

Abstract
Purpose of Review
The field of systemic lupus erythematosus (SLE) genetics has been advancing rapidly in recent years. This review will summarize recent progress.
Recent Findings
Genome-wide association and follow up studies have greatly expanded the list of associated polymorphisms, and much current work strives to integrate these polymorphisms into immune system biology and the pathogenic mediators involved in the disease. This review covers some current areas of interest, including genetic studies in non-European SLE patient populations, studies of pathogenic immune system subphenotypes such as type I interferon (IFN) and autoantibodies, and a rapidly growing body of work investigating the functional consequences of the genetic polymorphisms associated with SLE.
Summary
These studies provide a fascinating window into human SLE disease biology. As the work proceeds from genetic association signal to altered human biology, we move closer to tailoring interventions based upon an individual’s genetic substrate.
Keywords: systemic lupus erythematosus, genetics, interferon, autoantibodies
Introduction
Systemic lupus erythematosus (SLE) is a chronic autoimmune disease with a strong genetic component. Identical twin concordance rates for SLE range between 30–50%1, 2, and first-degree relatives of lupus patients have an approximately 20-fold increased risk of SLE as compared to the general population3. While much of the heritability of the disease remains to be explained, recent advances have significantly increased our knowledge regarding the genetic basis of SLE susceptibility. These include studies of multiple different world populations, genetic studies of immune system phenotypes such as type I interferon (IFN) and autoantibodies, and functional genetic studies examining the role of genetic variation in transcriptional regulation and immune cell functioning. This is a large body of work, and this review cannot be completely comprehensive, but I will try to illustrate some recent studies that illustrate the ideas outlined above and highlight some findings from the recent literature in SLE genetics. This exciting and rapidly evolving area of research has the potential to greatly inform our understanding of human lupus pathogenesis.
Case-control Genetic Association Studies
Genome-wide association studies have significantly expanded our list of SLE susceptibility loci in the last 10 years4, although these studies have largely been performed in European ancestry subjects. Strategies to enhance discovery of new SLE-associated genes have involved performing larger studies in European ancestry with greater statistical power for discovery5, and a growing number of studies have assessed the genetic basis of SLE in non-European ancestral backgrounds. There is a strong precedent that the genetic susceptibility alleles for lupus will differ between different world populations. The PTPN22 gene is an example of a well-established genetic risk factor for lupus and a number of other autoimmune conditions in European ancestry6. Studies to determine whether this allele is a risk factor in African populations have demonstrated that the allele assists essentially almost absent in sub-Saharan African populations7. The IRF5 gene provides an interesting example of another SLE-risk gene which is not shared across all ancestral backgrounds. A long haplotype of IRF5 has been definitively implicated as an SLE-risk allele8, 9, and interestingly this allele was not present in sub-Saharan African populations10. This risk allele promotes autoantibody formation in SLE and non-SLE affected individuals10, 11, and also has been shown to be associated with higher type I IFN levels in patients with SLE10, 12. It was initially unclear why this risk allele was not observed in African populations until recent studies of the Neanderthal genome have confirmed that this IRF5 SLE-risk allele is of Neanderthal origin13. Therefore, its distribution in the European population is related to the historical Neanderthal range.
Recent genome wide association studies in Asian SLE patients have demonstrated a number of novel associations that were unique to Asian populations, such as ETS1, IKZF1, RASGRP3, SLC15A4, and others14, 15. These studies also confirmed that many European ancestry risk loci were also relevant in this ancestral background14, 15. Studies of African-American SLE patients thus far have included an assessment of candidate loci discovered in European ancestry16, 17. This study found that some of the SLE-risk alleles discovered in European ancestry were relevant in African-American lupus, such as BANK1, ITGAM, IRF7, BLK, and others16, 17. As would be expected based on the discussion above, some European ancestry risk loci were not risk factors for lupus in African-Americans, such as PTPN22, MECP2, and others16. Genome wide association scans are currently underway in African-Americans to discover novel risk loci relevant in that population, and some candidate gene studies guided by biological hypotheses have identified new SLE-risk loci relevant to African-Americans18–20.
Type I Interferon: pathogenic immune system phenotype regulated by SLE-associated genetic polymorphisms
Multiple lines of evidence support the idea of the type one interferon is a primary pathogenic feature in lupus21. Administration of recombinant human interferon to treat viral infections and malignancies has resulted in de novo cases of lupus which improved when interferon is stopped22. Additionally many of the genetic factors noted in the section above function within the type I IFN pathway23. Previous studies of lupus families have indicated that at type I IFN levels are high and lupus family members, and cluster as a complex heritable trait24, 25. Familial aggregation of type I IFN levels may also occur in juvenile dermatomyositis26, another autoimmune condition characterized by high circulating type I IFN. Increased type I IFN levels and evidence for heritability of the IFN trait are observed in SLE patients of all ancestral backgrounds24, 25, 27. Gene expression studies performed in multiple ancestral backgrounds suggest that there are differences in IFN pathway activation and IFN-induced gene expression between ancestral backgrounds28–30. In African-American SLE patients, it seems that high type I IFN levels are more dependent autoantibodies than in European-American SLE patients, as the correlation between autoantibodies to RNA-binding proteins and high IFN is stronger in African-American SLE cohorts than in European-American SLE cohorts28, 30. Familial aggregation has also been observed in IL-10 and tumor necrosis factor alpha (TNF-α) cytokines in SLE families31, 32, although in each of these studies there was also correlation between SLE patients and their spouses (unrelated family members), suggesting an environmental contribution to the familial correlation. No spousal correlation was observed with type I IFN levels.
When lupus risk genes which function in the interferon pathway have been studied, generally gain-of-function within the IFN pathway in humans has been confirmed23, 33. This makes sense, as type I IFN is a pathogenic factor, and is heritable to some degree in a polygenic fashion. Follow up studies have demonstrated examples of polymorphisms which are associated with higher circulating levels of type I IFN, such as IRF5, IRF7, SPP1, and others12, 34, 35. Examples of polymorphisms that result in increased IFN-induced gene expression or increased sensitivity to interferon have been demonstrated, such as STAT4, IFIH1, and IRF836–38.
Given that type I IFN is a heritable immune system phenotype related to risk of lupus, it would make sense that studying interferon levels as a genetic trait could yield additional risk loci. Studies have been conducted to identify novel genetic risk factors for SLE using serum IFN as the genetic trait and determining which genetic polymorphisms are associated with high IFN levels in lupus patients39–41. These studies have demonstrated number of novel polymorphisms, including PNP and PRKG141, which have not been reported in case-control design studies, suggesting that the study of sub-phenotypes within SLE will be important to fully map the genetic architecture of the disease. The model would be that the genetic variations induce functional genetic changes, which then modulate important immune system phenotypes such as type I IFN or autoantibodies, and then these immune system phenotypes impact disease manifestations (See Figure 1).
Figure 1.
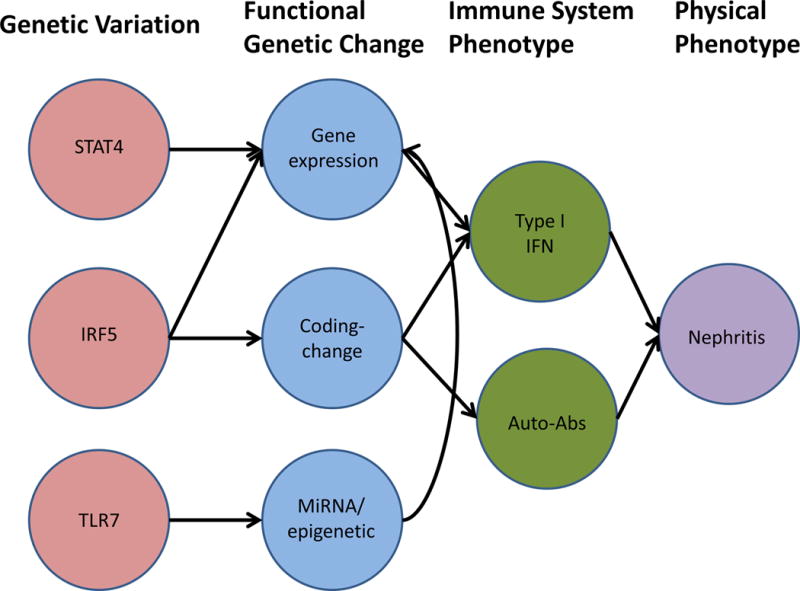
Schematic diagram illustrating potential pathways and causal chains linking genetic variations, functional genetic changes, immune system phenotypes, and physical phenotypes in SLE. Arrows are meant to indicate that one factor influences the other.
Associations with autoantibodies and nephritis
Genetic variations in the STAT4 gene have been associated with nephritis in SLE patients42, as have variants in the MYH9 and APOL1 genes18, 43. The same STAT4 variant has been associated with dsDNA autoantibodies, as well as increased sensitivity to type I IFN37, 42. A number of SLE-risk loci have been associated with autoantibody formation in SLE, including STAT4 and IRF5 as mentioned above, IRF7, IRF8, UBE2L3, CR2, and others35, 38, 44, 45. While the exact molecular mechanism by which these genetic variants influence autoantibody formation is not clear, it is interesting that many of these genes have functions within the TLR and type I IFN pathways. HLA alleles have also been associated with autoantibody production in SLE46, and in this case an attractive hypothesis is that the SLE-associated HLA allele might correspond to an altered MHC molecule which presents self-antigens of a particular type in an abnormal fashion, leading to autoantibody formation against those self-antigens.
Monogenic SLE and SLE-like syndromes
While family studies have indicated that most cases of lupus are polygenic, there are monogenic forms of SLE and lupus-like syndromes which are of monogenic origin. One of the earliest discovered monogenic causes of lupus with a very high penetrance is C1Q deficiency47. Homozygous deficiency of C1q is a single gene recessive inheritance cause of lupus with greater than 90% penetrance. With the evolution of modern sequencing techniques which can reliably detect rare genetic variations across the entire genome, a number of other monogenic causes of lupus and lupus-like syndromes are emerging. Interestingly deficiency of a DNase enzyme (DNASE1L3) was discovered in consanguineous families, and deficiency of this enzyme was associated with a lupus like syndrome and a recessive fashion48. Rare mutations in the TREX1 gene have been associated with a monogenic disease with some similarities to lupus called Aicardi-Gutierrez syndrome (AGS)49, 50. AGS is characterized by elevated circulating levels of interferon, CNS inflammation and calcification, and vasculitis. Mutations in TREX1 have also been reported in the clinical syndromes familial chilblain lupus50 and retinal vasculopathy with cerebral leukodystrophy51. These monogenic syndromes are characterized by a detectable interferon signature in serum and cerebrospinal fluid, neurological manifestations, and vasculitis, and have been termed as a group “monogenic interferonopathies”52. Other single gene defects that have been associated with interferonopathies include mutations in RNASEH2A, RNASEH2C, SAMHD1, ADAR, and IFIH152. This fascinating group of syndromes share some features of lupus such as vasculitis and an increase in circulating type I IFN, but are also somewhat distinct as they are not associated some other classical SLE features such as immune complex renal disease. Thus, these conditions may be considered “phenocopies” of SLE – monogenic syndromes that resemble the complex genetic disease SLE in some ways. The overlap between risk alleles for these conditions is also fascinating, as both IFIT1 and TREX1 are genes that harbor common polygenic risk alleles for SLE49, 53. In the case of IFIT1, increased IFN-induced gene expression has been observed in SLE patients who carry the risk allele36, 53. Another recently reported monogenic interferonopathy is STING- associated vasculopathy with onset in infancy (SAVI)54. These patients had early onset vasculopathy and pulmonary inflammation associated with mutations in TMEM173, which encodes the stimulator of IFN genes (STING) protein. Constitutive IFN pathway activation was observed in the context of the disease-associated mutations54, supporting this syndrome as a novel monogenic interferonopathy.
Functional genetics of SLE-associated polymorphisms
Genetic risk variants discovered in autoimmune diseases are frequently in regulatory regions instead of the coding region of the gene55. This is true for SLE, as many SLE-associated polymorphisms are found nearby genes in regulatory regions and likely impact cell biology by altering gene expression. Only a few lupus associated polymorphisms are coding-change polymorphisms that change the amino acid sequence of the protein. UBE2L3 provides an example of a functional haplotype tagged by a polymorphism in the promoter region which confers increased expression of UBE2L3 messenger RNA56 and protein57. This haplotype is also associated with increased NF-κB activation in B cells and monocytes in vitro57. In SLE patients in vivo, the UBE2L3 genotype is associated with increased circulating plasma cells and plasmablasts57, and increased circulating IFN in African-Americans in a recessive fashion44. The TNFAIP3 gene contains a number of different functional elements, however the best fit lupus-associated polymorphism occurs downstream of the gene in a functional regulatory element58. This regulatory element has been shown to interact with the TNFAIP3 promoter region via long-range looping and binding of NF-κB and SATB1 proteins, altering expression of TNFAIP359. In African-Americans, rare coding change variations in TNFAIP3 which are uniquely African in origin have been shown to alter the ubiquitination activity of the TNFAIP3 gene product19. This theme is also common, in which the same risk locus contains multiple different functional polymorphisms with different effects which result in disease susceptibility. The TNFAIP3 region seems to have a complex influence on autoimmunity, as different alleles have been associated with different autoimmune diseases60–62, and interestingly the SLE-risk haplotype appears to be protective in psoriasis62. The IL10 gene provides another example of a lupus risk polymorphism which has regulatory function. In this case, the SLE-risk allele is upstream of the IL10 gene, within a binding region for the Elk-1 transcription factor63. This study was able to demonstrate that the single base change encoded by the SLE-risk allele resulted in alternate binding of the Elk-1 transcription factor. It was also noted that expression of IL-10 and phosphorylated Elk-1 were elevated in SLE patients and correlated with disease activity63. Another interesting example of an SLE-risk regulatory polymorphism is in the ETS1 gene. Genetic variations in the near downstream region of the ETS1 gene have associated with SLE in Asian populations14. Follow up fine-mapping and functional studies confirm this region as being associated preferentially in Asian populations, and expression databases suggest that the polymorphism is associated with alternate ETS1 expression in Asian cell lines64. The SLE risk allele alters binding of phosphorylated STAT164, providing an attractive potential explanation for the differences observed in gene expression related to the allele. The SLE-risk allele is polymorphic across all studied populations, with a relatively similar allele frequency, and while some evidence exists for association of this allele with SLE in Europeans65, it seems that there is a larger effect in Asian populations with respect to SLE susceptibility. Thus, this study provides a fascinating example of an ancestry-specific functional genetic polymorphism, and reminds us of the complexity of the human biology being studied. The reasons for differences by ancestry are not clear, but gene-gene interactions and epistasis, ancestry-specific differences in protein co-factor expression, and differences in environmental factors would be possible explanations for why the same functional change in genomic DNA results in a differential effect on SLE susceptibility between populations.
While less common, coding change polymorphisms have also been associated with risk of SLE. PTPN22 is an example of a common coding change polymorphism, and many immunological and biological changes have been associated with carriage of this variant. Some of these include altered B cell maturation and activation66, alterations and interferon signaling in innate cells67, 68, as well as alterations in the T-cell compartment69, 70. This gene illustrates the difficulty in assigning functional immune system abnormalities to genetic variations, as it can be difficult when there are multiple effects and the gene is expressed in multiple cell lineages. In such a case, the gene could be having distinct effects in multiple different cell lines, or it could be possible that there is one major primary biological event and the other observed immunological abnormalities are secondary to the one primary event. In the case of PTPN22, it is clear that this gene impacts the immune system in a complex and multi-factorial fashion. Coding-change polymorphisms in the ILT3 gene have been shown to impact ILT3 protein expression on dendritic cells and cytokine levels in lupus patients71. ILT3 is a suppressive receptor expressed on innate cells, and is induced by type I IFN. These data suggest a potential defective feedback regulation loop with respect to type I IFN in innate cells – a suppressive receptor induced by type I IFN is reduced in expression in SLE patients, and cytokine levels are higher in these patients71. The MAVS gene provides an example of an ancestry-specific coding-change functional polymorphism. MAVS is a mitochondrial protein which is involved in signaling downstream of cytosolic nucleic acid sensors which respond to virus infection with type I IFN and inflammatory cytokine production72. African-derived coding change polymorphisms in MAVS which were not found in other ancestral backgrounds were shown to be loss-of-function with respect to cytokine production after viral infection20, and these same polymorphisms were associated with a low IFN, autoantibody negative SLE phenotype in African-Americans20.
Genetic variations have also been shown to impact microRNA expression and binding in systemic lupus erythematosus. MicroRNA 146a is a negative regulator of the type I IFN pathway. A polymorphism in the promoter region of this microRNA results in decreased Ets-1 transcription factor binding, and decreased levels of microRNA146a73. This would be expected to reduce the suppressive effect of microRNA 146a on the type I IFN pathway in SLE patients. Another study has confirmed this finding of SLE-associated genetic variation near microRNA 146a resulting in decreased expression of the microRNA in European populations74. Sequence variations which alter microRNA binding sites have also been implicated in lupus. TLR7 is an endosomal Toll-like receptor that has been strongly implicated in immune complex recognition and type I IFN production in SLE75. A genetic variant in the 3′ untranslated region of TLR7 has been associated with SLE76. Follow up studies of this allele demonstrated that the change in the 3′ UTR sequence altered the binding of microRNA 314877. This reduced binding of the microRNA resulted in increased transcript level of TLR7, and higher expression levels of TLR7 protein77. Greater TLR7 expression would be expected to increase TLR signaling, and it’s likely that this genetic variation impacts disease pathogenesis by increasing endosomal TLR signaling. The various mechanisms by which the genes discussed above alter gene function are shown in Figure 2.
Figure 2.
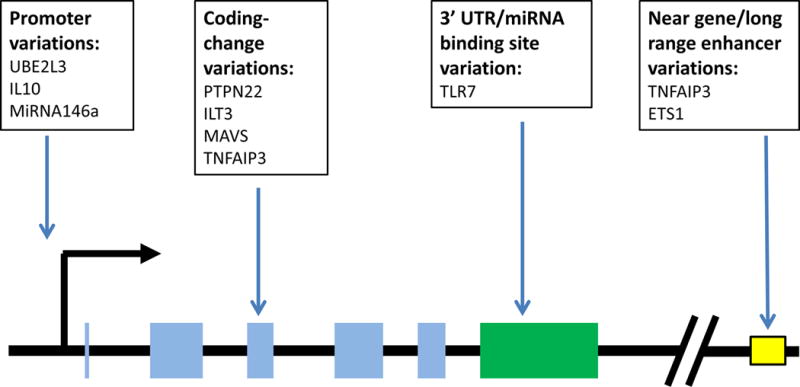
Summary of functional genetic changes discussed in the article, illustrating location relative to the gene structure. Transcriptional start site is indicated with the large black 90 degree arrow, gene exons are indicated by light blue rectangles, 3′ UTR region is a green rectangle, diagonal lines used to represent a large skipped interval, yellow outlined box indicates an enhancer element.
Conclusions
The above studies demonstrate the diversity of work and examples of some of the exciting findings thus far in the early days of “post-GWAS” SLE genetics. GWAS continues to be a useful tool, as genetic mapping via GWAS in non-European populations is greatly needed, and results from GWAS studies of additional world populations are expected in the near future. Large scale follow-up fine-mapping experiments such as the Immunochip consortium SLE results are also expected soon, and these data will likely extend the list of genetic variations definitively associated with SLE. As the review suggests, mapping and association is just the beginning in human genetics, and an enormous amount of work remains to translate the findings from association signals into functional human biology. Work has begun in this next frontier, which will bring us closer to biological understanding and the potential for intervention based upon an individual’s genetic substrate.
Key Points.
Genetic studies in non-European populations are critical, as these populations are frequently more commonly affected, and the genetic basis of disease differs to some degree
SLE-associated genetic variations coalesce to impact biological and immunological pathways – the type I IFN system is an important example with both polygenic and monogenic influences
By studying the function of SLE-associated polymorphisms we can move the genetic association signals into cell biology, and begin to understand the human biology of disease
Acknowledgments
Supported by grants from: the NIH (AR060861, AR057781, AR065964, AI071651), Rheumatology Research Foundation, CureJM Foundation, the Mayo Clinic Foundation, and the Lupus Foundation of Minnesota.
Footnotes
Conflict of interest: Dr. Niewold has received research grants from Janssen, Inc and EMD Serono, Inc.
References
- 1.Deapen D, et al. A revised estimate of twin concordance in systemic lupus erythematosus. Arthritis Rheum. 1992;35:311–318. doi: 10.1002/art.1780350310. [DOI] [PubMed] [Google Scholar]
- 2.Block SR, Winfield JB, Lockshin MD, D’Angelo WA, Christian CL. Studies of twins with systemic lupus erythematosus. A review of the literature and presentation of 12 additional sets. Am J Med. 1975;59:533–552. doi: 10.1016/0002-9343(75)90261-2. [DOI] [PubMed] [Google Scholar]
- 3.Alarcon-Segovia D, et al. Familial aggregation of systemic lupus erythematosus, rheumatoid arthritis, and other autoimmune diseases in 1,177 lupus patients from the GLADEL cohort. Arthritis Rheum. 2005;52:1138–1147. doi: 10.1002/art.20999. [DOI] [PubMed] [Google Scholar]
- 4.Harley JB, et al. Genome-wide association scan in women with systemic lupus erythematosus identifies susceptibility variants in ITGAM, PXK, KIAA1542 and other loci. Nature genetics. 2008;40:204–210. doi: 10.1038/ng.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gateva V, et al. A large-scale replication study identifies TNIP1, PRDM1, JAZF1, UHRF1BP1 and IL10 as risk loci for systemic lupus erythematosus. Nat Genet. 2009;41:1228–1233. doi: 10.1038/ng.468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Namjou B, et al. PTPN22 association in systemic lupus erythematosus (SLE) with respect to individual ancestry and clinical sub-phenotypes. PLoS One. 2013;8:e69404. doi: 10.1371/journal.pone.0069404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chapman SJ, et al. PTPN22 and invasive bacterial disease. Nat Genet. 2006;38:499–500. doi: 10.1038/ng0506-499. [DOI] [PubMed] [Google Scholar]
- 8.Graham RR, et al. A common haplotype of interferon regulatory factor 5 (IRF5) regulates splicing and expression and is associated with increased risk of systemic lupus erythematosus. Nat Genet. 2006;38:550–555. doi: 10.1038/ng1782. [DOI] [PubMed] [Google Scholar]
- 9.Graham RR, et al. Three functional variants of IFN regulatory factor 5 (IRF5) define risk and protective haplotypes for human lupus. Proc Natl Acad Sci U S A. 2007;104:6758–6763. doi: 10.1073/pnas.0701266104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Niewold TB, et al. IRF5 haplotypes demonstrate diverse serological associations which predict serum interferon alpha activity and explain the majority of the genetic association with systemic lupus erythematosus. Annals of the rheumatic diseases. 2012;71:463–468. doi: 10.1136/annrheumdis-2011-200463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cherian TS, et al. Brief Report: IRF5 systemic lupus erythematosus risk haplotype is associated with asymptomatic serologic autoimmunity and progression to clinical autoimmunity in mothers of children with neonatal lupus. Arthritis and rheumatism. 2012;64:3383–3387. doi: 10.1002/art.34571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Niewold TB, et al. Association of the IRF5 risk haplotype with high serum interferon-alpha activity in systemic lupus erythematosus patients. Arthritis Rheum. 2008;58:2481–2487. doi: 10.1002/art.23613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sankararaman S, et al. The genomic landscape of Neanderthal ancestry in present-day humans. Nature. 2014;507:354–357. doi: 10.1038/nature12961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Han JW, et al. Genome-wide association study in a Chinese Han population identifies nine new susceptibility loci for systemic lupus erythematosus. Nature genetics. 2009;41:1234–1237. doi: 10.1038/ng.472. [DOI] [PubMed] [Google Scholar]
- 15.Yang W, et al. Meta-analysis followed by replication identifies loci in or near CDKN1B, TET3, CD80, DRAM1, and ARID5B as associated with systemic lupus erythematosus in Asians. American journal of human genetics. 2013;92:41–51. doi: 10.1016/j.ajhg.2012.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sanchez E, et al. Identification of novel genetic susceptibility loci in African American lupus patients in a candidate gene association study. Arthritis and rheumatism. 2011;63:3493–3501. doi: 10.1002/art.30563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nath SK, et al. A nonsynonymous functional variant in integrin-alpha(M) (encoded by ITGAM) is associated with systemic lupus erythematosus. Nat Genet. 2008;40:152–154. doi: 10.1038/ng.71. [DOI] [PubMed] [Google Scholar]
- 18.Freedman BI, et al. End-stage renal disease in African Americans with lupus nephritis is associated with APOL1. Arthritis Rheumatol. 2014;66:390–396. doi: 10.1002/art.38220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lodolce JP, et al. African-derived genetic polymorphisms in TNFAIP3 mediate risk for autoimmunity. J Immunol. 2010;184:7001–7009. doi: 10.4049/jimmunol.1000324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pothlichet J, et al. A loss-of-function variant of the antiviral molecule MAVS is associated with a subset of systemic lupus patients. EMBO Mol Med. 2011;3:142–152. doi: 10.1002/emmm.201000120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Niewold TB. Interferon alpha as a primary pathogenic factor in human lupus. Journal of interferon & cytokine research : the official journal of the International Society for Interferon and Cytokine Research. 2011;31:887–892. doi: 10.1089/jir.2011.0071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Niewold TB, Swedler WI. Systemic lupus erythematosus arising during interferon-alpha therapy for cryoglobulinemic vasculitis associated with hepatitis C. Clin Rheumatol. 2005;24:178–181. doi: 10.1007/s10067-004-1024-2. [DOI] [PubMed] [Google Scholar]
- 23.Ghodke-Puranik Y, Niewold TB. Genetics of the type I interferon pathway in systemic lupus erythematosus. Int J Clin Rheumtol. 2013;8 doi: 10.2217/ijr.13.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Niewold TB, Hua J, Lehman TJ, Harley JB, Crow MK. High serum IFN-alpha activity is a heritable risk factor for systemic lupus erythematosus. Genes and immunity. 2007;8:492–502. doi: 10.1038/sj.gene.6364408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Niewold TB, et al. Age- and sex-related patterns of serum interferon-alpha activity in lupus families. Arthritis Rheum. 2008;58:2113–2119. doi: 10.1002/art.23619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Niewold TB, Wu SC, Smith M, Morgan GA, Pachman LM. Familial aggregation of autoimmune disease in juvenile dermatomyositis. Pediatrics. 2011;127:e1239–1246. doi: 10.1542/peds.2010-3022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Weckerle CE, et al. Network analysis of associations between serum interferon-alpha activity, autoantibodies, and clinical features in systemic lupus erythematosus. Arthritis Rheum. 2011;63:1044–1053. doi: 10.1002/art.30187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ko K, et al. Genetic ancestry, serum interferon-alpha activity, and autoantibodies in systemic lupus erythematosus. The Journal of rheumatology. 2012;39:1238–1240. doi: 10.3899/jrheum.111467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sharma S, et al. Widely divergent transcriptional patterns between SLE patients of different ancestral backgrounds in sorted immune cell populations. Journal of autoimmunity. 2015 doi: 10.1016/j.jaut.2015.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ko K, Koldobskaya Y, Rosenzweig E, Niewold TB. Activation of the Interferon Pathway is Dependent Upon Autoantibodies in African-American SLE Patients, but Not in European-American SLE Patients. Frontiers in immunology. 2013;4:309. doi: 10.3389/fimmu.2013.00309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Grondal G, et al. Increased number of interleukin-10-producing cells in systemic lupus erythematosus patients and their first-degree relatives and spouses in Icelandic multicase families. Arthritis Rheum. 1999;42:1649–1654. doi: 10.1002/1529-0131(199908)42:8<1649::AID-ANR13>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 32.Mangale D, et al. Familial aggregation of high tumor necrosis factor alpha levels in systemic lupus erythematosus. Clin Dev Immunol. 2013;2013:267430. doi: 10.1155/2013/267430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Niewold TB. Interferon alpha as a primary pathogenic factor in human lupus. Journal of interferon & cytokine research : the official journal of the International Society for Interferon and Cytokine Research. 2011;31:887–892. doi: 10.1089/jir.2011.0071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kariuki SN, et al. Age- and gender-specific modulation of serum osteopontin and interferon-alpha by osteopontin genotype in systemic lupus erythematosus. Genes Immun. 2009;10:487–494. doi: 10.1038/gene.2009.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Salloum R, et al. Genetic variation at the IRF7/PHRF1 locus is associated with autoantibody profile and serum interferon-alpha activity in lupus patients. Arthritis Rheum. 2010;62:553–561. doi: 10.1002/art.27182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Robinson T, et al. Autoimmune disease risk variant of IFIH1 is associated with increased sensitivity to IFN-alpha and serologic autoimmunity in lupus patients. Journal of immunology. 2011;187:1298–1303. doi: 10.4049/jimmunol.1100857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kariuki SN, et al. Cutting edge: Autoimmune disease risk variant of STAT4 confers increased sensitivity to IFN-alpha in lupus patients in vivo. J Immunol. 2009;182:34–38. doi: 10.4049/jimmunol.182.1.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chrabot BS, et al. Genetic variation near IRF8 is associated with serologic and cytokine profiles in systemic lupus erythematosus and multiple sclerosis. Genes and immunity. 2013;14:471–478. doi: 10.1038/gene.2013.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kariuki SN, et al. Trait-stratified genome-wide association study identifies novel and diverse genetic associations with serologic and cytokine phenotypes in systemic lupus erythematosus. Arthritis Res Ther. 2010;12:R151. doi: 10.1186/ar3101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Koldobskaya Y, et al. Gene-expression-guided selection of candidate loci and molecular phenotype analyses enhance genetic discovery in systemic lupus erythematosus. Clinical & developmental immunology. 2012;2012:682018. doi: 10.1155/2012/682018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kariuki SN, et al. Genetic analysis of the pathogenic molecular sub-phenotype interferon-alpha identifies multiple novel loci involved in systemic lupus erythematosus. Genes and immunity. 2015;16:15–23. doi: 10.1038/gene.2014.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Taylor KE, et al. Specificity of the STAT4 genetic association for severe disease manifestations of systemic lupus erythematosus. PLoS genetics. 2008;4:e1000084. doi: 10.1371/journal.pgen.1000084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lin CP, et al. Role of MYH9 and APOL1 in African and non-African populations with lupus nephritis. Genes and immunity. 2012;13:232–238. doi: 10.1038/gene.2011.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Agik S, et al. The autoimmune disease risk allele of UBE2L3 in African American patients with systemic lupus erythematosus: a recessive effect upon subphenotypes. The Journal of rheumatology. 2012;39:73–78. doi: 10.3899/jrheum.110590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhao J, et al. Preferential association of a functional variant in complement receptor 2 with antibodies to double-stranded DNA. Annals of the rheumatic diseases. 2014 doi: 10.1136/annrheumdis-2014-205584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Graham RR, et al. Specific combinations of HLA-DR2 and DR3 class II haplotypes contribute graded risk for disease susceptibility and autoantibodies in human SLE. European journal of human genetics : EJHG. 2007;15:823–830. doi: 10.1038/sj.ejhg.5201827. [DOI] [PubMed] [Google Scholar]
- 47.Botto M, et al. Homozygous C1q deficiency causes glomerulonephritis associated with multiple apoptotic bodies. Nat Genet. 1998;19:56–59. doi: 10.1038/ng0598-56. [DOI] [PubMed] [Google Scholar]
- 48.Al-Mayouf SM, et al. Loss-of-function variant in DNASE1L3 causes a familial form of systemic lupus erythematosus. Nature genetics. 2011;43:1186–1188. doi: 10.1038/ng.975. [DOI] [PubMed] [Google Scholar]
- 49.Namjou B, et al. Evaluation of the TREX1 gene in a large multi-ancestral lupus cohort. Genes and immunity. 2011;12:270–279. doi: 10.1038/gene.2010.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rice G, et al. Heterozygous mutations in TREX1 cause familial chilblain lupus and dominant Aicardi-Goutieres syndrome. Am J Hum Genet. 2007;80:811–815. doi: 10.1086/513443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rice GI, Rodero MP, Crow YJ. Human Disease Phenotypes Associated With Mutations in TREX1. J Clin Immunol. 2015;35:235–243. doi: 10.1007/s10875-015-0147-3. [DOI] [PubMed] [Google Scholar]
- 52.Crow YJ. Type I interferonopathies: mendelian type I interferon up-regulation. Current opinion in immunology. 2015;32:7–12. doi: 10.1016/j.coi.2014.10.005. [DOI] [PubMed] [Google Scholar]
- 53.Molineros JE, et al. Admixture mapping in lupus identifies multiple functional variants within IFIH1 associated with apoptosis, inflammation, and autoantibody production. PLoS genetics. 2013;9:e1003222. doi: 10.1371/journal.pgen.1003222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Liu Y, et al. Activated STING in a vascular and pulmonary syndrome. The New England journal of medicine. 2014;371:507–518. doi: 10.1056/NEJMoa1312625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nicolae DL, et al. Trait-associated SNPs are more likely to be eQTLs: annotation to enhance discovery from GWAS. PLoS Genet. 2010;6:e1000888. doi: 10.1371/journal.pgen.1000888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wang S, et al. A functional haplotype of UBE2L3 confers risk for systemic lupus erythematosus. Genes and immunity. 2012;13:380–387. doi: 10.1038/gene.2012.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lewis MJ, et al. UBE2L3 polymorphism amplifies NF-kappaB activation and promotes plasma cell development, linking linear ubiquitination to multiple autoimmune diseases. American journal of human genetics. 2015;96:221–234. doi: 10.1016/j.ajhg.2014.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Adrianto I, et al. Association of a functional variant downstream of TNFAIP3 with systemic lupus erythematosus. Nature genetics. 2011;43:253–258. doi: 10.1038/ng.766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wang S, Wen F, Wiley GB, Kinter MT, Gaffney PM. An enhancer element harboring variants associated with systemic lupus erythematosus engages the TNFAIP3 promoter to influence A20 expression. PLoS genetics. 2013;9:e1003750. doi: 10.1371/journal.pgen.1003750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Plenge RM, et al. Two independent alleles at 6q23 associated with risk of rheumatoid arthritis. Nature genetics. 2007;39:1477–1482. doi: 10.1038/ng.2007.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Raychaudhuri S, et al. Common variants at CD40 and other loci confer risk of rheumatoid arthritis. Nature genetics. 2008;40:1216–1223. doi: 10.1038/ng.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Nititham J, et al. Meta-analysis of the TNFAIP3 region in psoriasis reveals a risk haplotype that is distinct from other autoimmune diseases. Genes and immunity. 2015;16:120–126. doi: 10.1038/gene.2014.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sakurai D, et al. Preferential binding to Elk-1 by SLE-associated IL10 risk allele upregulates IL10 expression. PLoS genetics. 2013;9:e1003870. doi: 10.1371/journal.pgen.1003870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lu X, et al. Lupus Risk Variant Increases pSTAT1 Binding and Decreases ETS1 Expression. American journal of human genetics. 2015;96:731–739. doi: 10.1016/j.ajhg.2015.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wang C, et al. Genes identified in Asian SLE GWASs are also associated with SLE in Caucasian populations. European journal of human genetics : EJHG. 2013;21:994–999. doi: 10.1038/ejhg.2012.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Menard L, et al. The PTPN22 allele encoding an R620W variant interferes with the removal of developing autoreactive B cells in humans. The Journal of clinical investigation. 2011;121:3635–3644. doi: 10.1172/JCI45790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kariuki SN, Crow MK, Niewold TB. The PTPN22 C1858T polymorphism is associated with skewing of cytokine profiles toward high interferon-alpha activity and low tumor necrosis factor alpha levels in patients with lupus. Arthritis Rheum. 2008;58:2818–2823. doi: 10.1002/art.23728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wang Y, et al. The autoimmunity-associated gene PTPN22 potentiates toll-like receptor-driven, type 1 interferon-dependent immunity. Immunity. 2013;39:111–122. doi: 10.1016/j.immuni.2013.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Vang T, et al. Autoimmune-associated lymphoid tyrosine phosphatase is a gain-of-function variant. Nat Genet. 2005;37:1317–1319. doi: 10.1038/ng1673. [DOI] [PubMed] [Google Scholar]
- 70.Maine CJ, et al. PTPN22 alters the development of regulatory T cells in the thymus. Journal of immunology. 2012;188:5267–5275. doi: 10.4049/jimmunol.1200150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Jensen MA, et al. Functional genetic polymorphisms in ILT3 are associated with decreased surface expression on dendritic cells and increased serum cytokines in lupus patients. Annals of the rheumatic diseases. 2013;72:596–601. doi: 10.1136/annrheumdis-2012-202024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Shrivastav M, Niewold TB. Nucleic Acid Sensors and Type I Interferon Production in Systemic Lupus Erythematosus. Frontiers in immunology. 2013;4:319. doi: 10.3389/fimmu.2013.00319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Luo X, et al. A functional variant in microRNA-146a promoter modulates its expression and confers disease risk for systemic lupus erythematosus. PLoS genetics. 2011;7:e1002128. doi: 10.1371/journal.pgen.1002128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lofgren SE, et al. Genetic association of miRNA-146a with systemic lupus erythematosus in Europeans through decreased expression of the gene. Genes and immunity. 2012;13:268–274. doi: 10.1038/gene.2011.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Jensen MA, Niewold TB. Interferon regulatory factors: critical mediators of human lupus. Translational research : the journal of laboratory and clinical medicine. 2015;165:283–295. doi: 10.1016/j.trsl.2014.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Shen N, et al. Sex-specific association of X-linked Toll-like receptor 7 (TLR7) with male systemic lupus erythematosus. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:15838–15843. doi: 10.1073/pnas.1001337107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Deng Y, et al. MicroRNA-3148 modulates allelic expression of toll-like receptor 7 variant associated with systemic lupus erythematosus. PLoS genetics. 2013;9:e1003336. doi: 10.1371/journal.pgen.1003336. [DOI] [PMC free article] [PubMed] [Google Scholar]