome P450 reductase from NADPH to its acceptors is key for its function; impairment at any step along this pathway will inhibit enzyme activity (Murataliev et al., 2004). Using recombinant cytochrome P450 reductase, we analyzed the inhibitory effects of CEES at different sites of electron flow through the enzyme including NADPH utilization, as determined by disappearance of the pyridine cofactor, electron flow through the flavin cofactors, as measured by reduction of DCPIP, reduction of cytochrome c, menadione-mediated redox cycling, and electron flow to CYP1A1 (Vermilion and Coon, 1978). The most sensitive sites were cytochrome c reduction (IC50 = 0.9 mM) and CYP1A1 activity (IC50 = 0.9 mM), followed by NADPH utilization (IC50 = 3.5 mM), redox cycling (IC50 = 4.1 mM), and DCPIP reduction (IC50 = 8.6 mM). Generally similar results were obtained for cytochrome P450 reductase activity in type II lung epithelial cells. The increased sensitivity of the reduction of cytochrome c and CYP1A1-related activity to CEES over that of NADPH oxidation suggests that CEES may interfere with the final steps of electron transfer between the reductase and terminal electron acceptors including cytochrome P450.
Free radical derivatives of CEES and sulfur mustard are highly reactive, rapidly modifying proteins and DNA (Brookes and Lawley, 1961; Zhang et al., 1995). Ionization of a terminal chlorine of either CEES or sulfur mustard results in the formation of a reactive carbonium ion; alternatively, internal cyclization reactions result in the formation of cyclic sulfonium ions (Loveless and Revell, 1949; Lawley, 1966; Van Duuren et al., 1974; Fox and Scott, 1980). Reactions of the latter species with intracellular oxidoreductases including cytochrome P450 reductase, thioredoxin reductase, or neuronal nitric oxide synthase have been reported to form terminal carbon radicals (Brimfield et al., 2008). These radicals preferentially alkylate cysteine, although other amino acids are also targets including the carboxyl groups of acidic amino acids (Moore et al., 1946; Goodlad, 1957). Site-directed mutagenesis studies using human cytochrome P450 reductase have identified two amino acid residues in the substrate binding site of human cytochrome P450 reductase, aspartic acid 148 and glutamic acid 153, that are critical for activity (Zhao et al., 1999). Mutations at these sites, while not affecting FMN binding, strongly alter the ability of the enzyme to either reduce cytochrome c or support CYP2D6-mediated codeine O-demethylation (Zhao et al., 1999). The identification of these amino acids in the substrate binding pocket is consistent with crystallographic data showing that cytochrome P450 reductase has an unusual charge structure that overlaps with the substrate docking site (Zhao et al., 1999). In this regard, cross-linking studies between cytochrome P450 reductase and CYP2B4 indicate that the connecting domain of the reductase, a structure rich in sulfhydryl-containing amino acids, is critical in substrate binding (Bumpusa and Hollenberg, 2010). Further studies are required to determine if CEES selectively alkylates critical amino acids needed for enzyme activity or that alters the structure or charge of the cytochrome P450 reductase substrate binding site.
Under homeostatic conditions, cytochrome P450 reductase spontaneously generates superoxide anion, a process that results from the direct reduction of molecular oxygen by the flavin cofactors in the enzyme (Grover and Piette, 1981; Mishin et al., 2010). We found that, at the same time that CEES inhibited cytochrome P450 reductase-mediated substrate reduction, it stimulated spontaneous ROS formation. This reaction was NADPH-dependent and occurred under conditions where CEES had no effect on the transfer of electrons from NADPH to the flavin cofactors in the enzyme. These data suggest that CEES may induce toxicity, at least in part, by enhancing the production of ROS and inducing oxidative stress. This is supported by previous studies demonstrating that vesicants induce oxidative stress in lung tissue and that antioxidants can protect against vesicant-induced tissue injury (Kumar et al., 2001; Das et al., 2003).
The precise mechanisms by which CEES and sulfur mustard induce oxidative stress are not known. Alkylation and disruption of critical cellular components in cells and tissues elicits an inflammatory response characterized by the infiltration of leukocytes into sites of tissue injury (Das et al., 2003). Macrophages and neutrophils recruited to the lung following injury are known to release a variety of oxidants that can contribute to cell damage (Osterlund et al., 2005; Brown et al., 2008; Bucht, 2008). These cells, as well as damaged parenchymal cells, can also generate ROS as well as reactive nitrogen species and contribute to the oxidative burden of the lung (Rahman and MacNee, 2000; Das et al., 2003). Sulfur mustard is also known to react directly with cellular antioxidants such as glutathione as well as enzymes important in detoxifying ROS including superoxide dismutase and catalase (Black et al., 1992; Husain et al., 1996), a process that reduces their activity leading to oxidative stress (Gross et al., 1993; Rahman et al., 2006). Our work demonstrating that CEES can directly stimulate cytochrome P450 reductase to produce ROS represents a novel mechanism underlying vesicant-induced oxidative stress in target cells. The ubiquitous expression of this enzyme may make it an important target for vesicants in other tissues, including the eye and skin, as these tissues also undergo oxidative stress in response to vesicants (Utley and Mehendale, 1989). Depletion of intracellular antioxidants by CEES directly or increased ROS production by modified cytochrome P450 reductase damaged by CEES may make this tissue more susceptible to other inhaled oxidants such as ozone, or nitrogen dioxide, or to oxidants generated by endogenous inflammatory cells including macrophages and neutrophils (Rahman, 2003). Increased ROS production by CEES-modified cytochrome P450 reductase may also explain, in part, persistant pulmonary injury even in the absence of the original damaging agent.
In summary, our studies demonstrate that CEES is a selective inhibitor of substrate reduction by cytochrome P450 reductase. Cytochrome P450 reductase is important not only in drug metabolism, but also in mediating a number of key enzymatic reactions including those involved in sterol biosynthesis and metabolism, fatty acid metabolism, bile acid synthesis and heme catabolism (Henderson et al., 2003). Thus, inhibition of cytochrome P450 reductase activity is likely to have significant adverse consequences in tissues targeted by vesicants including maintaining homeostasis and responding to injury and oxidative stress. Of particular interest was our novel finding that CEES stimulates NADPH-dependent production of ROS, a process that may contribute to pulmonary oxidative stress. Further studies are required to elucidate the molecular mechanism by which CEES inhibits substrate reduction by cytochrome P450 reductase while at the same time, stimulating the production of ROS.
Acknowledgments
The authors thank Sohaib Jamil and Joanne Gentile for technical assistance. This work was supported in part by National Institutes of Health grants (AR055073, CA100994, CA093798, ES004738, CA132624, GM034310 and ES005022) and the Alexander Trust Fund. This work was also funded by the National Institutes of Health CounterACT Program through the National Institute of Arthritis and Musculoskeletal and Skin Diseases (award #U54AR055073).
Abbreviations
- CEES
2-chloroethyl ethyl sulfide
- EROD
ethoxyresorufin O-deethylation
- DCPIP
2,6-dichlorophenolindophenol
- ROS
reactive oxygen species
- PBS
phosphate buffered saline
Footnotes
Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the United States Coast Guard or the federal government.
Conflict of Interest Statement
The authors declare that there are no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Anderson DR, Byers SL, Vesely KR. Treatment of sulfur mustard (HD)-induced lung injury. J Appl Toxicol. 2000;20(Suppl 1):S129–132. doi: 10.1002/1099-1263(200012)20:1+<::aid-jat670>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- Aust SD, Roerig DL, Pederson TC. Evidence for superoxide generation by NADPH-cytochrome c reductase of rat liver microsomes. Biochem Biophys Res Commun. 1972;47:1133–1137. doi: 10.1016/0006-291x(72)90952-7. [DOI] [PubMed] [Google Scholar]
- Black RM, Brewster K, Clarke RJ, Hambrook JL, Harrison JM, Howells DJ. Biological fate of sulphur mustard, 1,1′-thiobis(2-chloroethane): isolation and identification of urinary metabolites following intraperitoneal administration to rat. Xenobiotica. 1992;22:405–418. doi: 10.3109/00498259209046652. [DOI] [PubMed] [Google Scholar]
- Brimfield AA, Mancebo AM, Mason RP, Jiang JJ, Siraki AG, Novak MJ. Free radical production from the interaction of 2-chloroethyl vesicants (mustard gas) with pyridine nucleotide-driven flavoprotein electron transport systems. Toxicol Appl Pharmacol. 2008;234:128–134. doi: 10.1016/j.taap.2008.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brookes P, Lawley PD. The reaction of mono- and di-functional alkylating agents with nucleic acids. Biochem J. 1961;80:496–503. doi: 10.1042/bj0800496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown RS, Jugg B, Smith A, Mann T, Fairhall S, Platt J, Sciuto AM. Inhalation toxicology of sulfur mustard: Dose ranging study in an anesthetized porcine model. Proceedings of the US Army Medical Defense Bioscience Review. 2008:191. [Google Scholar]
- Bucht A. Role of the immune system in the patholgenesis of respiratory disease caused by aklylating agents. Proceedings of the US Army Medical Defense Bioscience Review. 2008:174. [Google Scholar]
- Burke MD, Thompson S, Weaver RJ, Wolf CR, Mayer RT. Cytochrome P450 specificities of alkoxyresorufin O-dealkylation in human and rat liver. Biochem Pharmacol. 1994;48:923–936. doi: 10.1016/0006-2952(94)90363-8. [DOI] [PubMed] [Google Scholar]
- Butler J, Hoey BM. The one-electron reduction potential of several substrates can be related to their reduction rates by cytochrome P-450 reductase. Biochim Biophys Acta. 1993;1161:73–78. doi: 10.1016/0167-4838(93)90198-z. [DOI] [PubMed] [Google Scholar]
- Chatterjee D, Mukherjee S, Smith MG, Das SK. Signal transduction events in lung injury induced by 2-chloroethyl ethyl sulfide, a mustard analog. Journal of biochemical and molecular toxicology. 2003;17:114–121. doi: 10.1002/jbt.10068. [DOI] [PubMed] [Google Scholar]
- Das SK, Mukherjee S, Smith MG, Chatterjee D. Prophylactic protection by N-acetylcysteine against the pulmonary injury induced by 2-chloroethyl ethyl sulfide, a mustard analogue. Journal of biochemical and molecular toxicology. 2003;17:177–184. doi: 10.1002/jbt.10076. [DOI] [PubMed] [Google Scholar]
- Elsayed NM, Omaye ST. Biochemical changes in mouse lung after subcutaneous injection of the sulfur mustard 2-chloroethyl 4-chlorobutyl sulfide. Toxicology. 2004;199:195–206. doi: 10.1016/j.tox.2004.02.020. [DOI] [PubMed] [Google Scholar]
- Elsayed NM, Omaye ST, Klain GJ, Korte DW., Jr Free radical-mediated lung response to the monofunctional sulfur mustard butyl 2-chloroethyl sulfide after subcutaneous injection. Toxicology. 1992;72:153–165. doi: 10.1016/0300-483x(92)90109-r. [DOI] [PubMed] [Google Scholar]
- Emad A, Rezaian GR. Immunoglobulins and cellular constituents of the BAL fluid of patients with sulfur mustard gas-induced pulmonary fibrosis. Chest. 1999;115:1346–1351. doi: 10.1378/chest.115.5.1346. [DOI] [PubMed] [Google Scholar]
- Fox M, Scott D. The genetic toxicology of nitrogen and sulphur mustard. Mutat Res. 1980;75:131–168. doi: 10.1016/0165-1110(80)90012-3. [DOI] [PubMed] [Google Scholar]
- Goodlad GA. Esterification of protein and amino acid carboxyl groups by mustard gas and related compounds. Biochim Biophys Acta. 1957;24:645–646. doi: 10.1016/0006-3002(57)90264-0. [DOI] [PubMed] [Google Scholar]
- Gray JP, Heck DE, Mishin V, Smith PJ, Hong JY, Thiruchelvam M, Cory-Slechta DA, Laskin DL, Laskin JD. Paraquat increases cyanide-insensitive respiration in murine lung epithelial cells by activating an NAD(P)H:paraquat oxidoreductase: identification of the enzyme as thioredoxin reductase. J Biol Chem. 2007;282:7939–7949. doi: 10.1074/jbc.M611817200. [DOI] [PubMed] [Google Scholar]
- Gross CL, Innace JK, Hovatter RC, Meier HL, Smith WJ. Biochemical manipulation of intracellular glutathione levels influences cytotoxicity to isolated human lymphocytes by sulfur mustard. Cell biology and toxicology. 1993;9:259–267. doi: 10.1007/BF00755604. [DOI] [PubMed] [Google Scholar]
- Grover TA, Piette LH. Influence of flavin addition and removal on the formation of superoxide by NADPH-Cytochrome P-450 reductase: a spin-trap study. Arch Biochem Biophys. 1981;212:105–114. doi: 10.1016/0003-9861(81)90348-9. [DOI] [PubMed] [Google Scholar]
- Henderson CJ, Otto DM, Carrie D, Magnuson MA, McLaren AW, Rosewell I, Wolf CR. Inactivation of the hepatic cytochrome P450 system by conditional deletion of hepatic cytochrome P450 reductase. J Biol Chem. 2003;278:13480–13486. doi: 10.1074/jbc.M212087200. [DOI] [PubMed] [Google Scholar]
- Husain K, Dube SN, Sugendran K, Singh R, Das Gupta S, Somani SM. Effect of topically applied sulphur mustard on antioxidant enzymes in blood cells and body tissues of rats. J Appl Toxicol. 1996;16:245–248. doi: 10.1002/(SICI)1099-1263(199605)16:3<245::AID-JAT339>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- Kumar O, Sugendran K, Vijayaraghavan R. Protective effect of various antioxidants on the toxicity of sulphur mustard administered to mice by inhalation or percutaneous routes. Chem Biol Interact. 2001;134:1–12. doi: 10.1016/s0009-2797(00)00209-x. [DOI] [PubMed] [Google Scholar]
- Lawley PD. Effects of some chemical mutagens and carcinogens on nucleic acids. In: Davidson JN, Cohn WE, editors. Prog Mucl Acid Res Mol Biol. Academic Press; New York and London: 1966. pp. 89–131. [DOI] [PubMed] [Google Scholar]
- Loveless A, Revell S. New evidence on the mode of action of ‘metotic poisons’. Nature (London) 1949;164:938–944. doi: 10.1038/164938a0. [DOI] [PubMed] [Google Scholar]
- Mishin V, Gray JP, Heck DE, Laskin DL, Laskin JD. Application of the Amplex Red/Horseradish Peroxidase Assay to measure hydrogen peroxide generation by recombinant microsomal enzymes. J Free Radic Biol Med. 2010 doi: 10.1016/j.freeradbiomed.2010.02.030. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishin V, Pokrovsky A, Lyakhovich VV. Interactions of some acceptors with superoxide anion radicals formed by the NADPH-specific flavoprotein in rat liver microsomal fractions. Biochem J. 1976;154:307–310. doi: 10.1042/bj1540307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore S, Stein WH, Fruton JS. Chemical reactions of mustard gas and related compounds: II. The reaction of mustard gas with carboxyl groups and with the amino groups of amino acids and peptides. J Org Chem. 1946;11:675–680. doi: 10.1021/jo01176a008. [DOI] [PubMed] [Google Scholar]
- Murataliev MB, Feyereisen R, Walker FA. Electron transfer by diflavin reductases. Biochim Biophys Acta. 2004;1698:1–26. doi: 10.1016/j.bbapap.2003.10.003. [DOI] [PubMed] [Google Scholar]
- O’Neill HC, White CW, Veress LA, Hendry-Hofer TB, Loader JE, Min E, Huang J, Rancourt RC, Day BJ. Treatment with the catalytic metalloporphyrin AEOL 10150 reduces inflammation and oxidative stress due to inhalation of the sulfur mustard analog 2-chloroethyl ethyl sulfide. Free Radic Biol Med. doi: 10.1016/j.freeradbiomed.2010.01.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osterlund C, Billiehook B, Ekstrand-Hammarstrom B, Sandstrom T, Bucht A. The nitrogen mustard melphalan activates mitogen-activated phosphorylated kinases (MAPK), nuclear factor-KB and inflammatory response in lung epithelial cells. J Appl Toxicol. 2005;25(4):328–337. doi: 10.1002/jat.1070. [DOI] [PubMed] [Google Scholar]
- Paine MJI, Scrutton NS, Munro AW, Gutierrez A, Roberts GCK, Wolf CR. Electron Transfer Partners of Cytochrome P450. In: Ortiz de Montellano PR, editor. Cytochrome P450: Structure, Mechanism, and Biochemistry. Kluwer Academic/Planum Publishers; New York: 2005. pp. 115–148. [Google Scholar]
- Papirmeister B, Feister AJ, Robinson SI, Ford RD. Medical defense against mustard gas: Toxic mechanisms and pharmacological implications. CRC Press; Boca Raton, FL: 1991. [Google Scholar]
- Phillips AH, Langdon RG. Hepatic triphosphopyridine nucleotide-cytochrome c reductase: isolation, characterization, and kinetic studies. J Biol Chem. 1962;237:2652–2660. [PubMed] [Google Scholar]
- Rahman I. Oxidative stress, chromatin remodeling and gene transcription in inflammation and chronic lung diseases. Journal of biochemistry and molecular biology. 2003;36:95–109. doi: 10.5483/bmbrep.2003.36.1.095. [DOI] [PubMed] [Google Scholar]
- Rahman I, Biswas SK, Kode A. Oxidant and antioxidant balance in the airways and airway diseases. Eur J Pharmacol. 2006;533:222–239. doi: 10.1016/j.ejphar.2005.12.087. [DOI] [PubMed] [Google Scholar]
- Rahman I, MacNee W. Regulation of redox glutathione levels and gene transcription in lung inflammation: therapeutic approaches. Free Radic Biol Med. 2000;28:1405–1420. doi: 10.1016/s0891-5849(00)00215-x. [DOI] [PubMed] [Google Scholar]
- Smith GC, Tew DG, Wolf CR. Dissection of NADPH-cytochrome P450 oxidoreductase into distinct functional domains. Proc Natl Acad Sci U S A. 1994;91:8710–8714. doi: 10.1073/pnas.91.18.8710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thor H, Smith MT, Hartzell P, Bellomo G, Jewell SA, Orrenius S. The metabolism of menadione (2-methyl-1,4-naphthoquinone) by isolated hepatocytes. A study of the implications of oxidative stress in intact cells. J Biol Chem. 1982;257:12419–12425. [PubMed] [Google Scholar]
- Uhrig HT. Some medical aspects of chemical agents. J Med Assoc Ala. 1962;32:144–150. [PubMed] [Google Scholar]
- Utley WS, Mehendale HM. Phenobarbital-induced cytosolic cytoprotective mechanisms that offset increases in NADPH cytochrome P450 reductase activity in menadione-mediated cytotoxicity. Toxicology and applied pharmacology. 1989;99:323–333. doi: 10.1016/0041-008x(89)90014-8. [DOI] [PubMed] [Google Scholar]
- Van Duuren BL, Witz G, Sivak V. Chemical carcinogenesis. In: Homburger F, editor. The physiopathology of cancer. Karger; Basel: 1974. pp. 1–63. [Google Scholar]
- Vermilion JL, Ballou DP, Massey V, Coon MJ. Separate roles for FMN and FAD in catalysis by liver microsomal NADPH-cytochrome P-450 reductase. J Biol Chem. 1981;256:266–277. [PubMed] [Google Scholar]
- Vermilion JL, Coon MJ. Identification of the high and low potential flavins of liver microsomal NADPH-cytochrome P-450 reductase. J Biol Chem. 1978;253:8812–8819. [PubMed] [Google Scholar]
- Wikenheiser KA, Vorbroker DK, Rice WR, Clark JC, Bachurski CJ, Oie HK, Whitsett JA. Production of immortalized distal respiratory epithelial cell lines from surfactant protein C/simian virus 40 large tumor antigen transgenic mice. Proc Natl Acad Sci U S A. 1993;90:11029–11033. doi: 10.1073/pnas.90.23.11029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Peters BP, Monteiro-Riviere NA. Assessment of sulfur mustard interaction with basement membrane components. Cell Biol Toxicol. 1995;11:89–101. doi: 10.1007/BF00767494. [DOI] [PubMed] [Google Scholar]
- Zhao Q, Modi S, Smith G, Paine M, McDonagh PD, Wolf CR, Tew D, Lian LY, Roberts GC, Driessen HP. Crystal structure of the FMN-binding domain of human cytochrome P450 reductase at 1.93 A resolution. Protein Sci. 1999;8:298–306. doi: 10.1110/ps.8.2.298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou M, Diwu Z, Panchuk-Voloshina N, Haugland RP. A stable nonfluorescent derivative of resorufin for the fluorometric determination of trace hydrogen peroxide: applications in detecting the activity of phagocyte NADPH oxidase and other oxidases. Analytical biochemistry. 1997;253:162–168. doi: 10.1006/abio.1997.2391. [DOI] [PubMed] [Google Scholar]