

Abstract
We characterize sleep-like states in cultured neurons and glia during development in vitro, and after electrical stimulation, the addition of tumor necrosis factor alpha (TNF), and the combination of TNF plus electrical stimulation. We also characterize optogenetic stimulation-induced ATP release and neuronal interleukin-1 and TNF expression in vitro demonstrating the activity-dependence of these putative sleep regulatory substances. Action potential (AP) burstiness (BI; burstiness index), synchronization of slow electrical potentials between recording electrodes (SYN), and slow wave (SW) power (0.25 – 3.75Hz) determined using fast Fourier analyses emerged as network properties, maturing after two weeks in culture. Homologous in vivo measures are used to characterize sleep. Electrical stimulation reduced the BI, SYN, and SW power values during and/or after the stimulus period. One day later, homeostasis was evident by rebounds of SYN and SW power values to above baseline levels; the magnitude of the rebound was stimulus pattern-dependent. The addition of TNF enhanced BI, SYN, and SW power values suggesting the induction of a deeper sleep-like state. Electrical stimulation reversed these TNF effects suggesting the network state was more wake-like. The day after TNF plus electrical stimulation, the changes in SYN and SW power values were dependent upon the stimulus patterns the cells received the day before. We conclude that sleep- and wake states in cultured in vitro networks can be controlled and they share molecular regulatory mechanisms with local in vivo networks. Further, sleep is an activity-dependent emergent local network property.
Keywords: interleukin-1, homeostasis, optogenetics, ATP, slow wave synchronization
Graphical Abstract
Introduction
The minimal brain components necessary for, and capable of, sleep remain unknown. Several theories propose that sleep is a fundamental property of small neuronal/glial networks (reviewed Krueger et al., 2013). Our theory predicts that any viable neuronal/glial network, including in vitro networks, will exhibit self-organizing emergent network properties characteristic of sleep (Roy et al., 2008). Small networks within brain tissue possess sleep-like characteristics. Thus, isolated cortical islands in vivo spontaneously display local slow sleep-like electroencephalographic (EEG) oscillations (Kristiansen and Courtois, 1949; Lemieux et al., 2014). Individual cortical columns in vivo oscillate between sleep- and wake-like states (Rector et al., 2005, 2009; Phillips et al., 2011). Cortical column sleep-like states are dependent upon prior network activity and individual column state is capable of affecting whole animal behavior (Krueger et al., 2013). In vitro the default state of neuronal/glial cultures is sleep-like (Hinard et al., 2012). The burst-pause action potential (AP) patterns occurring in culture are reminiscent of firing patterns in thalamic and cortical neurons during sleep in vivo (Timofeev et al., 2001).
Chemical (Hinard et al., 2012) or electrical (Wagenaar et al., 2005) stimulation of cultured neuronal/glial networks promotes a wake-like state as characterized by more random (less bursty) AP firing and gene expression profiles. However, those experiments are limited to the use of wake-promoting chemicals or brief fixed pattern electrical stimuli. Herein, we contrast the effects of an electrical burst/pause (bp) stimulus pattern to the effects of fixed pattern stimulation. We use output measures that are correlates of sleep in vivo, e.g., burstiness (BI for burstiness index), cross-channel slow wave (SW) (0.25–3.75 Hz) synchronization (SYN), and SW power determined using fast Fourier transformations (FFT), to show that the duration and pattern of electrical stimulation differentially affect short-term network outputs and long-term network homeostasis in vitro.
Neuronal and glial activities induce the release or expression of putative sleep regulatory substances (Krueger et al., 2008); adenosine triphosphate (ATP) and interleukin-1 beta (IL1), (Ferrari et al., 2006; Solle et al., 2001) and tumor necrosis factor alpha (TNF), (Suzuki et al., 2004). Increased afferent input induces TNF (Churchill et al., 2008) and IL1 (Hallett et al., 2010) neuronal expression in somatosensory cortical columns in vivo. We demonstrate herein that optogenetic stimulation of neurons in vitro induces release of ATP into the incubation medium and neuronal expression of IL1 and TNF. Further, TNF directly applied to cortical columns increases the probability of the occurrence of the cortical column sleep-like state (Churchill et al., 2008). We now extend that work by showing that the addition of TNF to mature neuronal/glial cultures enhances burstiness, SYN values, and the magnitude of SW power suggesting a deeper sleep-like state. These effects are reversed by electrical stimulation. We also show that electrical simulation and the presence of TNF alters long-term spontaneous network activity.
Collectively, current data suggest that sleep- and wake-like states can be induced and controlled in cultured mature neuronal/glial networks in culture. Further, the mechanisms involved in network state oscillations in vitro parallel those occurring in vivo.
Materials & Methods
Primary Cultures
C57BL/6J mice were obtained from Jackson Labs, (Bar Harbor, ME) and bred for 2–6 generations at WSU. Dissociated cortical primary cultures were prepared from 0–3 day old pups. Mouse protocols were consistent with NIH guidelines for care and use of animals and approved by Washington State University’s Institutional Animal Care and Use Committee. Briefly, for each preparation, cortical tissues from 6 pups were dissected in ice-chilled Hibernate-E (HE, BrainBits, Springfield, IL) then digested with 2 mg/mL papain (Worthington Biochemical, Lakewood, NJ) for 20 min at 37°C. Next cells were dissociated by trituration using a 20 gauge needle (twice) followed by a 22 gauge needle (once) in Hibernate-E with 2% GlutaMAX (Invitrogen, Grand Island, NY). The suspended cells were separated with a 40 μm strainer to remove aggregate cells.
The cells were then centrifuged at 200g for 10 min and the pellet was dissolved in warm Dulbecco’s Modified Eagle Medium (DMEM) (Sigma, St. Louis, MO) with 10% fetal bovine serum (FBS), 1% GlutaMAX, and 2% penicillin/streptomycin (pen/strep). Cells were counted with a hemocytometer and plated 2 × 105 cells/20μL/well on 6-well multielectrode arrays (MEAs; Multi Channel Systems, Reutlingen, Germany) aseptically prepared for cell culture and coated with poly-D-lysine (Sigma) solution for 1 h, rinsed three times with deionized water, and then allowed to dry. Cultures were incubated with 5% CO2 at 37°C in a humidified incubator for 50 min. Then 350 μL of DMEM, supplemented with FBS, GlutaMAX, and pen/strep, were added to each well and a MEA Teflon cover membrane (ALA Scientific, Farmingdale, NY) was placed over the MEA to prevent loss of water. After 4 h, 350 μL of NbActiv4 (BrainBits; contains a mitotic inhibitor to suppress glial proliferation) with 2% pen/strep and 0.2% gentamicin (Invitrogen) was added to each well. Cultures were fed twice a week by replacing half the media in each well with fresh warm supplemented NbActiv4 (as described above).
MEA Recording
A MEA60-BC system (Multi Channel Systems) was used for multiunit extracellular recordings. Immediately before recording, the Teflon-covered MEA was transferred from the humidified incubator to amplifiers that were contained inside a 5% CO2, 37°C, dry incubator. The 6-well MEA sat in a MEA 1060 amplifier (1100x) with 9 titanium nitride electrodes per well (in a 3 × 3 grid) plus one ground electrode. Field potentials at each electrode relative to the ground electrode were recorded at 10 kHz. For data acquisition, MC_Rack software (Multi Channel Systems) was used. Data were stored on the acquisition computer until they were transferred for analyses and storage to external hard drives. After recording, the MEA was returned to the 5% CO2, 37°C, humidified incubator.
MEA Data Analysis
MC_Rack software (Multi Channel Systems) was used to extract APs and SW activity from raw data. For APs, a 2nd order Butterworth filter, 200 Hz high-pass, was used. A threshold of ±4 standard deviations was independently set for each channel; filtered electrical activity exceeding this threshold was counted as an AP. AP burst were defined by a minimum of 4 APs within the first 10 msec with no more than 10 msec between spikes throughout the burst, a minimum duration of 20 msec, and a minimum of 10 msec between individual bursts. A burstiness Index (BI) was calculated over 5 min periods as the total number of APs in the top 15% of the 300 1-sec epochs divided by the total number of spikes occurring in 5 min (Wagenaar et al., 2005). A higher BI indicates more burstiness; a lower BI indicates more random firing. Electrical activity from all channels that had an average firing rate greater than 0.5 APs/sec on culture day 10 were included in AP and BI data analyses of developmental data; a minimum cut-off of 0.25 APs/sec during baseline recordings on the day of the experiment was used for stimulation data. Channels were included in burst data analyses if they displayed at least one burst. Statistical significance between experimental periods (pre-, during, and post-stimulation) and between experimental conditions (e.g. 1 Hz vs. 10 Hz) was evaluated with Student’s t-tests and two-way and three-way ANOVAs.
For SW activity, two 2nd order Butterworth filters, 100 Hz low-pass and 0.1 Hz high-pass, were used and data were down-sampled to 100 Hz (10 msec bins). FFT spectra from signals at randomly sampled active network nodes were calculated using SleepSign for Animal (Kissei Comtec, Japan) and a custom MATLAB program for 5-min periods across developmental days or across pre-, during, and post- stimulation periods using a cosine tapered window. For developmental experiments, total spectral content for the 0–30 Hz range was binned by conventional EEG frequency bands: delta (0.25–3.75 Hz), theta (4–8 Hz), alpha (8–12 Hz), beta (12–30 Hz). For electrical stimulation experiments, spectral content for the 0.25 – 30.25 Hz range was binned by half Hz frequency bands then summed depending on the frequency range desired.
The data were also processed to determine SW cross-channel correlations from 0.25 – 3.75 Hz (called SYN herein). Specifically, during each period of interest, sample correlation coefficients were computed for each pair of adjacent channels in an MEA well using the following equation:
where and are the sample means, σV1 and σV2 are the standard deviations of the samples, and V1(t) and V2(t) are the voltages at time point t.
These pairwise correlation coefficients were averaged over all pairs of adjacent MEA channels as an aggregate measure of correlation. A high correlation coefficient reflects a high synchrony of slow waves between electrodes. Channels used for AP/BI data analyses were also used for the SW activity analyses. Statistical significance between experimental periods (pre-, during, and post-stimulation raw data) and between experimental conditions (e.g. 1 Hz fold change versus 10 Hz fold change) was evaluated with Student’s t-tests and mixed two-way (Recording Bin x Stimulus) and three-way (Recording Bin x Stimulus x Stimulus Pattern) ANOVAs, with α=0.01.
Culture Development
To characterize the development of neuronal activity in vitro five separate preparations (cells from 5 different litters) of primary cortical cells were plated on eight 6-well MEAs. Each MEA with at least one successfully plated well (cells growing over electrodes as determined by digital images) was recorded from every day starting with incubation day 4 – 7 and continuing through day 18 in vitro. Preliminary 48 h recordings did not suggest a circadian rhythm in APs/sec; regardless each MEA was recorded from for 1 h at the same time as it was recorded from the previous day. Data from 38 wells and 231 channels were used for AP, BI, SYN, and SW power analyses. Data from 27 wells and 192 channels were used for burst analyses.
Optogenetic Stimulation
Transfection
After cell counting but before plating on coverslips, cortical cells were transfected with synChR2-YFP (channelrhodopsin-2-YFP with a synapsin1 promoter) (Boyden et al., 2005). Endo-free plasmid preparation kit (Qiagen, Valencia, CA) was used for purification of the synChR2-YFP construct that was a gift from Karl Deisseroth (Stanford University). Cells were transfected using a Nucleofector II device (Lonza, Switzerland) following manufacturer’s protocol. To achieve an optimized and reproducible level of transfection, 10 μg of synChR2-YFP plasmid was used per 2 × 106 cells suspended in 100 μL nucleofection buffer (Lonza). Nucleofection pulse protocol #O-005 was used as it yielded good transfection efficiency (30% of neurons) with minimal cell death. For mock-transfected control preparations, cells went through the nucleofection procedure but without any DNA added to the nucleofection buffer.
Light stimulation
A multi-channel LED light stimulator was built in-house to deliver light pulses to cultured cortical networks. For excitation of ChR2, 470nm LEDs (Digi-Key Corporation, Thief River Falls, MN) were used. A graphical user interface software component was developed in-house to control the light stimulation parameters. The system delivered pulse trains with random stimulation patterns of 30 or 90 min duration (pulse ON time = 15 msec, minimum pulse OFF time = 10 msec, frequency = 10 Hz, maximum burst length = 1 sec). The stimulator was interfaced with a 24-well cell culture dish cover and kept inside a 37°C, 5% CO2 dehumidified incubator.
Mock transfected (ChR2-negative) and synChR2-YFP transfected (ChR2-positive) networks were optogenetically stimulated at day 8 – 9 in vitro. For light stimulation experiments, light pulses were delivered for 15, 30, or 90 min. For no stimulation control experiments, ChR2-positive or ChR2-negative networks did not receive any light pulses. Culture media of all networks were changed one day prior to the day of experiment.
ATP assay
Immediately after light stimulation, the culture medium (1 mL per coverslip) was assayed for extracellular ATP using ATPlite, a single addition luciferase bioluminescence based ATP detection kit, following the manufacturer’s protocol (Perkin Elmer, Waltham, MA). Supernatant from each network (1mL) was distributed into 4–6 wells (100μL per well) of a 96-well plate, 100μL of ATP-lite reagent was added per well, and luminescence was measured using a micro-plate reader (Perkin Elmer/Packard Fusion, Meriden, CT). Every well was measured 3 times (each time for 5 sec), and the average luminescence values were used for data analyses. Post-stimulation changes in extracellular ATP are presented as percent increase using time-matched non-stimulated samples as controls. Each network (coverslip) was treated as an independent sample for ATP measurement.
Immunohistochemistry
After light stimulation, networks were immediately fixed with 3.7% paraformaldehyde (PFA) for 20 min at room temperature. Samples were washed thrice with PBS/50mM glycine solution to neutralize PFA, and then with Tween-20 to make cell membranes permeable. A blocking solution made of 3% BSA, 5% goat serum and 50 mM glycine in PBS was used to minimize nonspecific labeling with antibodies. Cells were first incubated with primary antibody [c-fos: sc-253 (Santa Cruz Biotechnology, Dallas, TX); interleukin-1 beta (IL1): AF-501-NA, TNF: AF-410-NA (R&D Systems, Minneapolis, MN)] at room temperature for 1–2 h, washed thrice, and then treated with a suitable Alexa-633 labeled secondary antibody (Invitrogen) for 1–2 h. Samples were washed twice, labelled with 4′,6-diamidino-2-phenylindole (DAPI), and then mounted on glass slides using ProLong Gold anti-fade reagent (Invitrogen) following the manufacturer’s protocol. All samples were imaged within three days of preparation.
Microscopy
For fluorescence imaging experiments two different microscopes were used: an upright fluorescence microscope (Axio Imager M2, Zeiss) coupled to a X-Cite Series 120Q (EXFO) illumination system and a digital camera (Axio Cam MRm, Zeiss), and an inverted laser scanning confocal microscope (LSM 510Meta, Zeiss) equipped with multiple laser lines (405nm, 488nm, 633nm etc.) and photo multiplier tube (PMT) detectors. For all experiments the detector gain and the pixel time (or the channel exposure times) used for recording images from light-stimulated samples and time-matched non-stimulated control samples were kept constant. For every network, 4–6 fields (1000 μm × 800 μm for fluorescence microscope; 140 μm × 140 μm for confocal microscope) were imaged from different regions of the network. Higher fluorescence intensity was interpreted as a higher concentration of the protein being probed.
Image analysis
ImageJ software (National Institutes of Health, MD) was used for image analyses. For every image, neuronal cell bodies were selected (~50 cells per image, 4–6 images per network) using the Region of Interest (ROI) Manager tool, and average fluorescence intensity for each ROI was measured. Distributions of fluorescence intensity were obtained by histogram analysis of the single cell fluorescence intensity data for both time-matched non-stimulated control and light stimulation samples. Finally, to combine data from multiple preparations, the change in fluorescence intensity is presented as percent increase (or decrease) using the average values of time-matched non-stimulated samples as controls. Each network was treated as an independent sample. Statistical differences were analyzed using non-directional Student’s t-tests.
MEA Preparations with Chemical and Electrical Stimulation
Electrical Stimulation
Fourteen separate cortical cell cultures were plated on twenty-six 6-well MEAs. To determine the effects of stimulus frequency, a 5 min baseline (pre-stimulation) and an immediate 5 min post-stimulation period after 5 min of electrical stimulation were analyzed. For the 30 min stimulation experiments, after cultures were moved from incubation chambers to the recording apparatus they were allowed 20 min to acclimate because preliminary studies indicated that there was an exponential decrease in the number of APs/sec during the first 20 min after the moving the cultures to the recording apparatus. Then we stimulated for 30 min after which we continued to record for a post-stimulation 30 min period. The next day spontaneous activity was also recorded from the cells stimulated 24 h earlier. All stimulation experiments were done during day 10 – day 30 in vitro. Some MEAs were used for more than one stimulation experiment on a given day and some were used on multiple days; they were returned to the humidified incubator for at least 1 h immediately after recording and a new baseline was recorded before each experiment. Eighty-four stimulation experiments were conducted on the 26 MEAs after excluding wells not adequately plated and inactive channels, and excluding the one channel in each well used for stimulation data from 431 wells and 3337 channels were used for AP, BI, SW correlation, and SW power analyses.
Six different stimulation protocols were tested using MC_Stimulus II (Multi Channel Systems), 0.1 Hz, 1 Hz, and 10 Hz for 5 min; 1 Hz and 1 bp for 30 min and 10 Hz and 10 bp for 30 min with TNF. For the 1 and 10 Hz and bp stimulus patterns, we used a duty cycle of 10 sec and another stimulus pattern using the same number of stimuli only within each duty cycle all the stimuli (n=10, or 100, for bp) were given within 1 sec while the next 9 sec in each duty cycle lacked stimuli; we call this a burst/pause pattern (bp). The bp patterns, although not within the operational definition of a burst, were used to provide a more rapid stimulation pattern with intermittent periods of no stimulation. The fixed stimulation was used to provide an alternative pattern of stimulation containing an equal number of stimuli.
Stimulation parameters were optimized using the methods of Wagenaar et al., (2004). Positive-then-negative 300 μsec biphasic pulses of 300 mV were used. The blanking circuit (Multi Channel Systems) disconnected the recording amplifier during the stimulation pulse plus an additional 100 μsec after each pulse, significantly reducing recorded stimulus artifacts. Stimulation electrodes were randomly chosen from any one of the nine channels in each well showing APs during baseline recording. Stimulation electrodes (one in each well) in the same MEA all received the same stimulation protocol.
Long-term changes in network activity
The day after stimulation, MEAs were recorded from again without stimulation. Values obtained were compared to the baseline (pre-stimulation) activity recorded the prior day before the initiation of the stimulation experiment. Data from six 1 Hz-30 min (264 channels) and seven 1 bp-30 min (292 channels) experiments were used to characterize APs/sec, BI, SYN values, and SW power.
The effects of TNF on network activity
Eight separate cortical cell cultures were plated on eighteen 6-well MEAs. TNF stimulation experiments were done during day 13 – day 24 in vitro. For each experiment a baseline was recorded for 1 h. Then the recording was stopped and 0.01 or 0.1 ng of recombinant mouse tumor necrosis factor alpha (TNF; R & D Systems, 410-MT), dissolved in saline (5 μL), was added directly into the MEA wells. The recording was started again promptly and continued for 1 h. Three MEAs were returned to the humidified incubator after recording and then spontaneous activity was recorded once again 24 h later. Higher doses of TNF (1, 10, and 100 ng) were tried in preliminary experiments; with higher doses signs of long-term toxicity were evident, e.g. longer quiescent periods and higher interchannel variability in the measured parameters (Fig. 8).
Fig. 8.
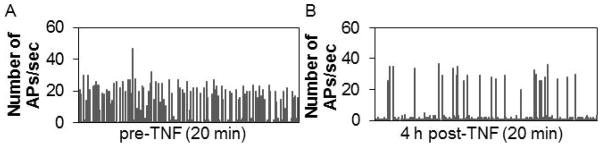
High doses of TNF disrupt the distribution of APs.
Number of APs/sec for (A.) control and (B.) 4 h after 1 ng TNF addition. This high dose of TNF induced long quiescent periods suggesting potential toxicity.
The effects TNF plus electrical stimulation on network activity
To examine the effect of electrical stimulation on network activity in the presence of TNF, three separate cortical cell cultures were plated on six 6-well MEAs. At day 13 – 16 in vitro, a baseline of 1 h before TNF addition was recorded, then 0.01 ng TNF was added directly into wells and recording continued. Thirty min after TNF addition, a pre-stimulation period of 30 min was recorded immediately before electrical stimulation (10 Hz or 10 bp) as well as a post-stimulation period of 30 min immediately after electrical stimulation. Some MEAs were returned to the humidified incubator after recording and then spontaneous activity was recorded once again 24 h later. The last 30 min directly preceding electrical stimulation (30 min after TNF addition) and the 30 min directly following stimulation are used in analyses of pre-stimulation and post-stimulation. The one channel in each well used for stimulation was excluded from analyses. Data from 36 wells (1 preparation, 3 MEAs, 114 channels) were used for AP, BI, SYN values, and FFT analyses.
Results
Characterization of network development in vitro
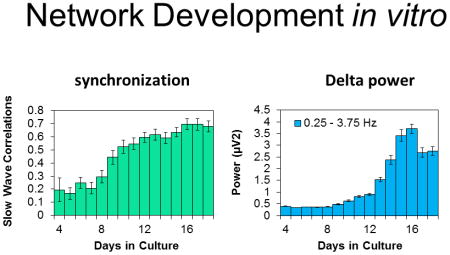
We determined the course of development over days of APs/sec, the BI, SYN values, and SW power. Literature findings were confirmed in that APs/sec and the BI suggested that networks matured by days 12 – 14 in culture (Fig. 1). Relatively few APs occurred during days 4–7 in vitro (Fig. 1A & B). The number of APs/sec increased from days 8–14 then was relatively stable through day 18 (Fig. 1A, C, & D). In parallel to the development of APs/sec, the mean number of bursts per hour was low until day 10 in culture then it increased, stabilizing on about day 14 (Fig. 1E). The mean burst duration (39.9 ± 0.57 msec for days 6–13) and number of APs within a burst (Fig. 1F) were relatively constant from days 6–13 then increased on day 14 reaching maximum values on days 17 and 18 (burst duration 57.6 ± 1.68 msec). The mean interburst interval decreased over development before stabilizing at about 50 sec on day 10. There was a decrease in the BI between days 10–14 (Fig. 1G). This corresponded with an increase in the mean number of APs occurring over the same days of development (Fig. 1A). The BI was higher before day 9 in vitro as a consequence of lower sporadic AP activity. Between days 9 and 12, APs increased but were not organized within bursts resulting in a lower BI. After day 12, APs/sec increased and became more organized as evidenced by the increasing number of bursts and BI (Fig. 1E & G). Some burst parameters changed after day 13, e.g. the mean number of bursts, suggesting continual subtle network reorganization; this likely manifested as subtle changes in the power in the lower frequency ranges and in SYN values. The correlation of AP activity between channels was previously described (Mazzoni et al., 2007) although the ontogenetic unfolding of SYN values in vitro had not been. After day 10 in culture, SYN values were relatively stable (Fig. 1H). SW power had not been previously described as a measure derived from co-cultures of neurons and glia; in intact animals, EEG SW power is a cardinal determinant of non-rapid eye movement sleep. During early post-natal development in vivo, EEG SW activity is very low in many mammalian species (Davis et al., 2011). In the cultured cells, SW power was low during the first 10 days of culture development (Fig. 2). By day 13 in culture, SW power significantly increased and reached relatively steady values between days 14–16. SW power (0.25–3.75 Hz) was largest in magnitude and power was progressively less in the theta, alpha, and beta frequencies (Fig. 2). The magnitudes of power in the various frequency bands were only 0.2 – 3 μV2, which is substantially less than that obtained from EEGs. The increases in SW power roughly paralleled the increases in the BI after day 12 (compare Fig. 2A to Fig. 1G), but these changes were preceded several days prior by increases in APs/sec and SYN values (Fig. 1A & H) suggesting that SW power in cultured cells is derivative of bursts instead of APs or SYN values. This is evident in the filtered data shown in Fig. 2F; each of the large downward deflections corresponds to a burst of APs and the periodicity of these downward deflections is in the 0.25 – 3.5 Hz range as quantified by the FFT analyses (Fig. 2A). Day 4 cultures lack AP bursts (Fig. 1E) and the large downward deflections (Fig. 2E). In conclusion, the four parameters used to characterize culture state, APs/sec, BI, SYN values, and SW power, suggest that the networks matured by two weeks in culture; this conclusion is consistent with other reports (Opitz et al., 2002; Cozzi et al., 2006; Wagenaar et al., 2006a; Chiappalone et al., 2007).
Figure 1. Development of spontaneous electrical activity within co-cultures of neurons and glia.
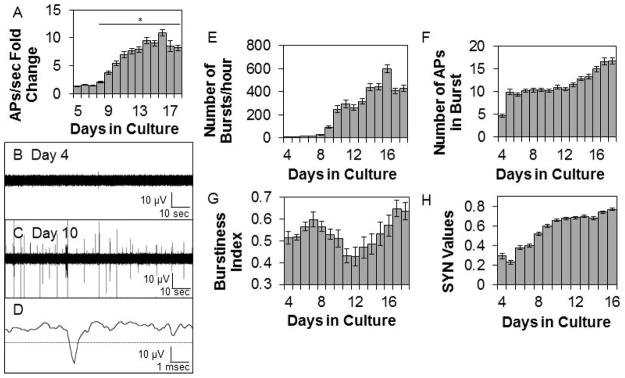
Means ± SE determined on sequential days in culture are shown. A. Fold change above the first day of recording in the average number of action potentials (APs)/sec (Preparations=5, Multi-electrode arrays (MEAs)=8, Wells=39, Channels=224). On days 10–18, at least one burst of APs occurred in 60–75% of the electrodes. * indicates a significant difference (p<0.01) from day 4 values. B. & C. Representative single channel raw data using a 200 Hz high-pass filter on day 4 and day 10 in vitro. D. Single action potential. The dashed line is the AP threshold of −4 standard deviations from the baseline average signal. E. Mean number of bursts (Preparations=5, Multi-electrode arrays (MEAs)=7, Wells=27, Channels=169). F. Mean number of action potentials in a burst (Preparations=5, MEAs=7, Wells=27, Channels=169). G. Mean burstiness index (Preparations=5, MEAs=7, Wells=26, Channels=186). H. Average SYN values between adjacent channels (Preparations=5, MEAs=7, Wells=26, Adjacent Channel Pairs=369).
Figure 2. Development of spontaneous spectral content within co-cultures of neurons and glia.

Means ± SE of fast Fourier transformation (FFT) power (μV2) in various frequency bands determined on sequential days in culture are shown. (Preparations=5, Multi-electrode arrays=7, Wells=26, Channels=186). A. 0.25 – 3.75 Hz. B. 3.75 – 7.75 Hz. C. 7.75 – 11.75 Hz. D. 11.75 – 30.25 Hz. E. & F. Representative single channel SW filtered data at day 4 and day 13 in vitro. The large waves evident in the filtered day 13 signals correspond to AP bursts.
Short-term Effects of Electrical Stimulation
In our initial studies, we stimulated for 5 min using 3 stimulus frequencies (Fig. 3). Briefly, rapid stimulation frequency induced a decrease in the BI and this replicated the results obtained by Wagenaar et al., (2005). The BI returned to baseline values immediately after the stimulus ended.
Figure 3. Electrical stimulation frequency affects APs/sec and the BI.
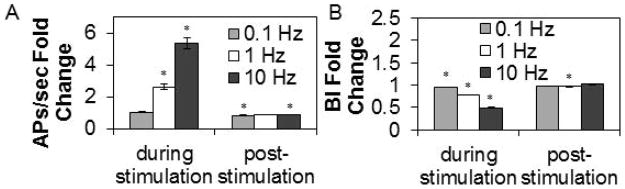
Values are means ± SE of fold changes from pre-stimulation values obtained during and after 5 min of stimulation. A. Fold change from pre-stimulation of action potentials (APs)/sec. Cultures stimulated with 0.1 Hz (Preparations=4, Multi-electrode arrays (MEAs)=7, Wells=63, Channels=493), 1 Hz (Preparations= 4, MEAs=9, Wells=63, Channels=489), or 10 Hz (Preparations=4, MEAs=7, Wells=64, Channels=499) electrical stimuli for 5 min are shown. B. Fold change from pre-stimulation values in burstiness index (BI). * indicates a significant difference (p<0.01) from pre-stimulation values.
We extended those studies by using longer (30 min) acclimation, stimulus, and post stimulus periods and two different stimulus patterns with the same number of stimuli. During 30 min stimulation periods using fixed 1 Hz or 1 bp (rapid stimulation with periods of no stimuli) stimulus patterns, APs/sec increased (Fig. 4A; Recording Bin-5min: F12, 8520=172.57, P<0.001; t-test, all values P<0.001 to P=0.003; Interaction between patterns: F1,710=512.91, P<0.001). The number of APs/sec returned to slightly less than pre-stimulation levels during the 30 min post-stimulation periods after both stimulation patterns (t-tests, all values P<0.001). During the 1 Hz stimulation, the BI decreased (Fig. 4B; t-tests, all values P<0.001). During the 1 bp stimulation period, the BI did not change although it was different than that observed during the 1 Hz stimulation period (Recording Bin-5min: F12,8520=42.62, P<0.001; t-tests, all values P<0.001). The post-stimulation BI, after 1 Hz stimulation, returned to pre-stimulation levels but the BI decreased below pre-stimulation levels for 25 min after the 1 bp stimulation pattern (Stimulus Pattern: F1,710=84.35, P<0.001; t-tests, all values P<0.001 to P=0.008; Interaction: F1,710= 11804.60, P<0.001). Stimulation had a significant effect on SYN values (Fig. 4C; Recording Bin-5min: F12,14916=210.00, P<0.001). During the first 5 min of 1 Hz stimulation, SYN values increased (t-test, P<0.001), then decreased below pre-stimulation levels by min 15 (t-test, P<0.001). The decrease in SYN values started in min 5–10 during the 1 bp stimulation period (t-test, P<0.001). These decreases persisted through the 30 min post-stimulation period after both stimulation patterns (t-tests, all values P<0.001). At various time points during stimulation and post-stimulation periods, the decreases in SYN values were greater with 1 bp than with 1 Hz (Stimulus Pattern: F1,1243=88.58, P<0.001). During the first 5 min of 1 Hz, and 1 bp stimulation, SW power increased compared to pre-stimulation values (Fig. 4D; Pre-stimulation Bin: F4,2828=76.23, P<0.001; t-test, P<0.001 for both patterns). During the 25 – 30 min stimulation period, SW values were different between the 1 Hz and 1 bp stimulation patterns (Stimulus Pattern: F1,707=12.08, P=0.001; t-test, P=0.006). During the post-stimulation periods after 1 Hz and 1 bp stimulation, SW power decreased below pre-stimulation values (Post-stimulation Bin: F4,2828=76.23, P<0.001; t-test, all values P<0.001). These results show that though the two patterns of stimulation provide the same number of stimuli (and a similar response in the number of APs/sec), the two patterns induce distinct BI, SYN values, and SW power responses. For instance, during the first 10 min of stimulation, SYN values increase with 1 Hz stimulation and decrease with 1 bp stimulation. Such results are likely a manifestation of temporal summation of short half-life substances released in response to stimulation that lead to longer term actions on SYN and SW power values.
Figure 4. Different patterns of electrical stimulation for 30 min differentially affect the BI, SYN, and SW power.

Values are means ± SE of fold changes (A & B) and absolute differences (C & D) from the pre-stimulation values obtained in the 5 min preceding the start of electrical stimulation (time 0 indicated by the left arrows). 24h:Bars are fold changes or absolute changes from previous day’s pre-stimulation values. A. Fold change from pre-stimulation in the mean number of action potentials (APs)/sec before, during, and after 30 min of 1 Hz electrical stimulation (time 30 indicated by the right arrows) (Preparations=6, Multi-electrode arrays (MEAs)=8, Wells=46, Channels=358) and the burst/pause pattern (1 bp)stimulation pattern (Preparations=6, MEAs=7, Wells=46, Channels=354). 24 h: Bars (Preparations=6, MEAs=8, Channels=218 and Channels=295, respectively). B. Fold change from pre-stimulation in the mean burstiness index (BI) before, during, and after 30 min electrical stimulation with the 1 Hz (Preparations=6, MEAs=8, Wells=46, Channels=357) and 1 bp (Preparations=6, MEAs=7, Wells=46, Channels=352) stimulation patterns. 24 h: Bars (Preparations=6, MEAs=8, Channels=264 and Channels=292, respectively). C. Change from pre-stimulation in mean SYN values before, during, and after 30 min of 1 Hz electrical stimulation (Preparations=6, MEAs=8, Wells=46, Channel pairs=642) and 1 bp (Preparations=6, MEAs=7, Wells=46, Channel pairs=603) stimulation patterns. 24 h: Bars (Preparations=6, MEAs=8, Channel pairs=494 and Channel pairs=505, respectively). D. Change in SW power (μV2) before, during, and after 30 min electrical stimulation with the 1 Hz (Preparations=6, MEAs=8, Wells=46, Channels=357) and 1 bp (Preparations=6, MEAs=7, Wells=46, Channels=352) stimulation patterns. 24 h: Bars (Preparations=6, MEAs=8, Channels=264 and Channels=292, respectively).
** indicates a significant difference (p<0.01) from pre-stimulation values for both stimulation patterns. * indicates a significant difference (p<0.01) from pre-stimulation values for one pattern of stimulation. # indicates the values between the 1 Hz and 1 bp stimulation patterns are significantly different (p<0.01).
Long-term Effects of Electrical Stimulation
Baseline values the day after stimulation were compared with the previous day’s baseline data (n=15) (Fig. 4) to determine if stimulus-induced changes were long lasting and dependent upon the stimulation pattern. The day after 30 min of stimulation using either the 1 Hz or 1 bp patterns, the number of APs/sec increased from the previous day’s pre-stimulation levels (Fig. 4A; Stimulus Condition: F3,1533=82.87, P<0.001; no effect of pattern). The BIs the day after stimulation with either pattern were not different from pre-stimulation values taken the day before (Fig. 4B). With 30 min of stimulation using either the 1 Hz or 1 bp patterns, SYN values were higher (Recording Condition: F1,1068=264.30, P<0.001) than pre-stimulation levels by the next day, and there was a difference between SYN values after 1 Hz and 1 bp stimulation (Stimulus Pattern: F1,1068=58.95, P<0.001) (Fig. 4C). SW power increased the day after 30 min of stimulation using either stimulus pattern (Recording Condition: F1,599=56.73, P<0.001) with a greater increase after 1 Hz stimulation (Fig. 4D; Stimulus Pattern: F1,599=38.06, P=0.001). The specific molecular events responsible for the differential long-term effects induced by the 1 bp vs 1 Hz stimulus patterns remain unknown although they likely begin as a consequence of temporal summation of substances released by stimulation which eventually manifest in the long-term differences observed 24 h after stimulation.
Collectively these results suggest that stimulation drove the cultures into a more wake-like state. Further, both stimulus conditions enhanced SYN values and SW power 24 h later suggesting a homeostatic rebound. The magnitude of these effects was pattern-dependent suggesting network activity-dependent plasticity.
Light Stimulation
Next we determined whether in vitro cell activity affected release or expression of sleep regulatory substances. In vivo ATP is released by neurons and glia (Fields and Stevens, 2000) and neurons express IL1 (Hallett et al., 2010) and TNF (Churchill et al., 2008) in response to cell activity (Krueger et al., 2008, 2013). Optogenetics were used to stimulate channelrhodopsin-2 (ChR2) transfected neurons. Dissociated cortical neurons were successfully transfected with ChR2-YFP (Fig. 5A). The cultures consisted of about 40% neurons and 60% non-neuronal cells (Fig. 5B). ChR2-YFP was expressed in 32 ± 4% of the neurons (Fig. 5C) with a transfection efficiency range from 10% to 40%.
Figure 5.
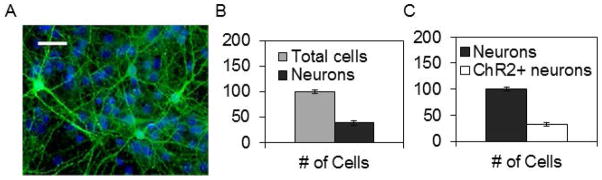
Fig. 5. Networks were successfully transfected with ChR2-YFP.
A. Fluorescence image of channelrhodopsin-2 yellow fluorescence protein (ChR2-YFP) transfected cortical network in culture fixed and stained at day 9 in vitro. Transfected neurons are green. DAPI staining (in blue) shows nuclei of all cells. (Scale bar 30 μm). B. To determine the fraction of cells that were neurons, non-transfected networks were stained with DAPI (for all nuclei) and NeuN conjugated to Alexa-488 (for neuronal nuclei). For this experiment; 39 ± 4% (mean ± SE) of the cells were neurons (n=2 separate preparations, 8 separate networks, at least 200 cells per network counted). C. After nucleofection with the synChR2-YFP construct (see Methods) 32 ± 5% (mean ± SE) neurons contained visible ChR2-YFP (n=3 separate preparations, 10 separate networks, at least 200 cells per network counted). Networks were stained with NeuN (for neuronal nuclei) and a secondary Alexa-633 antibody for this experiment (see Methods) and ChR2-YFP fluorescence positive cells were identified.
The baseline steady-state level of extracellular ATP for non-stimulated ChR2-positive networks was 240 ± 45 femtomoles (n = 32 networks). Extracellular ATP levels progressively elevated over the 90 min stimulation period (Fig. 6A; 90 min, t-test: P=0.006) compared to time-matched non-stimulated ChR2-positive networks: media ATP levels increased by 178 ± 32 femtomoles (n = 11 networks) and 270 ± 39 femtomoles (n = 21 networks) after 30 and 90 min of stimulation respectively, suggesting a reduction of ATP release or an increase in ATP catabolism or cellular uptake of ATP with prolonged stimulation. In contrast, light stimulation of ChR2-negative networks failed to induce increases in extracellular ATP (Fig. 6B). Finally, 90 min of light stimulation increased c-Fos expression in ChR2-positive networks (Fig. 6C; t-test: P=0.002) but not in ChR2-negative networks (Fig. 6D) confirming that the light activated neurons as in prior reports (Boyden et al., 2005).
Figure 6. Light stimulation of ChR2-positive networks progressively elevated levels of extracellular ATP and expression of c-Fos in neurons.

Graphs show means ± SE. Image dimensions: 115 μm by 75 μm. A. Optogenetic stimulation of channelrhodopsin-2(ChR2)-positive networks for 30 and 90 min increased levels of extracellular ATP compared to time-matched non-stimulated control networks. Number of networks with no stimulation/stimulation: 3/5 (15 min), 11/11 (30 min), 18/21 (90 min) from 4 preparations. B. Stimulation of ChR2-negative networks did not show any increase in levels of extracellular ATP over 30 or 90 min of light stimulation. Number of networks with no stimulation/stimulation: 3/3 (30 min), 3/4 (90 min). C. & D. Cells were fixed after light stimulation for determination of c-Fos expression (red), an early gene expressed in neurons in response to activation. Stimulation of ChR2-positive networks (C.), but not of ChR2-negative networks (D.), for 30 and 90 min enhanced c-Fos (red) expression compared to non-stimulated controls. YFP fluorescence (green) from ChR2-YFP was used to identify neurons. For ChR2-negative networks, neurons were stained with Alexa-488 (green) conjugated neuronal nuclei marker, NeuN. Fluorescence intensity for individual cells (200 or more per network) was measured. Bar graphs show relative change in average cellular fluorescence for stimulated networks relative to time-matched non-stimulated control networks. Number of networks with no stimulation/stimulation for ChR2-postive networks: 3/3 (30 min), 3/4 (90 min); no stimulation/stimulation for ChR2-negative networks: 3/3 (30 min), 3/3 (90 min). * indicates a significant difference (p<0.01) from pre-stimulation values.
Light stimulation for 30 min also increased TNF expression in ChR2-positive networks (Fig. 7A–C; t-test: P<0.001). After stimulating 90 min, TNF expression returned to baseline levels (Fig. 7D). In contrast, light stimulation for 30 min did not change IL1 expression in ChR2-positive networks (Fig. 7H), but IL1 expression increased after stimulating 90 min (Fig. 7E–G; t-test: P<0.001).
Figure 7. Light-stimulation of ChR2-positive networks enhanced cytokine expression.

Graphs show means ± SE. Image dimensions: 1000 μm × 800 μm. After light stimulation, channelrhodopsin-2(ChR2)-positive networks were fixed then stained for tumor necrosis factor alpha (TNF) (A. – D.; ChR2 – green, TNF – red) or interleukin-1 beta (IL1) (E. – H.; ChR2 – green, IL1 – red). Fluorescence intensity for each neuron (200 or more per network) was measured as described in Methods and histograms of intensity distribution (frequency of occurrence of cells measured at each intensity) were calculated by single-cell analysis for both stimulated networks (black) and time-matched non-stimulated control networks (gray). For each histogram, average intensity from cell bodies was measured. Histograms were normalized (to peak counts) for presentation. With 30 min of light stimulation, TNF expression increased then returned to pre-stimulation levels after 90 min of stimulation (D.). In contrast, only a modest increase in IL1 expression occurred after 30 min stimulation, and its expression was higher after 90 min of stimulation (H.). In ChR2-positive networks, the elevation of cytokine expression occurred in many cells irrespective of whether they expressed ChR2. Number of networks with no stimulation/stimulation stained for TNF: 8/8 (30 min), 16/16 (90 min) from 3 independent preparations; networks with stimulation/no stimulation stained for IL1: 10/10 (30 min), 12/17 (90 min) from 3 independent preparations. * indicates a significant difference (p<0.01) from pre-stimulation values.
Exogenous TNF Addition
As TNF is expressed as a consequence of cell activity in vivo, we determined whether exogenous TNF applied to the cortical tissue cultures would affect network state. TNF increased spontaneous APs/sec during the first h after the addition of 0.01 or 0.1 ng of TNF (Fig. 9A; TNF addition: F2,902=38.91, P<0.001). APs/sec remained elevated above baseline levels 24 h later after both doses of TNF. Concurrently, TNF enhanced the BI in the first h after both doses of TNF (Fig. 9B; TNF addition: F2,904=15.60, P<0.001). Twenty-four h later, the BI remained elevated after the 0.1 ng dose of TNF. SYN values increased during the first h following the addition of 0.01 or 0.1 ng of TNF (Fig. 9C; TNF addition: F2,1868=344.77, P<0.001). By 24 h after both TNF doses, SYN values remained higher than baseline values. SW power increased in the first h after the addition of 0.01 or 0.1 ng of TNF and this increase persisted 24 h later (Fig. 9D; TNF addition: F2,904=80.67, P<0.001). As mentioned (Fig. 8), higher doses of TNF induced long AP quiescent periods. The data from the low dose experiments suggest the cultures entered a deeper sleep-like state in that similar changes in the BI, EEG synchronization, and EEG SW power are associated with a deeper state of non-rapid eye movement sleep in vivo (Borbély et al., 1984; Hajnik et al., 2013).
Figure 9. TNF addition alters APs/sec, BI, SYN values, and SW power.

Values are means ± SE of absolute and fold changes from values obtained during the h before addition of 0.01 ng or 0.1 ng ng tumor necrosis factor alpha (TNF), 1 h after addition and 24 h after addition. A. Fold change from pre-TNF addition values in action potentials (APs)/sec (0.01 ng, n= 234 channels; 0.1 ng, n=219 channels). B. Fold change from pre-TNF addition values in burstiness index (BI) (0.01 ng, n=234 channels; 0.1 ng, n=219 channels). C. Change from pre-TNF addition values in SYN values (0.01 ng, n=520 channel pairs; 0.1 ng, n=416 channel pairs). D. Change (μV2) from pre-TNF addition values in SW power (0.01 ng, n=234 channels; 0.1 ng, n=221 channels).
* indicates a significant difference (p<0.01) from pre-TNF addition values.
TNF Addition Plus Electrical Stimulation
To determine if rapid electrical stimulation could reverse the actions of exogenous TNF we used 10 Hz (which produces a greater increase in APs, Fig. 3A, and reduction in the BI, Fig. 3B, than 1 Hz) and 10 bp stimulation protocols and used a 0.01 ng TNF dose. During 10 Hz stimulation for 30 min, APs/sec increased in the presence of TNF (Fig. 10A; Stimulation Condition: F3,84=56.52, P<0.001). Immediately after stimulation, APs/sec returned to pre-stimulation values and remained there 24 h later. The BI decreased during 10 Hz stimulation with TNF (Fig. 10B; Stimulation Condition: F3,84=142.54, P<0.001). The BI did not change directly post-stimulation but increased 24 h later after TNF treatment. During 10 Hz stimulation, SYN values decreased then returned to baseline values post-stimulation (Fig. 10C; Stimulation Condition: F3,135=16.87, P<0.001). There were no changes in SW power during or after 10 Hz stimulation in the presence of TNF (Fig. 10D).
Figure 10. Effects of 10 Hz and 10 bp electrical stimulation after 0.01 ng TNF addition.
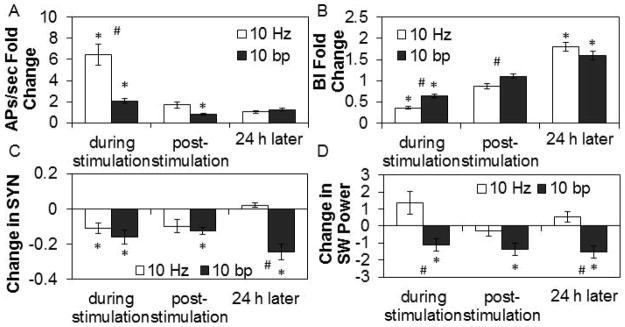
Values are means ± SE of absolute and fold changes from values obtained during the last 30 min of the baseline period after TNF addition but before electrical stimulation at 10 Hz or 10 burst/pause (bp). Tumor necrosis factor alpha (TNF) (0.01 ng) was added at 1 h prior to 30 min of electrical stimulation. A. Fold change from pre-stimulation values in action potentials (APs)/sec (10 Hz: n=16 channels; 10 bp: n=24 channels). B. Fold change from pre-stimulation values in burstiness index (BI) (10 Hz: n=16 channels; 10 bp: n=24 channels). C. Change from pre-stimulation values in SYN values (10 Hz: n=29 channel pairs; 10 bp: n=42 channel pairs). D. Change (μV2) from pre-stimulation values in SW power in the 0.25 – 3.75 Hz band (10 Hz: n=16 channels; 10 bp: n=24 channels).
* indicates a significant difference (p<0.01) from pre-stimulation values. # indicates a significant difference (p<0.01) between the 10 Hz and 10 bp patterns of stimulation.
During 10 bp stimulation for 30 min, APs/sec increased in the presence of TNF (Fig. 10A; Stimulation Condition: F3,126=82.01, P<0.001), though significantly less than during 10 Hz in the presence of TNF (t-test: P=0.009). After stimulation, APs/sec slightly decreased then returned to baseline values by 24 h later. During 10 bp stimulation in the presence of TNF, the BI decreased, though less than during 10 Hz in the presence of TNF (t-test: P<0.001) and then returned to pre-stimulation values immediately after stimulation (Fig. 10B; Stimulation Condition: F3,129=107.54, P<0.001). Twenty-four h later, the BI increased. SYN values during and after 10 bp stimulation and 24 h later decreased in the presence of TNF (Fig. 10C; Stimulation Condition: F3,198=11.30, P<0.001). During, immediately after, and 24 h after 10 bp stimulation, SW power decreased in the presence of TNF (Fig. 10D; Stimulation Condition: F3,129=11.96, P<0.001).
In summary, rapid electrical stimulation reversed the “sleep-promoting” effects of TNF as indicated by reductions in the BI, SYN values, and SW power during stimulation, suggesting a “waking” effect of electrical stimulation (compare Figs. 9 and 10). In the absence of TNF, we made similar conclusions as to the effects of electrical stimulation. Finally, the specific effects of rapid electrical stimulation in the presence of TNF are stimulus pattern-dependent. The stimulus-dependent long-term effects are more difficult to interpret with TNF present (Fig. 10); they likely involve an interaction between temporal summation of stimulus-induced release of substances and the feedback effects of the exogenous TNF added.
Discussion
An important issue is whether the emergent network properties, BI, SYN, and SW power, characterize sleep/wake states in small cultured neuronal/glia networks. Multiple findings suggest this is the case. The magnitudes of the BI, SYN and SW power values were very low the first few days of culture even though APs were present at these times. These parameters emerged as the networks matured as they do in intact neonates (Shimizu and Himwich, 1968; Jouvet-Mounier et al., 1969; Frank and Heller, 2003; Scher, 2008). Slow electrical stimulation (1Hz) transiently enhanced SYN and SW power values; in vivo slow electrical stimulation of cortical areas transiently enhances local EEG synchronization and SWs (Jouvet, 1967). In contrast, the long-term effects of electrical stimulation were to reduce the BI, SYN, and SW power values suggesting a more wake-like network state. The longer wake-like state was followed by a rebound in the sleep-like state thus paralleling the sleep homeostatic response to sleep loss. Further, TNF enhanced spontaneous BI, SYN, and SW power values suggesting the induction of a deeper sleep-like state. In vivo, TNF enhances non-REM sleep (Krueger et al., 2008). Electrical stimulation reversed the deeper TNF-induced sleep-like state. We conclude that small cultured neuronal/glial networks share sleep and wake states, and at least some of their regulatory components, with whole brain. Current studies are consistent with Hindard’s et al., (2012) conclusion that cultured neuronal/glia networks spontaneously have a default sleep-like state. We extend those studies by showing that a) TNF induces a deeper sleep-like state in cultured cells, b) electrical stimulation reverses these TNF effects, and c) prolonged wake-like states, induced by electrical stimulation, are followed by rebound of the sleep-like state suggesting network homeostasis in vitro. Collectively, current results are consistent with sleep theories proposing that sleep is an emergent property of small neuronal/glial networks (Krueger and Obal, 1993; Kavanau, 1994; Benington and Heller, 1995; Mahwald and Schenck, 2005; and Tononi and Cirelli, 2006).
The long-term changes in spontaneous APs/sec, SYN values, and SW power are different depending upon the stimulus pattern applied to the cells 24 h earlier suggesting that network dynamics and connectivity/excitability in vitro are dependent upon past patterns of activity. These findings are consistent with the interpretation that the cultured cells can achieve functional plasticity (Maeda et al., 1998, Jimbo et al., 1998, 1999; Tateno and Jimbo 1999; Shahaf and Marom, 2001; Ruaro et al., 2005; Chao et al., 2007). Unsuccessful attempts to elicit functional plasticity (Wagenaar et al., 2006b) cite an activity “drift” as the cause of others’ false positives as changes of similar magnitude occur spontaneously. We also observed an exponential decrease in spontaneous APs/sec at the start of our recordings. For this reason only the last 5 min prior to stimulation were used as baseline activity. Regardless, the long-term state of the network was dependent upon the stimulus pattern used 24 h earlier. It is hard to ascribe such differences to spontaneous drift.
The development and temporal responses of SW power had not been previously described in vitro. In vivo the EEG is thought to be derived from electrical activity in dendrites and axons that are perpendicular to the surface of the brain (Schaul, 1998). In vitro, cell geometry is very different – a monolayer neuronal network grows on top of a bed of glial cells. Regardless, the in vitro cellular geometry allows for the development of spontaneous SW power albeit the SW power values in culture are substantially lower (~100 fold) than those observed in vivo in mammals. That cultured networks spontaneously develop SW wave activity suggests that it is an intrinsic emergent property of cortical cells. Indeed, cortical slow oscillations (<1 Hz) persist after thalamectomy and are present in isolated cortical slabs (Steriade, 2003; Lemieux et al., 2013).
The developmental unfolding of SW EEG activity occurring in vivo in several species (Shimizu and Himwich et al., 1968; Jouvet-Mounier et al., 1969; Frank and Heller, 2003; Scher, 2008) parallels our in vitro results. These similarities suggest that in vitro ontogeny of spontaneous electrical activity mimics homologous measures in vivo during sleep ontogeny. Thus, EEG SW waves manifest only after several days of development in vivo e.g. post-natal day 11 in the rat (Seelke and Blumberg, 2008). Further, in vivo non-rapid eye movement sleep EEG gradually becomes synchronized during development (Nunes et al., 1997), as the SYN values increase in the mature cultured cells.
In ChR2-positive networks, the elevation of cytokine expression occurred in all neurons irrespective of whether they were ChR2-positive. This was likely due to indirect activation of ChR2-negative neurons by ChR2-positive neurons releasing ATP and neurotransmitters in response to the optical stimulation. These results have ramifications for in vivo optogenetic experimentation. Thus, if a specific neuronal population is transfected with ChR-2, their prolonged subsequent optogenetic stimulation likely would excite, or inhibit, those cells receiving projections from the ChR-2-stimulated cells and thereby the specificity provided by the targeted ChR-2 transfection would be lost.
Activity within the cultured networks affects the expression of ATP, IL1, and TNF, all implicated in sleep-wake state transitions in mammals and to a lesser extent in fruit flies (Wu et al., 2009). The addition of exogenous TNF drives the network into a deeper sleep-like state. Similar changes occur in vivo as the application of TNF to somatosensory cortical columns induces a sleep-like state (Churchill et al., 2010). The individual cortical column sleep-like state is associated with errors in learned behavior performance (Krueger et al., 2013). The effects of TNF, in vitro, persist for at least 24 h. TNF’s half-life in vitro is unknown but likely less than 24 h; TNF has a serum half-life of only 10.5 min in mice (Flick and Gifford, 1986). TNF is well-known as an effector of synaptic scaling and receptor trafficking (Krueger et al., 2008; 2013; Turrigiano, 2008) suggesting that the exogenous TNF addition produces long-term network changes. TNF potentiates AMPA-induced postsynaptic potentials (Beattie et al., 2002), AMPA-induced cytosolic Ca++ increases (De et al., 2003), as well as several voltage-dependent calcium channels (Wilkinson et al., 1996; Furukawa and Mattson 1998).
TNF has distinct dose-dependent actions; for instance, it is neuro-protective at low doses but neuro-toxic at high doses (Saha et al., 2006; Takeuchi et al., 2006). Current results are consistent with those findings. Whether TNFα is protective or damaging may depend upon the receptor type present, TNF-55 kD or the TNF 75 kD receptor (Peschon et al., 1998; Fontaine et al., 2002; Yang et al., 2002) and substances that modify TNFα activity (Carlson et al., 1998). The transmembrane form of TNF is likely the form visualized after optogenetic stimulation (Churchill et al., 2008). This form of TNF can act as a ligand for TNF receptors in cell-to-cell communication. The kinetics of transmembrane TNF cleavage to form soluble 17kD TNF in vitro are unknown. The soluble TNF could, for example, be responsible for the decline in TNF immuno-reactivity at the 90 min sampling time point (Fig. 7). Additionally, the membrane bound form of TNF may signal intracellularly upon binding the soluble form of TNF receptors (Horiuchi et al., 2010). Futher, the addition of exogenous TNF could interfere with such actions and/or directly signal through membrane TNF receptors to elicit its sleep-promoting actions. Some or all of these factors may be involved in the greater variability we observed with high doses of TNF.
Not knowing the minimal network size required for sleep-like states to emerge limits our understanding of sleep regulation and sleep function. Further, use of whole animals limits any statements regarding sleep because it is impossible to experimentally isolate sleep as an independent variable. During sleep almost every physiological parameter changes with respect to wake. Thus, if any changes are observed after sleep loss, one cannot know if they are due to sleep per se or due to changes in one of the other parameters that co-vary with sleep. In vitro cultures are simple in comparison to whole brains, yet the cultures exhibit homologous electrical properties to those used to characterize sleep in vivo. These properties spontaneously develop and emerge as networks mature and are influenced by stimulations that similarly affect whole animal state. We conclude that with appropriate stimuli such cultures can transition into a more wake-like state or deeper sleep-like state depending upon the specific stimuli. Further, after a stimulus-induced wake period, spontaneous sleep is more intense as indicated by SYN values and SW power indicating sleep homeostasis occurs in vitro. Collectively the results suggest that activity-dependent network state changes are a fundamental property of any viable neuronal/glial network. The in vitro system due to its simplicity and its state characterization and ability to control the intensity of the emergent state properties, offers a novel experimental platform to study the genetic, molecular, and electrical causality mechanisms of sleep states and perhaps even be applied to the elusive issue of sleep function.
Acknowledgments
This work was supported by NIH/NINDS NS025378 to JK and NSF CNS-1058124 to SR. We thank Dave Rector and Indika Rajapakse for their early contributions to the ideas behind this work. We also thank Karl Deisseroth for the gift of the ChR2 construct. Some of the work described herein was previously published in abstract form; Sleep 34:A14 and Sleep 34:A36, 2011.
Abbreviations
- AP
action potential
- ATP
adenosine triphosphate
- BI
burstiness index
- ChR2
channelrhodopsin-2
- EEG
electroencephalography
- FFT
fast Fourier transformation
- IL1
interleukin-1 beta
- MEA
multielectrode array
- SW
slow wave
- SYN
synchronization of slow electrical potentials between recording electrodes
- TNF
tumor necrosis factor alpha
Footnotes
The authors declare no competing financial interests.
References
- Beattie EC, Stellwagen D, Morishita W, Bresnahan JC, Ha BK, Von Zastrow M, Beattie MS, Malenka RC. Control of synaptic strength by glial TNF alpha. Science. 2002;295:2282–2285. doi: 10.1126/science.1067859. [DOI] [PubMed] [Google Scholar]
- Benington JH, Heller HC. Restoration of brain energy metabolism as the function of sleep. Prog Neurobiol. 1995;45:347–360. doi: 10.1016/0301-0082(94)00057-o. [DOI] [PubMed] [Google Scholar]
- Borbély AA, Tobler I, Hanagasioglu M. Effect of sleep deprivation on sleep and EEG power spectra in the rat. Behavioural Brain Res. 1984;14:171–182. doi: 10.1016/0166-4328(84)90186-4. [DOI] [PubMed] [Google Scholar]
- Boyden ES, Zhang F, Bamberg E, Nagel G, Deisseroth K. Millisecond-timescale, genetically targeted optical control of neural activity. Nat Neurosci. 2005;8:1263–1268. doi: 10.1038/nn1525. [DOI] [PubMed] [Google Scholar]
- Carlson NG, Bacchi A, Rogers SW, Gahring LC. Nicotine blocks TNF-alpha mediated neuroprotection to NMDA by an alpha-bungarotoxin-sensitive pathway. J Neurobiol. 1998;35:29–36. doi: 10.1002/(sici)1097-4695(199804)35:1<29::aid-neu3>3.0.co;2-d. [DOI] [PubMed] [Google Scholar]
- Chao ZC, Bakkum DJ, Potter SM. Region-specific network plasticity in simulated and living cortical networks: Comparison of the center of activity trajectory (CAT) with other statistics. J Neural Eng. 2007;4:1–15. doi: 10.1088/1741-2560/4/3/015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiappalone M, Vato A, Berdondini L, Koudelka-Hep M, Martinoia S. Network dynamics and synchronous activity in cultured cortical neurons. Int J Neural Syst. 2007;17:87–103. doi: 10.1142/S0129065707000968. [DOI] [PubMed] [Google Scholar]
- Churchill L, Rector DM, Yasuda K, Fix C, Rojas MJ, Yasuda T, Krueger JM. Tumor necrosis factor alpha: activity dependent expression and promotion of cortical column sleep in rats. Neuroscience. 2008;156:71–80. doi: 10.1016/j.neuroscience.2008.06.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cozzi L, D’Angelo P, Sanguineti V. Encoding of time-varying stimuli in populations of cultured neurons. Biol Cybern. 2006;94:335–349. doi: 10.1007/s00422-006-0051-2. [DOI] [PubMed] [Google Scholar]
- Davis CJ, Clinton JM, Jewett KA, Zielinski MR, Krueger JM. EEG delta wave power: an independent sleep phenotype or epiphenomenon? J Clin Sleep Med. 2011;7:S16–S18. doi: 10.5664/JCSM.1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De A, Krueger JM, Simasko SM. Tumor necrosis factor alpha increases cytosolic calcium response AMPA and KCl in primary cultures of rat hippocampal neurons. Brain Res. 2003;981:133–142. doi: 10.1016/s0006-8993(03)02997-4. [DOI] [PubMed] [Google Scholar]
- Ferrari D, Pizzirani C, Adinolfi E, Lemoli RM, Curti A, Idzko M, Panther E, DiVirgilio F. The P2X7 receptor: a key player in IL-1 processing and release. J Immuno. 2006;176:3877–3883. doi: 10.4049/jimmunol.176.7.3877. [DOI] [PubMed] [Google Scholar]
- Fields RD, Stevens B. ATP: an extracellular signaling molecule between neurons and glia. Trends Neurosci. 2000;23:625–633. doi: 10.1016/s0166-2236(00)01674-x. [DOI] [PubMed] [Google Scholar]
- Flick DA, Gifford GE. Pharmacokinetics of murine tumor necrosis factor. J Immunopharmacol. 1986;8:89–97. doi: 10.3109/08923978609031087. [DOI] [PubMed] [Google Scholar]
- Fontaine V, Mohand-Said N, Hanoteau N, Fuchs C, Pfizenmaier K, Eisel U. Neurodegenerative and neuroprotective effects of tumor necrosis factor (TNF) in retinal ischemia: opposite roles of TNF receptor 1 and TNF receptor 2. J Neurosci. 2002;22:RC216. doi: 10.1523/JNEUROSCI.22-07-j0001.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank MG, Heller HC. The ontogeny of mammalian sleep: a reappraisal of alternative hypotheses. J Sleep Res. 2003;12:25–34. doi: 10.1046/j.1365-2869.2003.00339.x. [DOI] [PubMed] [Google Scholar]
- Furukawa K, Mattson MP. The transcription factor NFkB mediates increases in calcium currents and decreases in NMDA- and AMPA/kainate-induced currents induced by tumor necrosis factor alpha in hippocampal neurons. J Neurochem. 1998;70:1876–1886. doi: 10.1046/j.1471-4159.1998.70051876.x. [DOI] [PubMed] [Google Scholar]
- Hajnik T, Toth A, Detari L. Characteristic changes in the slow cortical waves after a 6 h sleep deprivation in rat. Brain Res. 2013;1501:1–11. doi: 10.1016/j.brainres.2012.12.047. [DOI] [PubMed] [Google Scholar]
- Hallett H, Churchill L, Taishi P, De A, Krueger JM. Whisker stimulation increases expression of nerve growth factor-and interleukin-1β-immunoreactivity in the rat somatosensory cortex. Brain Res. 2010;1333:48–56. doi: 10.1016/j.brainres.2010.03.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinard V, Mikhail C, Pradervand S, Curie T, Houtkooper RH, Auwerx J, Franken P, Tafti M. Key electrophysiological, molecular, and metabolic signatures of sleep and wakefulness revealed in primary cortical cultures. J Neurosci. 2012;32:12506–12517. doi: 10.1523/JNEUROSCI.2306-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horiuchi T, Mitoma H, Harashima S, Tsukamoto H, Shimoda T. Transmembrane TNF-alpha: structure, function and interaction with anti-TNF agents. Rheumatology. 2010;49:1215–1228. doi: 10.1093/rheumatology/keq031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jimbo Y, Robinson HP, Kawana A. Strengthening of synchronized activity by tetanic stimulation in cortical cultures: application of planar electrode arrays. IEEE Trans Biomed Eng. 1998;45:1297–1304. doi: 10.1109/10.725326. [DOI] [PubMed] [Google Scholar]
- Jimbo Y, Tateno T, Robinson HPC. Simultaneous induction of pathway-specific potentiation and depression in networks of cortical neurons. Biophys J. 1999;76:670–678. doi: 10.1016/S0006-3495(99)77234-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jouvet M. Neurophysiology of the states of sleep. Physiol Rev. 1967;47:117–177. doi: 10.1152/physrev.1967.47.2.117. [DOI] [PubMed] [Google Scholar]
- Jouvet Mounier D, Astic L, Lacote D. Ontogenesis of the states of sleep in rat, cat, and guinea pig during the first postnatal month. Dev Psychobiol. 1969;2:216–239. doi: 10.1002/dev.420020407. [DOI] [PubMed] [Google Scholar]
- Kavanau JL. Sleep and dynamic stabilization of neural circuitry: a review and synthesis. Behav Brain Res. 1994;63:111–126. doi: 10.1016/0166-4328(94)90082-5. [DOI] [PubMed] [Google Scholar]
- Kristiansen K, Courtois G. Rhythmic electrical activity from isolated cerebral cortex. Electroen Clin Neuro. 1949;1:265–272. [PubMed] [Google Scholar]
- Krueger JM, Obál F., Jr A neuronal group theory of sleep function. J Sleep Res. 1993;2:63–69. doi: 10.1111/j.1365-2869.1993.tb00064.x. [DOI] [PubMed] [Google Scholar]
- Krueger JM, Huang YH, Rector DM, Buysse DJ. Sleep: a synchrony of cell activity-driven small network states. Eur J Neurosci. 2013;38:2199–2209. doi: 10.1111/ejn.12238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krueger JM, Rector DM, Roy S, Van Dongen HP, Belenky G, Panksepp J. Sleep as a fundamental property of neuronal assemblies. Nat Rev Neurosci. 2008;9:910–919. doi: 10.1038/nrn2521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemieux M, Chen JY, Lonjers P, Bazhenov M, Timofeev I. The Impact of Cortical Deafferentation on the Neocortical Slow Oscillation. J Neurosci. 2014;34:5689–5703. doi: 10.1523/JNEUROSCI.1156-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda E, Kuroda Y, Robinson HP, Kawana A. Modification of parallel activity elicited by propagating bursts in developing networks of rat cortical neurones. Eur J Neurosci. 1998;10:488–496. doi: 10.1046/j.1460-9568.1998.00062.x. [DOI] [PubMed] [Google Scholar]
- Mahowald MW, Schenck CH. Insights from studying human sleep disorders. Nature. 2005;437:1279–1285. doi: 10.1038/nature04287. [DOI] [PubMed] [Google Scholar]
- Mazzoni A, Broccard FD, Garcia-Perez E, Bonifazi P, Ruaro ME, Torre V. On the dynamics of the spontaneous activity in neuronal networks. PLoS ONE. 2007;2:e439. doi: 10.1371/journal.pone.0000439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunes ML, Costa JCD, Leme Moura-Ribeiro MV. Polysomnographic quantification of bioelectrical maturation in preterm and fullterm newborns at matched conceptional ages. Electroen Clin Neuro. 1997;102:186–191. doi: 10.1016/s0013-4694(96)95191-7. [DOI] [PubMed] [Google Scholar]
- Opitz T, de Lima AD, Voigt T. Spontaneous development of synchronous oscillatory activity during maturation of cortical networks in vitro. J Neurophysiol. 2002;88:2196–2206. doi: 10.1152/jn.00316.2002. [DOI] [PubMed] [Google Scholar]
- Papoulis A, Pillai SU. Probability, Random Variables, and Stochastic Processes. Tata McGraw-Hill Education; 2002. [Google Scholar]
- Peschon JJ, Toorance DS, Stocking KL, Glaccum MB, Otten C, Willis CR, Charrier K, Morrissey PJ, Ware CB, Mohler KM. TNF receptor-deficient mice reveal divergent role for p55 and p75 in several models of inflammation. J Immunol. 1998;160:943–952. [PubMed] [Google Scholar]
- Phillips DJ, Schei JL, Meighan PC, Rector DM. State-dependent changes in cortical gain control as measured by auditory evoked responses to varying intensity stimuli. Sleep. 2011;34:65–72. doi: 10.5665/sleep.1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rector DM, Schei JL, Van Dongen HPA, Belenky G, Krueger JM. Physiological markers of localized sleep. Eur J Neurosci. 2009;29:1771–1778. doi: 10.1111/j.1460-9568.2009.06717.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rector DM, Topchiy IA, Carter KM, Rojas MJ. Local functional state differences between rat cortical columns. Brain Res. 2005;1047:45–55. doi: 10.1016/j.brainres.2005.04.002. [DOI] [PubMed] [Google Scholar]
- Roy S, Krueger JM, Rector DM, Wan Y. Network models for activity-dependent sleep regulation. J Theor Biol. 2008;253:462–468. doi: 10.1016/j.jtbi.2008.03.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruaro ME, Bonifazi P, Torre V. Toward the neurocomputer: Image processing and pattern recognition with neuronal cultures. IEEE Trans Biomed Eng. 2005;52:371–383. doi: 10.1109/TBME.2004.842975. [DOI] [PubMed] [Google Scholar]
- Saha RN, Liu X, Pahan K. Up-regulation of BDNF in astrocytes by TNF-α: a case for the neuroprotective role of cytokine. J Neuroimmune Pharmacol. 2006;1:212–222. doi: 10.1007/s11481-006-9020-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaul N. The fundamental neural mechanisms of electroencephalography. Electroen Clin Neuro. 1998;106:101–107. doi: 10.1016/s0013-4694(97)00111-9. [DOI] [PubMed] [Google Scholar]
- Scher MS. Ontogeny of EEG-sleep from neonatal through infancy periods. Sleep Med. 2008;9:615–636. doi: 10.1016/j.sleep.2007.08.014. [DOI] [PubMed] [Google Scholar]
- Seelke AMH, Blumberg MS. The microstructure of active and quiet sleep as cortical delta activity emerges in infant rats. Sleep. 2008;31:691–699. doi: 10.1093/sleep/31.5.691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shahaf G, Marom S. Learning in networks of cortical neurons. J Neurosci. 2001;21:8782–8788. doi: 10.1523/JNEUROSCI.21-22-08782.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu A, Himwich HE. The ontogeny of sleep in kittens and young rabbits. Electroen Clin Neuro. 1968;24:307–318. [PubMed] [Google Scholar]
- Solle M, Labasi J, Perreguax DG, Stam E, Petrushova N, Koller BH, Griffiths RJ, Gabel CA. Altered cytokine production in mice lacking P2X7Receptors. J Biol Chem. 2001;276:125–132. doi: 10.1074/jbc.M006781200. [DOI] [PubMed] [Google Scholar]
- Steriade M. The corticothalamic system in sleep. Front Biosci. 2003;8:d878–d899. doi: 10.2741/1043. [DOI] [PubMed] [Google Scholar]
- Suzuki T, Hide I, Ido K, Kohsaka S, Inoue K, Nakata Y. Production and release of neuroprotective tumor necrosis factor by P2X7 receptor-activated microglia. J Neurosci. 2004;24:1–7. doi: 10.1523/JNEUROSCI.3792-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi H, Jin S, Wang J, Zhang G, Kawanokuchi J, Kuno R, Sonobe Y, Mizuno T, Suzumura A. Tumor necrosis factor-α induces neurotoxicity via glutamate release from hemichannels of activated microglia in an autocrine manner. J Biol Chem. 2006;281:21362–21368. doi: 10.1074/jbc.M600504200. [DOI] [PubMed] [Google Scholar]
- Tateno T, Jimbo Y. Activity-dependent enhancement in the reliability of correlated spike timings in cultured cortical neurons. Biol Cybern. 1999;80:45–55. doi: 10.1007/s004220050503. [DOI] [PubMed] [Google Scholar]
- Timofeev I, Grenier F, Steriade M. Disfacilitation and active inhibition in the neocortex during the natural sleep-wake cycle: an intracellular study. PNAS. 2001;98:1924–1929. doi: 10.1073/pnas.041430398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tononi G, Cirelli C. Sleep function and synaptic homeostasis. Sleep Med Rev. 2006;10:49–62. doi: 10.1016/j.smrv.2005.05.002. [DOI] [PubMed] [Google Scholar]
- Turrigiano GG. The self-tuning neuron: synaptic scaling of excitatory synapses. Cell. 2008;135:422–435. doi: 10.1016/j.cell.2008.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagenaar DA, Pine J, Potter SM. Effective parameters for stimulation of dissociated cultures using multi-electrode arrays. J Neurosci Methods. 2004;138:27–37. doi: 10.1016/j.jneumeth.2004.03.005. [DOI] [PubMed] [Google Scholar]
- Wagenaar DA, Madhavan R, Pine J, Potter SM. Controlling bursting in cortical cultures with closed-loop multi-electrode stimulation. J Neurosci. 2005;25:680–688. doi: 10.1523/JNEUROSCI.4209-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagenaar DA, Pine J, Potter SM. An extremely rich repertoire of bursting patterns during the development of cortical cultures. BMC Neurosci. 2006a;7:11. doi: 10.1186/1471-2202-7-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagenaar DA, Pine J, Potter SM. Searching for plasticity in dissociated cortical cultures on multi-electrode arrays. J Negat Results Biomed. 2006b;5:16. doi: 10.1186/1477-5751-5-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkinson MK, Earle ML, Triggle CR, Barnes S. Interleukin-1 beta, tumor necrosis factor alpha and LPS enhances calcium channel currents in isolated vascular smooth muscle cells of rat tail artery. FASEB J. 1996;10:785–791. doi: 10.1096/fasebj.10.7.8635696. [DOI] [PubMed] [Google Scholar]
- Wu MN, Ho K, Crocker A, Yue Z, Koh K, Sehgal A. The effects of caffeine on sleep in Drosophila require PKA activity, but not the adenosine receptor. J Neurosci. 2009;29:11029–11037. doi: 10.1523/JNEUROSCI.1653-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Lindholm K, Konishi Y, Li R, Shen Y. Target depletion of distinct tumor necrosis factor receptor subtypes reveals hippocampal neuron death and survival through different signal transduction pathways. J Neurosci. 2002;22:3025–3032. doi: 10.1523/JNEUROSCI.22-08-03025.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]